Chemical Reactivities of ortho-Quinones Produced in Living Organisms: Fate of Quinonoid Products Formed by Tyrosinase and Phenoloxidase Action on Phenols and Catechols



Abstract
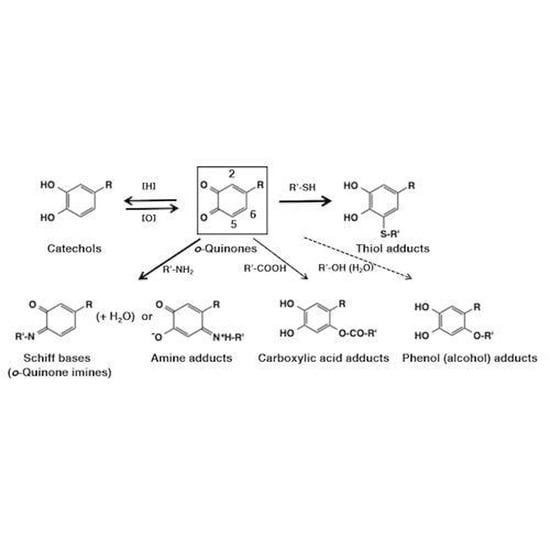
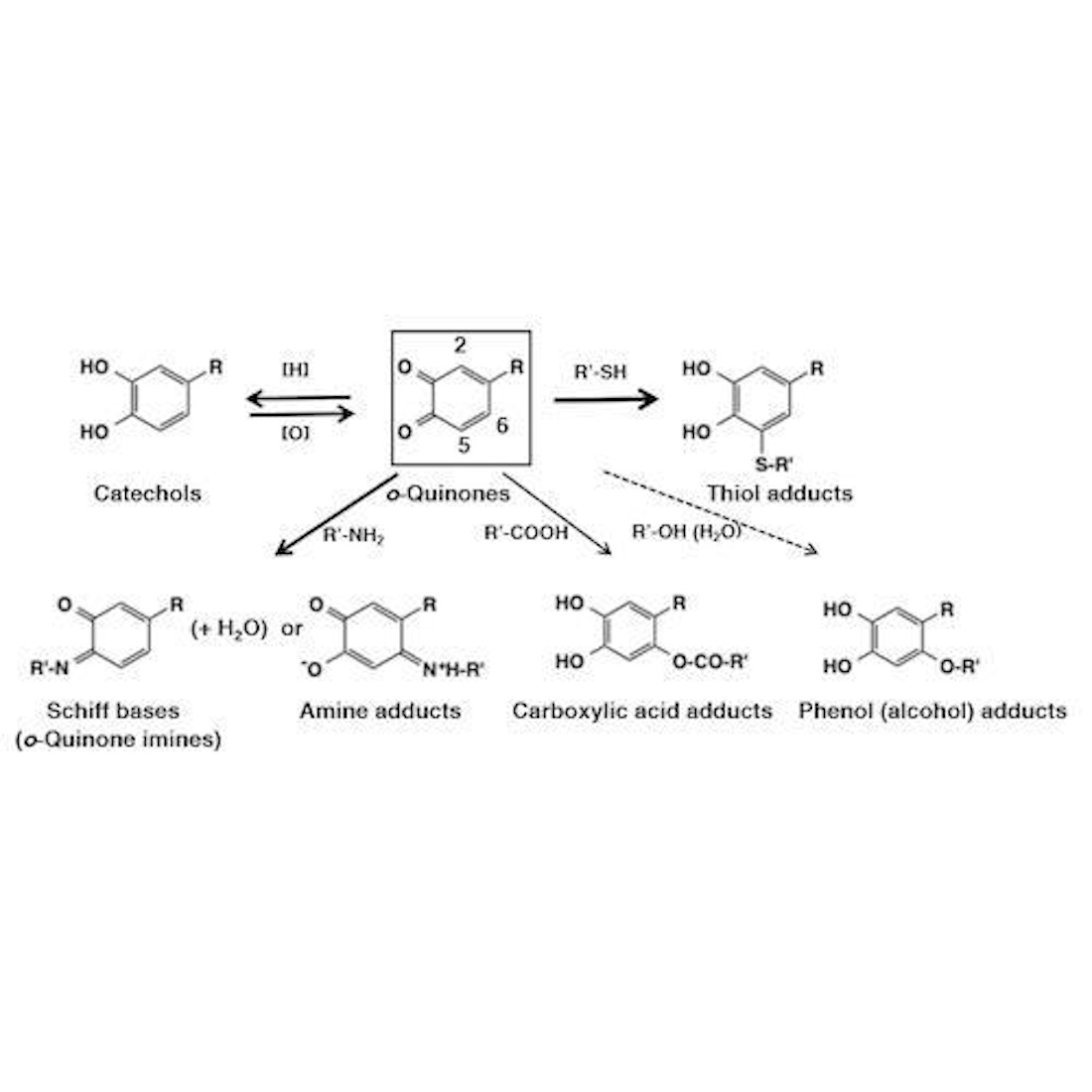
:
1. Introduction
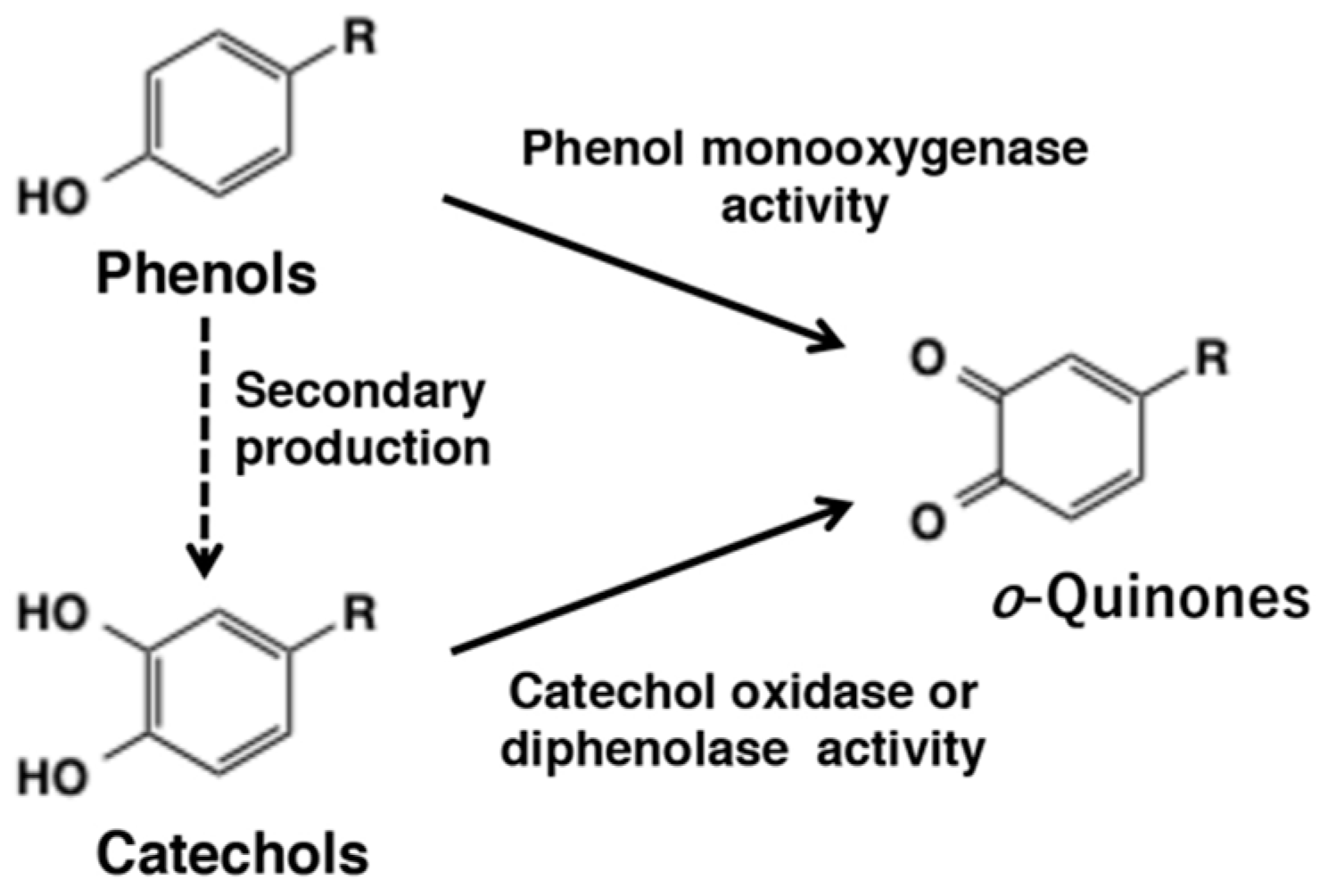
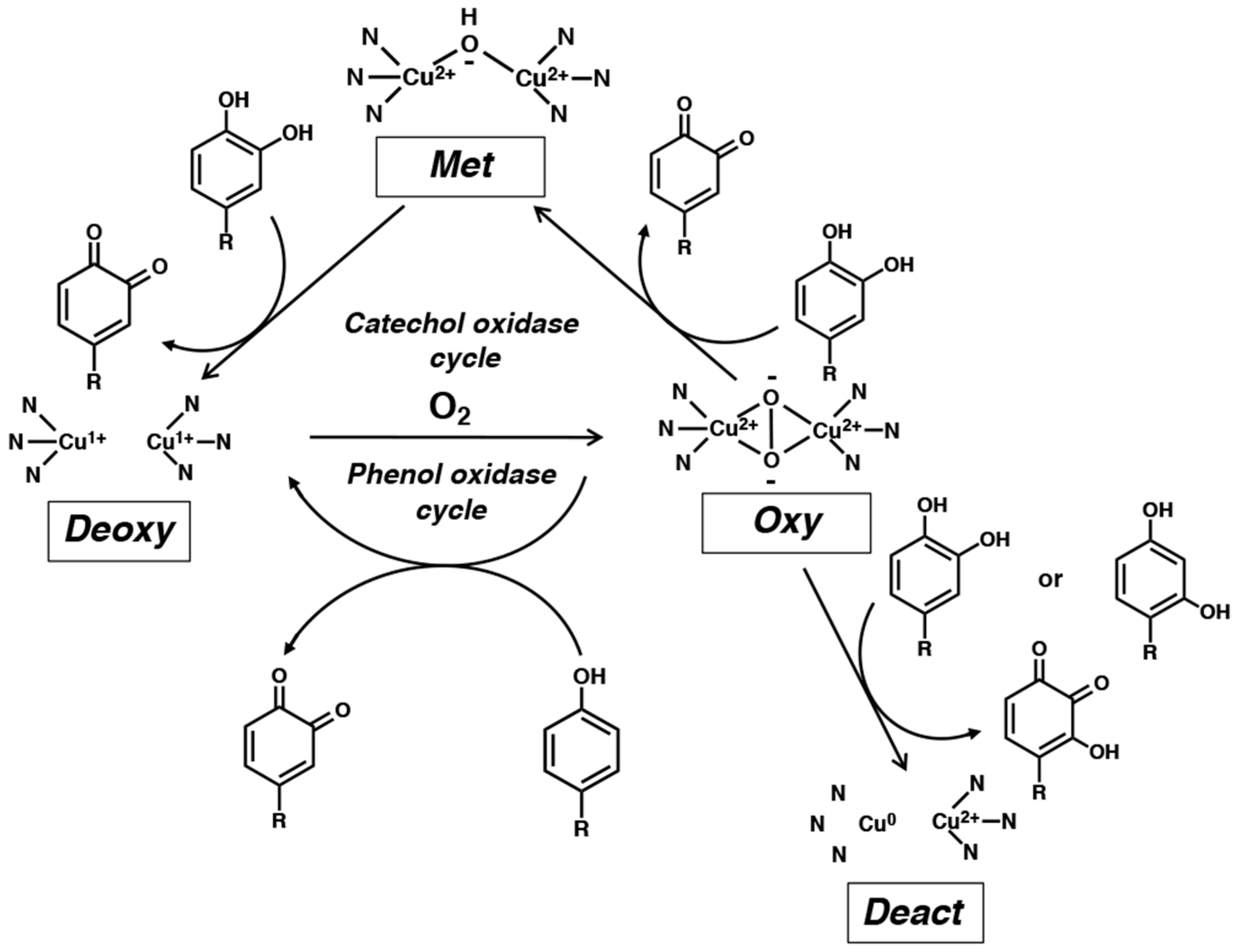
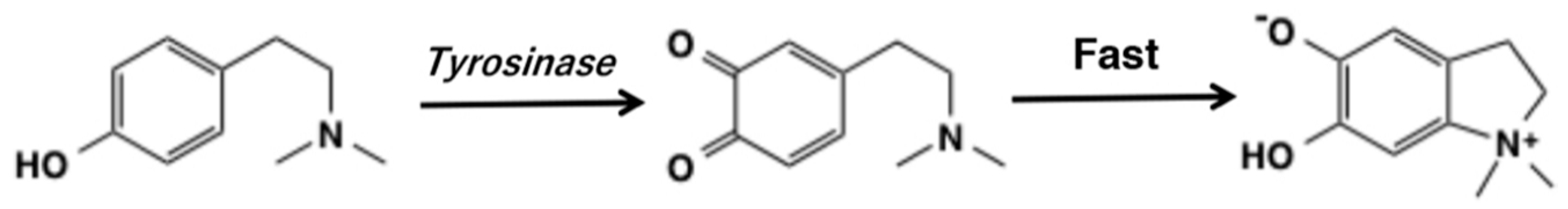
2. o-Quinone Formation from Phenols and Catechols by Tyrosinase
3. Reactivity of o-Quinones with Small Molecules Leading to Adducts Formation
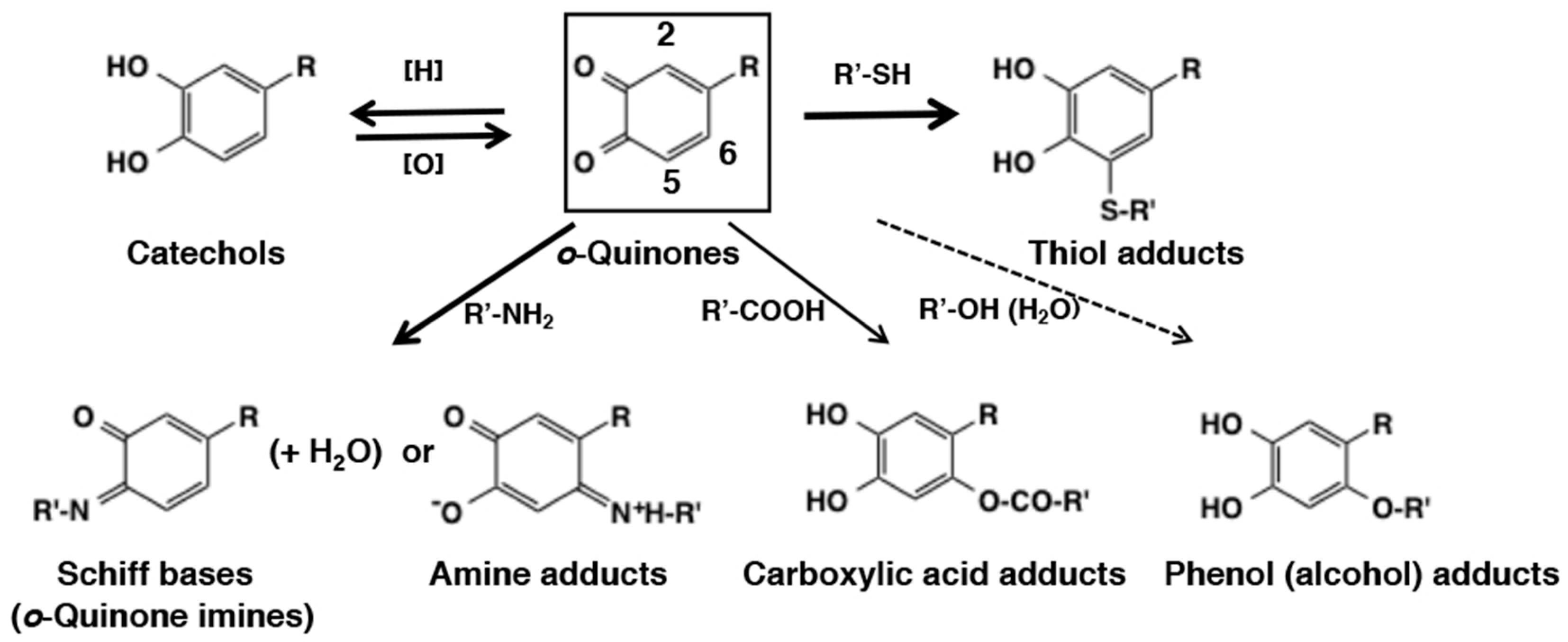
3.1. Chemical Fate of o-Quinones
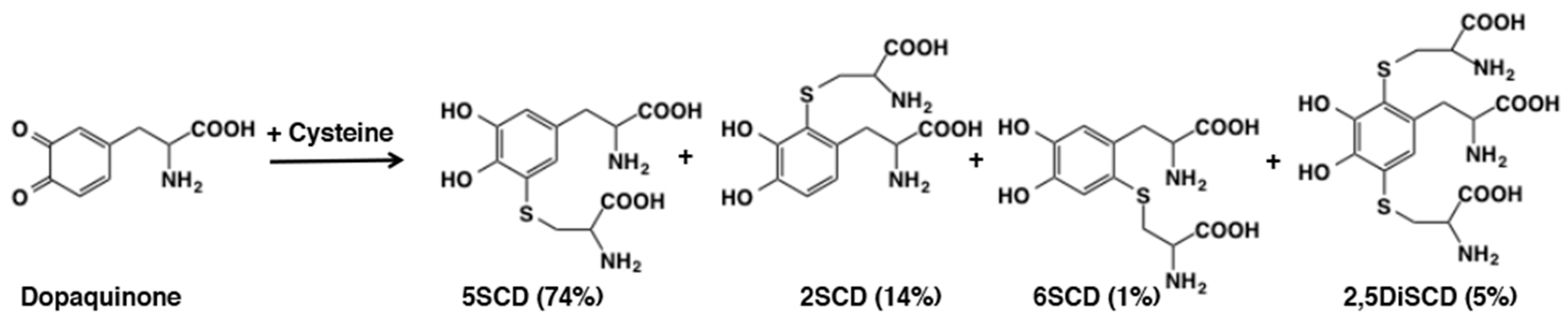
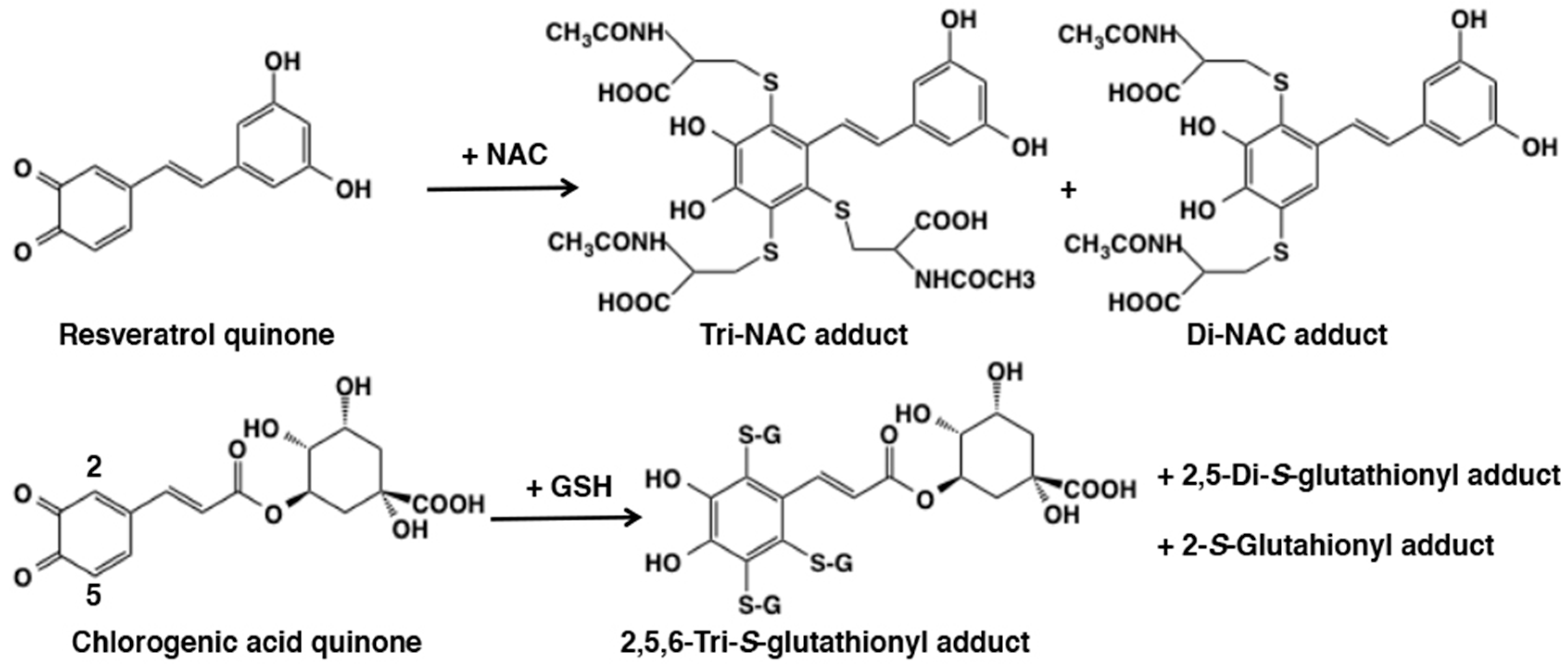
3.2. Reaction of o-Quinones with Thiols
3.3. Reaction of o-Quinones with Amines
3.4. Reaction of o-Quinones with Carboxylic Acids, Phenols, and Alcohols (Water)
3.5. Redox Exchange of o-Quinones with Reducing Agents
3.6. Reaction of o-Quinone from Hydroquinone
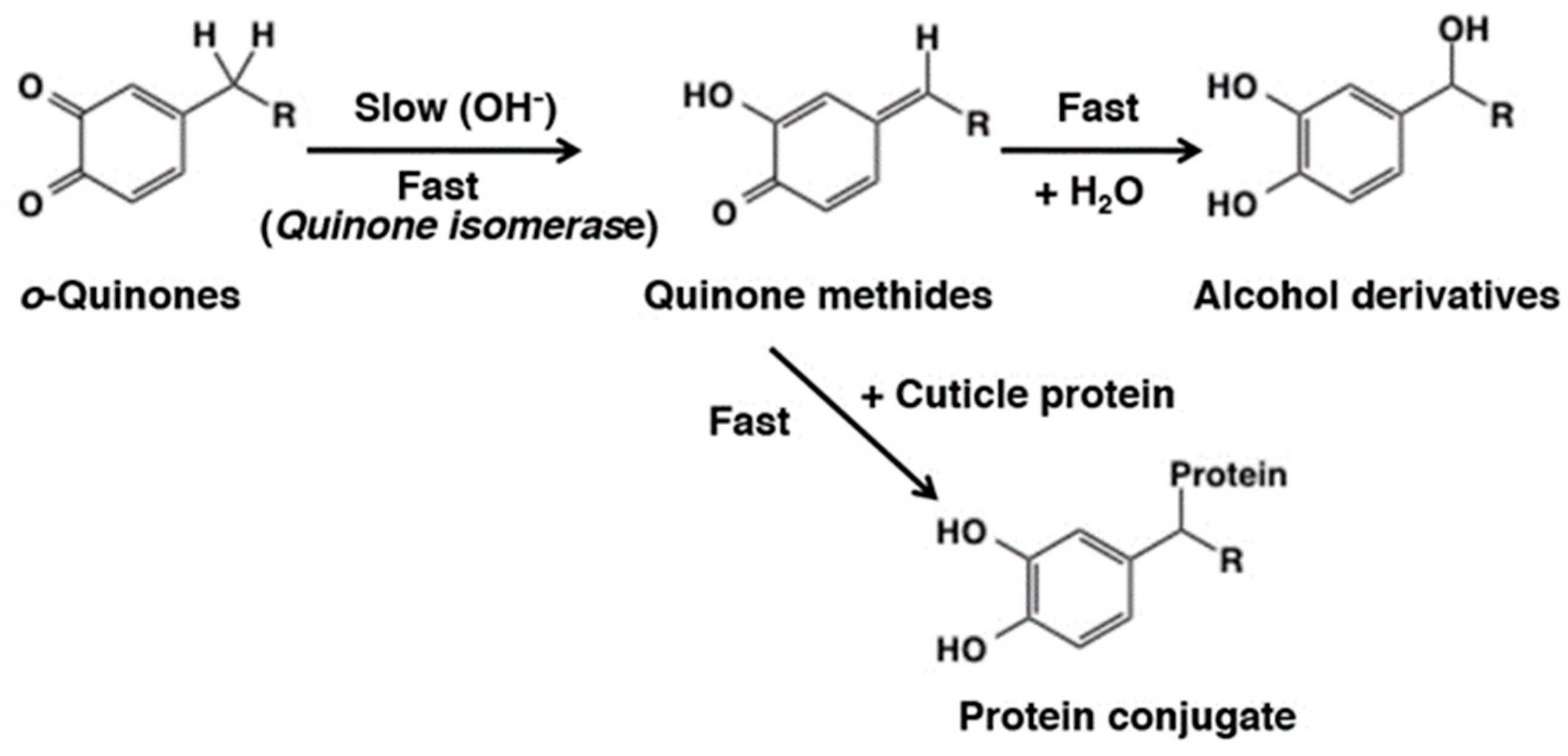
4. Reactivity of o-Quinones through Quinone Methide Tautomers
4.1. Addition of Water to Quinone Methide Tautomers
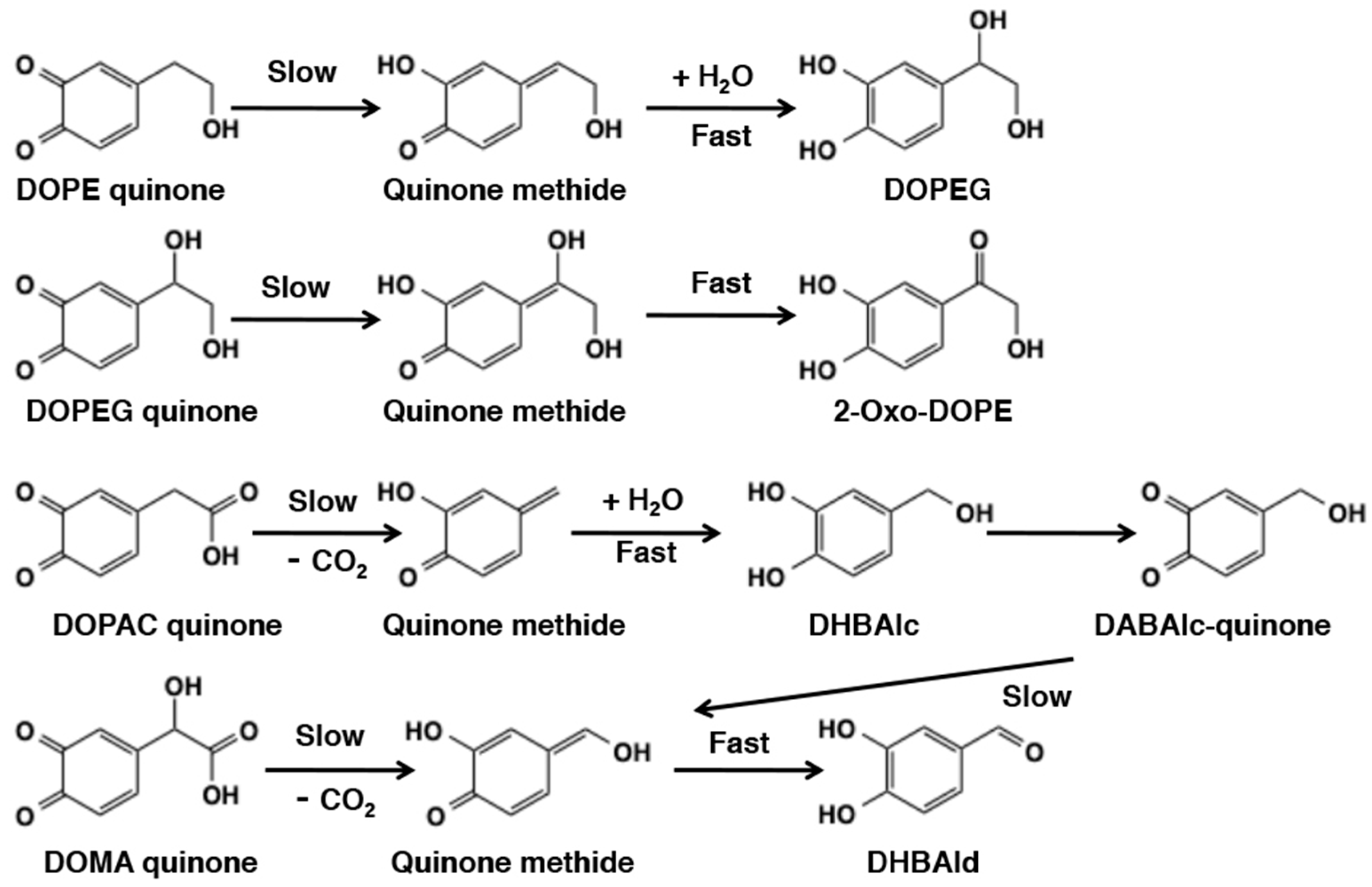
4.2. Reaction of Quinone Methides from Catecholamine Metabolites
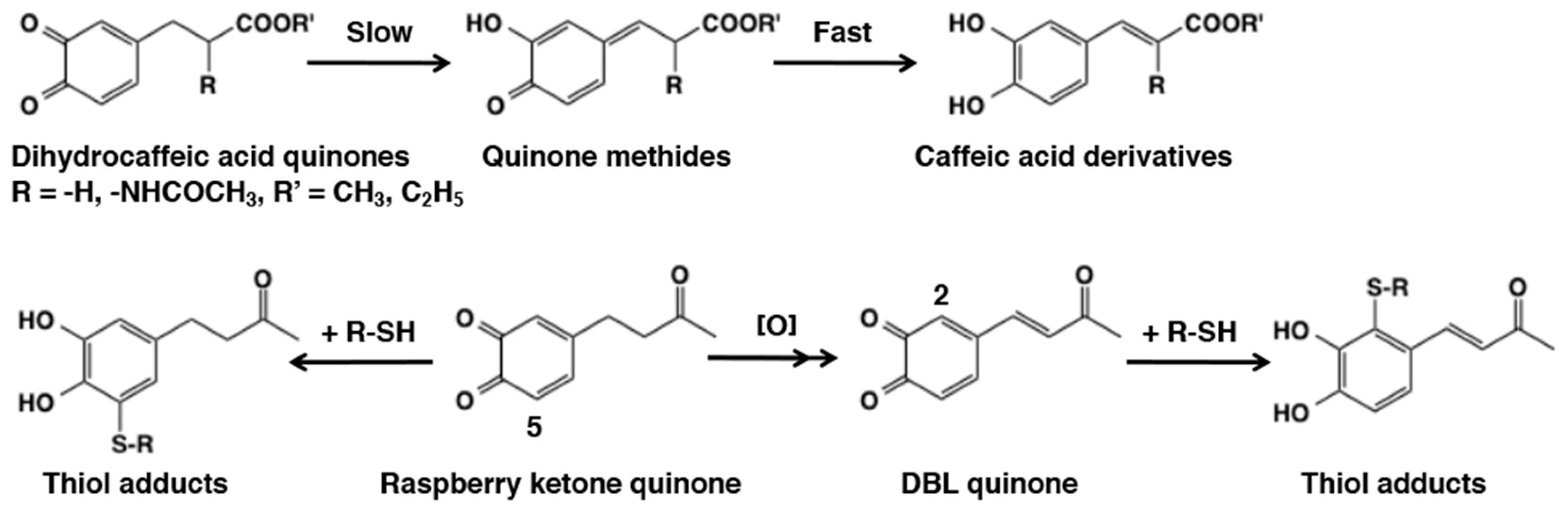
4.3. Side Chain Desaturation (Dehydrogenation) Reactions
4.4. Reactivity of Side Chain Desaturated Quinones
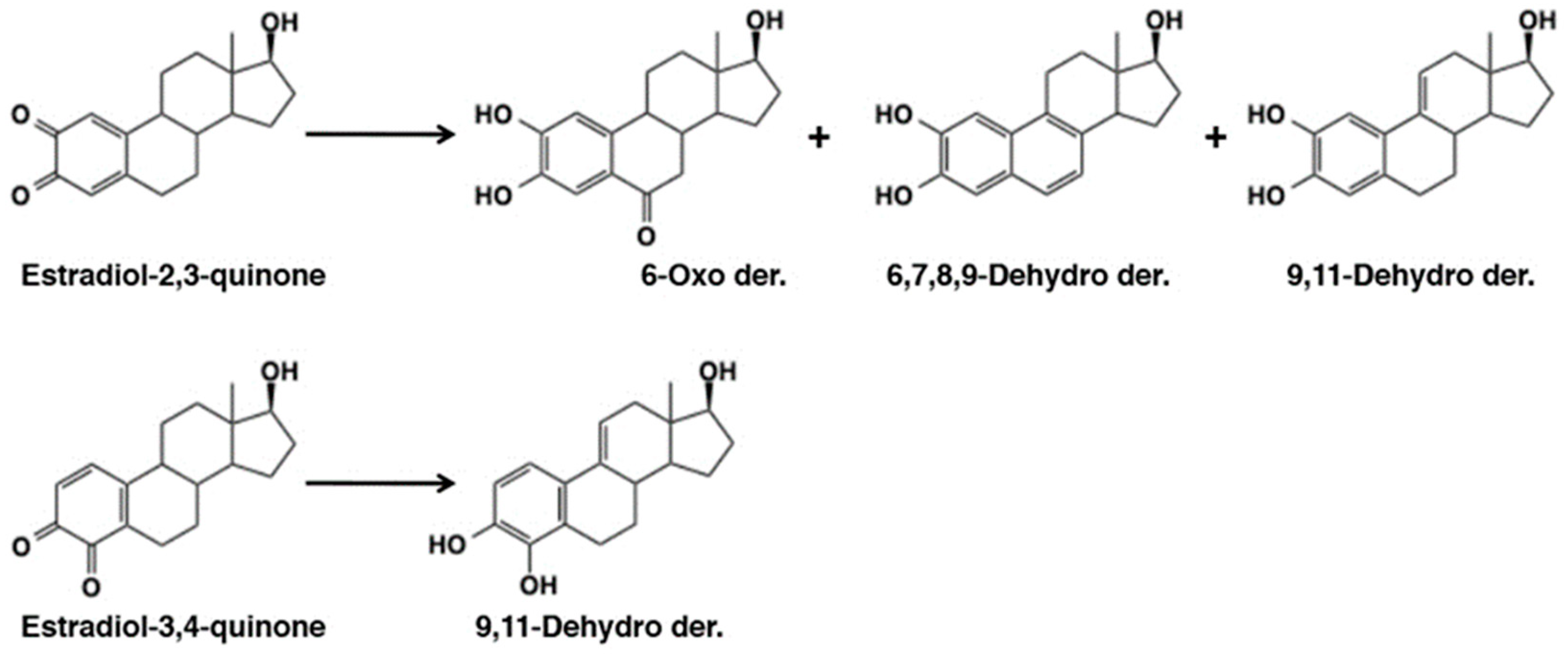
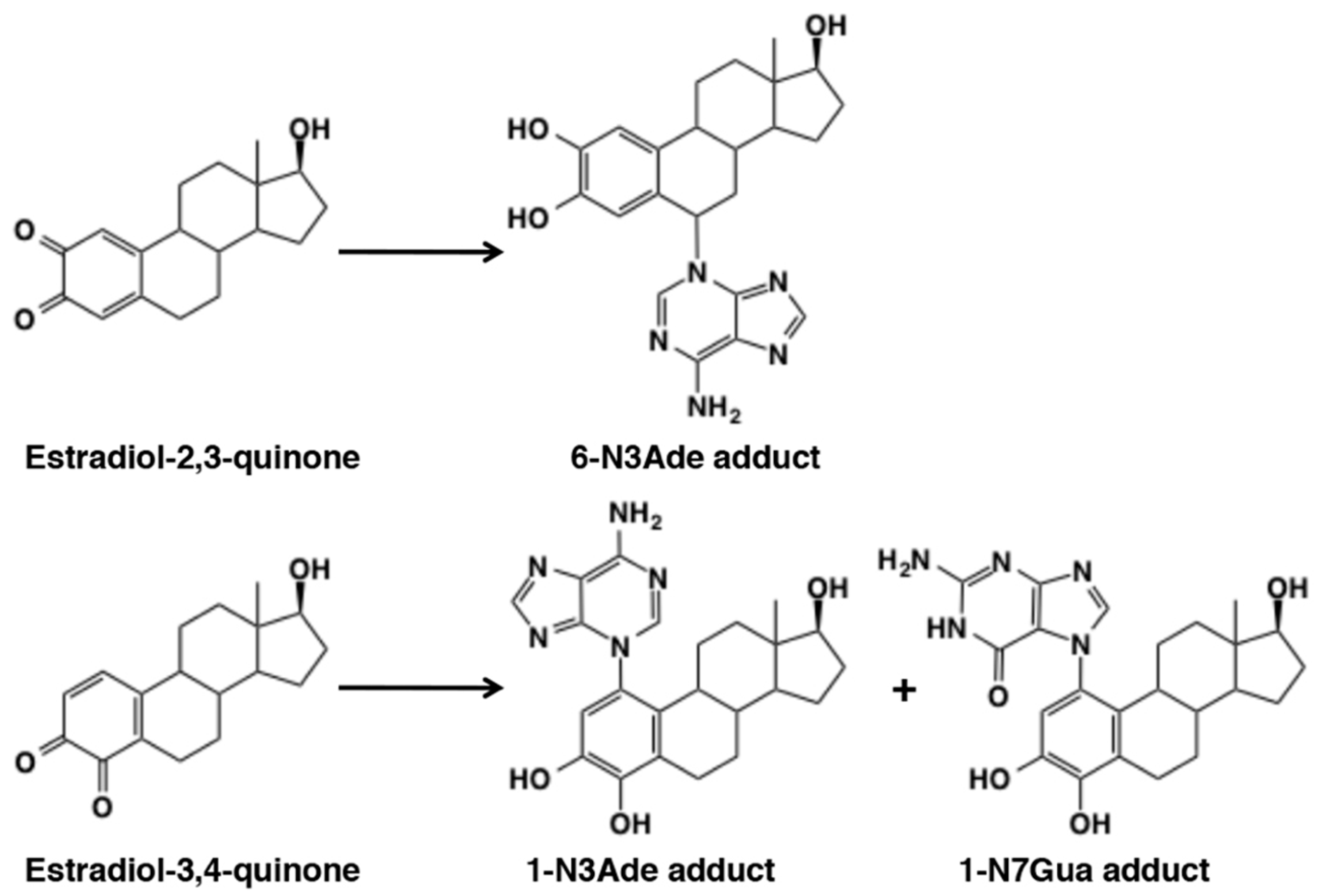
4.5. Reaction of Estradiol Quinones
5. Reactivity of o-Quinones with Macromolecules
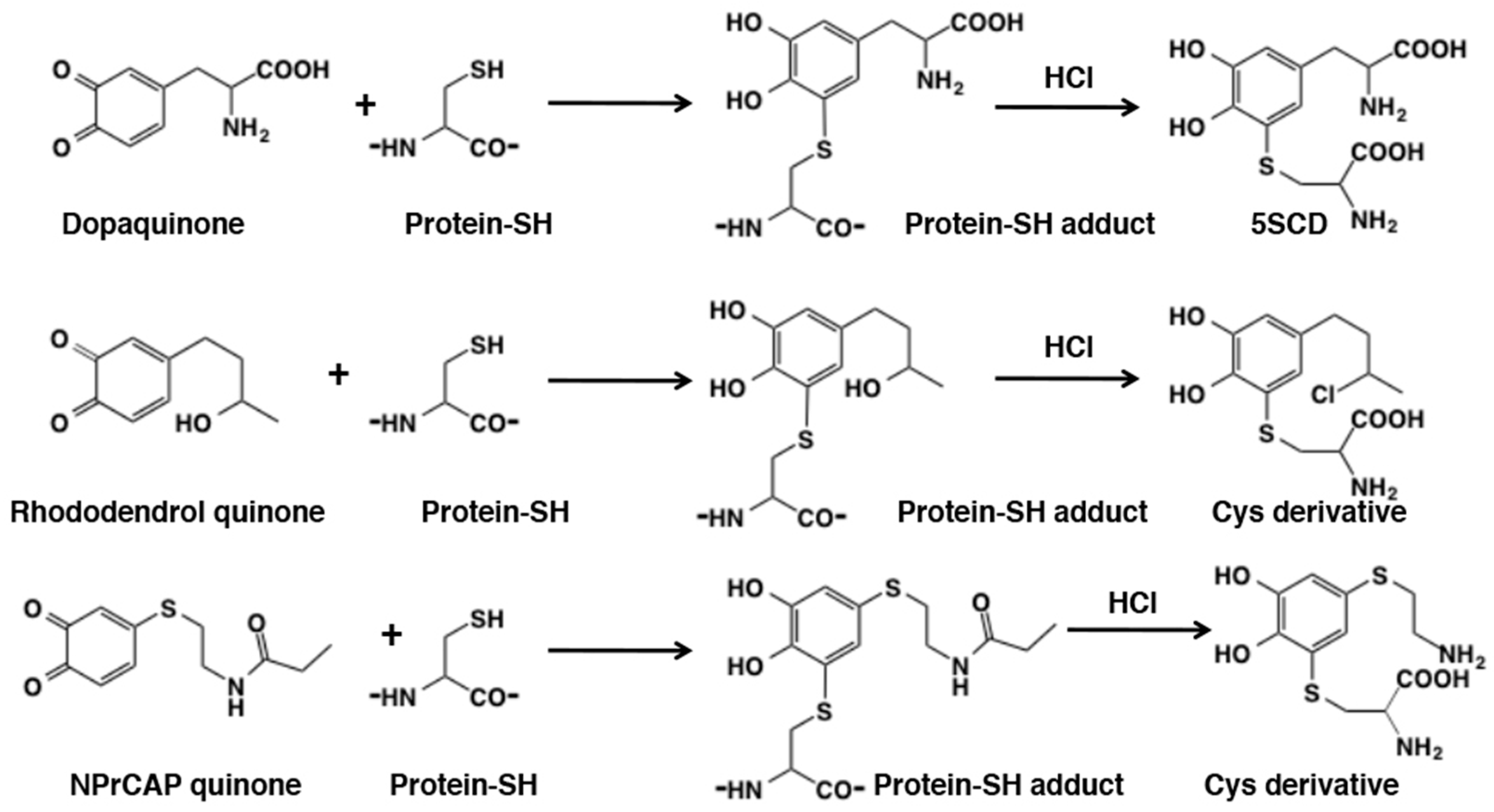
5.1. Covalent Binding with Proteins
5.2. Covalent Binding with DNA
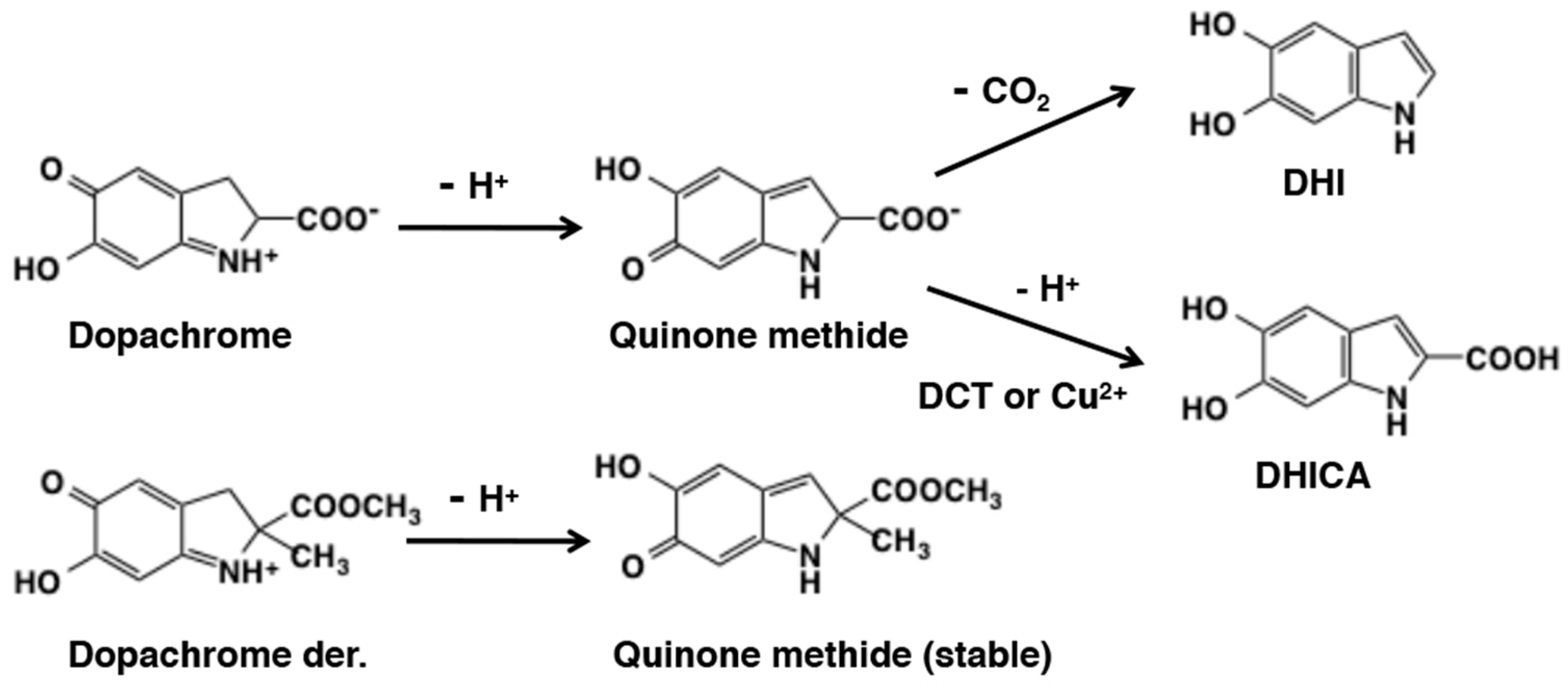
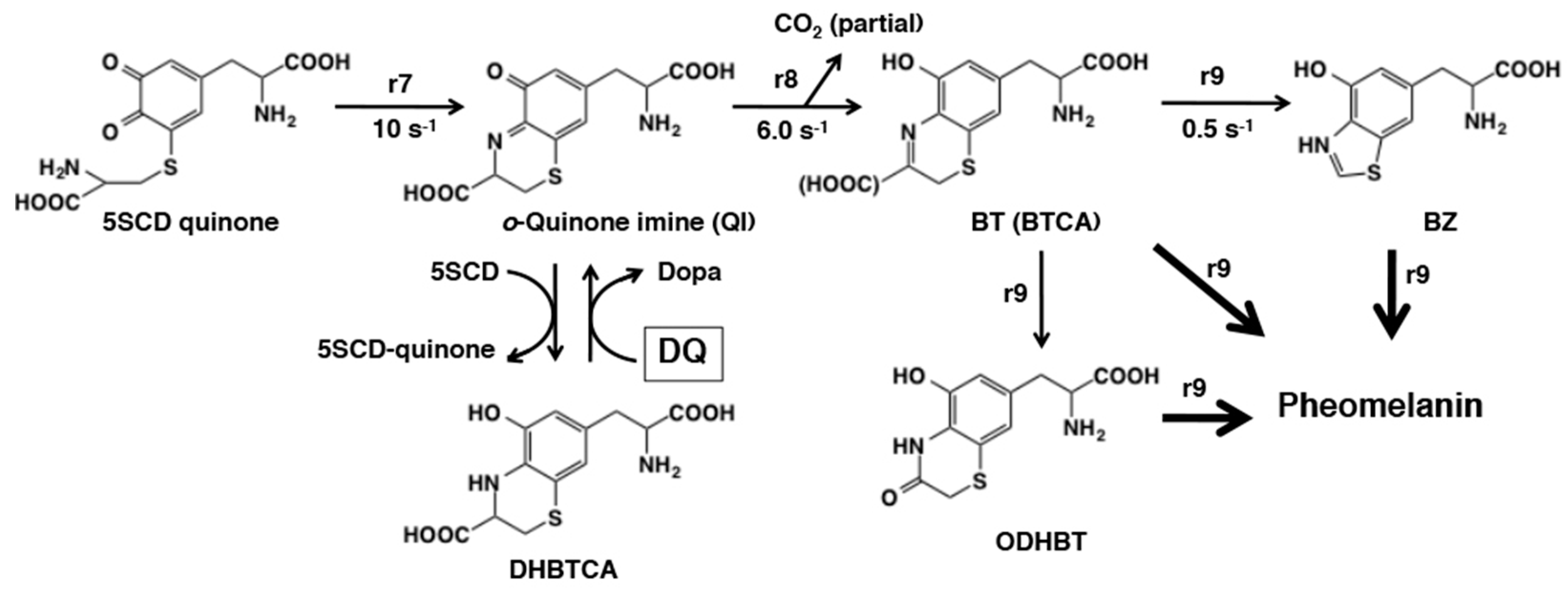
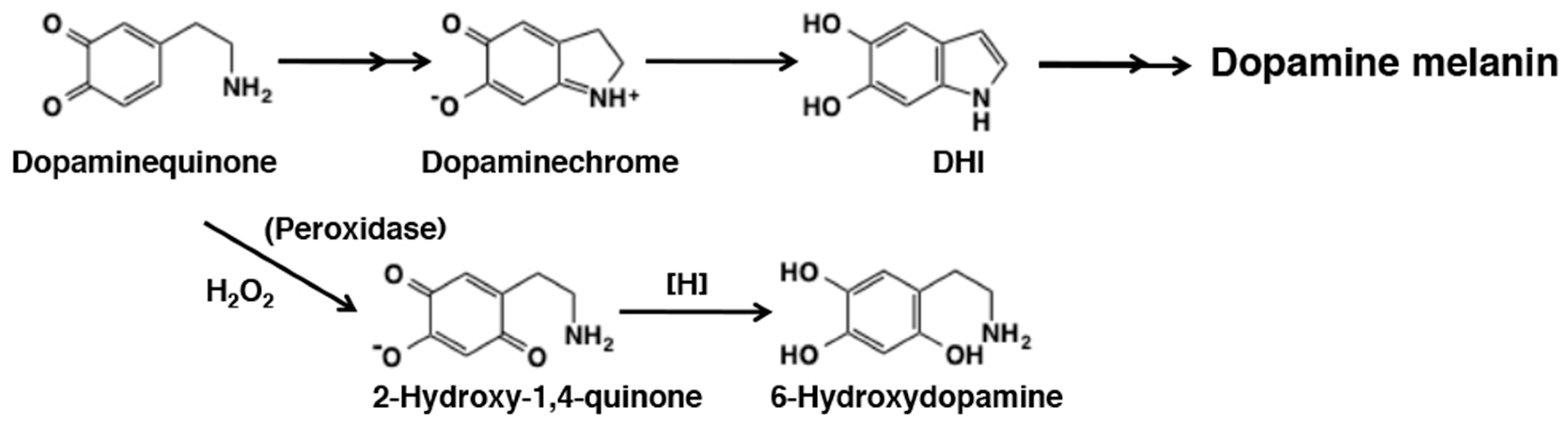
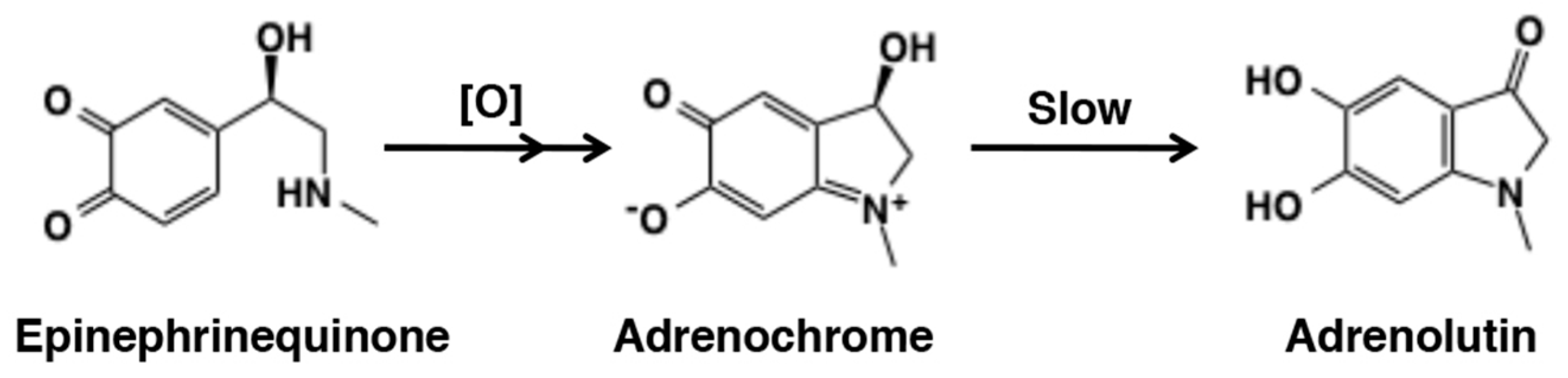
6. Melanogenesis in Relation to o-Quinone Chemistry
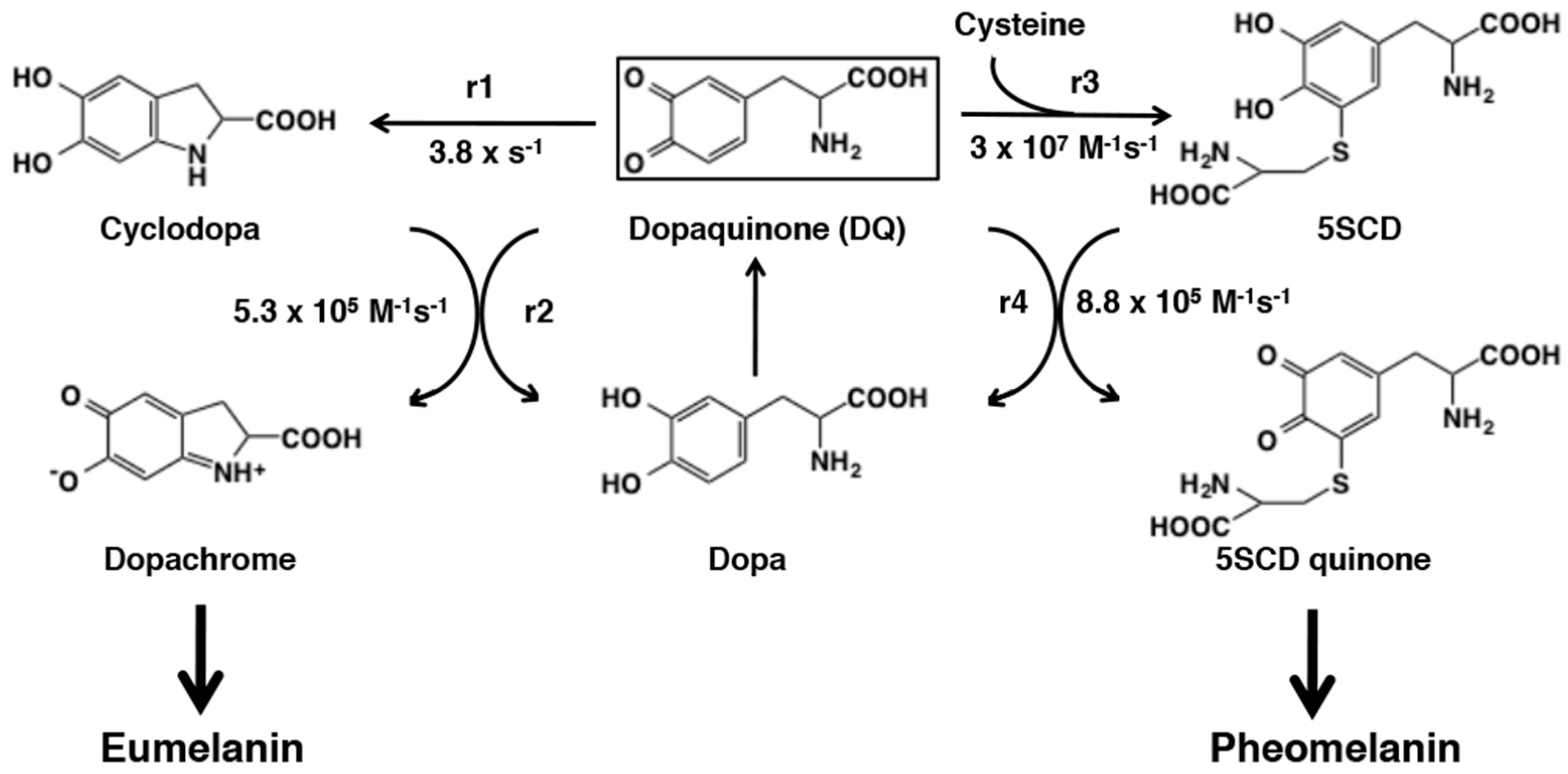
6.1. Early Stages of Mixed Melanogenesis
6.2. Late Stages of Mixed Melanogenesis
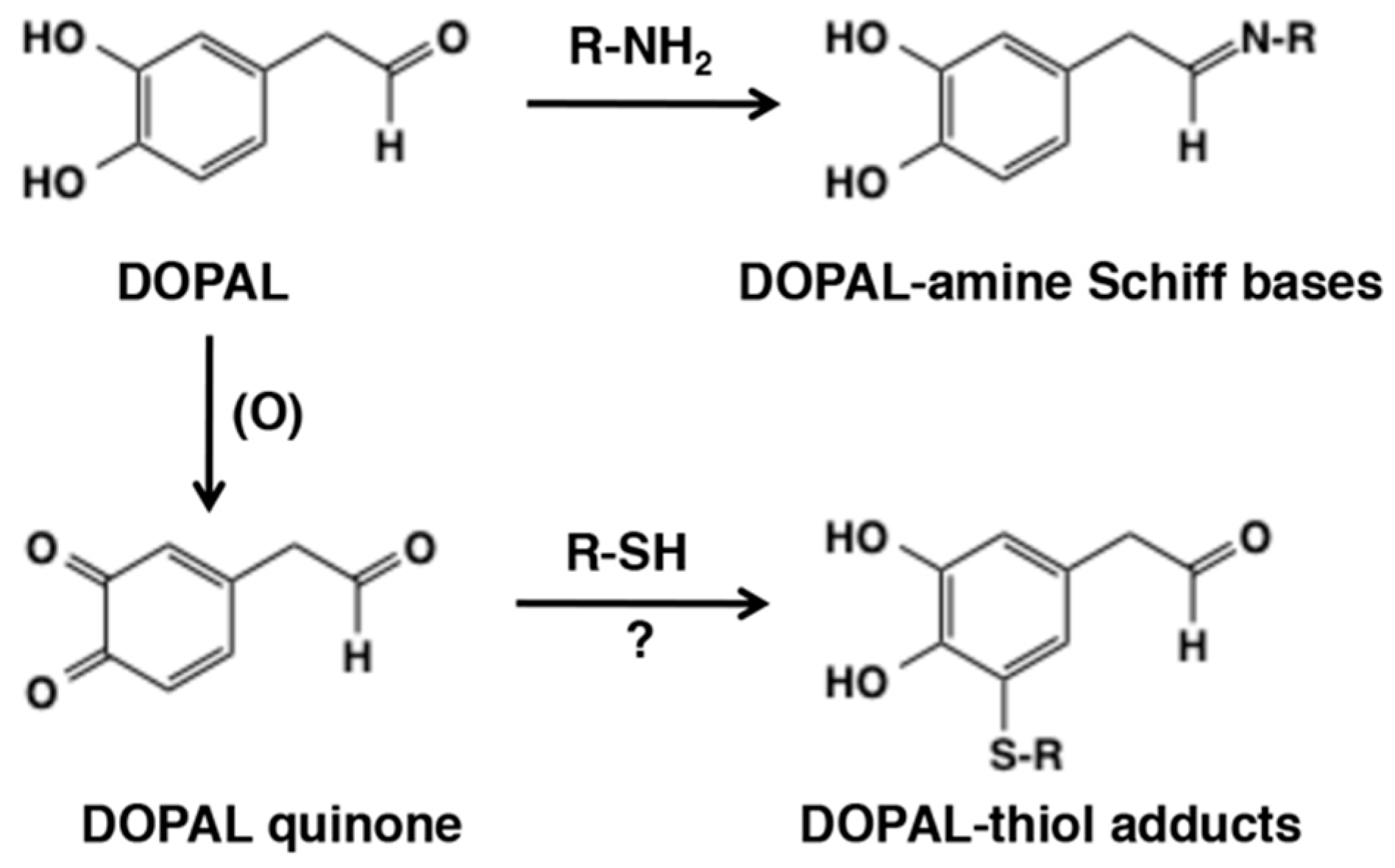
6.3. Metabolic Fates of Catecholamine Quinones
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BT | 1,4-Benzothiazine |
| BTCA | 1,4-Benzothiazine-3-carboxylic acid |
| BQ | Dihydro-1,4-benzothiazine-6,7-dione |
| BZ | Benzothiazole |
| CD quinone | Cysteinyldopaquinone |
| DBL | 3,4-Dihydroxybenzalacetone |
| DCT | Dopachrome tautomerase, Tyrp2 |
| DHBA | 3,4-Dihydroxybenzoic acid |
| DHBAlc | 3,4-Dihydroxybenzylalcohol |
| DHBAld | 3,4-dihydroxybenzaldehyde |
| DHBTCA | Dihydro-1,4-benzothiazine-3-carboxylic acid |
| DHI | 5,6-Dihydroxyindole |
| DHICA | 5,6-Dihydroxyindole-2-carboxylic acid |
| Dihydrocaffeic acid | 3-(3,4-Dihydroxyphenyl)propionic acid |
| DOMA | 3,4-Dihydromandelic acid |
| DOPAC | 3,4-Dihydroxyphenylacetic acid |
| DOPAL | 3,4-Dihydroxybenzaldehyde |
| DOPE | 3,4-Dihydroxyphenylethanol |
| DOPEG | 3,4-Dihydroxyphenylethyleneglycol |
| DQ | Dopaquinone |
| NAC | N-Acetylcysteine |
| ODHBT | 3-Oxo-3,4-dihydro-1,4-benzothiazine |
| 4SCAP | 4-S-Cysteaminylphenol |
| 2SCD | 2-S-cysteinyldopa |
| 5SCD | 5-S-cysteinyldopa |
References
- Ito, S.; Wakamatsu, K. Chemistry of mixed melanogenesis—pivotal roles of dopaquinone. Photochem. Photobiol. 2008, 84, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Bellei, C.; Costi, P.; Albertini, A.; Monzani, E.; Casella, L.; Gallorini, M.; Bergamaschi, L.; Moscatelli, A.; Turro, N.J.; et al. New melanic pigments in the human brain that accumulate in aging and block environmental toxic metals. Proc. Natl. Acad. Sci. USA 2008, 105, 17567–17572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucca, F.A.; Basso, E.; Francesca, A.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the human substantia nigra: An update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Waite, J.H. Mussel adhesion—Essential footwork. J. Exp. Biol. 2017, 220, 517–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, Q.F.; Abebe, A.; Evans, J.; Sugumaran, M. Oxidative transformation of tunichromes—Model studies with 1,2-dehydro-N-acetyldopamine and N-acetylcysteine. Bioorg. Chem. 2017, 73, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M. Chemistry of cuticular sclerotization. Adv. Insect Physiol. 2010, 39, 151–209. [Google Scholar]
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 2388–2395. [Google Scholar] [CrossRef]
- Ismaya, W.T.; Rozeboom, H.J.; Weijn, A.; Mes, J.J.; Fusetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef] [Green Version]
- Land, E.J.; Ramsden, C.A.; Riley, P.A. Tyrosinase autoactivation and the chemistry of ortho-quinone amines. Acc. Chem. Res. 2003, 36, 300–308. [Google Scholar] [CrossRef]
- Hernández-Romero, D.; Sanchez-Amat, A.; Solano, F. A tyrosinase with an abnormally high tyrosine hydroxylase/dopa oxidase ratio. Role of the seventh histidine and accessibility to the active site. FEBS J. 2006, 273, 257–270. [Google Scholar] [CrossRef]
- Cooksey, C.J.; Garratt, P.J.; Land, E.J.; Pavel, S.; Ramsden, C.A.; Riley, P.A.; Smit, N.P.M. Evidence of the indirect formation of the catecholic intermediate substrate responsible for the autoactivation kinetics of tyrosinase. J. Biol. Chem. 1997, 272, 26226–26235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Land, E.J.; Ramsden, C.A.; Riley, P.A. The mechanism of suicide-inactivation of tyrosinase: A substrate structure investigation. Tohoku J. Exp. Med. 2007, 212, 341–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Land, E.J.; Ramsden, C.A.; Riley, P.A.; Stratford, M.R.L. Evidence consistent with the requirement of cresolase activity for suicide inactivation of tyrosinase. Tohoku J. Exp. Med. 2008, 216, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratford, M.R.L.; Ramsden, C.A.; Riley, R.A. Mechanistic studies on the inactivation of tyrosinase by resorcinol. Bioorg. Med. Chem. 2013, 21, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S. When quinones meet amino acids: Chemical, physical and biological consequences. Amino Acids 2006, 30, 205–224. [Google Scholar] [CrossRef]
- Bolton, J.L.; Trush, M.A.; Penning, T.M.; Dryhurst, G.; Monks, T.J. Role of quinones in toxicology. Chem. Res. Toxicol. 2000, 13, 136–160. [Google Scholar] [CrossRef]
- Land, E.J.; Ramsden, C.A.; Riley, P.A. Quinone chemistry and melanogenesis. Methods Enzymol. 2004, 378, 88–109. [Google Scholar]
- Nair, V.; Menon, R.S.; Biju, A.T.; Abhilash, K.G. 1,2-Benzoquinones in Diels-Alder reactions, dipolar cycloadditions, nucleophilic additions, multicomponent reactions and more. Chem. Soc. Rev. 2012, 41, 1050–1059. [Google Scholar] [CrossRef]
- Sugumaran, M. Reactivities of quinone methides versus o-quinones in catecholamine metabolism and eumelanin biosynthesis. Int. J. Mol. Sci. 2016, 17, 1576. [Google Scholar] [CrossRef] [Green Version]
- Bolton, J.L.; Dunlap, T.L.; Dietz, B.M. Formation and biological targets of botanical o-quinones. Food Chem. Toxicol. 2018, 120, 700–707. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, R.; Wang, G. Impact of dopamine oxidation on dopaminergic neurodegeneration. ACS Chem. Neurosci. 2019, 10, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Glagoleva, A.Y.; Shoeva, O.Y.; Khlestkina, E.K. Melanin pigment in plants: Current knowledge and future perspectives. Front. Plant Sci. 2020, 11, 770. [Google Scholar] [CrossRef] [PubMed]
- Kishida, R.; Kasai, H. Mechanistic study of melanogenesis: Binding of cysteine to dopaquinone. Pigment Cell Melanoma Res. 2017, 30, e21. [Google Scholar]
- Tse, D.C.S.; McCreery, R.L.; Adams, R.N. Potential oxidative pathways of brain catecholamines. J. Med. Chem. 1976, 19, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Jameson, G.N.L.; Zhang, J.; Jameson, R.F.; Linert, W. Kinetic evidence that cysteine reacts with dopaminoquinone via reversible adduct formation to yield 5-cysteinyl-dopamine: An important precursor of neuromelanin. Org. Biomol. Chem. 2004, 2, 777–782. [Google Scholar] [CrossRef]
- Ito, S.; Prota, G. A facile one-step synthesis of cysteinyldopas using mushroom tyrosinase. Experientia 1977, 33, 1118–1119. [Google Scholar] [CrossRef]
- Agrup, G.; Agrup, P.; Andersson, T.; Hafström, L.; Hansson, C.; Jacobsson, S.; Jönsson, P.E.; Rorsman, H.; Rosengren, A.M.; Rosengren, E. 5 years’ experience of 5-S-cysteinyldopa in melanoma diagnosis. Acta Derm. Vernereol. 1979, 59, 381–388. [Google Scholar]
- Wakamatsu, K.; Fukushima, S.; Minagawa, A.; Omodaka, T.; Hida, T.; Hatta, N.; Takata, M.; Uhara, H.; Okuyama, R.; Ihn, H. Significance of 5-S-cysteinyldopa as a marker for melanoma. Int. J. Mol. Sci. 2020, 21, 432. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Nicol, J.A.C. A new amino acid, 3-(2,5-SS-dicysteinyl-3,4-dihydroxyphenyl)alanine, from the tapetum lucidum of the gar (Lepisosteidae) and its enzymic synthesis. Biochem. J. 1977, 161, 499–507. [Google Scholar] [CrossRef]
- Ito, S.; Fujita, K.; Yoshioka, M.; Sienko, D.; Nagatsu, T. Identification of 5-S- and 2-S-cysteinyldopamine and 5-S-glutathionyldopamine formed from dopamine by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. 1986, 375, 134–140. [Google Scholar] [CrossRef]
- Ito, S.; Palumbo, A.; Prota, G. Tyrosinase-catalyzed conjugation of dopa with glutathione. Experientia 1985, 41, 960–961. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, E.; Linder-Eliasson, E.; Carlsson, A. Detection of 5-S-cysteinyldopamine in human brain. J. Neural. Transm. 1985, 63, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Fujikawa, K.; Zucca, F.A.; Zecca, L.; Ito, S. The structure of neuromelanin as studied by chemical degradative methods. J. Neurochem. 2003, 86, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Cooksey, C.J.; Land, E.J.; Rushton, E.A.P.; Ramsden, C.A.; Riley, P.A. Tyrosinase-mediated cytotoxicity of 4-substituted phenols: Use of QSAR to forecast reactivities of thiols towards the derived ortho-quinones. Quant. Struct. Act. Relatsh. 1996, 15, 498–503. [Google Scholar] [CrossRef]
- Ito, S.; Fujiki, Y.; Matsui, N.; Ojika, M.; Wakamatsu, K. Tyrosinase-catalyzed oxidation of resveratrol produces a highly reactive ortho-quinone: Implications for melanocyte toxicity. Pigment Cell Melanoma Res. 2019, 32, 766–776. [Google Scholar] [CrossRef]
- De Lucia, M.; Panzella, L.; Pezzella, A.; Napolitano, A.; d’Ischia, M. Plant catechols and their S-glutathionyl conjugates as antinitrosating agents; Expedient synthesis and remarkable potency of 5-S-glutathionylpiceatannol. Chem. Res. Toxicol. 2008, 21, 2407–2413. [Google Scholar] [CrossRef]
- Panzella, L.; Napolitano, A.; d’Ischia, M. Oxidative conjugation of chlorogenic acid with glutathione. Structural characterization of addition products and a new nitrite-promoted pathway. Bioorg. Med. Chem. 2003, 11, 4797–4805. [Google Scholar] [CrossRef]
- Sugumaran, M.; Dali, H.; Semensi, V. Chemical and cuticular phenoloxidase—Mediated synthesis of cysteinyl-catechol adducts. Arch. Insect Biochem. Physiol. 1989, 11, 127–137. [Google Scholar] [CrossRef]
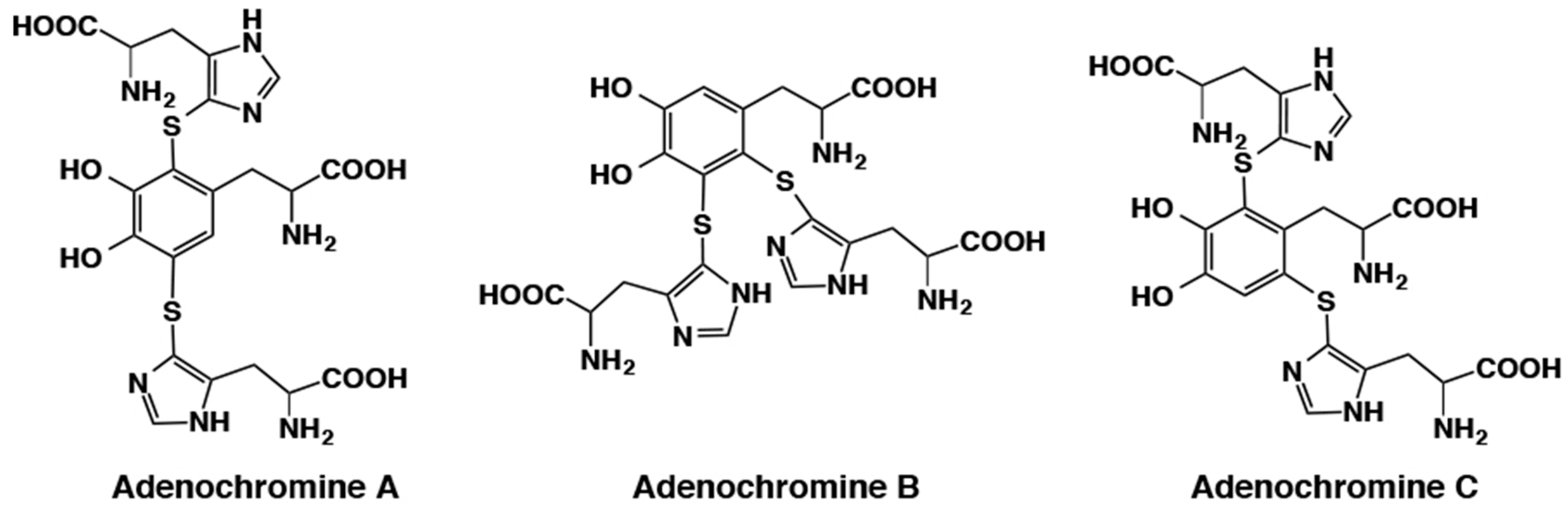
- Ito, S.; Nardi, G.; Palumbo, A.; Prota, G. Isolation and characterization of adenochrome, a unique iron (III)-binding peptide from Octopus vulgaris. J. Chem. Soc. Perkin Trans. I 1979, 1, 2617–2623. [Google Scholar] [CrossRef]
- Ito, S.; Nardi, G.; Palumbo, A.; Prota, G. A possible pathway for the biosynthesis of adenochromines. Experientia 1979, 35, 14–15. [Google Scholar] [CrossRef]
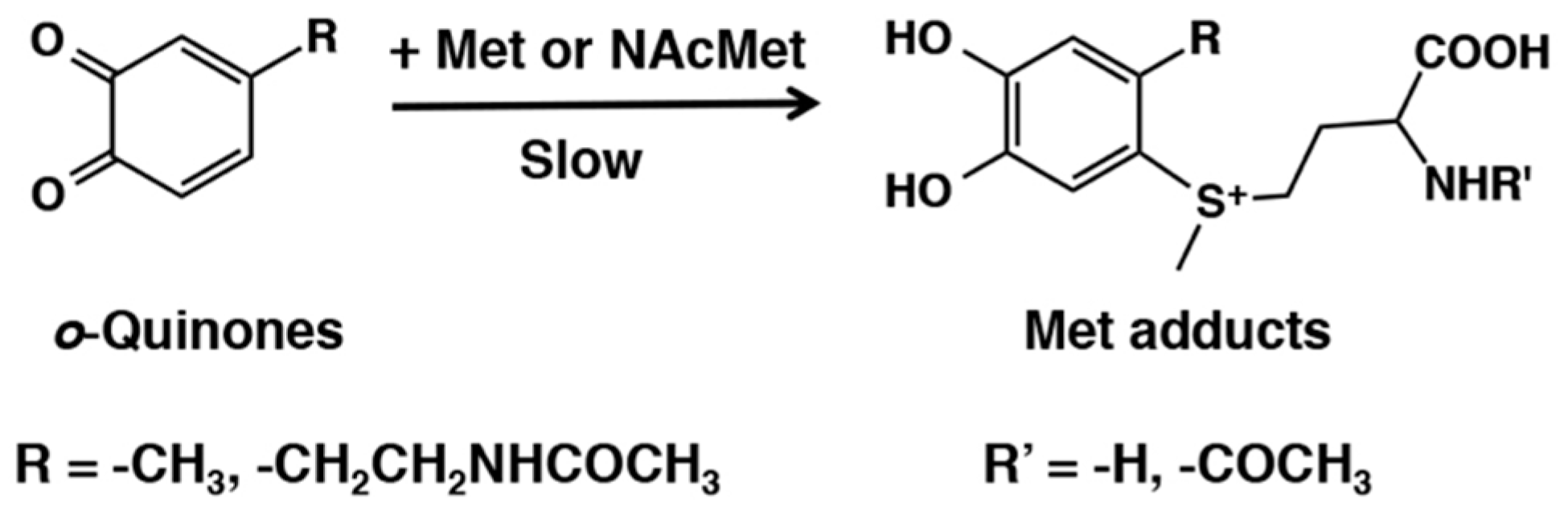
- Gupta, M.N.; Vithayathil, P.J. Chemical modification of methionines of ribonuclease A with o-benzoquinone. Int. J. Pept. Protein Res. 1980, 15, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.N.; Vithayathil, P.J. Isolation and characterization of a methionine adduct of dopa-o-quinone. Bioorg. Chem. 1982, 11, 101–107. [Google Scholar] [CrossRef]
- Gupta, M.N.; Murthy, G.S.; Vithayathil, P.J. o-Benzoquinone – A reagent for determining the conformational differences in related proteins. Int. J. Pept. Protein Res. 1980, 15, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Vithayathil, P.J.; Murthy, G.S. New reaction of o-benzoquinone at the thioester group of methionine. Nature 1972, 236, 101–103. [Google Scholar]
- Vithayathil, P.J.; Gupta, M.N. Reaction of methionine with some biologically important o-quinones. Indian J. Biochem. Biophys. 1981, 18, 82–83. [Google Scholar]
- Sugumaran, M.; Nelson, E. Model sclerotization studies. 4. Generation of N-acetyl methionyl catechol adducts during tyrosinase-catalyzed oxidation of catechols in the presence of N-acetylmethionine. Arch. Insect Biochem. Physiol. 1998, 38, 44–52. [Google Scholar] [CrossRef]
- Hawley, M.D.; Tatawawadi, S.V.; Piekarski, S.; Adams, R.N. Electrochemical studies of the oxidation pathways of catecholamines. J. Am. Chem. Soc. 1967, 89, 447–450. [Google Scholar] [CrossRef]
- Graham, D.G.; Tiffany, S.M.; Bell, W.R., Jr.; Gutknecht, W.F. Autoxidation versus covalent binding of quinones as the mechanism of toxicity of dopamine, 6-hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol. Pharmacol. 1978, 14, 644–653. [Google Scholar]
- Li, J.; Christensen, B.M. Effect of pH on the oxidation pathway of dopamine and dopa. J. Electroanal. Chem. 1994, 375, 219–231. [Google Scholar] [CrossRef]
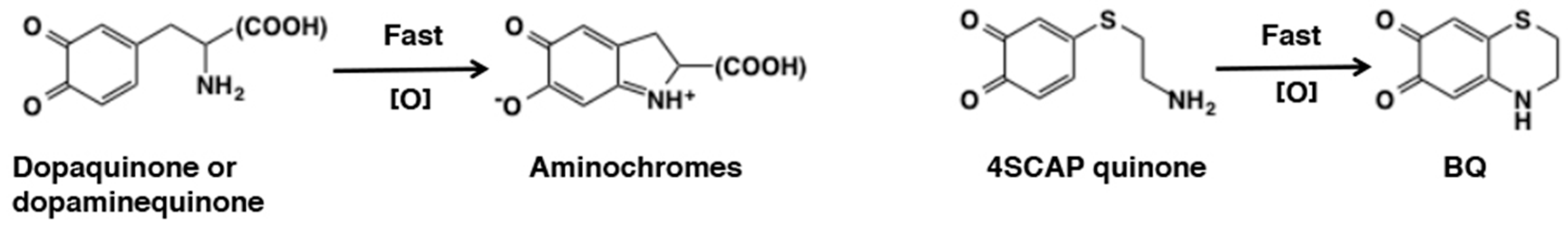
- Mascagna, D.; Costantini, C.; d’Ischia, M.; Prota, G. Biomimetic oxidation of the antimelanoma agent 4-S-cysteaminylphenol and related catechol thioethers: Isolation and reaction behavior of novel dihydrobenzothiazinequinones. Tetrahedron 1994, 50, 8757–8764. [Google Scholar] [CrossRef]
- Inoue, S.; Hasegawa, K.; Wakamatsu, K.; Ito, S. Comparison of antimelanoma effects of 4-S-cysteaminylphenol and its homologues. Melanoma Res. 1998, 8, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Jimbow, K.; Ito, S. The in vivo antimelanoma effect of 4-S-cysteaminylphenol and its N-acetyl derivative. Int. J. Cancer 1990, 46, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Ito, S.; Inoue, S.; Wakamatsu, K.; Ozeki, H.; Ishiguro, I. Dihydro-1,4-benzothiazine-6,7-dione, the ultimate toxic metabolite of 4-S-cysteaminylphenol and 4-S-cysteaminylcatechol. Biochem. Pharmacol. 1997, 53, 1435–1444. [Google Scholar] [CrossRef]
- Li, Y.; Jongberg, S.; Andersen, M.L.; Davies, M.J.; Lund, M.N. Quinone-induced protein modifications: Kinetic preference for reaction of 1,2-benzoquinones with thiol groups in proteins. Free Radic. Biol. Med. 2016, 97, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qi, H.; Fan, M.; Zhu, Z.; Zhan, S.; Li, L.; Li, B.; Zhang, X.; Zhao, X.; Ma, J.; et al. Quantifying the efficacy of o-benzoquinones reaction with amino acids and related nucleophiles by cyclic voltammetry. Food Chem. 2020, 317, 126454. [Google Scholar] [CrossRef]
- Sugumaran, M.; Lipke, H. Crosslink precursor for dipteran puparium. Proc. Natl. Acad. Sci. USA 1982, 79, 2480–2484. [Google Scholar] [CrossRef] [Green Version]
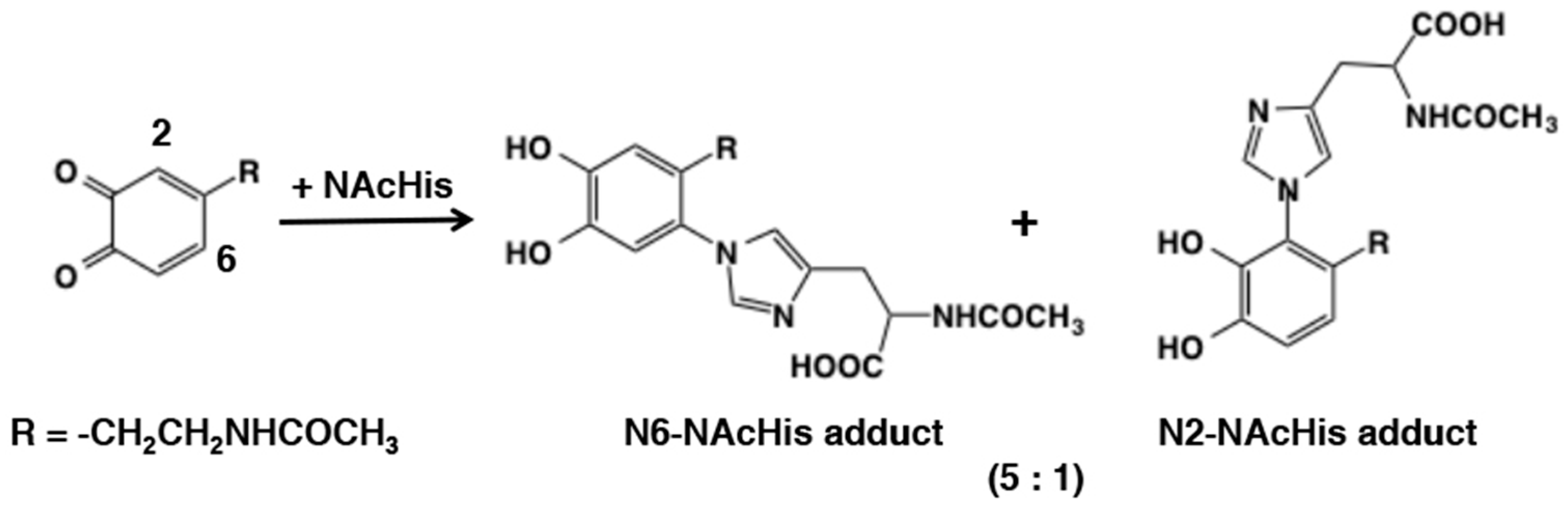
- Xu, R.; Huang, X.; Morgan, T.D.; Prakash, O.; Kramer, K.; Hawley, M.D. Characterization of products from the reactions of N-acetyldopamine quinone with the N-acetylhistidine. Arch. Biochem. Biophys. 1996, 329, 56–64. [Google Scholar] [CrossRef]
- Cavalieri, E.L.; Li, K.-M.; Balu, N.; Saeed, M.; Devanesan, P.; Higginbotham, S.; Zhao, J.; Gross, M.L.; Rogan, E.G. Catechol ortho-quinones: The electrophilic compounds that form depurinating DNA adducts and could initiate cancer and other diseases. Carcinogenesis 2002, 23, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Barek, H.; Evans, J.; Sugumaran, M. Unraveling complex molecular transformations of N-β-alanyldopamine that accounts for brown coloration of insect cuticle. Rapid Commun. Mass Spectrom. 2017, 31, 1363–1373. [Google Scholar] [CrossRef]
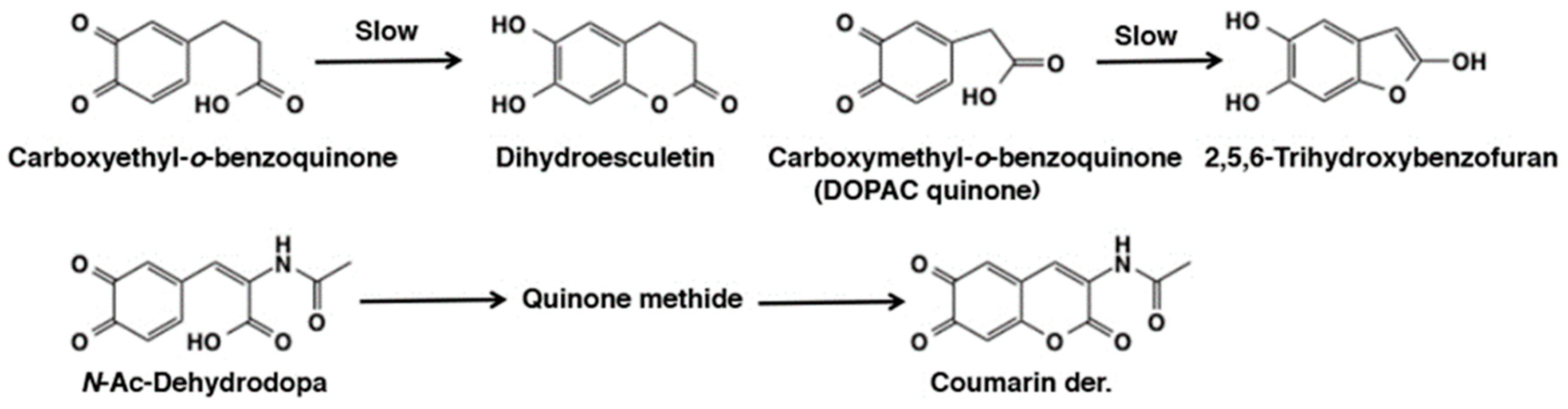
- Sugumaran, M.; Dali, H.; Kundzicz, H.; Semensi, V. Unusual, intramolecular cyclization and side chain desaturation of carboxyethyl-o-benzoquinone derivatives. Bioorg. Chem. 1989, 17, 443–453. [Google Scholar] [CrossRef]
- Sugumaran, M.; Semensi, V.; Dali, H.; Mitchell, W. Novel oxidative transformations of enzymatically generated carboxymethyl-o-benzoquinone to 2,5,6-trihydroxybenzofuran and 3,4-dihydroxy mandelic acid. Bioorg. Chem. 1989, 17, 86–95. [Google Scholar] [CrossRef]
- Abebe, A.; Kuang, Q.F.; Evans, J.; Sugumaran, M. Mass spectrometric studies shed light on unusual oxidative transformations of 1,2-dehydro-N-acetyldopa. Rapid Comm. Mass Spectrom. 2013, 27, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Duggaraju, P.; Jayachandran, E.; Kirk, K. Formation of a new quinone methide intermediate during the oxidative transformation of 3,4-dihydroxyphenylacetic acids: Implications for eumelanin biosynthesis. Arch. Biochem. Biophys. 1999, 371, 98–106. [Google Scholar] [CrossRef] [PubMed]
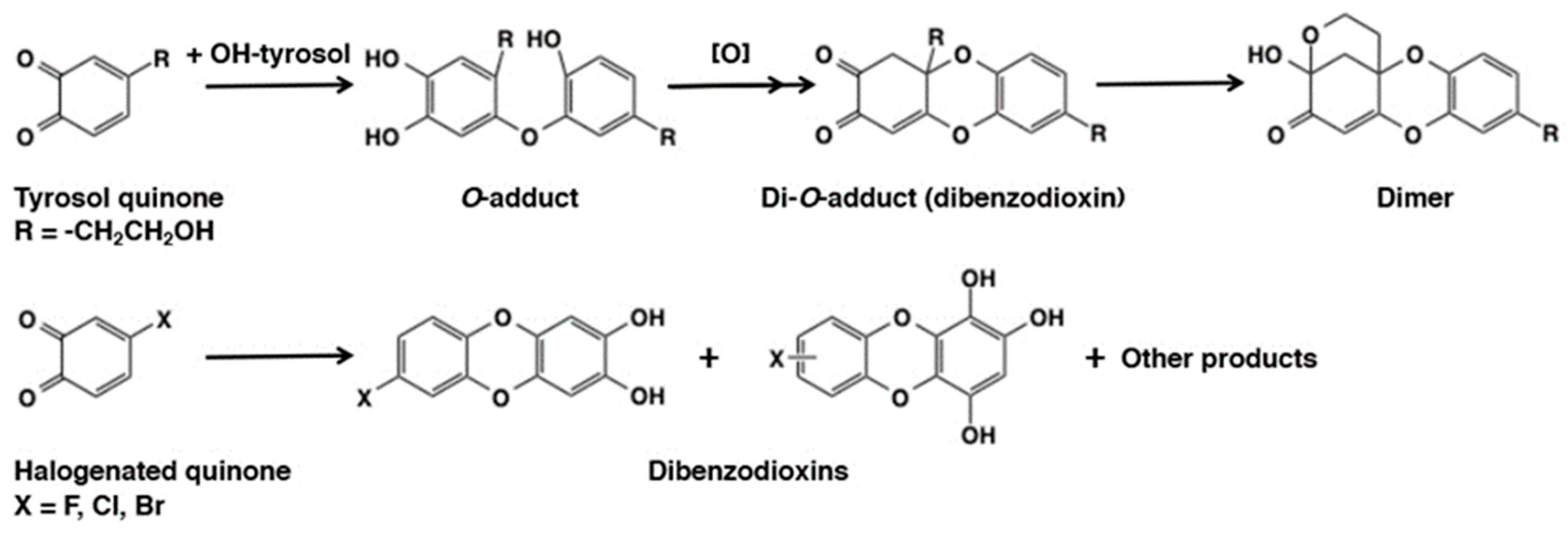
- Vogna, D.; Pezzella, A.; Panzella, L.; Napolitano, A.; d’Ischia, M. Oxidative chemistry of hydroxytyrosol: Isolation and characterization of novel methanooxocinobenzodioxinone derivatives. Tetrahedron Lett. 2003, 44, 8239–8292. [Google Scholar] [CrossRef]
- Stratford, M.R.L.; Riley, R.A.; Ramsden, C.A. Rapid halogen substitution and dibenzodioxin formation during tyrosinase-catalyzed oxidation of 4-halocatechols. Chem. Res. Toxicol. 2011, 24, 350–356. [Google Scholar] [CrossRef]
- Jane, S.M.; Mu, D.; Wemmer, D.; Smith, A.I.; Kaur, S.; Malby, D.; Burlingame, A.I.; Klinman, J.P. A new redox cofactor in eukaryotic enzymes: 6-Hydroxydopa at the active site of bovine amine oxidase. Science 1990, 248, 981–987. [Google Scholar] [CrossRef]
- Napolitano, A.; Crescenzi, O.; Pezzella, A.; Prota, G. Generation of the neurotoxin 6-hydroxydopamine by peroxidase/H2O2 oxidation of dopamine. J. Med. Chem. 1995, 38, 917–922. [Google Scholar] [CrossRef]
- De Lucia, M.; Panzella, L.; Pezzella, A.; Napolitano, A.; d’Ischia, M. Oxidative chemistry of the natural antioxidant hydroxytyrosol: Hydrogen peroxide-dependent hydroxylation and hydroquinone/o-quinone coupling pathway. Tetrahedron 2006, 62, 1273–1278. [Google Scholar] [CrossRef]
- Nishigori, C.; Aoyama, Y.; Ito, A.; Suzuki, K.; Suzuki, T.; Tanemura, A.; Ito, M.; Katayama, I.; Oiso, N.; Kagohashi, Y.; et al. Guide for medical professionals (i.e., dermatologists) for the management of Rhododenol-induced leukoderma. J. Dermatol. 2015, 42, 113–128. [Google Scholar] [CrossRef]
- Sasaki, M.; Kondo, M.; Sato, K.; Umeda, M.; Kawabata, K.; Takahashi, Y.; Suzuki, T.; Matsunaga, K.; Inoue, S. Rhododendrol, a depigmentation-inducing phenolic compound, exerts melanocyte cytotoxicity via a tyrosinase-dependent mechanism. Pigment Cell Melanoma Res. 2014, 27, 754–763. [Google Scholar] [CrossRef]
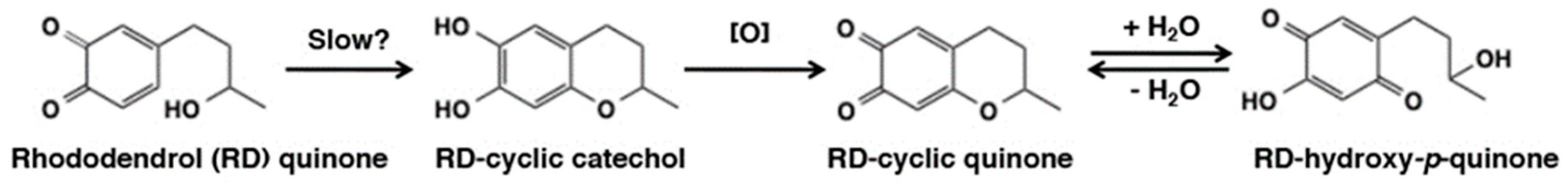
- Ito, S.; Ojika, M.; Yamashita, T.; Wakamatsu, K. Tyrosinase-catalyzed oxidation of rhododendrol produces 2-methylchromane-6,7-dione, the putative ultimate toxic metabolite: Implications for melanocyte toxicity. Pigment Cell Melanoma Res. 2014, 27, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. Biochemical mechanism of rhododendrol-induced leukoderma. Int. J. Mol. Sci. 2018, 19, 552. [Google Scholar]
- Ito, S.; Gerwat, W.; Kolbe, L.; Yamashita, T.; Ojika, M.; Wakamatsu, K. Human tyrosinase is able to oxidize both enantiomers of rhododendrol. Pigment Cell Melanoma Res. 2014, 27, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Kishida, R.; Kasai, H.; Aspera, S.M.; Arevalo, R.L.; Nakanishi, H. Density functional theory-based First Principles calculations of rhododendrol-quinone reactions: Preference to thiol binding over cyclization. J. Phys. Soc. Jpn. 2017, 86, 024804. [Google Scholar] [CrossRef]
- Lee, N.; Lee, S.H.; Baek, K.; Kim, B.G. Heterologous expression of tyrosinase (MelC2) from Streptomyces avermitilis MA4680 in E. coli and its application for ortho-hydroxylation of resveratrol to produce piceatannol. Appl. Microbiol. Biotechnol. 2015, 99, 7915–7924. [Google Scholar] [CrossRef]
- Marín-Zamora, M.E.; Rojas-Melgarejo, F.; García-Cánovas, F.; García-Ruit, P.A. Production of o-diphenols by immobilized mushroom tyrosinase. J. Biotechnol. 2009, 139, 163–168. [Google Scholar] [CrossRef]
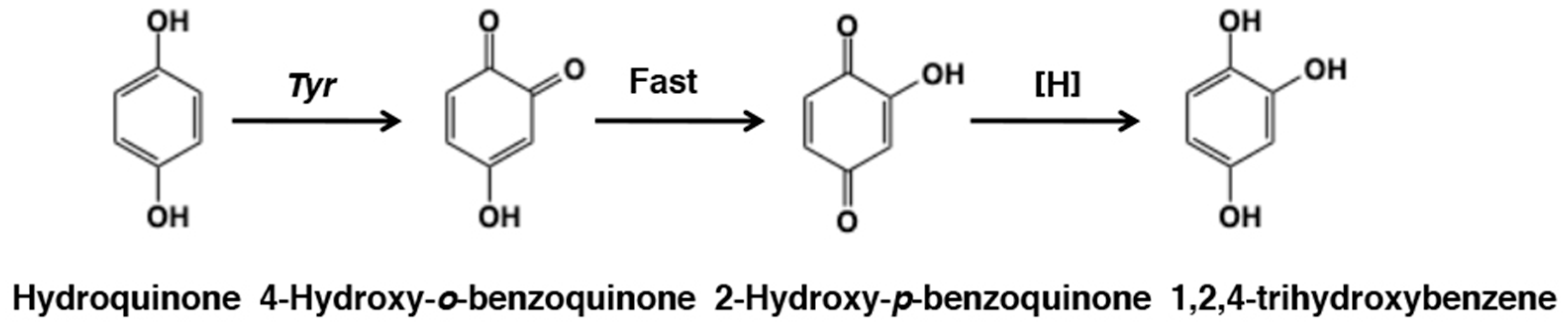
- Palumbo, A.; d’Ischia, M.; Misuraca, G.; Prota, G. Mechanism of inhibition of melanogenesis by hydroquinone. Biochim. Biophys. Acta 1991, 1073, 85–90. [Google Scholar] [CrossRef]
- Solano, F.; Briganti, S.; Picardo, M.; Ghanem, G. Hypopigmenting agents: An updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006, 19, 550–571. [Google Scholar] [CrossRef]
- Jimbow, K.; Obata, H.; Pathak, M.A.; Fitzpatrick, T.B. Mechanisms of depigmentation by hydroquinone. J. Investig. Dermatol. 1974, 62, 436–449. [Google Scholar] [CrossRef] [Green Version]
- Del García-Molina, M.M.; Muñoz Muñoz, J.L.; Martinez-Ortiz, F.; Martinez, J.R.; García-Ruiz, P.A.; Rodriguez-López, J.N.; García-Cánovas, F. Tyrosinase-catalyzed hydroxylation of hydroquinone, a depigmenting agent, to hydroxyhydroquinone: A kinetic study. Bioorg. Med. Chem. 2014, 22, 3360–3369. [Google Scholar] [CrossRef]
- Ramsden, C.A.; Riley, P.A. Mechanistic aspects of the tyrosinase oxidation of hydroquinone. Bioorg. Med. Chem. Lett. 2014, 24, 2463–2464. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Wu, H.W.; Hu, L.Q. Mechanism of isomerization of 4-propyl-o-quinone to its tautomeric p-quinone methide. Chem. Res. Toxicol. 1996, 9, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M. Unified mechanism for sclerotization of insect cuticle. Adv. Insect Physiol. 1998, 27, 229–334. [Google Scholar]
- Sugumaran, M.; Semensi, V.; Dali, H.; Nellaiappan, K. Oxidation of 3,4-dihydroxybenzyl alcohol: A sclerotizing precursor for cockroach ootheca. Arch. Insect Biochem. Physiol. 1991, 16, 31–44. [Google Scholar] [CrossRef]
- Sugumaran, M. Oxidation of 3,4-dihydroxybenzylamine affords 3,4-dihydroxy benzaldehyde via the quinone methide intermediate. Pigment Cell. Res. 1995, 8, 250–254. [Google Scholar] [CrossRef]
- Sugumaran, M.; Tan, S.; Sun, H.L. Tyrosinase catalyzed oxidation of 3,4-dihydroxyphenylglycine. Arch. Biochem. Biophys. 1996, 329, 175–180. [Google Scholar] [CrossRef]
- Brooks, S.J.; Nikodinovic, J.; Martin, L.; Doyle, E.M.; O’Sullivan, T.; Guiry, P.J.; Coulombel, L.; Li, Z.; O’Connor, K.E. Production of a chiral alcohol, 1-(3,4-dihydroxyphenyl)ethanol, by mushroom tyrosinase. Biotechnol. Lett. 2013, 35, 779–783. [Google Scholar] [CrossRef]
- Senoh, S.; Witkop, B. Non-enzymatic conversions of dopamine to norepinephrine and trihydroxyphenethylamines. J. Am. Chem. Soc. 1959, 81, 6222–6231. [Google Scholar] [CrossRef]
- Kaufman, S.; Bridgers, W.F.; Eisenberg, F.; Friedman, F. The source of oxygen in the phenylalanine hydroxylase and the dopamine-β-hydroxylase catalyzed reactions. Biochem. Biophys. Res. Commun. 1962, 9, 497–502. [Google Scholar] [CrossRef]
- Sugumaran, M.; Lipke, H. Quinone methide formation from 4-alkylcatechols. A novel reaction catalyzed by cuticular polyphenol oxidase. FEBS Lett. 1983, 155, 65–68. [Google Scholar] [CrossRef] [Green Version]
- Saul, S.J.; Sugumaran, M. 4-Alkyl-o-quinone/2-hydroxy-p-quinone methide isomerase from the larval hemolymph of Sarcophaga bullata. I. Purification and characterization of enzyme catalyzed reaction. J. Biol. Chem. 1990, 265, 16992–16999. [Google Scholar] [PubMed]
- Wakamatsu, K.; Tanaka, H.; Tabuchi, K.; Ojika, M.; Zucca, F.A.; Zecca, L.; Ito, S. Reduction of the nitro group to amine by hydroiodic acid to synthesize o-aminophenol derivatives as putative degradative makers of neuromelanin. Molecules 2014, 19, 8039–8050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugumaran, M.; Semensi, V.; Saul, S.J. On the oxidation of 3,4-dihydroxyphenyl alcohol and 3,4-dihydroxyphenyl glycol by cuticular enzyme(s) from Sarcophaga bullata. Arch. Insect Biochem. Physiol. 1989, 10, 13–27. [Google Scholar] [CrossRef]
- Sugumaran, M.; Dali, H.; Semensi, V. Mechanistic studies on tyrosinase-catalyzed oxidative decarboxylation of 3,4-dihydroxymandelic acid. Biochem. J. 1992, 281, 353–357. [Google Scholar] [CrossRef] [Green Version]
- Czapla, T.H.; Claeys, M.R.; Morgan, T.D.; Kramer, K.J.; Hopkins, T.L.; Hawley, M.D. Oxidative decarboxylation of 3,4-dihydroxymandelic acid to 3,4-dihydroxybenzaldehyde: Electrochemical and HPLC analysis of the reaction mechanism. Biochim. Biophys. Acta 1991, 1077, 400–406. [Google Scholar] [CrossRef]
- Mefford, I.N.; Kincl, L.; Dykstra, K.H.; Simpson, J.T.; Markey, S.P.; Diez, S.; Wighman, R.M. Facile oxidative decarboxylation of 3,4-dihydroxyphenylacetic acid catalyzed by copper and manganese ions. Biochim. Biophys. Acta 1996, 1290, 224–230. [Google Scholar] [CrossRef]
- Ito, S.; Yamanaka, Y.; Ojika, M.; Wakamatsu, K. The metabolic fate of ortho-quinones derived from catecholamine metabolites. Int. J. Mol. Sci. 2016, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Dali, H.; Semensi, V. The mechanism of tyrosinase catalyzed oxidative decarboxylation of α-(3,4-dihydroxyphenyl) lactic acid. Biochem. J. 1991, 277, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Umebachi, Y. Papiliochrome, a new pigment group of butterfly. Zool. Sci. 1985, 2, 163–174. [Google Scholar]
- Umebachi, Y. The third way of dopamine. Trends Comp. Biochem. Physiol. 1993, 1, 709–720. [Google Scholar]
- Saul, S.J.; Sugumaran, M. Quinone methide as a reactive intermediate formed during the biosynthesis of papiliochrome II, a yellow wing pigment of papilionid butterflies. FEBS Lett. 1991, 279, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Sugumaran, M.; Semensi, V. Quinone methide as a new intermediate in eumelanin biosynthesis. J. Biol. Chem. 1991, 266, 6073–6078. [Google Scholar] [PubMed]
- Sugumaran, M.; Dali, H.; Semensi, V. Formation of a stable quinone methide during tyrosinase catalyzed oxidation of a-methyldopa methyl ester and its implication in melanin biosynthesis. Bioorg. Chem. 1990, 18, 144–153. [Google Scholar] [CrossRef]
- Sugumaran, M.; Semensi, V.; Dali, H.; Saul, S.J. Nonenzymatic transformations of enzymatically generated N-acetyldopamine quinone and isomeric dihydrocaffeiyl methyl amide quinone. FEBS Lett. 1989, 255, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.W.; Molinski, T.F.; Rzepecki, L.M.; Waite, J.H. Oxidation of peptidyl 3,4-dihydroxyphenylalanine analogues: Implications for the biosynthesis of tunichromes and related oligopeptides. J. Nat. Prod. 1991, 54, 918–922. [Google Scholar] [CrossRef]
- Sugumaran, M.; Ricketts, D. Model sclerotization studies. 3. Cuticular enzyme catalyzed oxidation of peptidyl model tyrosine and dopa derivatives. Arch. Insect Biochem. Physiol. 1995, 28, 17–32. [Google Scholar] [CrossRef]
- Sugumaran, M.; Robinson, W.E. Structure, biosynthesis and possible function of tunichromes and related compounds. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2012, 163, 1–25. [Google Scholar] [CrossRef]
- Saul, S.J.; Sugumaran, M. Characterization of a new enzyme system that desaturates the side chain of N-acetyldopamine. FEBS Lett. 1989, 251, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Saul, S.J.; Sugumaran, M. N-Acetyldopamine quinone methide/1,2-dehydro-N-acetyldopamine tautomerase—A new enzyme involved in sclerotization of insect cuticle. FEBS Lett. 1989, 255, 340–344. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, Y.; Nagano, M.; Futatsuka, M. Occupational leukoderma in workers engaged in 4-(p-hydroxyphenyl)-2-butanone manufacturing. J. Occup. Health 1998, 40, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Hinoshita, M.; Suzuki, E.; Ojika, M.; Wakamatsu, K. Tyrosinase-catalyzed oxidation of the leukoderma-inducing agent Raspberry ketone produces (E)-4-(3-oxo-1-butenyl)-1,2-benzoquinone: Implications for melanocyte toxicity. Chem. Res. Toxicol. 2017, 30, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Abebe, A.; Zheng, D.; Evans, J.; Sugumaran, M. Reexamination of the mechanisms of oxidative transformation of the insect cuticular sclerotizing precursor, 1,2-dehydro-N-acetyldopamine. Insect Biochem. Mol. Biol. 2010, 40, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, D.; Sugumaran, M. 1,2-Dehydro-N-β-alanyldopamine as a new intermediate in insect cuticular sclerotization. J. Biol. Chem. 1994, 269, 22217–22221. [Google Scholar] [PubMed]
- Abebe, A.; Zheng, D.; Evans, J.; Sugumaran, M. Novel post-translational oligomerization of peptidyl dehydrodopa model compound, 1,2-dehydro-N-acetyldopa methyl ester. Bioorg. Chem. 2016, 66, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Sugumaran, M.; Robinson, W.E. The crosslinking and antimicrobial properties of tunichrome. Comp. Biochem. Physiol. B 2008, 151, 110–117. [Google Scholar] [CrossRef]
- Abebe, A.; Kuang, Q.F.; Evans, J.; Robinson, W.E.; Sugumaran, M. Oxidative transformation of a tunichrome model compound provides new insight into the crosslinking and defense reaction of tunichromes. Bioorg. Chem. 2017, 71, 219–229. [Google Scholar] [CrossRef]
- Sugumaran, M. Oxidation chemistry of 1,2-dehydro-N-acetyldopamines: Direct evidence for the formation of 1,2-dehydro-N-acetyldopamine quinone. Arch. Biochem. Biophys. 2000, 378, 404–419. [Google Scholar] [CrossRef]
- Pezzella, A.; Lista, L.; Napolitano, A.; d’Ischia, M. Tyrosinase-catalyzed oxidation of 17β-estradiol: Structure elucidation of the products formed beyond catechol estrogen quinones. Chem. Res. Toxicol. 2005, 18, 1413–1419. [Google Scholar] [CrossRef]
- Kato, T.; Ito, S.; Fujita, K. Tyrosinase-catalyzed binding of 3,4-dihydroxyphenylalanine with proteins through the sulfhydryl group. Biochim. Biophys. Acta 1986, 881, 415–421. [Google Scholar] [CrossRef]
- Ito, S.; Okura, M.; Nakanishi, Y.; Ojika, M.; Wakamatsu, K.; Yamashita, T. Tyrosinase-catalyzed metabolism of rhododendrol (RD) in B16 melanoma cells: Production of RD-pheomelanin and covalent binding with thiol proteins. Pigment Cell Melanoma Res. 2015, 28, 295–306. [Google Scholar] [CrossRef]
- Ito, S.; Nishigaki, A.; Ishii-Osai, Y.; Ojika, M.; Wakamatsu, K.; Yamashita, T.; Tamura, Y.; Ito, A.; Honda, H.; Nakayama, E.; et al. Mechanism of putative neo-antigen formation from N-Propionyl-4-S-cysteaminylphenol, a tyrosinase substrate, in melanoma models. Biochem. Pharmacol. 2012, 84, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Okamura, K.; Kawaguchi, M.; Hozumio, Y.; Aoki, H.; Kunisada, T.; Ito, S.; Wakamatsu, K.; Matsunaga, K.; Suzuki, T. Rhododendrol-induced leucoderma in a mouse model mimicking Japanese skin. J. Dermatol. Sci. 2016, 81, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Kato, T.; Fujita, K. Covalent binding of catechols to proteins through the sulphydryl group. Biochem. Pharmacol. 1988, 37, 1707–1710. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatsu, K. A convenient screening method to differentiate phenolic skin whitening tyrosinase inhibitors from leukoderma-inducing phenols. J. Dermatol. Sci. 2015, 80, 18–24. [Google Scholar] [CrossRef]
- Manini, P.; Napolitano, A.; Westerhof, W.; Riley, P.A.; d’Ischia, M. A reactive ortho-quinone generated by tyrosinase-catalyzed oxidation of the skin depigmenting agent monobenzone: Self-coupling and thiol-conjugation reactions and possible implications for melanocyte toxicity. Chem. Res. Toxicol. 2009, 22, 1398–1405. [Google Scholar] [CrossRef]
- Westerhof, W.; Manini, P.; Napolitano, A.; d’Ischia, M. The haptenation theory of vitiligo and melanoma rejection: A close-up. Exp. Dermatol. 2011, 20, 92–96. [Google Scholar] [CrossRef]
- Van den Boorn, J.G.; Picavet, D.I.; van Swieten, P.F.; van Veen, H.A.; Konijnenberg, D.; van Veelen, P.A.; van Capel, T.; de Jong, E.C.; Reits, E.A.; Drijfhout, J.W.; et al. Skin-depigmenting agent monobenzone induces potent T-cell autoimmunity toward pigmented cells by tyrosinase haptenation and melanosome autophagy. J. Investig. Dermatol. 2011, 131, 1240–1251. [Google Scholar] [CrossRef] [Green Version]
- Jimbow, K.; Miura, T.; Ito, S.; Ishikawa, K. Phenolic melanin precursors provide a rational approach to the design of antitumor agents for melanoma. Pigment Cell Res. 1989, 2, 34–39. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Nakao, K.; Tanaka, H.; Kitahori, Y.; Tanaka, Y.; Ojika, M.; Ito, S. The oxidative pathway to dopamine-protein conjugates and their pro-oxidant activities: Implications for the neurodegeneration of Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 2575. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.S.; Kopin, I.J.; Sharabi, Y. Catecholamines autotoxicity. Implications for pharmacology and therapeutics of Parkinson disease and related disorders. Pharmacol. Ther. 2014, 144, 268–282. [Google Scholar] [CrossRef] [Green Version]
- Rees, J.N.; Florang, V.R.; Eckert, L.L.; Doorn, J.A. Protein modification of 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolites, is dependent on both the aldehyde and the catechol. Chem. Res. Toxicol. 2009, 22, 1256–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinsmaa, Y.; Isonaka, R.; Sharabi, Y.; Goldstein, D.S. 3,4-Dihydroxyphenylacetaldehyde is more efficient than dopamine in oligomerizing and quinonizing α-synuclein. J. Pharmacol. Exp. Ther. 2020, 372, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.; Saeed, M.; Yang, L.; Beseler, C.; Rogan, E.; Cavalieri, E.L. Formation of dopamine quinone-DNA adducts and their potential role in the etiology of Parkinson’s disease. IUBMB Life 2011, 63, 1087–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahid, M.; Kohli, E.; Saeed, M.; Rogan, E.; Cavalieri, E. The greater reactivity of estradiol-3,4-quinone vs estradiol-2,3-quinone with DNA in the formation of depurinating adducts: Implications for tumor-initiating activity. Chem. Res. Toxicol. 2006, 19, 164–172. [Google Scholar] [CrossRef]
- Prota, G. Progress in the chemistry of melanins and related metabolites. Med. Res. Rev. 1988, 8, 525–556. [Google Scholar] [CrossRef]
- Prota, G. Melanins and Melanogenesis; Academic Press: San Diego, CA, USA, 1992; pp. 1–290. [Google Scholar]
- Prota, G. The chemistry of melanins and melanogenesis. Fortschr. Chem. Org. Nat. 1995, 64, 93–148. [Google Scholar]
- D’Ischia, M.; Wakamatsu, K.; Napolitano, A.; Briganti, S.; Garcia-Borron, J.-C.; Kovacs, D.; Meredith, P.; Pezzella, A.; Picardo, M.; Sarna, T.; et al. Melanins and melanogenesis: Methods, standards, protocols. Pigment Cell Melanoma Res. 2013, 26, 616–633. [Google Scholar] [CrossRef]
- D’Ischia, M.; Wakamatsu, K.; Cicoira, F.; di Mauro, E.; Garcia-Borron, J.-C.; Commo, S.; Galván, I.; Ghanem, G.; Koike, K.; Meredith, P.; et al. Melanins and melanogenesis: From pigment cells to human health and technological applications. Pigment Cell Melanoma Res. 2015, 28, 520–544. [Google Scholar] [CrossRef] [Green Version]
- Micillo, R.; Panzella, L.; Koike, K.; Monfrecola, G.; Napolitano, A.; d’Ischia, M. “Fifty shades” of black and red or how carboxyl groups fine tune eumelanin and pheomelanin properties. Int. J. Mol. Sci. 2016, 17, 746. [Google Scholar] [CrossRef]
- Sugumaran, M.; Barek, H. Critical analysis of the melanogenic pathway in insects and higher animals. Int. J. Mol. Sci. 2016, 17, 1753. [Google Scholar] [CrossRef] [Green Version]
- Raper, H.S. The tyrosinase-tyrosine reaction. VI. Production from tyrosine of 5,6-dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic acid—The precursors of melanin. Biochem. J. 1927, 21, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Mason, H.S. The chemistry of melanin. III. Mechanism of the oxidation of dihydroxyphenylalanine by tyrosinase. J. Biol. Chem. 1948, 172, 83–99. [Google Scholar] [PubMed]
- Thomson, R.H. The pigments of reddish hair and feathers. Angew. Chem. Int. Ed. Engl. 1974, 13, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. Quantitative analysis of eumelanin and pheomelanin in humans, mice, and other animals: A comparative review. Pigment Cell Res. 2003, 16, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.D.; Peles, D.; Wakamatsu, K.; Ito, S. Current challenges in understanding melanogenesis: Bridging chemistry, biological control, morphology, and function. Pigment Cell Melanoma Res. 2009, 22, 563–579. [Google Scholar] [CrossRef]
- Land, E.J.; Ramsden, C.A.; Riley, P.A. Pulse radiolysis studies of ortho-quinone chemistry relevant to melanogenesis. J. Photochem. Photobiol. B Biol. 2001, 64, 123–135. [Google Scholar] [CrossRef]
- Land, E.J.; Riley, P.A. Spontaneous redox reactions of dopaquinone and the balance between the eumelanic and phaeomelanic pathways. Pigment Cell Res. 2000, 13, 273–277. [Google Scholar] [CrossRef]
- Thompson, A.; Land, E.J.; Chedekel, M.R.; Subbarao, K.V.; Truscott, T.G. A pulse radiolysis investigation of the oxidation of the melanin precursors 3,4-dihydroxyphenylalanine (dopa) and the cysteinyldopas. Biochim. Biophys. Acta 1985, 843, 49–57. [Google Scholar] [CrossRef]
- Land, E.J.; Ito, S.; Wakamatsu, K.; Riley, P.A. Rate constants for the first two chemical steps of eumelanogenesis. Pigment Cell Res. 2003, 13, 487–493. [Google Scholar] [CrossRef]
- Palumbo, A.; d’Ischia, M.; Misuraca, G.; Prota, G. Effect of metal ions on the rearrangement of dopachrome. Biochim. Biophys. Acta 1987, 925, 203–209. [Google Scholar] [CrossRef]
- Jackson, I.J.; Chambers, D.M.; Tsukamoto, K.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Hearing, V.J. A second tyrosinase-related protein, TRP-2, maps to and is mutated at the mouse slaty locus. EMBO J. 1992, 11, 527–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, K.; Jackson, I.J.; Urabe, K.; Montaque, P.M.; Hearing, V.J. A second tyrosinase-related protein, TRP-2, is a melanogenic enzyme termed DOPAchrome tautomerase. EMBO J. 1992, 11, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Ito, S. Preparation of eumelanin-related metabolites, 5,6-dihydroxyindole, 5,6-dihydroxyindole-2-carboxylic acid, and their O-methyl derivatives. Anal. Biochem. 1988, 170, 335–340. [Google Scholar] [CrossRef]
- Ito, S.; Suzuki, N.; Takebayashi, S.; Commo, S.; Wakamatsu, K. Neutral pH and copper ions promote eumelanogenesis after the dopachrome stage. Pigment Cell Melanoma Res. 2013, 26, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Edge, R.; d’Ischia, M.; Land, E.J.; Napolitano, A.; Navaratnam, S.; Panzella, L.; Pezzella, A.; Ramsden, C.A.; Riley, P.A. Dopaquinone redox exchange with dihydroxyindole and dihydroxyindole-2-carboxylic acid. Pigment Cell Res. 2006, 19, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Olivares, C.; Jiménez-Cervantes, C.; Lozano, J.A.; Solano, F.; Gacía-Borrón, J.C. The 5,6-dihydroxyindole-2-carboxylic acid (DHICA) oxidase activity of human tyrosinase. Biochem. J. 2001, 354, 131–139. [Google Scholar] [CrossRef]
- Jiménez-Cervantes, C.; Solano, F.; Kobayashi, T.; Urabe, K.; Hearing, V.J.; Lozano, J.A.; García-Borrón, J.C. A new enzymatic function in the melanogenic pathway. The 5,6-dihydroxyindole-2-carboxylic acid oxidase activity of tyrosinase-related protein-1 (TRP1). J. Biol. Chem. 1994, 269, 17993–18000. [Google Scholar]
- Kobayashi, T.; Urabe, K.; Winder, A.J.; Jiménez -Cervantes, C.; Imokawa, G.; Brewington, T.; Solano, F.; García-Borrón, J.C.; Hearing, V.J. Tyrosinase related protein 1 (TRP1) functions as a DHICA oxidase in melanin biosynthesis. EMBO J. 1994, 13, 5818–5825. [Google Scholar] [CrossRef]
- Napolitano, A.; Crescenzi, O.; Prota, G. Copolymerization of 5,6-dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic acid in melanogenesis: Isolation of a cross-coupling product. Tetrahedron Lett. 1993, 34, 885–888. [Google Scholar] [CrossRef]
- Panzella, L.; Pezzella, A.; Napolitano, A.; d’Ischia, M. The first 5,6-dihydroxyindole tetramer by oxidation of 5,5′,6,6′-tetrahydroxy-2,4′-biindolyl and an unexpected issue of positional reactivity en route to eumelanin-related polymers. Org. Lett. 2007, 9, 1411–1414. [Google Scholar] [CrossRef]
- Pezzella, A.; Vogna, D.; Prota, G. Synthesis of optically active tetrameric melanin intermediates by oxidation of the melanogenic precursor 5,6-dihydroxyindole-2-carboxylic acid under biomimetic conditions. Tetrahedron Asymmetry 2003, 14, 1133–1140. [Google Scholar] [CrossRef]
- Panzella, L.; Ebato, A.; Napolitano, A.; Koike, K. The late stages of melanogenesis: Exploring the chemical facets and the application opportunities. Int. J. Mol. Sci. 2018, 19, 1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prota, G.; Crescenzi, S.; Misuraca, G.; Nicolaus, R.A. New intermediates of pheomelanogenesis in vitro. Experientia 1970, 26, 1058–1059. [Google Scholar] [CrossRef] [PubMed]
- Costantini, C.; Crescenzi, C.; Prota, G.; Palumbo, A. New intermediates of phaeomelanogenesis in vitro beyond the 1,4-benzothiazine stage. Tetrahedron 1990, 46, 6831–6838. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Ohtara, K.; Ito, S. Chemical analysis of late stages of pheomelanogenesis: Conversion of dihydrobenzothiazine to a benzothiazole structure. Pigment Cell Melanoma Res. 2009, 22, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.; Di Donato, P.; Prota, G.; Land, E.J. Transient quinonimines and 1,4-benzothiazines of pheomelanogenesis: New pulse radiolytic and spectrophotometric evidence. Free Radic. Biol. Med. 1999, 27, 521–528. [Google Scholar] [CrossRef]
- Napolitano, A.; Costantini, C.; Crescenzi, O.; Prota, G. Characterization of 1,4-benzothiazine intermediates in the oxidative conversion of 5-S-cysteinyldopa to pheomelanins. Tetrahedron Lett. 1994, 35, 6365–6368. [Google Scholar] [CrossRef]
- Di Donato, P.; Napolitano, A. 1,4-Benzothiazines as key intermediates in the biosynthesis of red hair pigment pheomelanins. Pigment Cell Res. 2003, 16, 532–539. [Google Scholar] [CrossRef]
- Napolitano, A.; De Lucia, M.; Panzella, L.; d’Ischia, M. The “benzothiazine” chromophore of pheomelanins: A reassessment. Photochem. Photobiol. 2008, 84, 593–599. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Ito, S.; Rees, J.L. The usefulness of 4-amino-3-hydroxyphenylalanine as a specific marker of pheomelanin. Pigment Cell Res. 2002, 15, 225–232. [Google Scholar] [CrossRef]
- Ito, S.; Nakanishi, Y.; Valenzuela, R.K.; Brilliant, M.H.; Kolbe, L.; Wakamatsu, K. Usefulness of alkaline hydrogen peroxide oxidation to analyze eumelanin and pheomelanin in various tissue samples: Application to chemical analysis of human hair melanins. Pigment Cell Melanoma Res. 2011, 24, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Ancas, J.; Tobin, J.; Hoogdujin, M.J.; Smit, N.P.; Wakamatsu, K.; Thody, A.J. Melanosomal pH controls rate of melanogenesis, eumelanin /pheomelanin ratio and melanosome maturation in melanocytes and melanoma cells. Exp. Cell Res. 2001, 268, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Fuller, B.B.; Spaulding, D.T.; Smith, D.R. Regulation of the catalytic activity of preexisting tyrosinase in black and Caucasian human melanocyte cell cultures. Exp. Cell Res. 2001, 262, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Nagao, A.; Watanabe, M.; Nakao, K.; Ito, S. Pheomelanogenesis is promoted at a weakly acidic pH. Pigment Cell Melanoma Res. 2017, 30, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, K.; Tabuchi, K.; Ojika, M.; Zucca, F.A.; Zecca, L.; Ito, S. Norepinephrine and its metabolites are involved in the synthesis of neuromelanin derived from the locus coeruleus. J. Neurochem. 2015, 135, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Peñuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, M.; Garcia-Carmona, F.; Garcia-Canovas, F.; Iborra, J.L.; Lozano, J.A.; Martinez, F. Chemical intermediates in dopamine oxidation by tyrosinase, and kinetic studies of the process. Arch. Biochem. Biophys. 1984, 235, 438–448. [Google Scholar] [CrossRef]
- Jimenez, M.; Garcia-Canovas, F.; Garcia-Carmona, F.; Lozano, J.A.; Iborra, J.L. Kinetic study and intermediates identification of noradrenaline oxidation by tyrosinase. Biochem. Pharmacol. 1984, 33, 3689–3697. [Google Scholar] [CrossRef]
- Liebscher, J. Chemistry of polydopamine—Scope, variation, and limitation. Eur. J. Org. Chem. 2019, 31–32, 4976–4994. [Google Scholar] [CrossRef]
- Hauser, D.; Septiadi, D.; Turner, J.; Petri-Fink, A.; Rothen-Rutishauser, B. From bioinspired glue to medicine: Polydopamine as a biomedical material. Materials 2020, 13, 1730. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, D.R.; Miller, D.J.; Freeman, R.D.; Paul, D.R.; Bielawski, C.W. Elucidating the structure of poly(dopamine). Langmuir 2012, 28, 6428–6435. [Google Scholar] [CrossRef]
- Alfieri, M.L.; Micillo, R.; Panzella, L.; Crescenzi, C.; Oscurato, S.L.; Maddalena, P.; Napolitano, A.; Ball, V.; d’Ischia, M. Structural basis of polydopamine film formation: Probing 5,6-dihydroxyindole-based eumelanin type units and the porphyrin issue. ACS Appl. Mater. Interfaces 2018, 10, 7670–7680. [Google Scholar] [CrossRef]
- Terland, O.; Almås, B.; Flatmark, T.; Andersson, K.K.; Sørlie, M. One-electron oxidation of catecholamines generates free radicals with an in vitro toxicity correlating with their lifetime. Free Radic. Biol. Med. 2006, 41, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Manini, P.; Panzella, L.; Napolitano, A.; d’Ischia, M. Oxidation chemistry of norepinephrine: Partitioning of the o-quinone between competing cyclization and chain breakdown pathways and their roles in melanin formation. Chem. Res. Toxicol. 2007, 20, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, J. Enzymic oxidation of epinephrine to adrenochrome by the salivary gland. Biochim. Biophys. Acta 1964, 85, 247–254. [Google Scholar] [CrossRef]
- Behonick, G.S.; Novak, M.J.; Nealley, E.W.; Baskin, S.I. Toxicology update: The cardiotoxicity of the oxidative stress metabolites of catecholamines (aminochromes). J. Appl. Toxicol. 2001, 21, S15–S22. [Google Scholar] [CrossRef]
- Remiāo, F.; Mihazes, N.; Borges, F.; Carvalho, F.; Bastos, M.; Lemos-Amado, F.; Domingues, P.; Ferrer-Correia, A. Synthesis and analysis of aminochromes by HPLC-photodiode array. Adrenochrome evaluation in rat blood. Biomed. Chromatogr. 2003, 17, 6–13. [Google Scholar] [CrossRef]
- Giulivi, C.; Cadenas, E. Oxidation of adrenaline by ferrylmyoglobin. Free Radic. Biol. Med. 1998, 25, 175–183. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, S.; Sugumaran, M.; Wakamatsu, K. Chemical Reactivities of ortho-Quinones Produced in Living Organisms: Fate of Quinonoid Products Formed by Tyrosinase and Phenoloxidase Action on Phenols and Catechols. Int. J. Mol. Sci. 2020, 21, 6080. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176080
Ito S, Sugumaran M, Wakamatsu K. Chemical Reactivities of ortho-Quinones Produced in Living Organisms: Fate of Quinonoid Products Formed by Tyrosinase and Phenoloxidase Action on Phenols and Catechols. International Journal of Molecular Sciences. 2020; 21(17):6080. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176080
Chicago/Turabian StyleIto, Shosuke, Manickam Sugumaran, and Kazumasa Wakamatsu. 2020. "Chemical Reactivities of ortho-Quinones Produced in Living Organisms: Fate of Quinonoid Products Formed by Tyrosinase and Phenoloxidase Action on Phenols and Catechols" International Journal of Molecular Sciences 21, no. 17: 6080. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176080