Gut Microbiota and Colon Cancer: A Role for Bacterial Protein Toxins?



Abstract
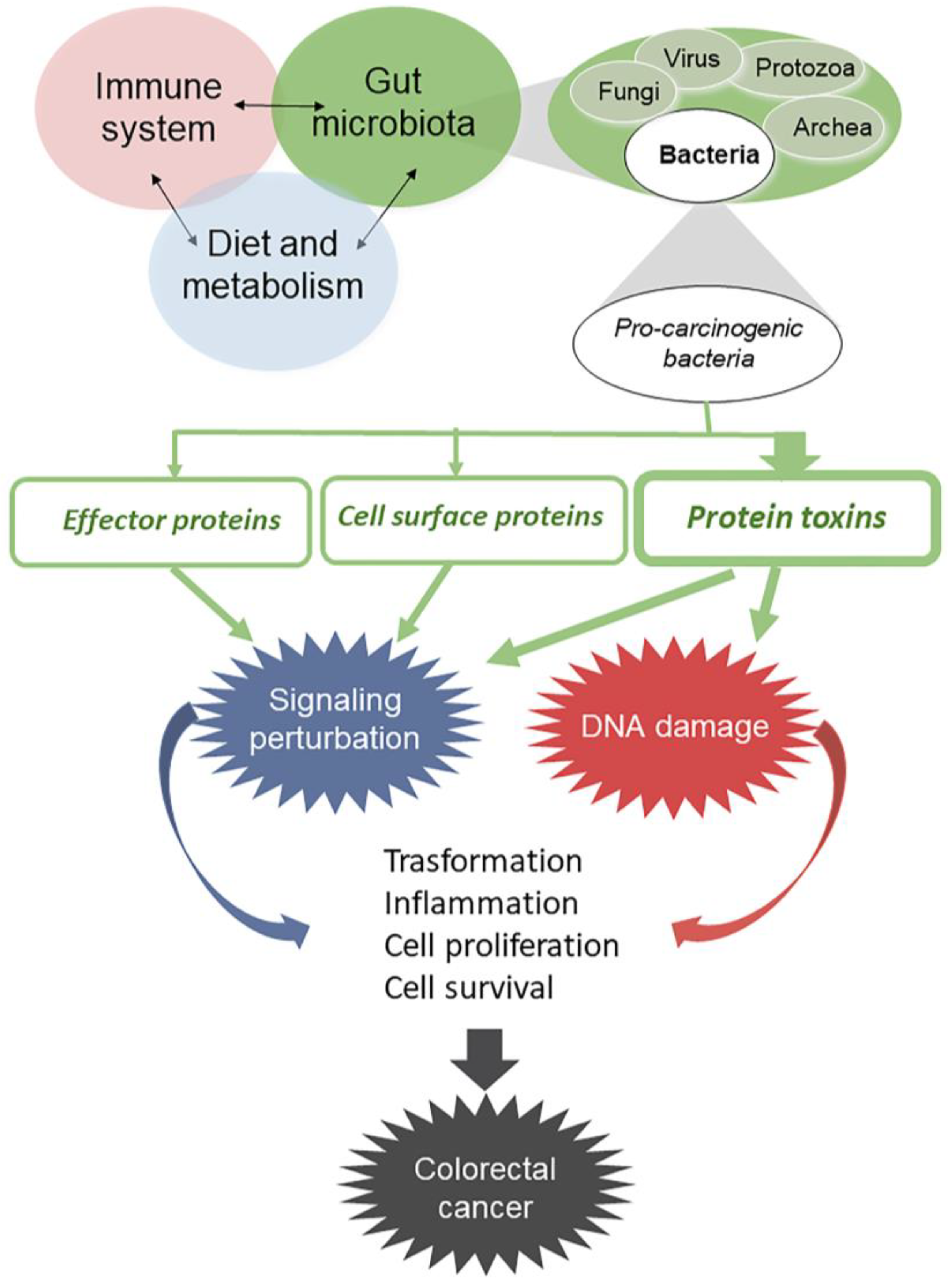
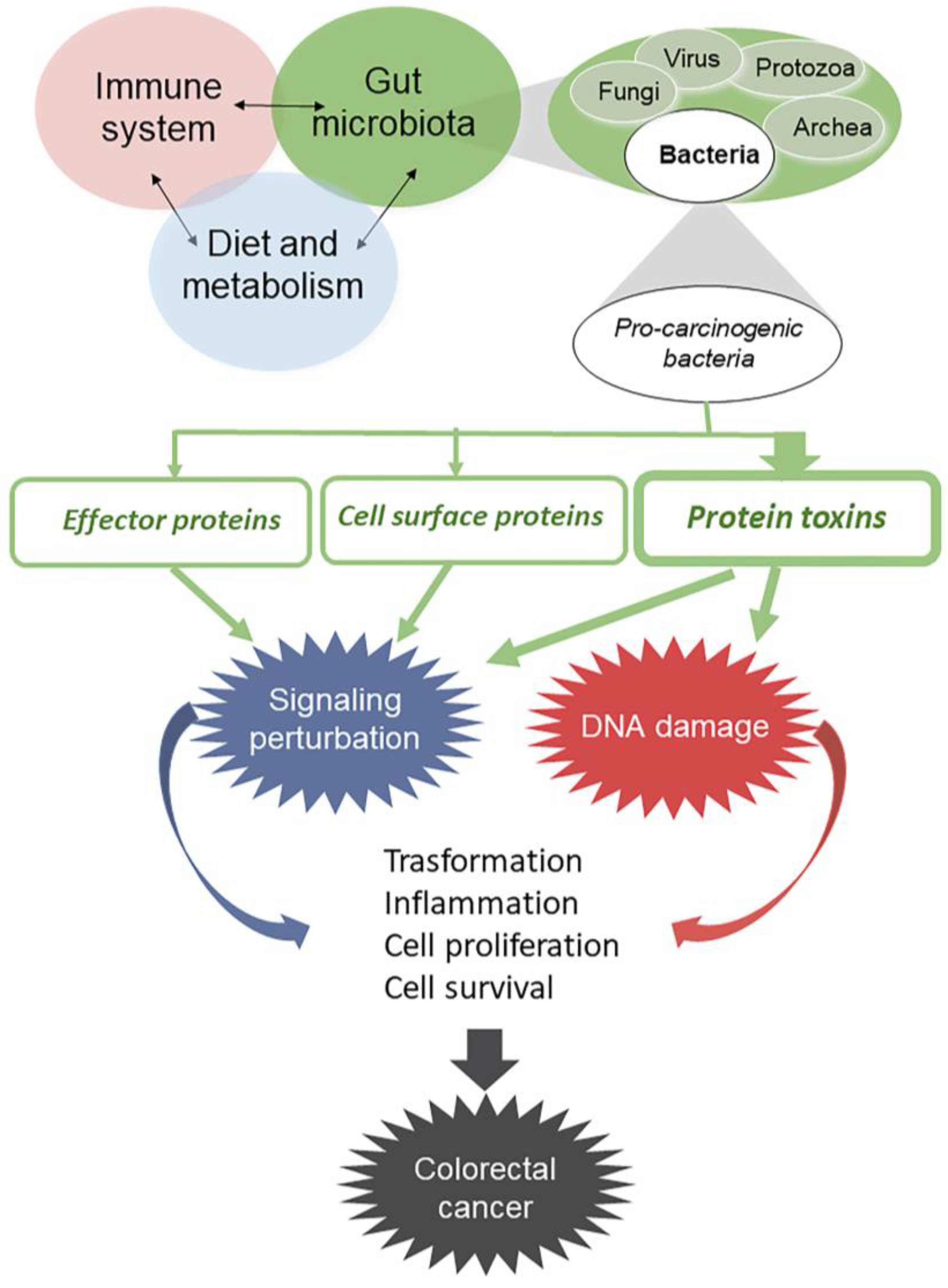
:1. Introduction
2. Bacterial Protein Toxins Causing Genome Instability
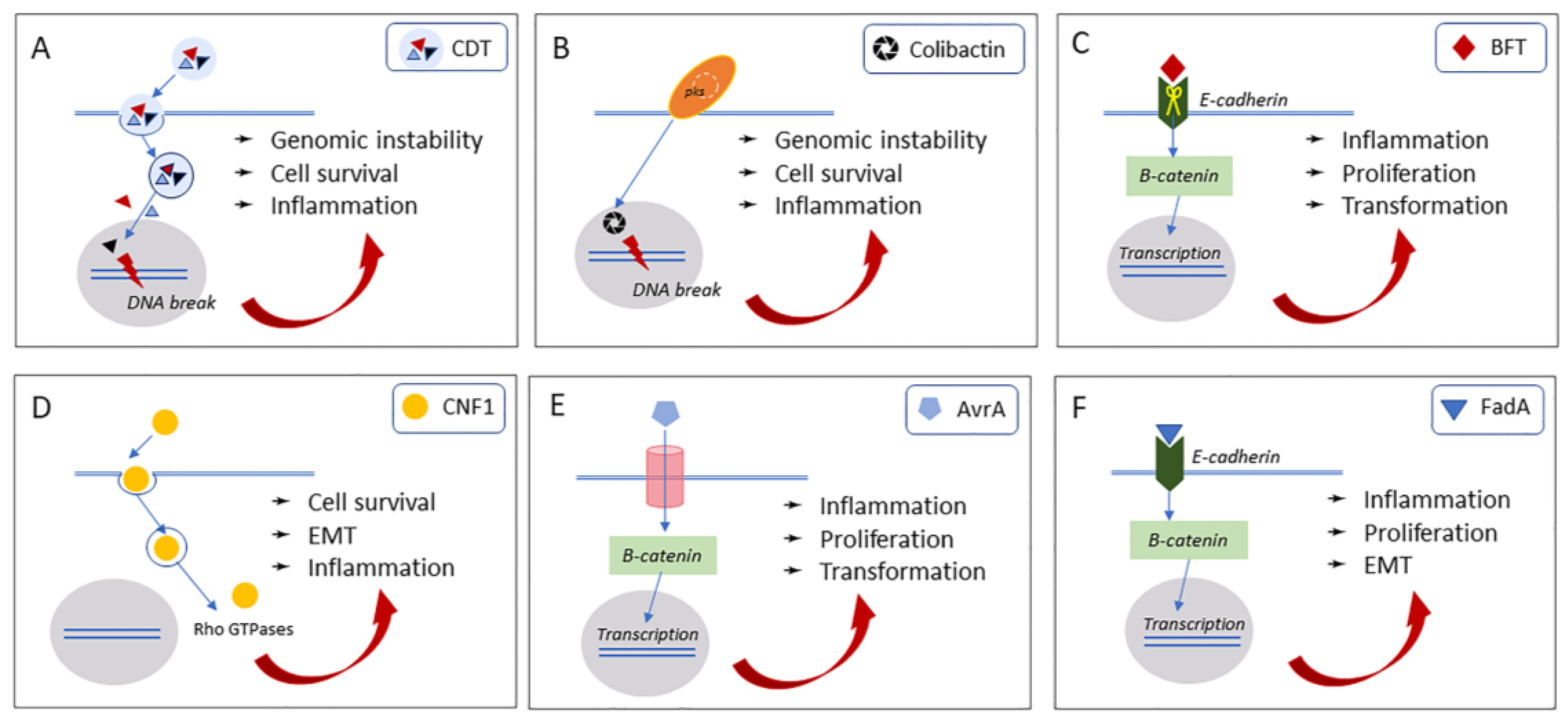
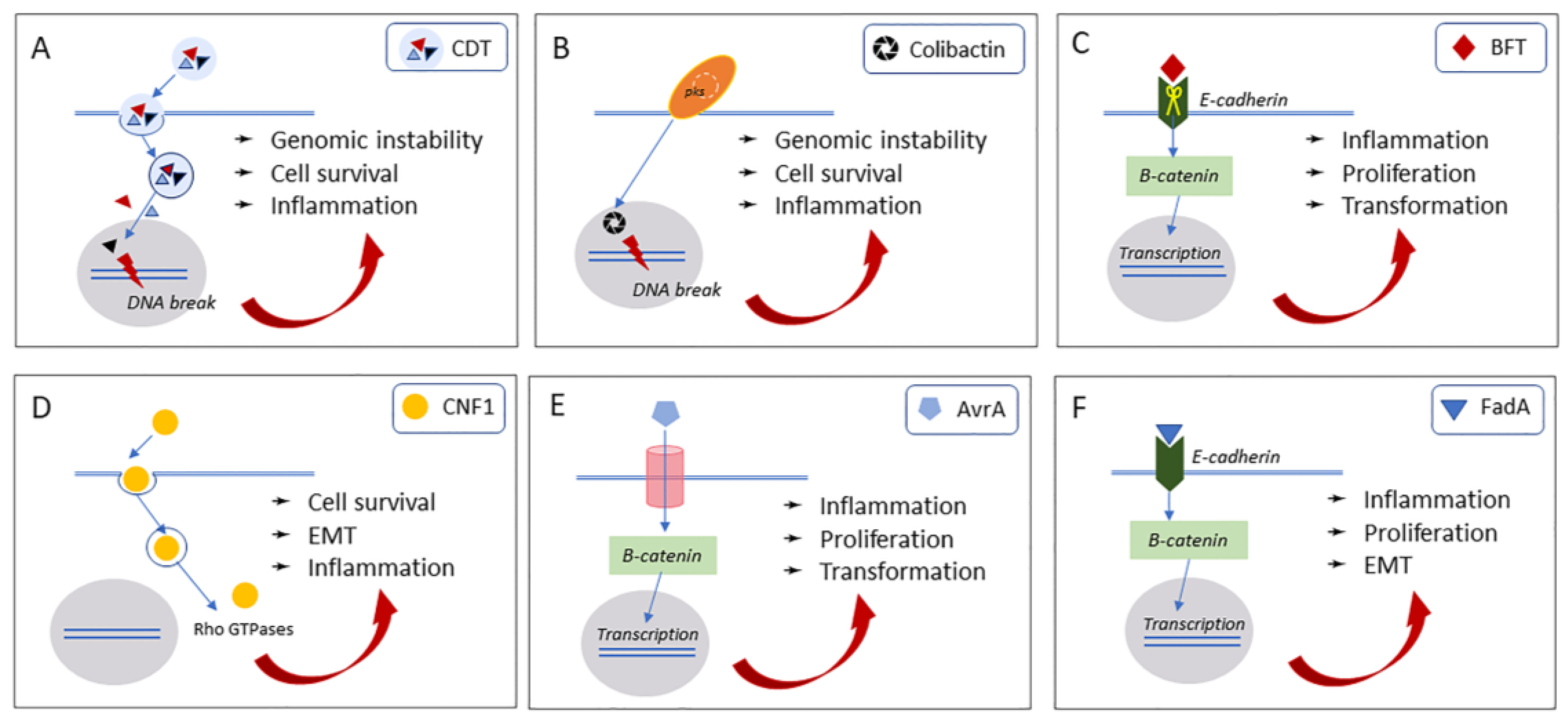
2.1. Cytolethal Distending Toxin (Cdt)
2.2. Colibactin
3. Bacterial Protein Toxins Causing Cell Signaling Alterations
3.1. Bacteroides Fragilis Toxin (BFT)
3.2. Cytotoxic Necrotizing Factor 1 (CNF1)
4. Other Bacterial Factors That May Influence Host Cell Transformation
4.1. Avirulence Protein A (AvrA)
4.2. Fusobacterium Nucleatum Adhesin A (FadA)
5. Bacterial Protein Toxins in Cancer Therapy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | ATM-and Rad3 related |
| APC | Adenomatous polyposis coli |
| AvrA | Avirulence protein A |
| BFT | Bacteroides fragilis toxin |
| CNF1 | Cytotoxic necrotizing factor 1 |
| CoPEC | Colibactin-producing Escherichia coli bacteria |
| CTCL | Cutaneous T-cell lymphoma |
| DT | Diphtheria toxin |
| EMT | Epithelial-to-mesenchymal |
| ETBF | Enterotoxigenic Bacteroides fragilis |
| FadA | Fusobacterium nucleatum adhesin A |
| FAP | Familial adenomatous polyposis |
| HCECs | Human colon epithelial cell lines |
| IT | Immunotoxin |
| KRAS | Kirsten-ras |
| MAPKs | Mitogen-activated protein kinases |
| SE | Staphylococcal enterotoxin |
| SEA | Staphylococcal enterotoxin A |
| STAT3 | Signal transducer and activator of transcription 3 |
References
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaser, M.J.; Falkow, S. What are the consequences of the disappearing human microbiota? Nat. Rev. Microbiol. 2009, 7, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The microbiota-gut-brain axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef] [PubMed]
- Cenit, M.C.; Sanz, Y.; Codoñer-Franch, P. Influence of gut microbiota on neuropsychiatric disorders. World J. Gastroenterol. 2017, 23, 5486. [Google Scholar] [CrossRef] [PubMed]
- Zink, A.; Samadelli, M.; Gostner, P.; Piombino-Mascali, D. Possible evidence for care and treatment in the Tyrolean Iceman. Int. J. Paleopathol. 2018, 25, 110–117. [Google Scholar] [CrossRef]
- Zink, A.R.; Maixner, F. The Current Situation of the Tyrolean Iceman. Gerontology 2019, 65, 699–706. [Google Scholar] [CrossRef]
- Maixner, F.; Krause-Kyora, B.; Turaev, D.; Herbig, A.; Hoopmann, M.R.; Hallows, J.L.; Kusebauch, U.; Vigl, E.E.; Malfertheiner, P.; Megraud, F.; et al. The 5300-year-old Helicobacter pylori genome of the Iceman. Science 2016, 351, 162–165. [Google Scholar] [CrossRef] [Green Version]
- Lugli, G.A.; Milani, C.; Mancabelli, L.; Turroni, F.; Ferrario, C.; Duranti, S.; van Sinderen, D.; Ventura, M. Ancient bacteria of the Ötzi’s microbiome: A genomic tale from the Copper Age. Microbiome 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Santiago-Rodriguez, T.M.; Fornaciari, G.; Luciani, S.; Dowd, S.E.; Toranzos, G.A.; Marota, I.; Cano, R.J. Natural mummification of the human gut preserves bacteriophage DNA. FEMS Microbiol. Lett. 2016, 363, fnv219. [Google Scholar] [CrossRef] [Green Version]
- Schnorr, S.L.; Candela, M.; Rampelli, S.; Centanni, M.; Consolandi, C.; Basaglia, G.; Turroni, S.; Biagi, E.; Peano, C.; Severgnini, M.; et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 2014, 5, 3654. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.A.; Leach, J.; Sonnenburg, E.D.; Gonzalez, C.G.; Lichtman, J.S.; Reid, G.; Knight, R.; Manjurano, A.; Changalucha, J.; Elias, J.E.; et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 2017, 357, 802–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Manfredo Vieira, S.; Hiltensperger, M.; Kumar, V.; Zegarra-Ruiz, D.; Dehner, C.; Khan, N.; Costa, F.R.C.; Tiniakou, E.; Greiling, T.; Ruff, W.; et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 2018, 359, 1156–1161. [Google Scholar] [CrossRef] [Green Version]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 2008, 105, 20858–20863. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Naik, S. Compartmentalized and systemic control of tissue immunity by commensals. Nat. Immunol. 2013, 14, 646–653. [Google Scholar] [CrossRef] [Green Version]
- Magnúsdóttir, S.; Ravcheev, D.; De Crécy-Lagard, V.; Thiele, I. Systematic genome assessment of B-vitamin biosynthesis suggests cooperation among gut microbes. Front. Genet. 2015, 6, 148. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Sinha, R.; Pei, Z.; Dominianni, C.; Wu, J.; Shi, J.; Goedert, J.J.; Hayes, R.B.; Yang, L. Human Gut Microbiome and Risk for Colorectal Cancer. JNCI J. Natl. Cancer Inst. 2013, 105, 1907–1911. [Google Scholar] [CrossRef] [Green Version]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut Microbiota in Health and Disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [Green Version]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787. [Google Scholar] [CrossRef]
- Candela, M.; Guidotti, M.; Fabbri, A.; Brigidi, P.; Franceschi, C.; Fiorentini, C. Human intestinal microbiota: Cross-talk with the host and its potential role in colorectal cancer. Crit. Rev. Microbiol. 2011, 37, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Candela, M.; Turroni, S.; Biagi, E.; Carbonero, F.; Rampelli, S.; Fiorentini, C.; Brigidi, P. Inflammation and colorectal cancer, when microbiota-host mutualism breaks. World J. Gastroenterol. 2014, 20, 908. [Google Scholar] [CrossRef] [PubMed]
- Rosadi, F.; Fiorentini, C.; Fabbri, A. Bacterial protein toxins in human cancers. Pathog. Dis. 2016, 74, ftv105. [Google Scholar] [CrossRef]
- Gargi, A.; Reno, M.; Blanke, S.R. Bacterial toxin modulation of the eukaryotic cell cycle: Are all cytolethal distending toxins created equally? Front. Cell. Infect. Microbiol. 2012, 2, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenker, B.J.; McKay, T.; Datar, S.; Miller, M.; Chowhan, R.; Demuth, D. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J. Immunol. 1999, 162, 4773–4780. [Google Scholar] [PubMed]
- Lara-Tejero, M.; Galán, J.E. CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect. Immun. 2001, 69, 4358–4365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, V.B.; Knox, K.A.; Pratt, J.S.; Cortez, J.S.; Mansfield, L.S.; Rogers, A.B.; Fox, J.G.; Schauer, D.B. In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect. Immun. 2004, 72, 2521–2527. [Google Scholar] [CrossRef] [Green Version]
- Thelestam, M.; Frisan, T. Cytolethal distending toxins. Rev. Physiol. Biochem. Pharmacol. 2004, 152, 111–133. [Google Scholar] [CrossRef]
- DiRienzo, J.M. Breaking the Gingival Epithelial Barrier: Role of the Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin in Oral Infectious Disease. Cells 2014, 3, 476–499. [Google Scholar] [CrossRef]
- DiRienzo, D.B. Effect of probiotics on biomarkers of cardiovascular disease: Implications for heart-healthy diets. Nutr. Rev. 2014, 72, 18–29. [Google Scholar] [CrossRef]
- Scuron, M.D.; Boesze-Battaglia, K.; Dlakic, M.; Shenker, B.J. The cytolethal distending toxin contributes to microbial virulence and disease pathogenesis by acting as a tri-perditious toxin. Front. Cell. Infect. Microbiol. 2016, 6, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Z.; Feng, Y.; Ge, L.; Parry, N.; Muthupalani, S.; Fox, J.G. Helicobacter hepaticus cytolethal distending toxin promotes intestinal carcinogenesis in 129Rag2-deficient mice. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.A.; Kaper, J.B. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect. Immun. 1994, 62, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Bratti, X.; Chaves-Olarte, E.; Lagergård, T.; Thelestam, M. Cellular internalization of cytolethal distending toxin from Haemophilus ducreyi. Infect. Immun. 2000, 68, 6903–6911. [Google Scholar] [CrossRef] [Green Version]
- Guerra, L.; Nemec, K.N.; Massey, S.; Tatulian, S.A.; Thelestam, M.; Frisan, T.; Teter, K. A novel mode of translocation for cytolethal distending toxin. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 489–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisan, T. Bacterial genotoxins: The long journey to the nucleus of mammalian cells. Biochim. Biophys. Acta 2016, 1858, 567–575. [Google Scholar] [CrossRef]
- Hu, X.; Nesic, D.; Stebbins, C.E. Comparative structure-function analysis of cytolethal distending toxins. Proteins Struct. Funct. Bioinform. 2006, 62, 421–434. [Google Scholar] [CrossRef]
- Fedor, Y.; Vignard, J.; Nicolau-Travers, M.-L.; Boutet-Robinet, E.; Watrin, C.; Salles, B.; Mirey, G. From single-strand breaks to double-strand breaks during S-phase: A new mode of action of the Escherichia coli Cytolethal Distending Toxin. Cell. Microbiol. 2013, 15, 1–15. [Google Scholar] [CrossRef]
- Fahrer, J.; Huelsenbeck, J.; Jaurich, H.; Dörsam, B.; Frisan, T.; Eich, M.; Roos, W.P.; Kaina, B.; Fritz, G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair 2014, 18, 31–43. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.; Rodriguez, A.; Sugimoto, K. Activation of ATR-related protein kinase upon DNA damage recognition. Curr. Genet. 2019, 66, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal distending toxin: A conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 2011, 157, 1851–1875. [Google Scholar] [CrossRef] [Green Version]
- Okuda, J.; Fukumoto, M.; Takeda, Y.; Nishibuchi, M. Examination of diarrheagenicity of cytolethal distending toxin: Suckling mouse response to the products of the cdtABC genes of Shigella dysenteriae. Infect. Immun. 1997, 65, 428–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickey, T.E.; McVeigh, A.L.; Scott, D.A.; Michielutti, R.E.; Bixby, A.; Carroll, S.A.; Bourgeois, A.L.; Guerry, P. Campylobacter jejuni cytolethal distending toxin mediates release of interleukin-8 from intestinal epithelial cells. Infect. Immun. 2000, 68, 6535–6541. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.G.; Rogers, A.B.; Whary, M.T.; Ge, Z.; Taylor, N.S.; Xu, S.; Horwitz, B.H.; Erdman, S.E. Gastroenteritis in NF-κB-Deficient Mice Is Produced with Wild-Type Camplyobacter jejuni but Not with C. jejuni Lacking Cytolethal Distending Toxin despite Persistent Colonization with Both Strains. Infect. Immun. 2004, 72, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Jain, D.; Prasad, K.N.; Sinha, S.; Husain, N. Differences in virulence attributes between cytolethal distending toxin positive and negative Campylobacter jejuni strains. J. Med. Microbiol. 2008, 57, 267–272. [Google Scholar] [CrossRef]
- Shenker, B.J.; Walker, L.P.; Zekavat, A.; Dlakić, M.; Boesze-Battaglia, K. Blockade of the PI-3K signalling pathway by the Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces macrophages to synthesize and secrete pro-inflammatory cytokines. Cell. Microbiol. 2014, 16, 1391–1404. [Google Scholar] [CrossRef] [Green Version]
- Péré-Védrenne, C.; Cardinaud, B.; Varon, C.; Mocan, I.; Buissonnière, A.; Izotte, J.; Mégraud, F.; Ménard, A. The Cytolethal Distending Toxin Subunit CdtB of Helicobacter Induces a Th17-related and Antimicrobial Signature in Intestinal and Hepatic Cells In Vitro. J. Infect. Dis. 2016, 213, 1979–1989. [Google Scholar] [CrossRef] [Green Version]
- Ando-Suguimoto, E.S.; Da Silva, M.P.; Kawamoto, D.; Chen, C.; DiRienzo, J.M.; Mayer, M.P.A. The cytolethal distending toxin of Aggregatibacter actinomycetemcomitans inhibits macrophage phagocytosis and subverts cytokine production. Cytokine 2014, 66, 46–53. [Google Scholar] [CrossRef]
- Pons, B.J.; Vignard, J.; Mirey, G. Cytolethal distending toxin subunit B: A review of structure-function relationship. Toxins 2019, 11, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsland, D.; Neefjes, J. Bacterial infections and cancer. EMBO Rep. 2018, 19, e46632. [Google Scholar] [CrossRef] [PubMed]
- Graillot, V.; Dormoy, I.; Dupuy, J.; Shay, J.W.; Huc, L.; Mirey, G.; Vignard, J. Genotoxicity of Cytolethal Distending Toxin (CDT) on Isogenic Human Colorectal Cell Lines: Potential Promoting Effects for Colorectal Carcinogenesis. Front. Cell. Infect. Microbiol. 2016, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Rogers, A.B.; Feng, Y.; Lee, A.; Xu, S.; Taylor, N.S.; Fox, J.G. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell. Microbiol. 2007, 9, 2070–2080. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Feng, Y.; Sheh, A.; Muthupalani, S.; Gong, G.; Chawanthayatham, S.; Essigmann, J.M.; Fox, J.G. Mutagenicity of Helicobacter hepaticus infection in the lower bowel mucosa of 129/SvEv Rag2-/- Il10-/- gpt delta mice is influenced by sex. Int. J. Cancer 2019, 145, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High Prevalence of Mucosa-Associated E. coli Producing Cyclomodulin and Genotoxin in Colon Cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef] [Green Version]
- Nougayrède, J.-P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef]
- Homburg, S.; Oswald, E.; Hacker, J.; Dobrindt, U. Expression analysis of the colibactin gene cluster coding for a novel polyketide in Escherichia coli. FEMS Microbiol. Lett. 2007, 275, 255–262. [Google Scholar] [CrossRef]
- Olier, M.; Marcq, I.; Salvador-Cartier, C.; Secher, T.; Dobrindt, U.; Boury, M.; Bacquié, V.; Pénary, M.; Gaultier, E.; Nougayrède, J.-P.; et al. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes 2012, 3, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Putze, J.; Hennequin, C.; Nougayrède, J.-P.; Zhang, W.; Homburg, S.; Karch, H.; Bringer, M.-A.; Fayolle, C.; Carniel, E.; Rabsch, W.; et al. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect. Immun. 2009, 77, 4696–4703. [Google Scholar] [CrossRef] [Green Version]
- Martin, O.C.B.; Frisan, T. Bacterial Genotoxin-Induced DNA Damage and Modulation of the Host Immune Microenvironment. Toxins 2020, 12, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, A.R.; Herzon, S.B. Molecular Basis of Gut Microbiome-Associated Colorectal Cancer: A Synthetic Perspective. J. Am. Chem. Soc. 2017, 139, 14817–14824. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Kim, C.S.; Healy, A.R.; Wernke, K.M.; Wang, Z.; Frischling, M.C.; Shine, E.E.; Wang, W.; Herzon, S.B.; Crawford, J.M. Structure elucidation of colibactin and its DNA cross-links. Science 2019, 365, eaax2685. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Berezo, T.; Pujo, J.; Martin, P.; Le Faouder, P.; Galano, J.M.; Guy, A.; Knauf, C.; Tabet, J.C.; Tronnet, S.; Barreau, F.; et al. Identification of an analgesic lipopeptide produced by the probiotic Escherichia coli strain Nissle. Nat. Commun. 2017, 8, 1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faïs, T.; Delmas, J.; Barnich, N.; Bonnet, R.; Dalmasso, G. Colibactin: More Than a New Bacterial Toxin. Toxins 2018, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Massip, C.; Branchu, P.; Bossuet-Greif, N.; Chagneau, C.V.; Gaillard, D.; Martin, P.; Boury, M.; Sécher, T.; Dubois, D.; Nougayrède, J.P.; et al. Deciphering the interplay between the genotoxic and probiotic activities of Escherichia coli Nissle 1917. PLoS Pathog. 2019, 15, e1008029. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363. [Google Scholar] [CrossRef]
- Bossuet-Greif, N.; Vignard, J.; Taieb, F.; Mirey, G.; Dubois, D.; Petit, C.; Oswald, E.; Nougayrède, J.-P. The Colibactin Genotoxin Generates DNA Interstrand Cross-Links in Infected Cells. MBio 2018, 9, e02393–e02417. [Google Scholar] [CrossRef] [Green Version]
- Guerra, L.; Cortes-Bratti, X.; Guidi, R.; Frisan, T. The Biology of the Cytolethal Distending Toxins. Toxins 2011, 3, 172–190. [Google Scholar] [CrossRef]
- Iyadorai, T.; Mariappan, V.; Vellasamy, K.M.; Wanyiri, J.W.; Roslani, A.C.; Lee, G.K.; Sears, C.; Vadivelu, J. Prevalence and association of pks+ Escherichia coli with colorectal cancer in patients at the University Malaya Medical Centre, Malaysia. PLoS ONE 2020, 15, e0228217. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Huber, A.R.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalmasso, G.; Cougnoux, A.; Delmas, J.; Darfeuille-Michaud, A.; Bonnet, R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2014, 5, 675–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopès, A.; Billard, E.; Casse, A.H.; Villéger, R.; Veziant, J.; Roche, G.; Carrier, G.; Sauvanet, P.; Briat, A.; Pagès, F.; et al. Colibactin-positive Escherichia coli induce a procarcinogenic immune environment leading to immunotherapy resistance in colorectal cancer. Int. J. Cancer 2020, 146, 3147–3159. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.C.; Gharaibeh, R.Z.; Mühlbauer, M.; Perez-Chanona, E.; Uronis, J.M.; McCafferty, J.; Fodor, A.A.; Jobin, C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat. Commun. 2014, 5, 4724. [Google Scholar] [CrossRef] [Green Version]
- Wassenaar, T.M.E. coli and colorectal cancer: A complex relationship that deserves a critical mindset. Crit. Rev. Microbiol. 2018, 44, 619–632. [Google Scholar] [CrossRef] [Green Version]
- Sears, C.L.; Islam, S.; Saha, A.; Arjumand, M.; Alam, N.H.; Faruque, A.S.G.; Salam, M.A.; Shin, J.; Hecht, D.; Weintraub, A.; et al. Association of Enterotoxigenic Bacteroides fragilis Infection with Inflammatory Diarrhea. Clin. Infect. Dis. 2008, 47, 797–803. [Google Scholar] [CrossRef] [Green Version]
- Thiele Orberg, E.; Fan, H.; Tam, A.J.; Dejea, C.M.; Destefano Shields, C.E.; Wu, S.; Chung, L.; Finard, B.B.; Wu, X.; Fathi, P.; et al. The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol. 2017, 10, 421–433. [Google Scholar] [CrossRef]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [Green Version]
- Chung, L.; Thiele Orberg, E.; Geis, A.L.; Chan, J.L.; Fu, K.; DeStefano Shields, C.E.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Shin, J.; Zhang, G.; Cohen, M.; Franco, A.; Sears, C.L. The Bacteroides fragilis toxin binds to a specific intestinal epithelial cell receptor. Infect. Immun. 2006, 74, 5382–5390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Lim, K.C.; Huang, J.; Saidi, R.F.; Sears, C.L. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc. Natl. Acad. Sci. USA 1998, 95, 14979–14984. [Google Scholar] [CrossRef] [Green Version]
- Kharlampieva, D.; Manuvera, V.; Podgorny, O.; Grafskaia, E.; Kovalchuk, S.; Pobeguts, O.; Altukhov, I.; Govorun, V.; Lazarev, V. Recombinant fragilysin isoforms cause E-cadherin cleavage of intact cells and do not cleave isolated E-cadherin. Microb. Pathog. 2015, 83–84, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.J.; Zhang, M.; Franco, A.; Sears, C.L. Bacteroides fragilis toxin stimulates intestinal epithelial cell shedding and γ-secretase-dependent E-cadherin cleavage. J. Cell Sci. 2007, 120, 1944–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.H.M.; Fang, C.M.; Chuah, L.H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018, 121, 11–22. [Google Scholar] [CrossRef]
- Wu, S.; Morin, P.J.; Maouyo, D.; Sears, C.L. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology 2003, 124, 392–400. [Google Scholar] [CrossRef]
- Kim, J.M.; Lee, J.Y.; Kim, Y.J. Inhibition of apoptosis in Bacteroides fragilis enterotoxin-stimulated intestinal epithelial cells through the induction of c-IAP-2. Eur. J. Immunol. 2008, 38, 2190–2199. [Google Scholar] [CrossRef]
- Ko, S.H.; Rho, D.J.; Jeon, J.I.; Kim, Y.J.; Woo, H.A.; Lee, Y.K.; Kim, J.M. Bacteroides fragilis enterotoxin upregulates heme oxygenase-1 in intestinal epithelial cells via a mitogen-activated protein kinase- and NF-κB-dependent pathway, leading to modulation of apoptosis. Infect. Immun. 2016, 84, 2541–2554. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.; Hao, S.; Sears, C.L.; Timp, W. Epigenetic changes induced by bacteroides fragilis toxin. Infect. Immun. 2019, 87, e00447–e00518. [Google Scholar] [CrossRef] [Green Version]
- Rhee, K.J.; Wu, S.; Wu, X.; Huso, D.L.; Karim, B.; Franco, A.A.; Rabizadeh, S.; Golub, J.E.; Mathews, L.E.; Shin, J.; et al. Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect. Immun. 2009, 77, 1708–1718. [Google Scholar] [CrossRef] [Green Version]
- Rabizadeh, S.; Rhee, K.J.; Wu, S.; Huso, D.; Gan, C.M.; Golub, J.E.; Wu, X.; Zhang, M.; Sears, C.L. Enterotoxigenic Bacteroides fragilis: A potential instigator of colitis. Inflamm. Bowel Dis. 2007, 13, 1475–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, E.C.; Rabizadeh, S.; Albesiano, E.; Wu, X.Q.; Wu, S.; Chan, J.; Rhee, K.J.; Ortega, G.; Huso, D.L.; Pardoll, D.; et al. Stat3 activation in murine colitis induced by enterotoxigenic bacteroides fragilis. Inflamm. Bowel Dis. 2014, 20, 821–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J.D. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef]
- Ulger Toprak, N.; Yagci, A.; Gulluoglu, B.M.; Akin, M.L.; Demirkalem, P.; Celenk, T.; Soyletir, G. A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin. Microbiol. Infect. 2006, 12, 782–786. [Google Scholar] [CrossRef] [Green Version]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [Green Version]
- Haghi, F.; Goli, E.; Mirzaei, B.; Zeighami, H. The association between fecal enterotoxigenic B. fragilis with colorectal cancer. BMC Cancer 2019, 19, 879. [Google Scholar] [CrossRef] [Green Version]
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C.; et al. The bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin. Infect. Dis. 2015, 60, 208–215. [Google Scholar] [CrossRef]
- Viljoen, K.S.; Dakshinamurthy, A.; Goldberg, P.; Blackburn, J.M. Quantitative profiling of colorectal cancer-associated bacteria reveals associations between Fusobacterium spp., enterotoxigenic Bacteroides fragilis (ETBF) and clinicopathological features of colorectal cancer. PLoS ONE 2015, 10, e0119462. [Google Scholar] [CrossRef] [Green Version]
- Purcell, R.V.; Pearson, J.; Aitchison, A.; Dixon, L.; Frizelle, F.A.; Keenan, J.I. Colonization with enterotoxigenic Bacteroides fragilis is associated with early-stage colorectal neoplasia. PLoS ONE 2017, 12, e0171602. [Google Scholar] [CrossRef] [Green Version]
- Zamani, S.; Taslimi, R.; Sarabi, A.; Jasemi, S.; Sechi, L.A.; Feizabadi, M.M. Enterotoxigenic Bacteroides fragilis: A Possible Etiological Candidate for Bacterially-Induced Colorectal Precancerous and Cancerous Lesions. Front. Cell. Infect. Microbiol. 2019, 9, 449. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Fabbri, A.; Matarrese, P.; Falzano, L.; Boquet, P.; Malorni, W. Hinderance of Apoptosis and Phagocytic Behaviour Induced byEscherichia coliCytotoxic Necrotizing Factor 1: Two Related Activities in Epithelial Cells. Biochem. Biophys. Res. Commun. 1997, 241, 341–346. [Google Scholar] [CrossRef]
- Schmidt, G.; Sehr, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997, 387, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 1997, 387, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Lerm, M.; Selzer, J.; Hoffmeyer, A.; Rapp, U.R.; Aktories, K.; Schmidt, G. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: Activation of c-Jun N-terminal kinase in HeLa cells. Infect. Immun. 1999, 67, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. Rho GTpases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Fiorentini, C.; Falzano, L.; Fabbri, A.; Stringaro, A.; Logozzi, M.; Travaglione, S.; Contamin, S.; Arancia, G.; Malorni, W.; Fais, S. Activation of Rho GTPases by Cytotoxic Necrotizing Factor 1 Induces Macropinocytosis and Scavenging Activity in Epithelial Cells. Mol. Biol. Cell 2001, 12, 2061–2073. [Google Scholar] [CrossRef] [Green Version]
- Doye, A.; Mettouchi, A.; Bossis, G.; Clément, R.; Buisson-Touati, C.; Flatau, G.; Gagnoux, L.; Piechaczyk, M.; Boquet, P.; Lemichez, E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002, 111, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Fiorentini, C.; Arancia, G.; Caprioli, A.; Falbo, V.; Ruggeri, F.M.; Donelli, G. Cytoskeletal changes induced in HEp-2 cells by the cytotoxic necrotizing factor of Escherichia coli. Toxicon 1988, 26, 1047–1056. [Google Scholar] [CrossRef]
- Malorni, W.; Fiorentini, C. Is the Rac GTPase-activating toxin CNF1 a smart hijacker of host cell fate? FASEB J. 2006, 20, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Nougayrède, J.-P.; Taieb, F.; De Rycke, J.; Oswald, E. Cyclomodulins: Bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 2005, 13, 103–110. [Google Scholar] [CrossRef] [PubMed]
- El-Aouar Filho, R.A.; Nicolas, A.; De Paula Castro, T.L.; Deplanche, M.; De Carvalho Azevedo, V.A.; Goossens, P.L.; Taieb, F.; Lina, G.; Le Loir, Y.; Berkova, N. Heterogeneous family of cyclomodulins: Smart weapons that allow bacteria to hijack the eukaryotic cell cycle and promote infections. Front. Cell. Infect. Microbiol. 2017, 7, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacerda, H.M.; Pullinger, G.D.; Lax, A.J.; Rozengurt, E. Cytotoxic necrotizing factor 1 from Escherichia coli and dermonecrotic toxin from Bordetella bronchiseptica induce p21(rho)-dependent tyrosine phosphorylation of focal adhesion kinase and paxillin in Swiss 3T3 cells. J. Biol. Chem. 1997, 272, 9587–9596. [Google Scholar] [CrossRef] [Green Version]
- Giamboi-Miraglia, A.; Travaglione, S.; Filippini, P.; Fabbri, A.; Fiorentini, C.; Falzano, L. A multinucleating Escherichia coli cytotoxin perturbs cell cycle in cultured epithelial cells. Toxicol. In Vitro 2007, 21, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Falzano, L.; Filippini, P.; Travaglione, S.; Miraglia, A.G.; Fabbri, A.; Fiorentini, C. Escherichia coli cytotoxic necrotizing factor 1 blocks cell cycle G2/M transition in uroepithelial cells. Infect. Immun. 2006, 74, 3765–3772. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Aung, K.M.; Uhlin, B.E.; Wai, S.N. Reversible senescence of human colon cancer cells after blockage of mitosis/cytokinesis caused by the CNF1 cyclomodulin from Escherichia coli. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Falzano, L.; Quaranta, M.G.; Travaglione, S.; Filippini, P.; Fabbri, A.; Viora, M.; Donelli, G.; Fiorentini, C. Cytotoxic necrotizing factor 1 enhances reactive oxygen species-dependent transcription and secretion of proinflammatory cytokines in human uroepithelial cells. Infect. Immun. 2003, 71, 4178–4181. [Google Scholar] [CrossRef] [Green Version]
- Boyer, L.; Travaglione, S.; Falzano, L.; Gauthier, N.C.; Popoff, M.R.; Lemichez, E.; Fiorentini, C.; Fabbri, A. Rac GTPase Instructs Nuclear Factor-κB Activation by Conveying the SCF Complex and IkBα to the Ruffling Membranes. Mol. Biol. Cell 2004, 15, 1124–1133. [Google Scholar] [CrossRef] [Green Version]
- Travaglione, S.; Loizzo, S.; Rizza, T.; Del Brocco, A.; Ballan, G.; Guidotti, M.; Vona, R.; Di Nottia, M.; Torraco, A.; Carrozzo, R.; et al. Enhancement of mitochondrial ATP production by the Escherichia coli cytotoxic necrotizing factor 1. FEBS J. 2014, 281, 3473–3488. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Matarrese, P.; Straface, E.; Falzano, L.; Fabbri, A.; Donelli, G.; Cossarizza, A.; Boquet, P.; Malorni, W. Toxin-Induced Activation of Rho GTP-Binding Protein Increases Bcl-2 Expression and Influences Mitochondrial Homeostasis. Exp. Cell Res. 1998, 242, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Matarrese, P.; Straface, E.; Falzano, L.; Donelli, G.; Boquet, P.; Malorni, W. Rho-dependent cell spreading activated by E.coli cytotoxic necrotizing factor 1 hinders apoptosis in epithelial cells. Cell Death Differ. 1998, 5, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Thomas, W.; Ascott, Z.K.; Harmey, D.; Slice, L.W.; Rozengurt, E.; Lax, A.J. Cytotoxic necrotizing factor from Escherichia coli induces RhoA-dependent expression of the cyclooxygenase-2 gene. Infect. Immun. 2001, 69, 6839–6845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, A.; Travaglione, S.; Ballan, G.; Loizzo, S.; Fiorentini, C.; Fabbri, A.; Travaglione, S.; Ballan, G.; Loizzo, S.; Fiorentini, C. The Cytotoxic Necrotizing Factor 1 from E. Coli: A Janus Toxin Playing with Cancer Regulators. Toxins 2013, 5, 1462–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Zhang, Z.; Wei, H.; Wang, J.; Lv, J.; Zhang, K.; Keller, E.T.; Yao, Z.; Wang, Q. Cytotoxic necrotizing factor 1 promotes prostate cancer progression through activating the Cdc42-PAK1 axis. J. Pathol. 2017, 243, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, A.; Travaglione, S.; Rosadi, F.; Ballan, G.; Maroccia, Z.; Giambenedetti, M.; Guidotti, M.; Ødum, N.; Krejsgaard, T.; Fiorentini, C. The Escherichia coli protein toxin cytotoxic necrotizing factor 1 induces epithelial mesenchymal transition. Cell. Microbiol. 2019. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.; Bringer, M.A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, S.; Kumar, S. Infection as a risk factor for gallbladder cancer. J. Surg. Oncol. 2006, 93, 633–639. [Google Scholar] [CrossRef]
- Gradel, K.O.; Nielsen, H.L.; Schønheyder, H.C.; Ejlertsen, T.; Kristensen, B.; Nielsen, H. Increased Short- and Long-Term Risk of Inflammatory Bowel Disease After Salmonella or Campylobacter Gastroenteritis. Gastroenterology 2009, 137, 495–501. [Google Scholar] [CrossRef]
- Lu, R.; Wu, S.; Liu, X.; Xia, Y.; Zhang, Y.-G.; Sun, J. Chronic effects of a Salmonella type III secretion effector protein AvrA in vivo. PLoS ONE 2010, 5, e10505. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Petrof, E.O.; Boone, D.; Claud, E.C.; Sun, J. Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am. J. Pathol. 2007, 171, 882–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Liu, X.; Wu, S.; Xia, Y.; Zhang, Y.-G.; Petrof, E.O.; Claud, E.C.; Sun, J. Consistent activation of the β-catenin pathway by Salmonella type-three secretion effector protein AvrA in chronically infected intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1113–G1125. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Hobert, M.E.; Rao, A.S.; Neish, A.S.; Madara, J.L. Bacterial activation of beta-catenin signaling in human epithelia. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G220–G227. [Google Scholar] [CrossRef] [Green Version]
- Liao, A.P.; Petrof, E.O.; Kuppireddi, S.; Zhao, Y.; Xia, Y.; Claud, E.C.; Sun, J. Salmonella type III effector AvrA stabilizes cell tight junctions to inhibit inflammation in intestinal epithelial cells. PLoS ONE 2008, 3, e2369. [Google Scholar] [CrossRef]
- Du, F.; Galán, J.E. Selective Inhibition of Type III Secretion Activated Signaling by the Salmonella Effector AvrA. PLoS Pathog. 2009, 5, e1000595. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.-Y.; Chen, W.; Lu, L.; Zheng, Z.; Xu, X. Involvement of RhoA/ROCK1 signaling pathway in hyperglycemia-induced microvascular endothelial dysfunction in diabetic retinopathy. Int. J. Clin. Exp. Pathol. 2014, 7, 7268–7277. [Google Scholar]
- Lu, R.; Wu, S.; Zhang, Y.G.; Xia, Y.; Zhou, Z.; Kato, I.; Dong, H.; Bissonnette, M.; Sun, J. Salmonella Protein AvrA Activates the STAT3 Signaling Pathway in Colon Cancer. Neoplasia 2016, 18, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Bosland, M.; Xia, Y.; Zhang, Y.G.; Kato, I.; Sun, J. Presence of Salmonella AvrA in colorectal tumor and its precursor lesions in mouse intestine and human specimens. Oncotarget 2017, 8, 55104–55115. [Google Scholar] [CrossRef] [Green Version]
- Brook, I. Fusobacterial infections in children. Curr. Infect. Dis. Rep. 2013, 15, 288–294. [Google Scholar] [CrossRef]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momen-Heravi, F.; Babic, A.; Tworoger, S.S.; Zhang, L.; Wu, K.; Smith-Warner, S.A.; Ogino, S.; Chan, A.T.; Meyerhardt, J.; Giovannucci, E.; et al. Periodontal disease, tooth loss and colorectal cancer risk: Results from the Nurses’ Health Study. Int. J. Cancer 2017, 140, 646–652. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.N.; Araújo-Pérez, F.; Azcárate-Peril, A.; Yeh, J.J.; Sandler, R.S.; Keku, T.O. Fusobacterium Is Associated with Colorectal Adenomas. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Hashemi Goradel, N.; Heidarzadeh, S.; Jahangiri, S.; Farhood, B.; Mortezaee, K.; Khanlarkhani, N.; Negahdari, B. Fusobacterium nucleatum and colorectal cancer: A mechanistic overview. J. Cell. Physiol. 2019, 234, 2337–2344. [Google Scholar] [CrossRef]
- Wu, J.; Li, Q.; Fu, X. Fusobacterium nucleatum Contributes to the Carcinogenesis of Colorectal Cancer by Inducing Inflammation and Suppressing Host Immunity. Transl. Oncol. 2019, 12, 846–851. [Google Scholar] [CrossRef]
- Han, Y.W.; Ikegami, A.; Rajanna, C.; Kawsar, H.I.; Zhou, Y.; Li, M.; Sojar, H.T.; Genco, R.J.; Kuramitsu, H.K.; Deng, C.X. Identification and characterization of a novel adhesin unique to oral fusobacteria. J. Bacteriol. 2005, 187, 5330–5340. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Yamada, M.; Li, M.; Liu, H.; Chen, S.G.; Han, Y.W. FadA from Fusobacterium nucleatum utilizes both secreted and nonsecreted forms for functional oligomerization for attachment and invasion of host cells. J. Biol. Chem. 2007, 282, 25000–25009. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Dalerba, P.; Wang, T.C.; Han, Y.W. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Zang, L.; Kondengaden, S.M.; Che, F.; Wang, L.; Heng, X. Potential Epigenetic-Based Therapeutic Targets for Glioma. Front. Mol. Neurosci. 2018, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Zahaf, N.I.; Schmidt, G. Bacterial toxins for cancer therapy. Toxins 2017, 9, 236. [Google Scholar] [CrossRef] [Green Version]
- Akbari, B.; Farajnia, S.; Ahdi Khosroshahi, S.; Safari, F.; Yousefi, M.; Dariushnejad, H.; Rahbarnia, L. Immunotoxins in cancer therapy: Review and update. Int. Rev. Immunol. 2017, 36, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Laliani, G.; Ghasemian Sorboni, S.; Lari, R.; Yaghoubi, A.; Soleimanpour, S.; Khazaei, M.; Hasanian, S.M.; Avan, A. Bacteria and cancer: Different sides of the same coin. Life Sci. 2020, 246, 117398. [Google Scholar] [CrossRef]
- Weerakkody, L.R.; Witharana, C. The role of bacterial toxins and spores in cancer therapy. Life Sci. 2019, 235, 116839. [Google Scholar] [CrossRef] [PubMed]
- Karpiński, T.M.; Adamczak, A. Anticancer activity of bacterial proteins and peptides. Pharmaceutics 2018, 10, 54. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, A.; Travaglione, S.; Falzano, L.; Fiorentini, C. Bacterial Protein Toxins: Current and Potential Clinical Use. Curr. Med. Chem. 2008, 15, 1116–1125. [Google Scholar] [CrossRef]
- Kumar, R.; Feltrup, T.M.; Kukreja, R.V.; Patel, K.B.; Cai, S.; Singh, B.R. Evolutionary features in the structure and function of bacterial toxins. Toxins 2019, 11, 15. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Chang, T.W.; Singh, B.R. Evolutionary Traits of Toxins. In Toxinology; Springer: Dordrecht, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Rudkin, J.K.; McLoughlin, R.M.; Preston, A.; Massey, R.C. Bacterial toxins: Offensive, defensive, or something else altogether? PLoS Pathog. 2017, 13, e1006452. [Google Scholar] [CrossRef] [Green Version]
- Scanu, T.; Spaapen, R.M.; Bakker, J.M.; Pratap, C.B.; Wu, L.E.; Hofland, I.; Broeks, A.; Shukla, V.K.; Kumar, M.; Janssen, H.; et al. Salmonella Manipulation of Host Signaling Pathways Provokes Cellular Transformation Associated with Gallbladder Carcinoma. Cell Host Microbe 2015, 17, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannemann, S.; Gao, B.; Galán, J.E. Salmonella Modulation of Host Cell Gene Expression Promotes Its Intracellular Growth. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijl, C.; Savage, N.D.L.; Marsman, M.; Tuin, A.W.; Janssen, L.; Egan, D.A.; Ketema, M.; Van Den Nieuwendijk, R.; Van Den Eeden, S.J.F.; Geluk, A.; et al. Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature 2007, 450, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Villéger, R.; Lopès, A.; Veziant, J.; Gagnière, J.; Barnich, N.; Billard, E.; Boucher, D.; Bonnet, M. Microbial markers in colorectal cancer detection and/or prognosis. World J. Gastroenterol. 2018, 24, 2327–2347. [Google Scholar] [CrossRef]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut microbiota and cancer: From pathogenesis to therapy. Cancers 2019, 11, 38. [Google Scholar] [CrossRef] [Green Version]
- Rea, D.; Coppola, G.; Palma, G.; Barbieri, A.; Luciano, A.; Del Prete, P.; Rossetti, S.; Berretta, M.; Facchini, G.; Perdonà, S.; et al. Microbiota effects on cancer: From risks to therapies. Oncotarget 2018, 9, 17915–17927. [Google Scholar] [CrossRef] [Green Version]
- Haag, L.M.; Fischer, A.; Otto, B.; Plickert, R.; Kühl, A.A.; Göbel, U.B.; Bereswill, S.; Heimesaat, M.M. Intestinal microbiota shifts towards elevated commensal escherichia coli loads abrogate colonization resistance against campylobacter jejuni in mice. PLoS ONE 2012, 7, e35988. [Google Scholar] [CrossRef]
- Cani, P.D.; Jordan, B.F. Gut microbiota-mediated inflammation in obesity: A link with gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 671–682. [Google Scholar] [CrossRef]
- Marshall, B.J. Helicobacter pylori. Am. J. Gastroenterol. 1994, 89, S116–S128. [Google Scholar]
- Dutilh, B.E.; Backus, L.; Van Hijum, S.A.F.T.; Tjalsma, H. Screening metatranscriptomes for toxin genes as functional drivers of human colorectal cancer. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 85–99. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzkowitz, S.H.; Yio, X. Inflammation and cancer—IV Colorectal cancer in inflammatory bowel disease: The role of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G7–G17. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.P.; Poutahidis, T.; Ge, Z.; Nambiar, P.R.; Boussahmain, C.; Wang, Y.Y.; Horwitz, B.H.; Fox, J.G.; Erdman, S.E. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res. 2006, 66, 7395–7400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulkens, J.; Vergauwen, G.; Van Deun, J.; Geeurickx, E.; Dhondt, B.; Lippens, L.; De Scheerder, M.A.; Miinalainen, I.; Rappu, P.; De Geest, B.G.; et al. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 2020, 69, 191–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wispelwey, N.; Hansen, E.J.; Scheld, W.M. Haemophilus influenzae outer membrane vesicle-induced blood-brain barrier permeability during experimental meningitis. Infect. Immun. 1989, 57, 2559–2562. [Google Scholar] [CrossRef] [Green Version]
- Nasef, N.A.; Mehta, S. Role of inflammation in pathophysiology of colonic disease: An update. Int. J. Mol. Sci. 2020, 21, 4748. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef]
- Monteiro, H.P.; Rodrigues, E.G.; Amorim Reis, A.K.C.; Longo, L.S.; Ogata, F.T.; Moretti, A.I.S.; da Costa, P.E.; Teodoro, A.C.S.; Toledo, M.S.; Stern, A. Nitric oxide and interactions with reactive oxygen species in the development of melanoma, breast, and colon cancer: A redox signaling perspective. Nitric Oxide 2019, 89, 1–13. [Google Scholar] [CrossRef]
- Bessède, E.; Staedel, C.; Acuña Amador, L.A.; Nguyen, P.H.; Chambonnier, L.; Hatakeyama, M.; Belleannée, G.; Mégraud, F.; Varon, C. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene 2014, 33, 4123–4131. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Kim, N.; Park, J.H.; Nam, R.H.; Choi, Y.J.; Park, S.M.; Choi, Y.J.; Yoon, H.; Shin, C.M.; Lee, D.H. Helicobacter pylori Might Induce TGF-β1-Mediated EMT by Means of cagE. Helicobacter 2015, 20, 438–448. [Google Scholar] [CrossRef]
- Sougleri, I.S.; Papadakos, K.S.; Zadik, M.P.; Mavri-Vavagianni, M.; Mentis, A.F.; Sgouras, D.N. Helicobacter pylori CagA protein induces factors involved in the epithelial to mesenchymal transition (EMT) in infected gastric epithelial cells in an EPIYA- phosphorylation-dependent manner. FEBS J. 2016, 283, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, C.; Liu, J.; Geng, F.; Shi, X.; Li, Q.; Lu, Z.; Pan, Y. Fusobacterium nucleatum promotes epithelial-mesenchymal transiton through regulation of the lncRNA MIR4435-2HG/miR-296-5p/Akt2/SNAI1 signaling pathway. FEBS J. 2020, febs.15233. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Bacterium | Type of Virulence Factor | Virulence Factor | Activity and Pro-Carcinogenic Outcomes | References |
|---|---|---|---|---|
| Gram-negative bacteria | Protein toxin | Cytolethal distending toxin (Cdt) | DNA damage, cell cycle arrest, survival, mitogen-activated protein kinase (MAPK) and STAT3 pathways, transformation | [24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55] |
| Escherichia coli | Protein toxin | Colibactin | DNA damage, apoptosis and senescence, proliferation | [57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] |
| Bacteroides fragilis | Protein toxin | B. fragilis toxin (BFT) | Cell proliferation, activation of MAPK and NF-κB pathway and of COX2, delay of apoptosis | [77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101] |
| Escherichia coli | Protein toxin | Cytotoxic necrotizing factor 1 (CNF1) | Activation of Rho GTPases, transformation, NF-κB activation and survival, EMT | [56,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128] |
| Salmonella sp. | Effector protein | Avirulence protein A (AvrA) | β-catenin and STAT-3 induction, proliferation and colonic tumorigenesis | [52,129,130,131,132,133,134,135,136,137,138,139] |
| Fusobacterium nucleatum | Cell surface adhesion protein | F. nucleatum Adhesin A (FadA) | Wnt/β-catenin activation, inflammatory genes induction and CRC cells proliferation, EMT | [52,140,141,142,143,144,145,146,147,148,149,150,151,152] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorentini, C.; Carlini, F.; Germinario, E.A.P.; Maroccia, Z.; Travaglione, S.; Fabbri, A. Gut Microbiota and Colon Cancer: A Role for Bacterial Protein Toxins? Int. J. Mol. Sci. 2020, 21, 6201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176201
Fiorentini C, Carlini F, Germinario EAP, Maroccia Z, Travaglione S, Fabbri A. Gut Microbiota and Colon Cancer: A Role for Bacterial Protein Toxins? International Journal of Molecular Sciences. 2020; 21(17):6201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176201
Chicago/Turabian StyleFiorentini, Carla, Francesca Carlini, Elena Angela Pia Germinario, Zaira Maroccia, Sara Travaglione, and Alessia Fabbri. 2020. "Gut Microbiota and Colon Cancer: A Role for Bacterial Protein Toxins?" International Journal of Molecular Sciences 21, no. 17: 6201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176201