Air Pollution and Atopic Dermatitis (AD): The Impact of Particulate Matter (PM10) on an AD Mouse-Model

 , ,
, , 
Abstract
:
1. Introduction
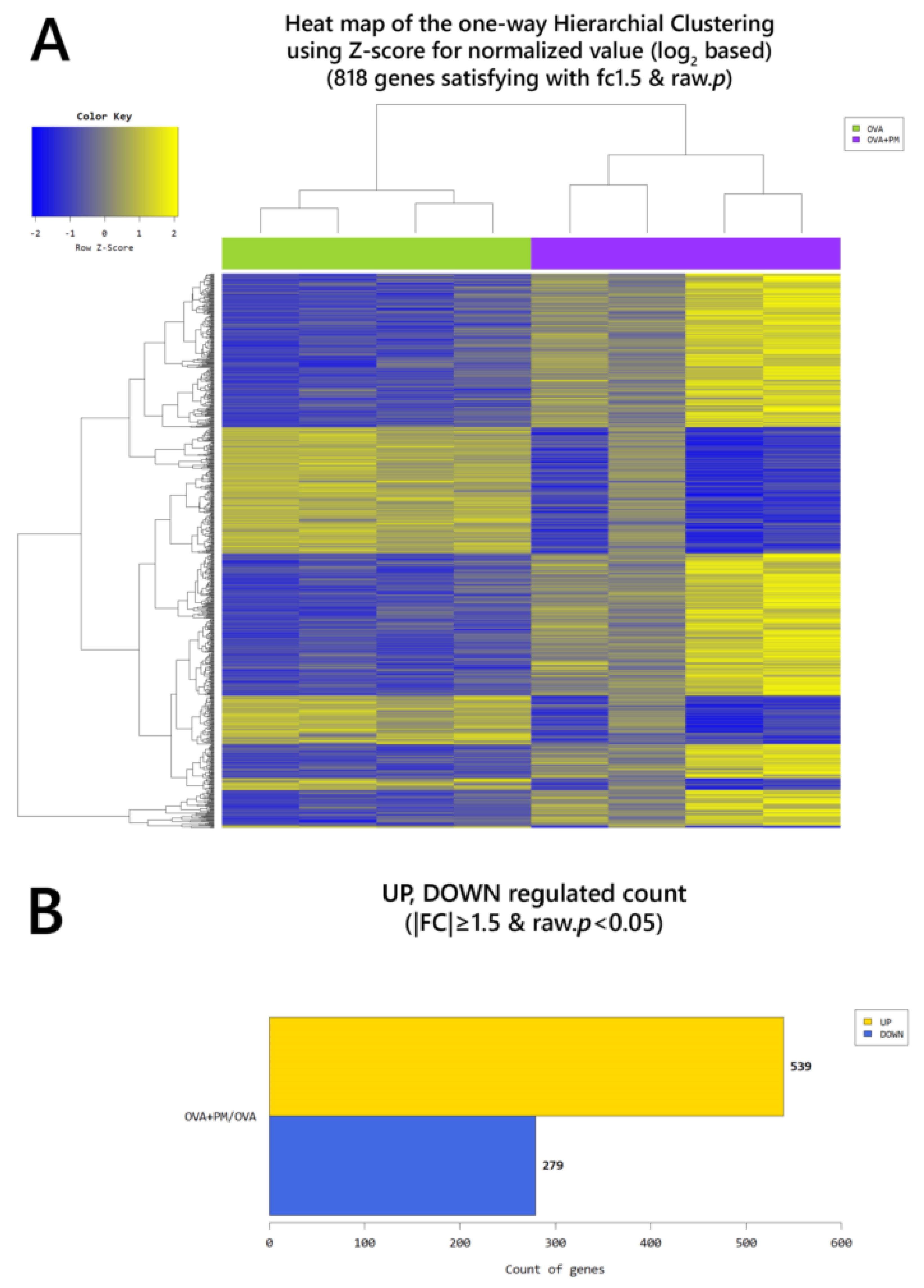
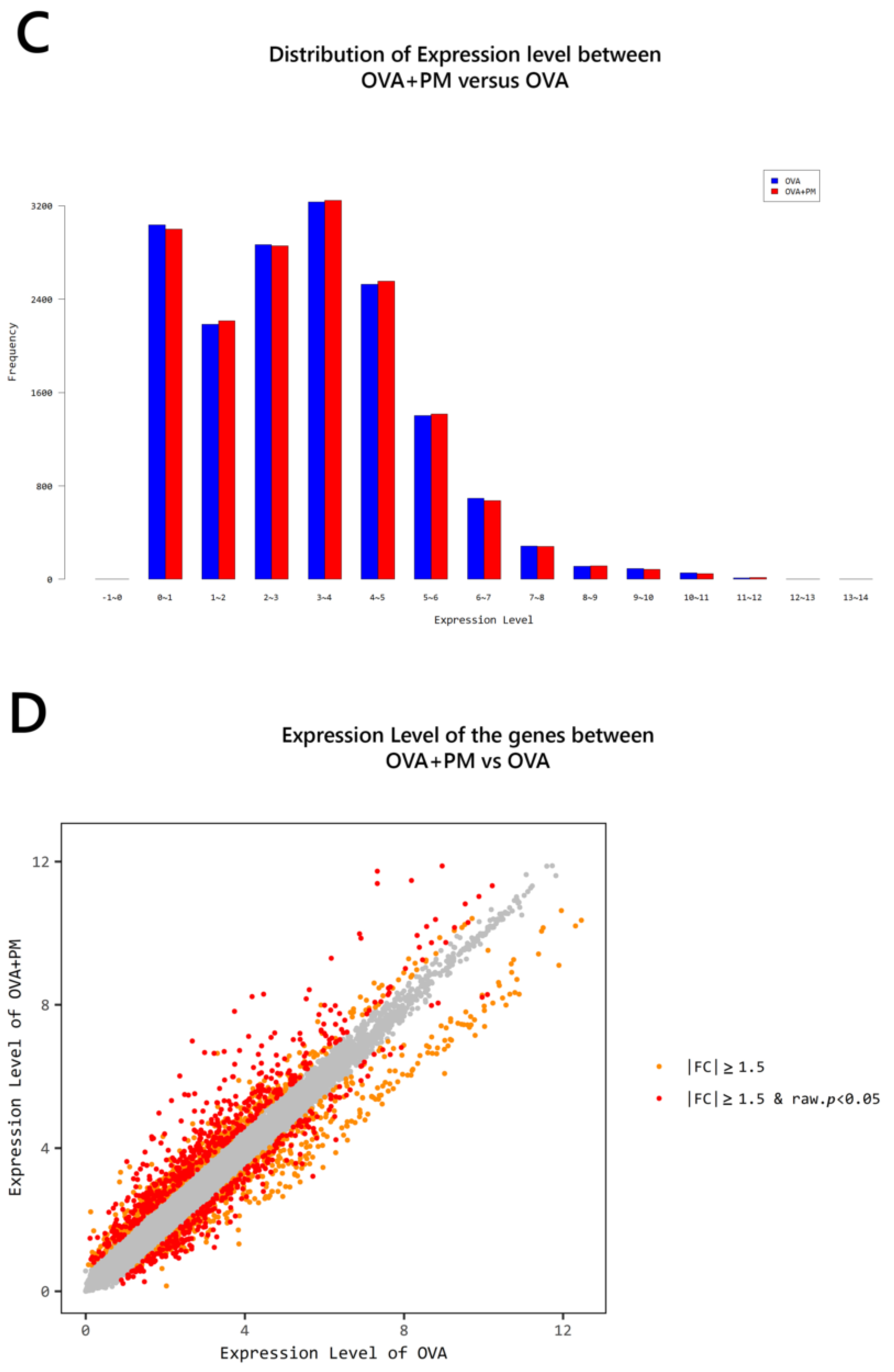
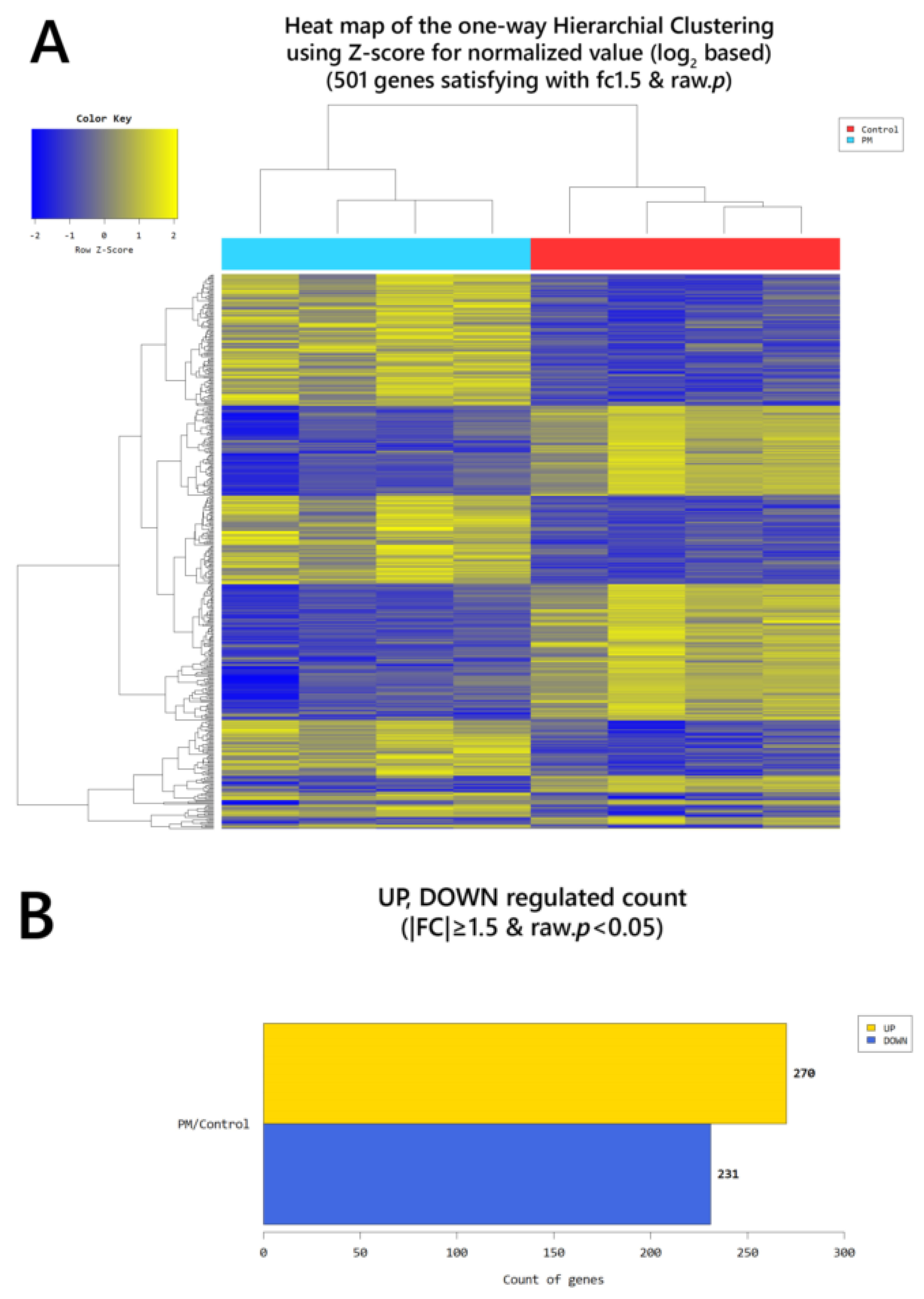
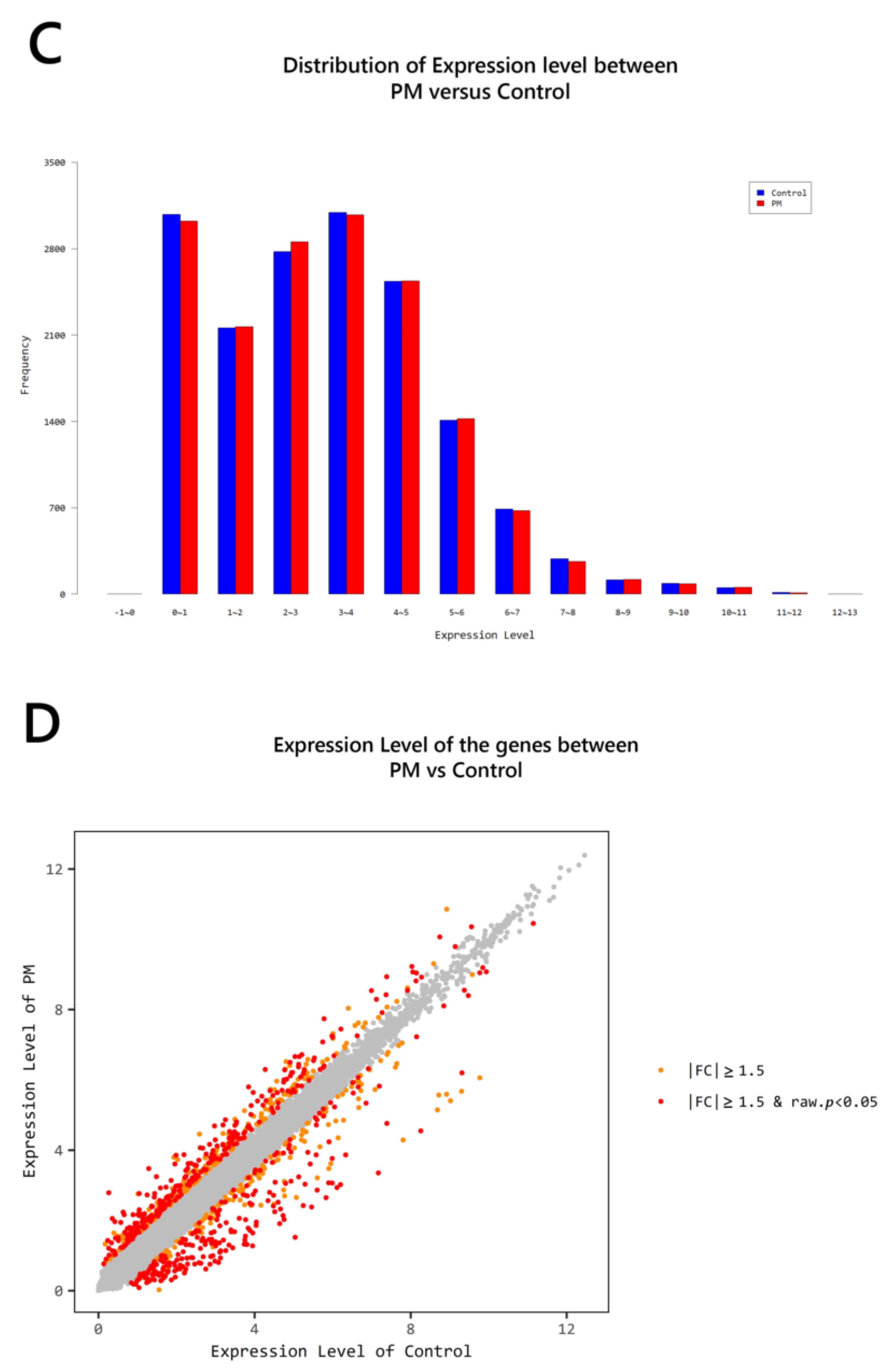
2. Results
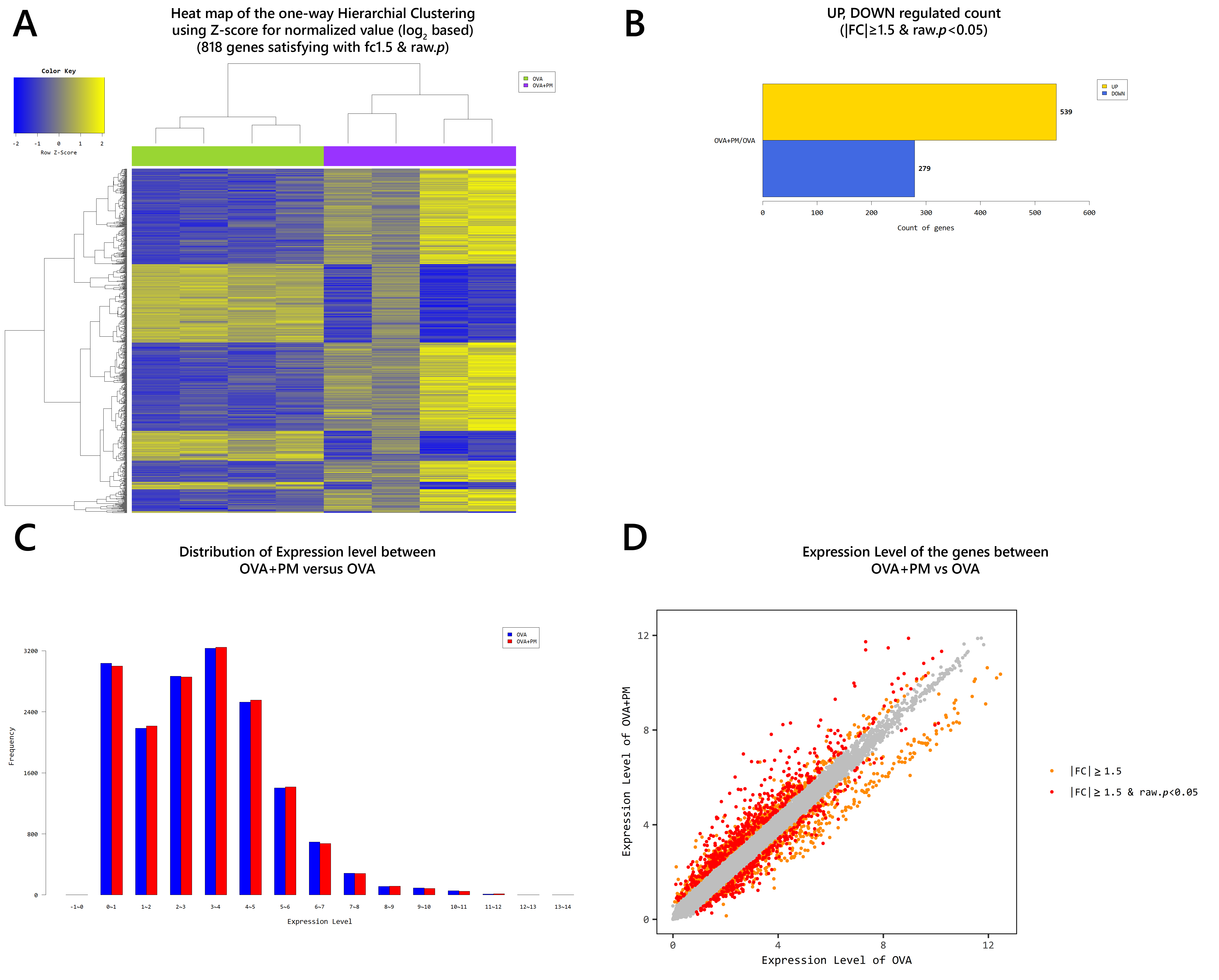
2.1. Gross Observation and Physiologic Parameters
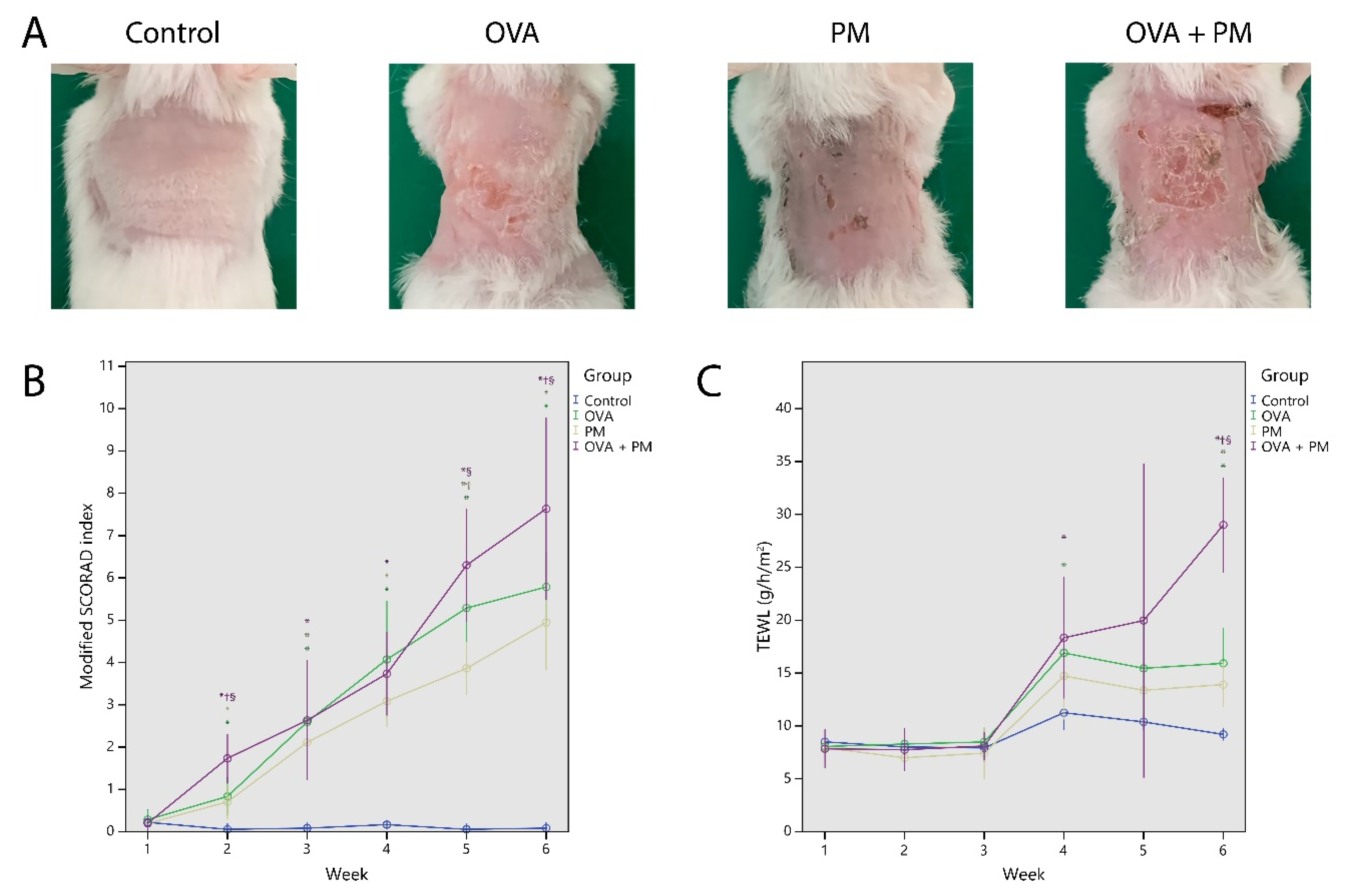
2.2. Hisopathologic Findings
2.3. Total Serum IgE
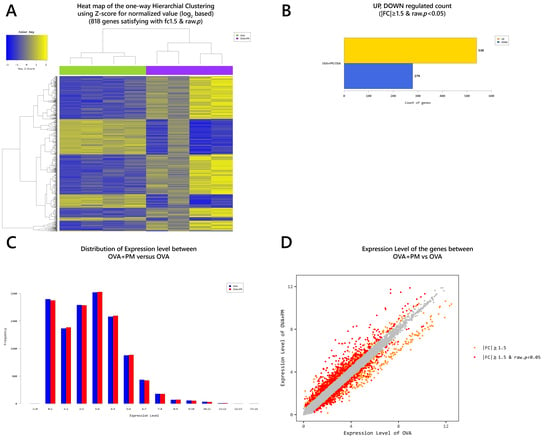
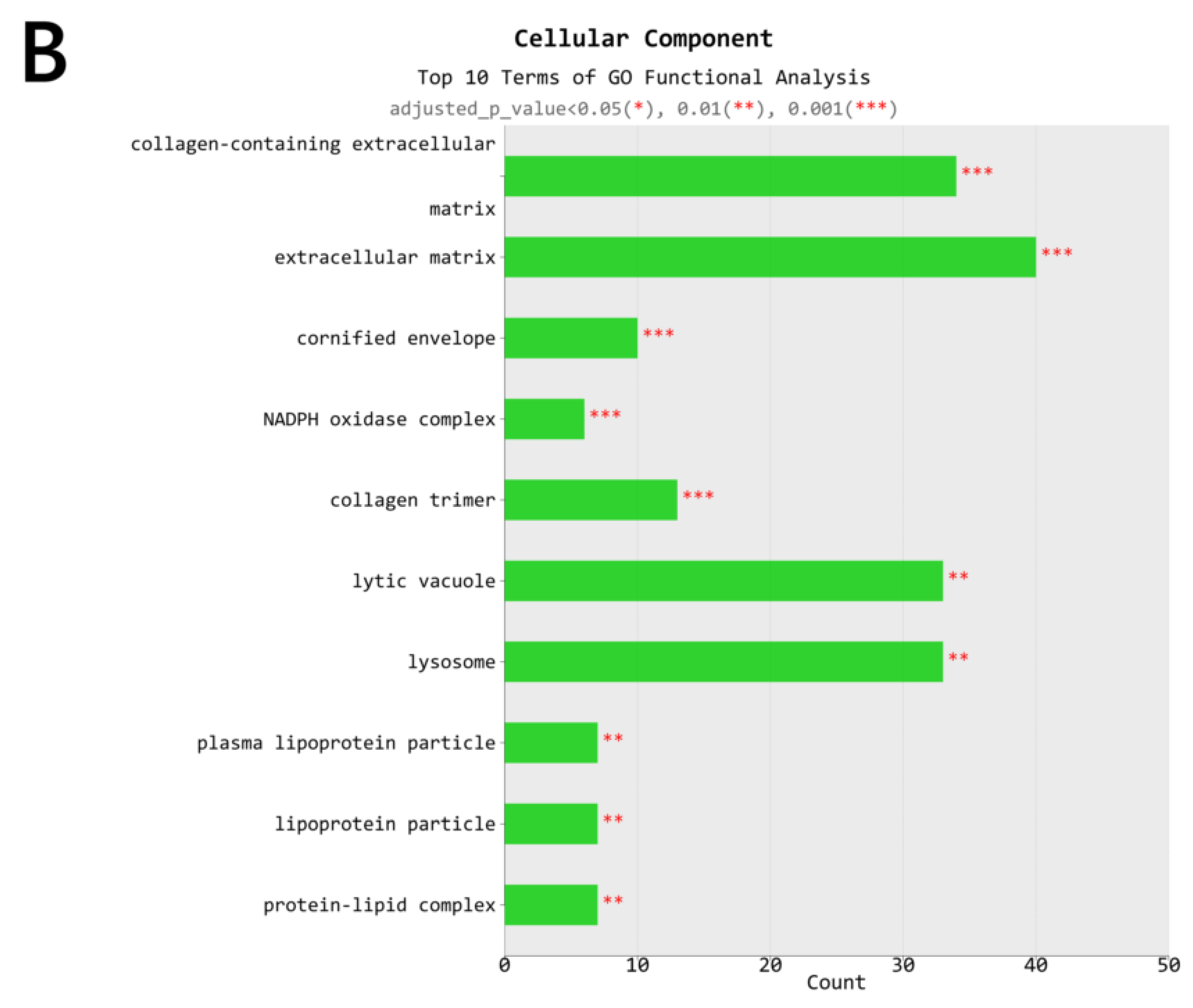
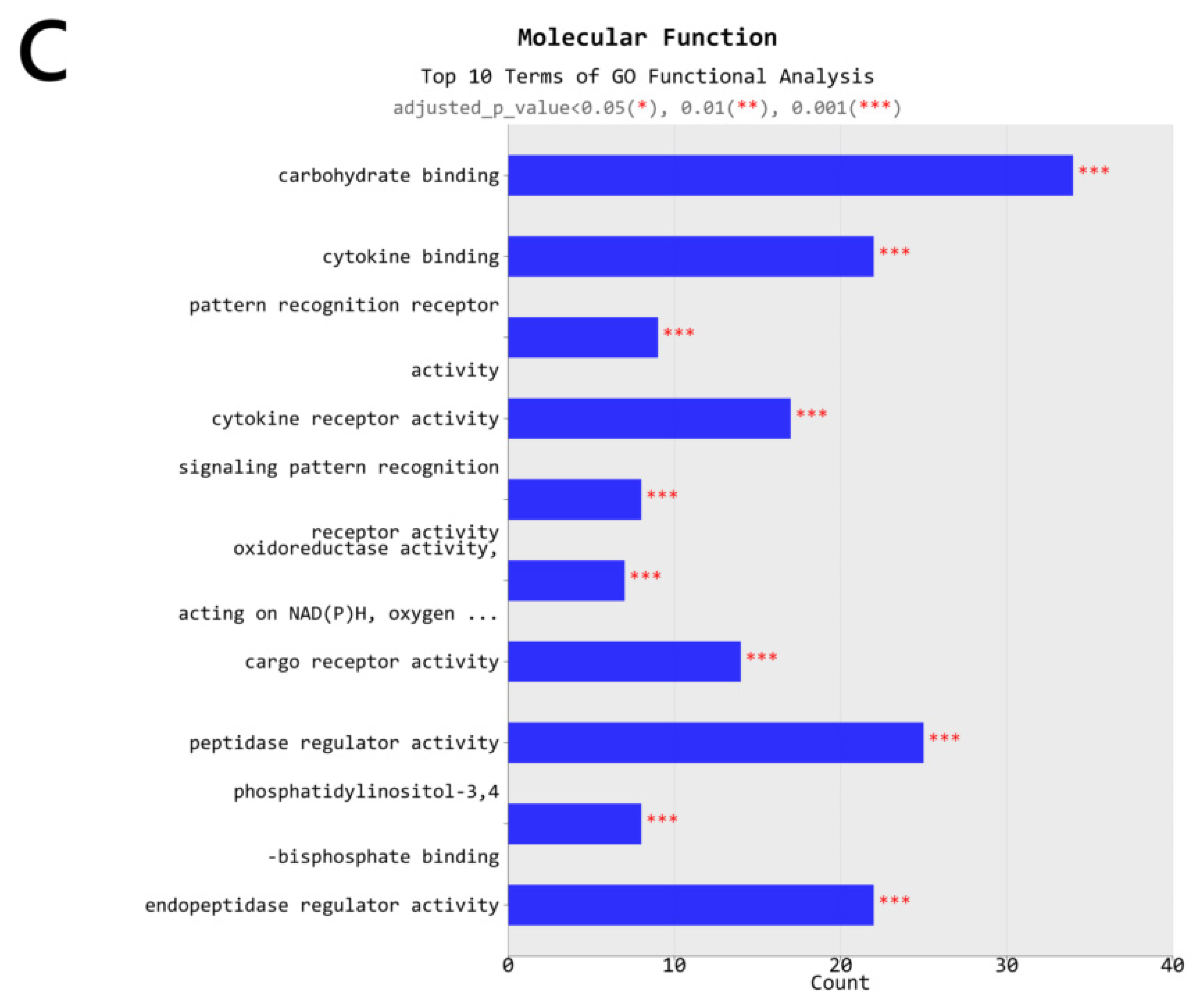
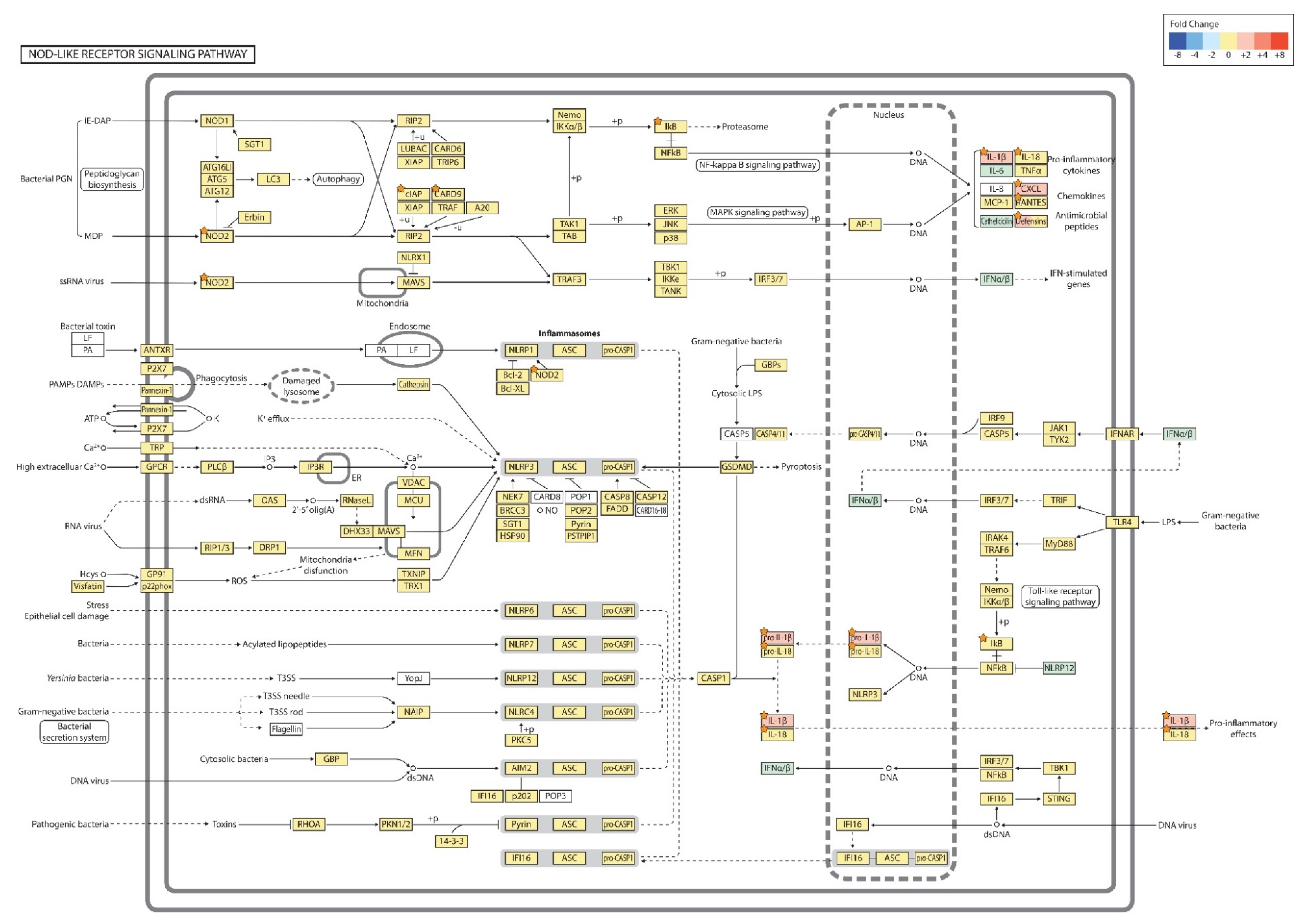
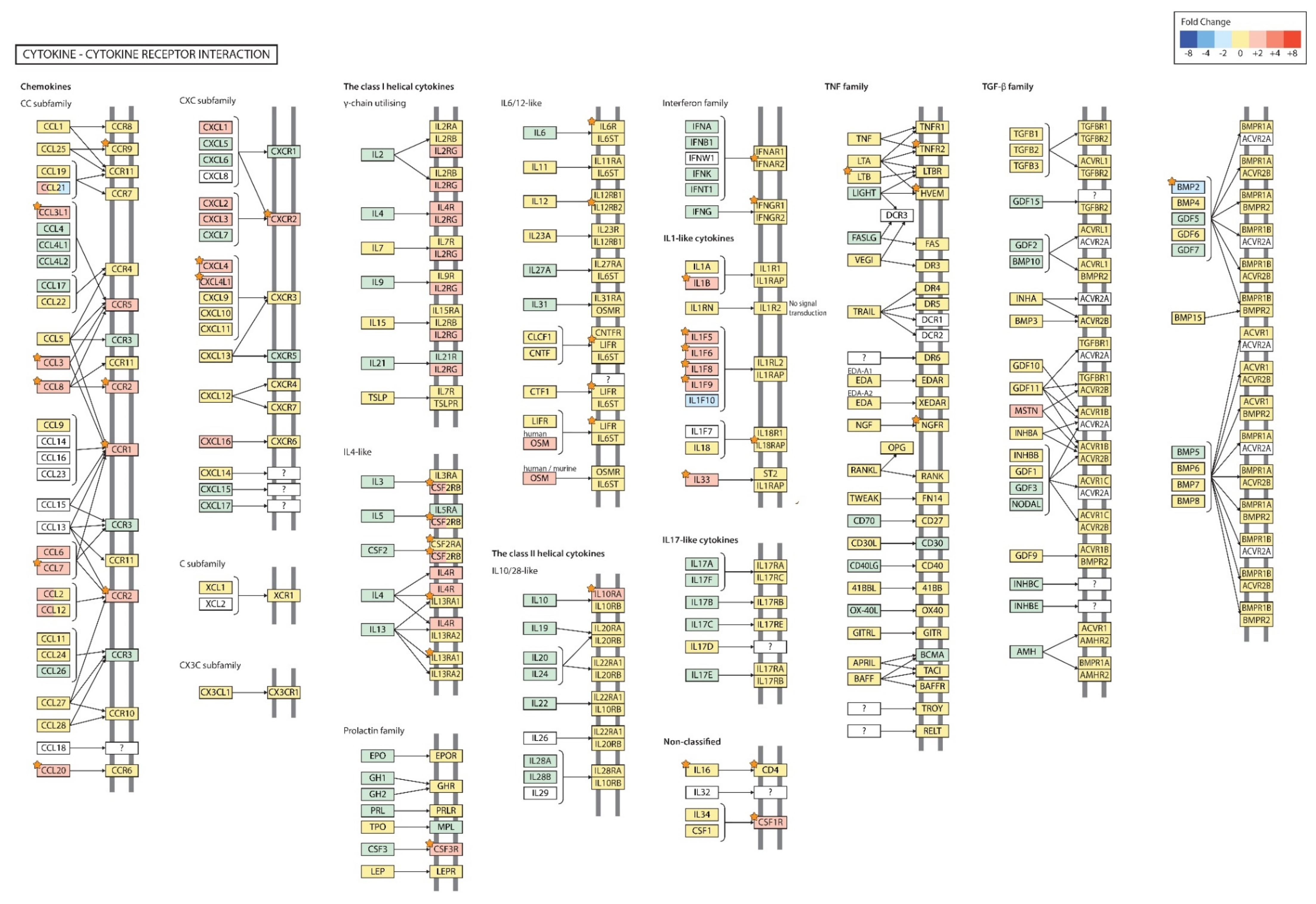
2.4. Gene Transcription Profile
3. Discussion
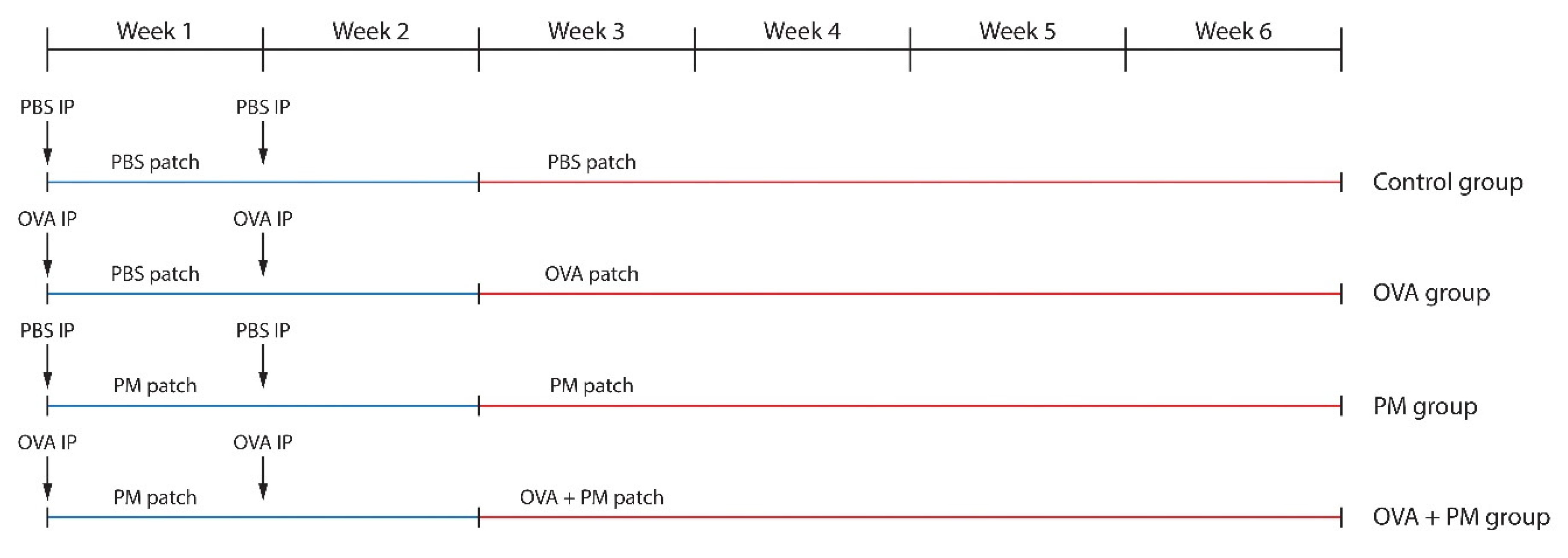
4. Materials and Methods
4.1. Particulate Matter
4.2. Animals
4.3. Sensitization and Challenge
4.4. Assessment of Clinical Parameters
4.5. Histopathology
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. mRNA-Seq
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Atopic dermatitis |
| PM | Particulate matter |
| OVA | Ovalbumin |
| TEWL | Trans-epidermal water loss |
| PAHs | Polyaromatic hydrocarbons |
| ROS | Reactive oxygen species |
| SCORAD | Scoring atopic dermatitis |
| H&E | Hematoxylin and eosin |
| DEG | Differentially expressed genes |
| SPRR | Small proline rich proteins |
| STFA | Stefin A |
| CHIL | Chitinase-like |
| DBP | D site-binding protein |
| LCE | Late cornified envelope |
| RNASE | Ribonuclease |
| THRSP | Thyroid hormone responsive |
| SERPIN | Serine proteinase inhibitor |
| C1QTNF | C1q and tumor necrosis factor |
| CXCL | C-X-C motif chemokine ligand |
| SAA | Serum amyloid A |
| NPY | Neuropeptide Y |
| FAM3B | Family with sequence similarity 3, member B |
| GUCA | Guanylate cyclase activator |
| WFDC | WAP four-disulfide core domain |
| UGT | UDP glucuronosyltransferase |
| TESC | Tescalcin |
| CRABP | Cellular retinoic acid binding protein |
| GO | Gene ontology |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| KEGG | Kyoto encyclopedia of genes and genomes |
| RAP | Ras-related protein |
| MAPK | Mitogen-activated protein kinase |
| JAK-STAT | Janus kinases-signal transducer and activator of transcription |
| NF-kB | Nuclear factor-kappa B |
| TNF | Tumor necrosis factor |
| HIF | Hypoxia-inducible factor |
| NOD | Nodulation |
| NFKBIA | NF-kappa-B inhibitor alpha |
| CARD | Caspase activation and recruitment domains |
| CCL | CC chemokine ligand |
| DEFB | Defensin beta |
| BIRC3 | Baculovial IAP repeat containing 3 |
| CCR | C-C chemokine receptor |
| CXCR | C-X-C chemokine receptor |
| CSF1R | Colony stimulation factor-1 receptor |
| PF4 | Platelet factor 4 |
| LTB | Lymphotoxin-beta |
| TNFRSF | Tumor necrosis factor receptor superfamily |
| IFNAR2 | Interferon alpha and beta receptor subunit 2 |
| BMP | Bone morphogenetic protein |
| NGFR | Nerve growth factor receptor |
| FCER | Fc fragment of IgE receptor |
| KRT | Keratin |
| KLK | Kallikreins |
| AHR | Aryl hydrocarbon receptor |
| XME | Xenobiotic metabolizing enzyme |
| CYP | Cytochrome P450 |
| NQO | NADPH quinone oxidoreductase |
| GSTA | Glutathione S-transferase |
| UGT | UDP-glucuronosyltransferase |
| IP | Intraperitoneal |
| PBS | Phosphate-buffered saline |
References
- Hendricks, A.J.; Eichenfield, L.F.; Shi, V.Y. The impact of airborne pollution on atopic dermatitis: A literature review. Br. J. Dermatol. 2020, 183, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.L.; MacIntyre, E.; Young, S.; Warden, H.; Drudge, C.; Kim, J.; Candido, E.; Demers, P.; Copes, R. An approach to estimating the environmental burden of cancer from known and probable carcinogens: Application to Ontario, Canada. BMC Public Health 2020, 20, 1–3. [Google Scholar] [CrossRef]
- Mueller, W.; Loh, M.; Vardoulakis, S.; Johnston, H.J.; Steinle, S.; Precha, N.; Kliengchuay, W.; Tantrakarnapa, K.; Cherrie, J.W. Ambient particulate matter and biomass burning: An ecological time series study of respiratory and cardiovascular hospital visits in northern Thailand. Environ. Health 2020, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Moelling, K.; Broecker, F. Air Microbiome and Pollution: Composition and Potential Effects on Human Health, Including SARS Coronavirus Infection. J. Environ. Public Health 2020, 2020, 1646943. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.E.; Cho, D.; Park, H.J. Air pollution and skin diseases: Adverse effects of airborne particulate matter on various skin diseases. Life Sci. 2016, 152, 126–134. [Google Scholar] [CrossRef]
- Park, S.Y.; Byun, E.J.; Lee, J.D.; Kim, S.; Kim, H.S. Air Pollution, Autophagy, and Skin Aging: Impact of Particulate Matter (PM(10)) on Human Dermal Fibroblasts. Int. J. Mol. Sci. 2018, 19, 2727. [Google Scholar] [CrossRef] [Green Version]
- Araviiskaia, E.; Berardesca, E.; Bieber, T.; Gontijo, G.; Sanchez Viera, M.; Marrot, L.; Chuberre, B.; Dreno, B. The impact of airborne pollution on skin. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1496–1505. [Google Scholar] [CrossRef]
- Ahn, K. The role of air pollutants in atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 993–999. [Google Scholar] [CrossRef]
- Pénard-Morand, C.; Raherison, C.; Charpin, D.; Kopferschmitt, C.; Lavaud, F.; Caillaud, D.; Annesi-Maesano, I. Long-term exposure to close-proximity air pollution and asthma and allergies in urban children. Eur. Respir. J. 2010, 36, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.L.; Su, H.J.; Sheu, H.M.; Yu, H.S.; Guo, Y.L. Traffic-related air pollution, climate, and prevalence of eczema in Taiwanese school children. J. Investig. Dermatol. 2008, 128, 2412–2420. [Google Scholar] [CrossRef]
- Oh, I.; Lee, J.; Ahn, K.; Kim, J.; Kim, Y.M.; Sun Sim, C.; Kim, Y. Association between particulate matter concentration and symptoms of atopic dermatitis in children living in an industrial urban area of South Korea. Environ. Res. 2018, 160, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.T.; Ku, K.C.; Chen, D.Y.; Lin, C.H.; Tsuang, B.J.; Chen, Y.H. Adult atopic dermatitis and exposure to air pollutants-a nationwide population-based study. Ann. Allergy Asthma Immunol. 2017, 118, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Kim, J.; Han, Y.; Jeon, B.H.; Cheong, H.K.; Ahn, K. Short-term effects of weather and air pollution on atopic dermatitis symptoms in children: A panel study in Korea. PLoS ONE 2017, 12, e0175229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassoun, Y.; James, C.; Bernstein, D.I. The Effects of Air Pollution on the Development of Atopic Disease. Clin. Rev. Allergy Immunol. 2019, 57, 403–414. [Google Scholar] [CrossRef]
- Hidaka, T.; Ogawa, E.; Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Fujimura, T.; Aiba, S.; Nakayama, K.; Okuyama, R.; et al. The aryl hydrocarbon receptor AhR links atopic dermatitis and air pollution via induction of the neurotrophic factor artemin. Nat. Immunol. 2017, 18, 64–73. [Google Scholar] [CrossRef]
- Guarnieri, M.; Balmes, J.R. Outdoor air pollution and asthma. Lancet 2014, 383, 1581–1592. [Google Scholar] [CrossRef] [Green Version]
- Schantz, M.M.; Cleveland, D.; Heckert, N.A.; Kucklick, J.R.; Leigh, S.D.; Long, S.E.; Lynch, J.M.; Murphy, K.E.; Olfaz, R.; Pintar, A.L.; et al. Development of two fine particulate matter standard reference materials (<4 μm and <10 μm) for the determination of organic and inorganic constituents. Anal. Bioanal. Chem. 2016, 408, 4257–4266. [Google Scholar] [CrossRef]
- Valacchi, G.; Sticozzi, C.; Pecorelli, A.; Cervellati, F.; Cervellati, C.; Maioli, E. Cutaneous responses to environmental stressors. Ann. N. Y. Acad. Sci. 2012, 1271, 75–81. [Google Scholar] [CrossRef]
- Magnani, N.D.; Muresan, X.M.; Belmonte, G.; Cervellati, F.; Sticozzi, C.; Pecorelli, A.; Miracco, C.; Marchini, T.; Evelson, P.; Valacchi, G. Skin Damage Mechanisms Related to Airborne Particulate Matter Exposure. Toxicol. Sci. 2016, 149, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Bieber, T. Atopic dermatitis. N. Engl. J. Med. 2008, 358, 1483–1494. [Google Scholar] [CrossRef]
- Suárez-Fariñas, M.; Tintle, S.J.; Shemer, A.; Chiricozzi, A.; Nograles, K.; Cardinale, I.; Duan, S.; Bowcock, A.M.; Krueger, J.G.; Guttman-Yassky, E. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 2011, 127, 954–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez-Fariñas, M.; Ungar, B.; Correa da Rosa, J.; Ewald, D.A.; Rozenblit, M.; Gonzalez, J.; Xu, H.; Zheng, X.; Peng, X.; Estrada, Y.D.; et al. RNA sequencing atopic dermatitis transcriptome profiling provides insights into novel disease mechanisms with potential therapeutic implications. J. Allergy Clin. Immunol. 2015, 135, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Ewald, D.A.; Noda, S.; Oliva, M.; Litman, T.; Nakajima, S.; Li, X.; Xu, H.; Workman, C.T.; Scheipers, P.; Svitacheva, N.; et al. Major differences between human atopic dermatitis and murine models, as determined by using global transcriptomic profiling. J. Allergy Clin. Immunol. 2017, 139, 562–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.P.; Li, Z.; Choi, E.K.; Lee, S.; Kim, Y.K.; Seo, E.Y.; Chung, J.H.; Cho, S. Urban particulate matter in air pollution penetrates into the barrier-disrupted skin and produces ROS-dependent cutaneous inflammatory response in vivo. J. Dermatol. Sci. 2018, 91, 175–183. [Google Scholar] [CrossRef]
- Li, R.; Ning, Z.; Majumdar, R.; Cui, J.; Takabe, W.; Jen, N.; Sioutas, C.; Hsiai, T. Ultrafine particles from diesel vehicle emissions at different driving cycles induce differential vascular pro-inflammatory responses: Implication of chemical components and NF-kappaB signaling. Part. Fibre Toxicol. 2010, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Totlandsdal, A.I.; Herseth, J.I.; Bølling, A.K.; Kubátová, A.; Braun, A.; Cochran, R.E.; Refsnes, M.; Ovrevik, J.; Låg, M. Differential effects of the particle core and organic extract of diesel exhaust particles. Toxicol. Lett. 2012, 208, 262–268. [Google Scholar] [CrossRef]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef] [Green Version]
- Esser, C.; Bargen, I.; Weighardt, H.; Haarmann-Stemmann, T.; Krutmann, J. Functions of the aryl hydrocarbon receptor in the skin. Semin. Immunopathol. 2013, 35, 677–691. [Google Scholar] [CrossRef]
- Mitchell, K.A.; Elferink, C.J. Timing is everything: Consequences of transient and sustained AhR activity. BioChem. Pharmacol. 2009, 77, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. Int. J. Mol. Sci. 2019, 20, 5424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhle, C.; Bock, K.W. Coordinate regulation of Phase I and II xenobiotic metabolisms by the Ah receptor and Nrf2. BioChem. Pharmacol. 2007, 73, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Roe, A.L.; Dieter, M.Z.; Solis, W.A.; Yang, Y.; Dalton, T.P. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. BioChem. Pharmacol. 2000, 59, 65–85. [Google Scholar] [CrossRef]
- Yu, J.; Luo, Y.; Zhu, Z.; Zhou, Y.; Sun, L.; Gao, J.; Sun, J.; Wang, G.; Yao, X.; Li, W. A tryptophan metabolite of the skin microbiota attenuates inflammation in patients with atopic dermatitis through the aryl hydrocarbon receptor. J. Allergy Clin. Immunol. 2019, 143, 2108–2119. [Google Scholar] [CrossRef] [Green Version]
- Tauchi, M.; Hida, A.; Negishi, T.; Katsuoka, F.; Noda, S.; Mimura, J.; Hosoya, T.; Yanaka, A.; Aburatani, H.; Fujii-Kuriyama, Y.; et al. Constitutive expression of aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol. Cell. Biol. 2005, 25, 9360–9368. [Google Scholar] [CrossRef] [Green Version]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef]
- Schiering, C.; Vonk, A.; Das, S.; Stockinger, B.; Wincent, E. Cytochrome P4501-inhibiting chemicals amplify aryl hydrocarbon receptor activation and IL-22 production in T helper 17 cells. BioChem. Pharmacol. 2018, 151, 47–58. [Google Scholar] [CrossRef]
- Anderson, C.; Hehr, A.; Robbins, R.; Hasan, R.; Athar, M.; Mukhtar, H.; Elmets, C.A. Metabolic requirements for induction of contact hypersensitivity to immunotoxic polyaromatic hydrocarbons. J. Immunol. 1995, 155, 3530–3537. [Google Scholar]
- Davila, D.R.; Davis, D.P.; Campbell, K.; Cambier, J.C.; Zigmond, L.A.; Burchiel, S.W. Role of alterations in Ca(2+)-associated signaling pathways in the immunotoxicity of polycyclic aromatic hydrocarbons. J. Toxicol. Environ. Health 1995, 45, 101–126. [Google Scholar] [CrossRef]
- Yamamoto, O.; Tokura, Y. Photocontact dermatitis and chloracne: Two major occupational and environmental skin diseases induced by different actions of halogenated chemicals. J. Dermatol. Sci. 2003, 32, 85–94. [Google Scholar] [CrossRef]
- Bonvallot, V.; Baeza-Squiban, A.; Baulig, A.; Brulant, S.; Boland, S.; Muzeau, F.; Barouki, R.; Marano, F. Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 expression. Am. J. Respir Cell Mol. Biol. 2001, 25, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepley, C.L.; Lauer, F.T.; Oliver, J.M.; Burchiel, S.W. Environmental polycyclic aromatic hydrocarbons, benzo(a) pyrene (BaP) and BaP-quinones, enhance IgE-mediated histamine release and IL-4 production in human basophils. Clin. Immunol. 2003, 107, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Pei, X.H.; Nakanishi, Y.; Inoue, H.; Takayama, K.; Bai, F.; Hara, N. Polycyclic aromatic hydrocarbons induce IL-8 expression through nuclear factor kappaB activation in A549 cell line. Cytokine 2002, 19, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Hatanaka, S.; Kimura, Y.; Hara, Y.; Nishiwaki, K.; Quan, Y.S.; Kamiyama, F.; Oiso, N.; Kawada, A.; Kabashima, K.; et al. A CCR4 antagonist ameliorates atopic dermatitis-like skin lesions induced by dibutyl phthalate and a hydrogel patch containing ovalbumin. BioMed. Pharmacother. 2019, 109, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Choi, H.S.; Seo, H.S.; Choi, Y.K.; Shin, Y.C.; Ko, S.G. Topical application of herbal mixture extract inhibits ovalbumin- or 2,4-dinitrochlorobenzene-induced atopic dermatitis. Evid. Based Complement. Altern. Med. 2012, 2012, 545497. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PM vs. Control | OVA vs. Control | OVA + PM vs. Control | OVA + PM vs. OVA | OVA + PM vs. PM | PM vs. OVA | |
|---|---|---|---|---|---|---|
| 1 | SPRR2D | RNASE2A | S100A9 | S100A9 | S100A9 | NPY |
| Log2FC | 4.537781 | 15.353418 | 175.102606 | 21.032206 | 43.232510 | 2.912855 |
| 2 | S100A9 | S100A9 | SPRR2D | SPRR2B | S100A8 | FAM3B |
| Log2FC | 4.050253 | 8.325451 | 122.348283 | 19.765663 | 36.247161 | 2.237210 |
| 3 | STFA3 | SPRR2D | SPRR2B | SAA3 | SAA3 | GUCA2A |
| Log2FC | 3.884888 | 7.414729 | 99.190350 | 16.871093 | 33.226551 | 2.082410 |
| 4 | CHIL1 | THRSP | S100A8 | S100A8 | SPRR2B | WFDC3 |
| Log2FC | 3.852430 | 6.074408 | 86.466724 | 16.615182 | 30.495545 | 2.057602 |
| 5 | DBP | SPRR2A1 | SPRR2A3 | SPRR2D | SPRR2D | IL22RA2 |
| Log2FC | 3.637145 | 5.429848 | 68.747026 | 16.500708 | 26.962138 | 1.771707 |
| 6 | IL1B | S100A8 | SEPINB3A | SPRR2A3 | SPRR2A3 | UGT1A1 |
| Log2FC | 3.375845 | 5.204079 | 65.385001 | 14.139278 | 21.554361 | 1.765702 |
| 7 | SPRR2A1 | SERPINB3A | STFA3 | SERPINB3A | SERPINB3A | TESC |
| Log2FC | 3.293317 | 5.127475 | 51.600623 | 12.751891 | 21.542993 | 1.728145 |
| 8 | LCE1H | SPRR2B | SPRR2A1 | SPRR2E | SPRR2E | SERPINE2 |
| Log2FC | 3.272727 | 5.018316 | 50.181213 | 12.506733 | 16.809473 | 1.705879 |
| 9 | SPRR2B | C1QTNF3 | SPRR2E | GM5416 | GM5416 | CRABP1 |
| Log2FC | 3.252618 | 4.536723 | 43.315581 | 10.651026 | 16.545459 | 1.702428 |
| 10 | 2610528A11RIK | CXCL1 | BC100530 | STFA3 | SPRR2A1 | PTGS1 |
| Log2FC | 3.202117 | 3.865055 | 29.494805 | 9.717934 | 15.237285 | 1.689424 |
| Genes | PM vs. Control | OVA vs. Control | OVA + PM vs. Control | OVA + PM vs. OVA | OVA + PM vs. PM | PM vs. OVA |
|---|---|---|---|---|---|---|
| Xenobiotic Metabolizing Enzyme | ||||||
| CYP1A1 | 2.364800 | |||||
| UGT1A1 | 1.968411 | 1.765702 | ||||
| UGT1A7C | 1.563135 | 4.020299 | 2.571945 | 2.447144 | ||
| Immune Response | ||||||
| IL1B | 3.375845 | 2.144279 | 15.898589 | 7.414423 | 4.709514 | |
| IL1F6 | 2.459084 | 2.677326 | 10.362620 | 3.870511 | 4.214016 | |
| IL1F8 | 1.682234 | 1.930127 | 5.440192 | 2.818568 | 3.220411 | |
| IL1F9 | 2.309176 | 1.647147 | 6.738345 | 4.090919 | ||
| IL-13ra1 | 2.339857 | 1.786709 | 1.753326 | |||
| IL-13ra2 | 2.445235 | 1.997591 | ||||
| IL-33 | 1.652810 | 5.151400 | 3.116752 | 3.571956 | ||
| CXCL1 | 2.034245 | 3.865055 | 15.266021 | 7.504513 | ||
| CCL2 | 2.414083 | 3.966503 | ||||
| CCL7 | 3.176686 | 6.829295 | 2.149817 | 3.330636 | ||
| CCL8 | 3.262495 | 9.964881 | 3.054375 | 7.572985 | ||
| CCR1 | 1.977892 | 7.166230 | 3.623166 | 5.588996 | ||
| CXCR2 | 5.409111 | 3.934086 | 4.409069 | |||
| TNFAIP2 | 2.981176 | 2.211034 | 2.536282 | |||
| TNFAIP6 | 3.665933 | 3.788700 | ||||
| FCER1A | 3.066804 | 5.700756 | 3.742706 | |||
| FCER1G | 1.866897 | 4.525466 | 2.424057 | 3.528595 | ||
| CHIL1 | 3.852430 | 3.413080 | 23.799435 | 6.973008 | 6.177772 | |
| RNASE2A | 15.353418 | 17.480094 | 5.250776 | −4.611952 | ||
| Skin Barrier | ||||||
| Epidermal Differentiation Complex | ||||||
| KRT1 | 2.012479 | 5.013296 | 3.007899 | 2.4911105 | ||
| KRT6b | 9.746359 | 7.671900 | 10.906282 | |||
| KRT16 | 14.027059 | 8.550596 | 9.550772 | |||
| LCE1F | 2.848601 | 1.856882 | 2.793710 | |||
| LCE1G | 3.202117 | 2.625508 | 3.709355 | |||
| LCE1H | 3.272727 | 2.649690 | 3.867855 | |||
| LCE3A | 1.985872 | 11.751650 | 5.917627 | 7.581050 | ||
| LCE3B | 12.008217 | 6.312606 | 9.559686 | |||
| LCE3E | 1.877260 | 9.465151 | 5.314134 | 5.042002 | ||
| LCE3F | 1.999833 | 11.052984 | 4.905179 | 5.526953 | ||
| S100A8 | 2.385476 | 5.204079 | 86.466724 | 16.615182 | 36.247161 | |
| S100A9 | 4.050253 | 8.325451 | 175.102606 | 21.032206 | 43.232510 | |
| SPRR2A1 | 3.293317 | 5.429848 | 50.181213 | 9.241734 | 15.237285 | |
| SPRR2A3 | 3.189472 | 68.747026 | 14.139278 | 21.554361 | ||
| SPRR2B | 3.252618 | 5.018316 | 99.190350 | 19.765663 | 30.495545 | |
| SPRR2D | 4.537781 | 7.414729 | 122.348283 | 16.500708 | 26.962138 | |
| SPRR2E | 2.576855 | 3.463381 | 43.315581 | 12.506733 | 16.809473 | |
| SPRR2I | 1.924438 | 2.981781 | 26.165916 | 8.775266 | 13.596652 | |
| FLG | −1.749558 | −2.481263 | ||||
| Protease | ||||||
| MMP3 | 2.031330 | 10.906441 | 5.369114 | 6.745412 | ||
| SERPINB3A | 3.035094 | 5.127475 | 65.385001 | 12.751891 | 21.542993 | |
| SERPINB3B | 2.497089 | 2.768278 | 17.026990 | 6.150751 | 6.818736 | |
| STFA1 | 2.008981 | 17.513492 | 6.855981 | 8.717599 | ||
| STFA3 | 3.884888 | 51.600623 | 9.717934 | 13.282395 | ||
| BC100530 | 2.912126 | 29.494805 | 7.555283 | 10.128274 | ||
| KLK6 | 3.837728 | |||||
| KLK8 | 2.456366 | 1.996790 | 6.233008 | 3.121514 | ||
| KLK9 | 2.747518 | 1.846616 | 9.784243 | 5.298470 | ||
| KLK13 | 2.201600 | 2.251518 | 13.571149 | 6.027555 | 6.164222 | |
| Antimicrobial Response | ||||||
| DEFB6 | 1.852542 | 1.683457 | 3.312694 | |||
| DEFB14 | 2.278557 | 5.154124 | 2.628721 | |||
| Other | ||||||
| 2610528A11RIK | 3.128210 | 3.748969 | 27.119970 | 7.233981 | 8.669484 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woo, Y.R.; Park, S.-Y.; Choi, K.; Hong, E.S.; Kim, S.; Kim, H.S. Air Pollution and Atopic Dermatitis (AD): The Impact of Particulate Matter (PM10) on an AD Mouse-Model. Int. J. Mol. Sci. 2020, 21, 6079. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176079
Woo YR, Park S-Y, Choi K, Hong ES, Kim S, Kim HS. Air Pollution and Atopic Dermatitis (AD): The Impact of Particulate Matter (PM10) on an AD Mouse-Model. International Journal of Molecular Sciences. 2020; 21(17):6079. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176079
Chicago/Turabian StyleWoo, Yu Ri, Seo-Yeon Park, Keonwoo Choi, Eun Sun Hong, Sungjoo Kim, and Hei Sung Kim. 2020. "Air Pollution and Atopic Dermatitis (AD): The Impact of Particulate Matter (PM10) on an AD Mouse-Model" International Journal of Molecular Sciences 21, no. 17: 6079. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176079