Neutralization of Lipocalin-2 Diminishes Stroke-Reperfusion Injury

Abstract
:1. Introduction
2. Results
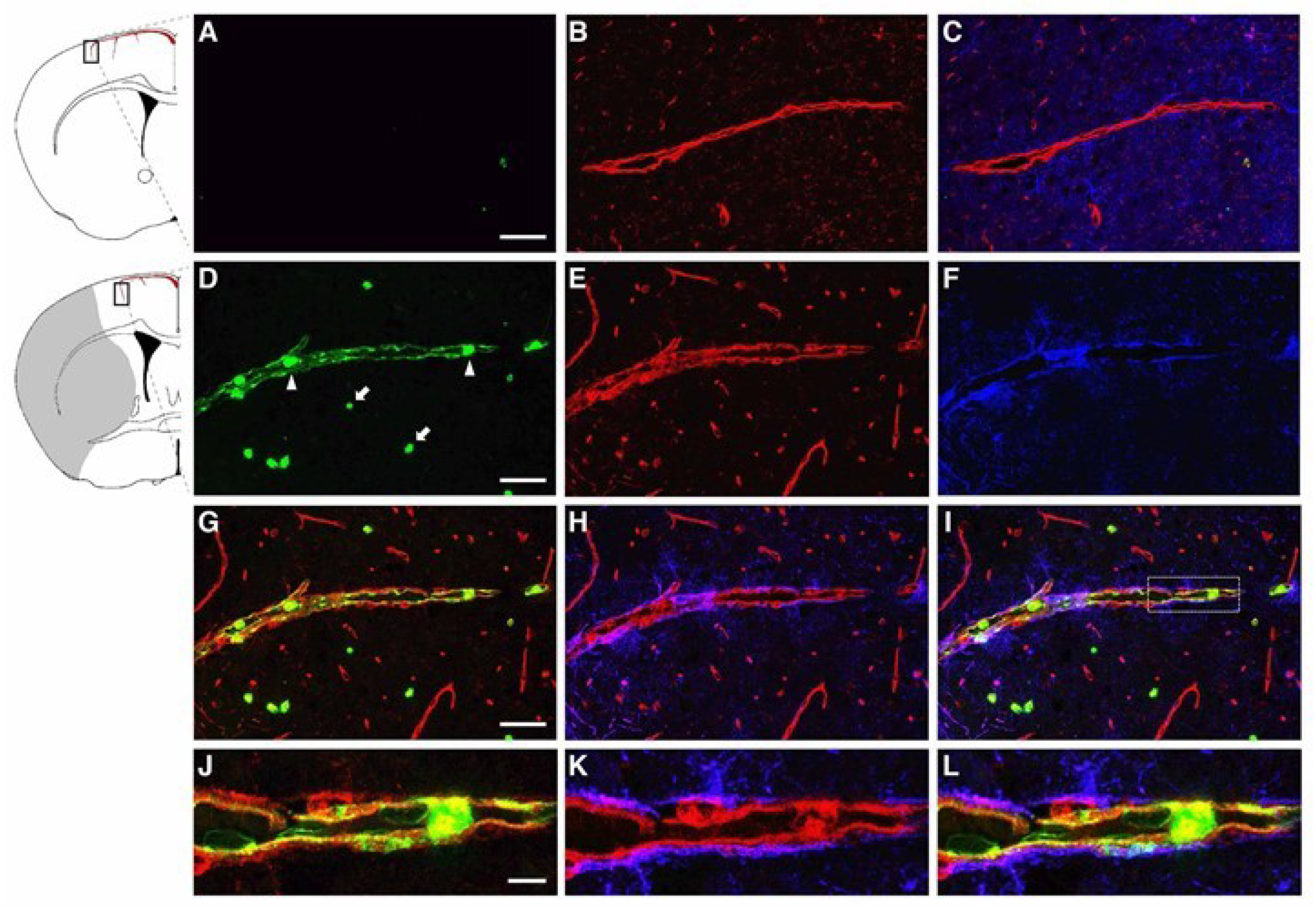
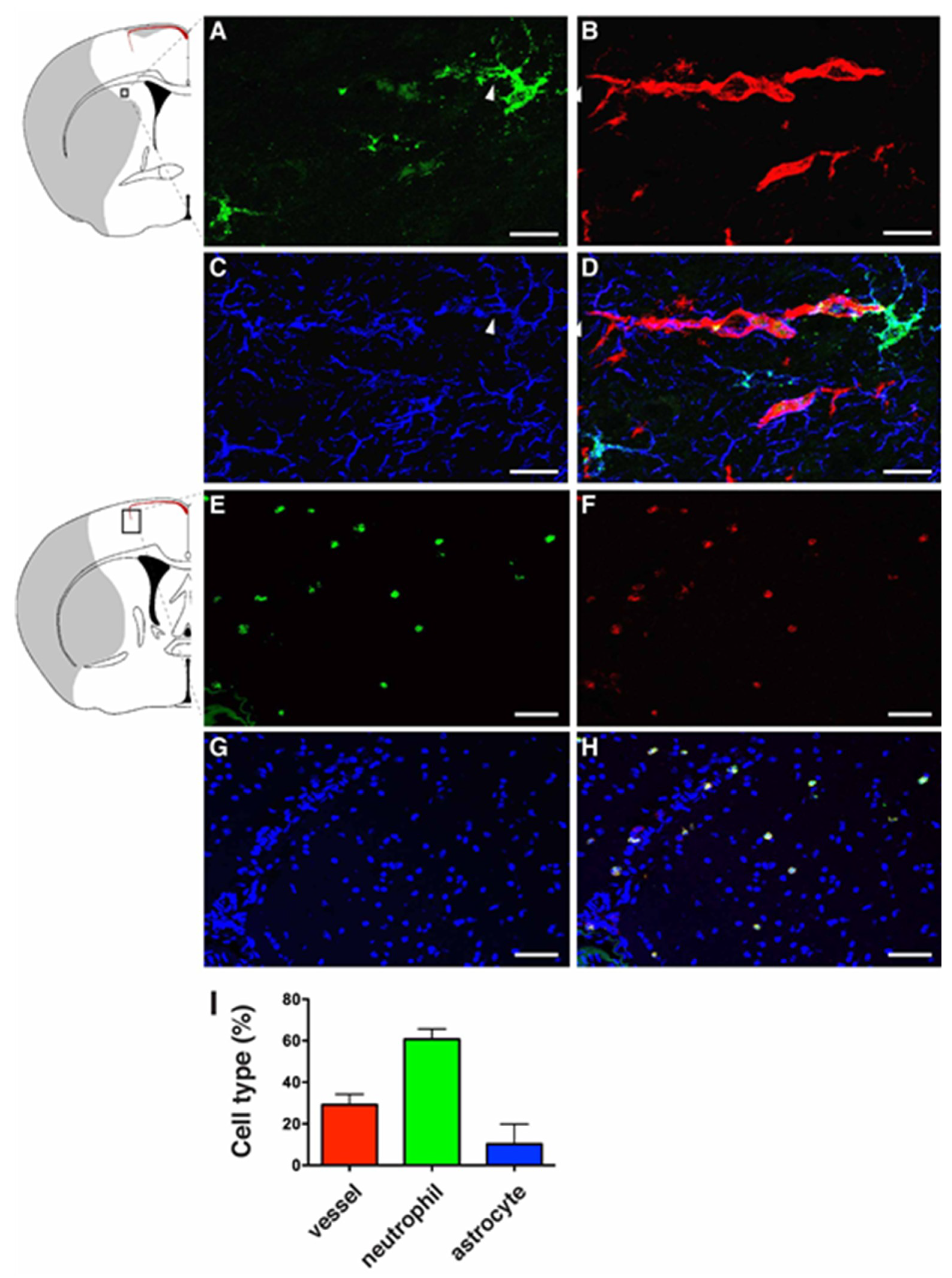
2.1. LCN2 was Detected in Cerebral Endothelial Cells, Astrocytes, and Infiltrating Neutrophils in the Ipsilateral Hemisphere after tMCAo
2.2. LCN2 Monoclonal Antibody (mAb) Specifically Targeted Recombinant and Endogenous LCN2 Proteins
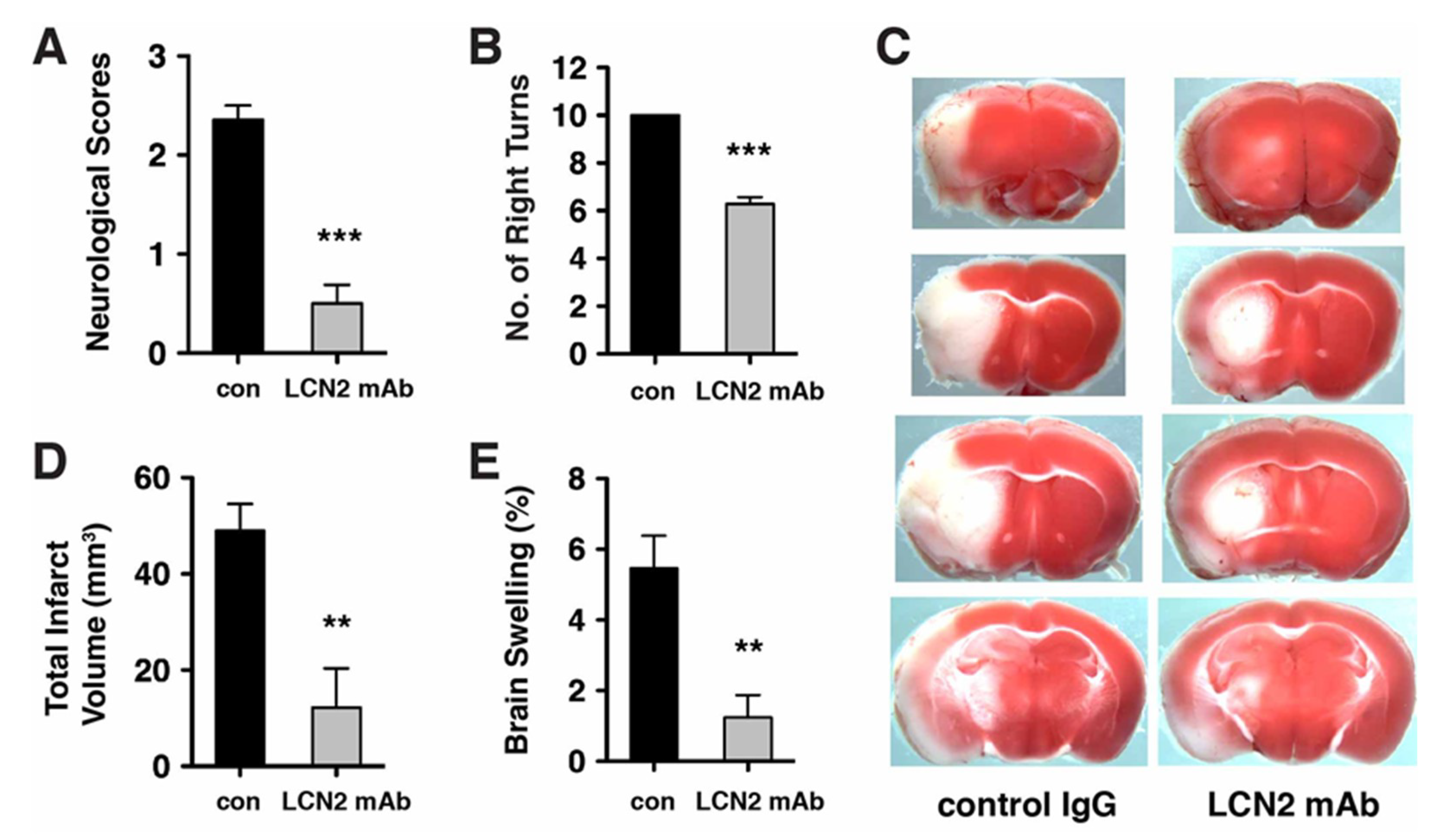
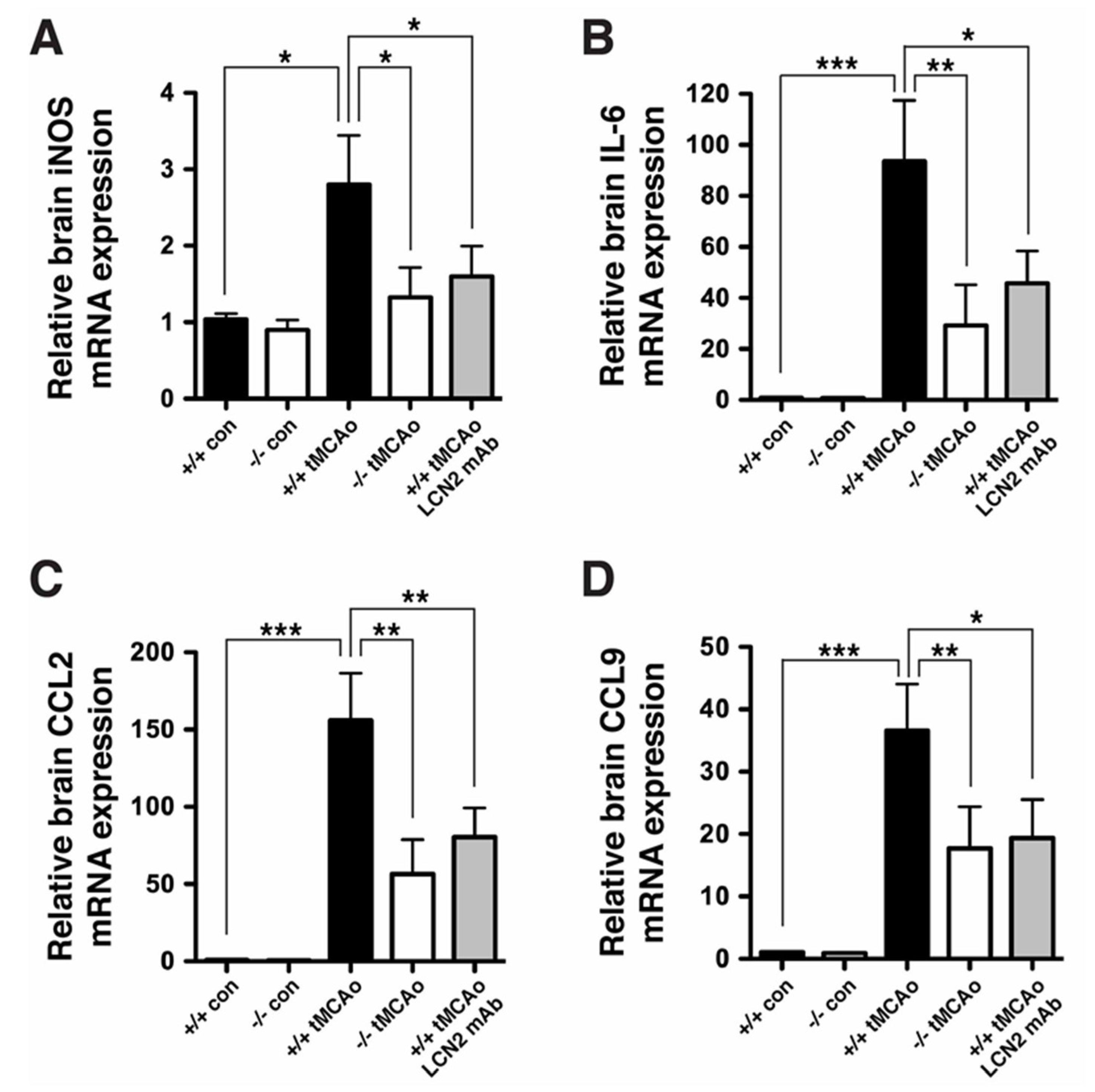
2.3. Neurological Deficits and Cerebral Infarction after tMCAo were Reduced after Treatment with LCN2 mAb
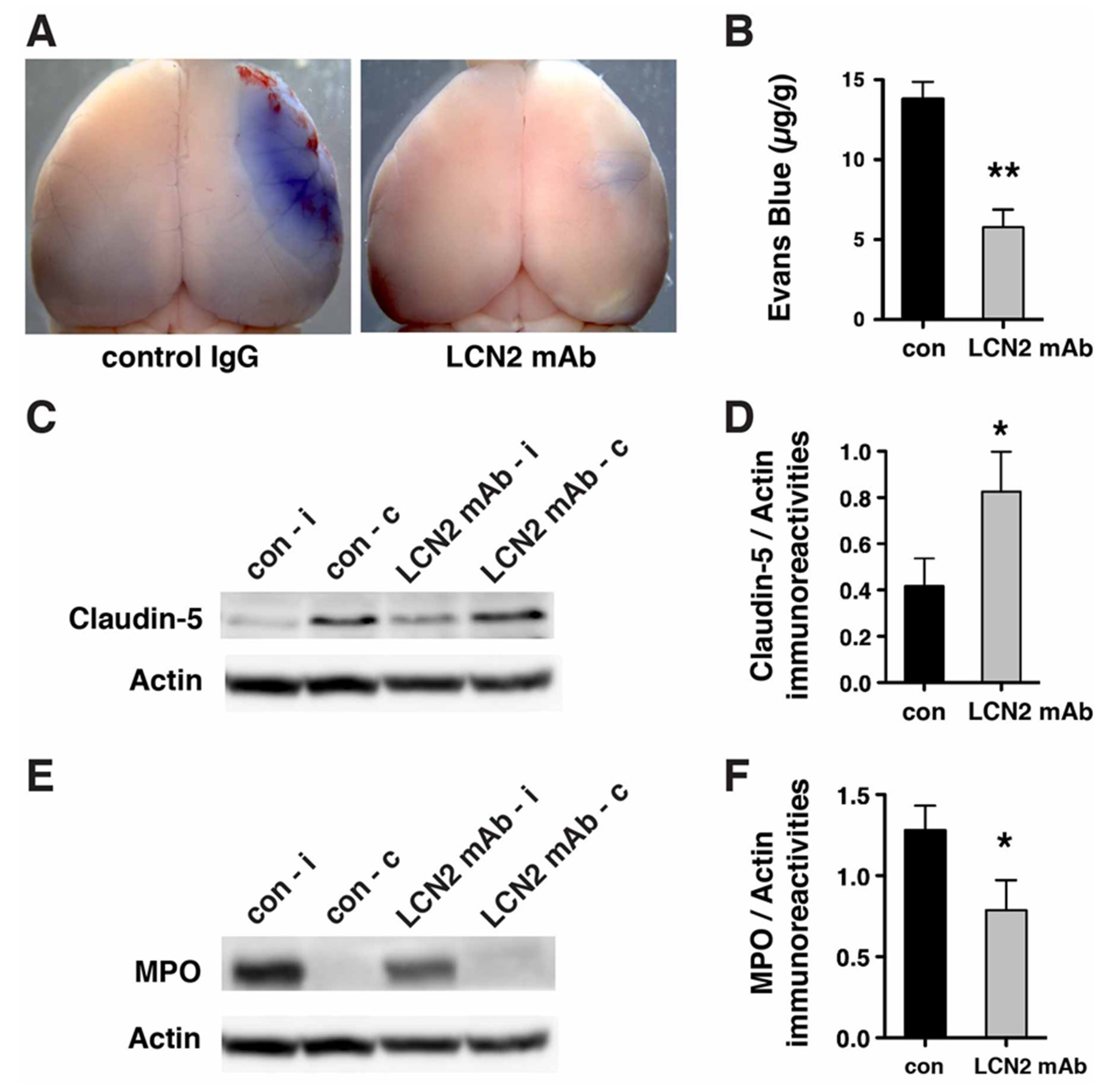
2.4. Blood–Brain Barrier Breakdown and Infiltration of Neutrophils after tMCAo were Reduced after Treatment with LCN2 mAb
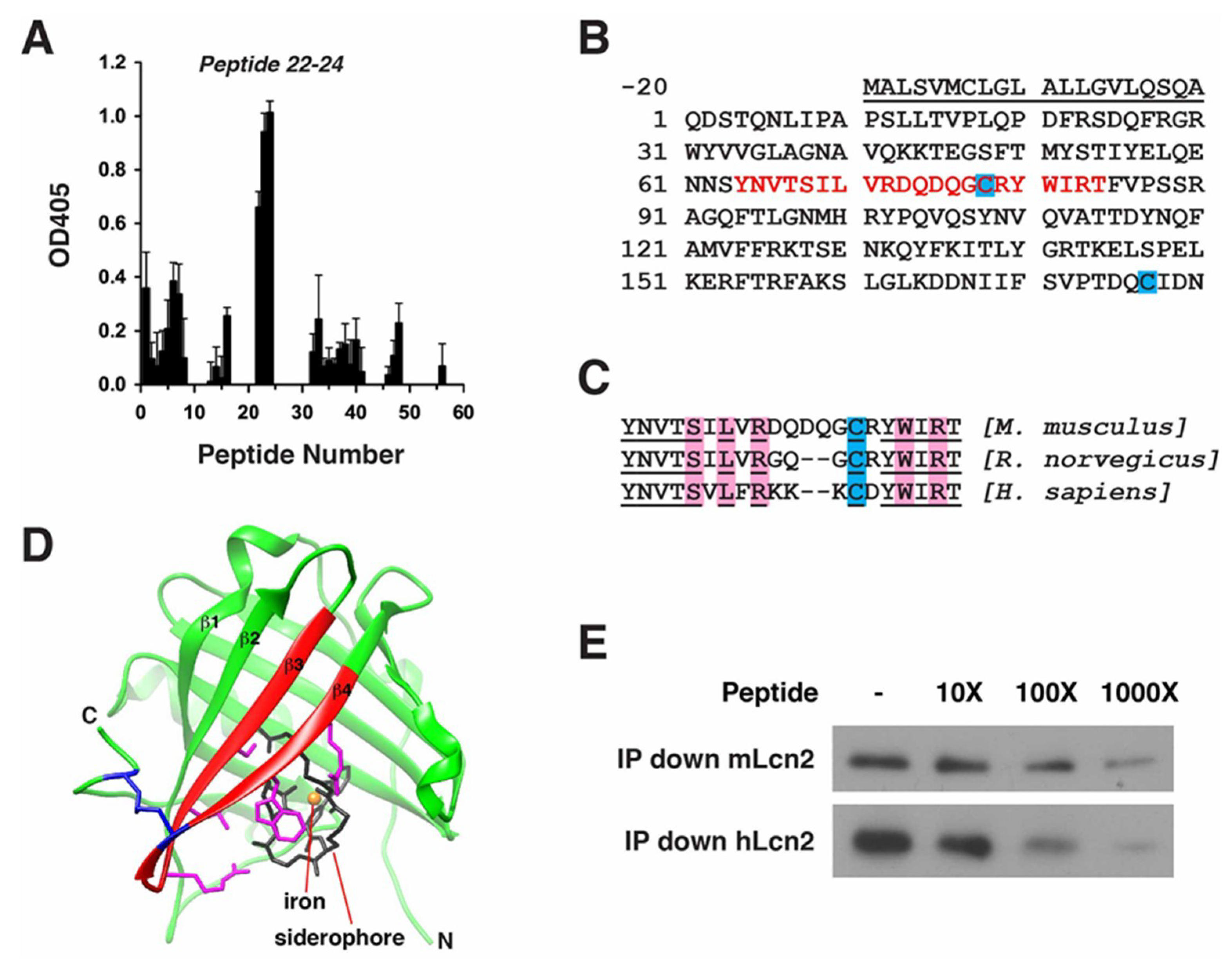
2.5. Epitope Mapping of LCN2 mAb
3. Discussion
4. Materials and Methods
4.1. Ischemic Stroke Model and LCN2 mAb Treatment
4.2. Immunofluorescence Staining
4.3. Immunoprecipitation and Western Blot Analysis of Recombinant LCN2 Protein
4.4. Immunoprecipitation, Western Blot, and ELISA Analysis of Endogenous LCN2 Protein in Mouse Brain and Blood Serum
4.5. Real-Time RT-PCR
4.6. Neurological Deficits and Corner Tests
4.7. Determination of Infarct Volume and Brain Swelling
4.8. Determination of Blood–Brain Barrier Leakage
4.9. Western Blot Analysis of Brain Homogenate after tMCAo
4.10. Epitope Mapping of LCN2 mAb
4.11. Immunoprecipitation with Competing Epitope Peptides
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LCN2 | Lipocalin-2 |
| NGAL | neutrophil gelatinase-associated lipocalin |
| tMCAo | transient middle cerebral artery occlusion |
| TTC | 2,3,5-triphenyltetrazolium chloride |
| BBB | blood–brain barrier |
| iNOS | inducible nitric oxide synthase |
| IL-6 | interleukin 6 |
| CCL2 | C-C motif chemokine ligand 2 |
| MCP-1 | monocyte chemoattractant protein-1 |
| CCL9 | C-C motif chemokine ligand 9 |
| MIP-1γ | macrophage inflammatory peptide gamma |
| tPA | tissue plasminogen activator |
| ROS | reactive oxygen species |
| i.p. | intraperitoneal injection |
| rCBF | regional cerebral blood flow |
| DAPI | 4′, 6-diamidino-2-phenylindole |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GFAP | glial fibrillary acidic protein |
| MPO | myeloperoxidase |
| PDB | Protein Data Bank archive |
| NMDA | N-methyl-d-aspartate |
| ICAM-1 | intercellular adhesion molecule-1 |
| TEER | transendothelial electrical resistance |
| CCR1 | C-C chemokine receptor type 1 |
References
- Krishnamurthi, R.V.; Feigin, V.L.; Forouzanfar, M.H.; Mensah, G.A.; Connor, M.; Bennett, D.A.; Moran, A.E.; Sacco, R.L.; Anderson, L.M.; Truelsen, T.; et al. Global and regional burden of first-ever ischaemic and haemorrhagic stroke during 1990–2010: Findings from the Global Burden of Disease Study 2010. Lancet Glob. Health 2013, 1, e259–e281. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, S.; Stanwell, P.; Cordato, D.; Attia, J.; Levi, C. Reperfusion therapy in acute ischemic stroke: Dawn of a new era? BMC Neurol. 2018, 18, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, B.C.V.; Donnan, G.A.; Lees, K.R.; Hacke, W.; Khatri, P.; Hill, M.D.; Goyal, M.; Mitchell, P.J.; Saver, J.L.; Diener, H.C.; et al. Endovascular stent thrombectomy: The new standard of care for large vessel ischaemic stroke. Lancet Neurol. 2015, 14, 846–854. [Google Scholar] [CrossRef]
- Mizuma, A.; Yenari, M.A. Anti-Inflammatory Targets for the Treatment of Reperfusion Injury in Stroke. Front. Neurol. 2017, 8, 467. [Google Scholar] [CrossRef] [Green Version]
- Chou, W.H.; Messing, R.O. Protein kinase C isozymes in stroke. Trends Cardiovasc. Med. 2005, 15, 47–51. [Google Scholar] [CrossRef]
- Sandoval, K.E.; Witt, K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef]
- De Felice, C.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Durand, T.; Valacchi, G.; Ciccoli, L.; Hayek, J. The role of oxidative stress in Rett syndrome: An overview. Ann. N. Y. Acad. Sci. 2012, 1259, 121–135. [Google Scholar] [CrossRef]
- Chou, W.H.; Choi, D.S.; Zhang, H.; Mu, D.; McMahon, T.; Kharazia, V.N.; Lowell, C.A.; Ferriero, D.M.; Messing, R.O. Neutrophil protein kinase Cdelta as a mediator of stroke-reperfusion injury. J. Clin. Investig. 2004, 114, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting neutrophils in ischemic stroke: Translational insights from experimental studies. J. Cereb. Blood Flow Metab. 2015, 35, 888–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjeldsen, L.; Cowland, J.B.; Borregaard, N. Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochim. Biophys. Acta 2000, 1482, 272–283. [Google Scholar] [CrossRef]
- Suk, K. Lipocalin-2 as a therapeutic target for brain injury: An astrocentric perspective. Prog. Neurobiol. 2016, 144, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Roudkenar, M.H.; Kuwahara, Y.; Baba, T.; Roushandeh, A.M.; Ebishima, S.; Abe, S.; Ohkubo, Y.; Fukumoto, M. Oxidative stress induced lipocalin 2 gene expression: Addressing its expression under the harmful conditions. J. Radiat. Res. 2007, 48, 39–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anwaar, I.; Gottsater, A.; Ohlsson, K.; Mattiasson, I.; Lindgarde, F. Increasing levels of leukocyte-derived inflammatory mediators in plasma and cAMP in platelets during follow-up after acute cerebral ischemia. Cereb. Dis. 1998, 8, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Hochmeister, S.; Engel, O.; Adzemovic, M.Z.; Pekar, T.; Kendlbacher, P.; Zeitelhofer, M.; Haindl, M.; Meisel, A.; Fazekas, F.; Seifert-Held, T. Lipocalin-2 as an Infection-Related Biomarker to Predict Clinical Outcome in Ischemic Stroke. PLoS ONE 2016, 11, e0154797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Cadenas, I.; Del Rio-Espinola, A.; Domingues-Montanari, S.; Mendioroz, M.; Fernandez-Morales, J.; Penalba, A.; Rubiera, M.; Hernandez-Guillamon, M.; Rosell, A.; Delgado, P.; et al. Genes involved in hemorrhagic transformations that follow recombinant t-PA treatment in stroke patients. Pharmacogenomics 2013, 14, 495–504. [Google Scholar] [CrossRef]
- Falke, P.; Elneihoum, A.M.; Ohlsson, K. Leukocyte activation: Relation to cardiovascular mortality after cerebrovascular ischemia. Cereb. Dis. 2000, 10, 97–101. [Google Scholar] [CrossRef]
- Haase, M.; Bellomo, R.; Haase-Fielitz, A. Novel biomarkers, oxidative stress, and the role of labile iron toxicity in cardiopulmonary bypass-associated acute kidney injury. J. Am. Coll. Cardiol. 2010, 55, 2024–2033. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Kim, J.H.; Jang, E.; Lee, Y.M.; Soo Han, H.; Woo, D.K.; Park, D.H.; Kook, H.; Suk, K. Lipocalin-2 deficiency attenuates neuroinflammation and brain injury after transient middle cerebral artery occlusion in mice. J. Cereb. Blood Flow. Metab. 2014, 34, 1306–1314. [Google Scholar] [CrossRef]
- Wang, G.; Weng, Y.C.; Han, X.; Whaley, J.D.; McCrae, K.R.; Chou, W.H. Lipocalin-2 released in response to cerebral ischaemia mediates reperfusion injury in mice. J. Cell. Mol. Med. 2015, 19, 1637–1645. [Google Scholar] [CrossRef]
- Yu, C.Y.; Ng, G.; Liao, P. Therapeutic antibodies in stroke. Transl. Stroke Res. 2013, 4, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Lambertsen, K.L.; Finsen, B.; Clausen, B.H. Post-stroke inflammation-target or tool for therapy? Acta Neuropathol. 2019, 137, 693–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, W.H.; Messing, R.O. Hypertensive encephalopathy and the blood-brain barrier: Is deltaPKC a gatekeeper? J. Clin. Investig. 2008, 118, 17–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, R.T.; Levine, S.T.; Haynes, S.M.; Gutierrez, P.; Baratta, J.L.; Tan, Z.; Longmuir, K.J. Use of labeled tomato lectin for imaging vasculature structures. Histochem. Cell Biol. 2015, 143, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef]
- Lee, S.; Park, J.Y.; Lee, W.H.; Kim, H.; Park, H.C.; Mori, K.; Suk, K. Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J. Neurosci. 2009, 29, 234–249. [Google Scholar] [CrossRef]
- Breckwoldt, M.O.; Chen, J.W.; Stangenberg, L.; Aikawa, E.; Rodriguez, E.; Qiu, S.; Moskowitz, M.A.; Weissleder, R. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc. Natl. Acad. Sci. USA 2008, 105, 18584–18589. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, J.H.; Kim, J.H.; Seo, J.W.; Han, H.S.; Lee, W.H.; Mori, K.; Nakao, K.; Barasch, J.; Suk, K. Lipocalin-2 Is a chemokine inducer in the central nervous system: Role of chemokine ligand 10 (CXCL10) in lipocalin-2-induced cell migration. J. Biol. Chem. 2011, 286, 43855–43870. [Google Scholar] [CrossRef] [Green Version]
- Holmes, M.A.; Paulsene, W.; Jide, X.; Ratledge, C.; Strong, R.K. Siderocalin (Lcn 2) also binds carboxymycobactins, potentially defending against mycobacterial infections through iron sequestration. Structure 2005, 13, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.C.; Wang, G.; Messing, R.O.; Chou, W.H. Identification of lipocalin-2 as a PKCdelta phosphorylation substrate in neutrophils. J. Biomed. Sci. 2015, 22, 21. [Google Scholar] [CrossRef] [Green Version]
- Olejniczak, E.T.; Ruan, Q.; Ziemann, R.N.; Birkenmeyer, L.G.; Saldana, S.C.; Tetin, S.Y. Rapid determination of antigenic epitopes in human NGAL using NMR. Biopolymers 2010, 93, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Chen, Z.Q.; Mou, R.T.; Feng, D.X.; Wang, Z.; Chen, G. The role of nitric oxide in stroke. Med. Gas Res. 2017, 7, 194–203. [Google Scholar] [PubMed] [Green Version]
- Garcia-Bonilla, L.; Moore, J.M.; Racchumi, G.; Zhou, P.; Butler, J.M.; Iadecola, C.; Anrather, J. Inducible nitric oxide synthase in neutrophils and endothelium contributes to ischemic brain injury in mice. J. Immunol. 2014, 193, 2531–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, W.M.; Rinker, L.G.; Lessov, N.S.; Hazel, K.; Eckenstein, F. Time course of IL-6 expression in experimental CNS ischemia. Neurol. Res. 1999, 21, 287–292. [Google Scholar] [CrossRef]
- de Vries, H.E.; Blom-Roosemalen, M.C.; van Oosten, M.; de Boer, A.G.; van Berkel, T.J.; Breimer, D.D.; Kuiper, J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J. Neuroimmunol. 1996, 64, 37–43. [Google Scholar] [CrossRef]
- Garcia-Berrocoso, T.; Giralt, D.; Llombart, V.; Bustamante, A.; Penalba, A.; Flores, A.; Rib’o, A.; Molina, C.A.; Rosell, A.; Montaner, J. Chemokines after human ischemic stroke: From neurovascular unit to blood using protein arrays. Transl. Proteom. 2014, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.; Bao, W.; Hong, X.; Jiang, H.; Yu, Z. Identification and functional analysis of differentially expressed genes associated with cerebral ischemia/reperfusion injury through bioinformatics methods. Mol. Med. Rep. 2018, 18, 1513–1523. [Google Scholar] [CrossRef]
- Conductier, G.; Blondeau, N.; Guyon, A.; Nahon, J.L.; Rovere, C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J. Neuroimmunol. 2010, 224, 93–100. [Google Scholar] [CrossRef]
- Hughes, P.M.; Allegrini, P.R.; Rudin, M.; Perry, V.H.; Mir, A.K.; Wiessner, C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J. Cereb. Blood Flow. Metab. 2002, 22, 308–317. [Google Scholar] [CrossRef] [Green Version]
- Dimitrijevic, O.B.; Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 2007, 38, 1345–1353. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.X.; Arumugam, T.V.; Gelderblom, M.; Magnus, T.; Drummond, G.R.; Sobey, C.G. Role of CCR2 in inflammatory conditions of the central nervous system. J. Cereb. Blood Flow. Metab. 2014, 34, 1425–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichel, C.A.; Khandoga, A.; Anders, H.J.; Schlondorff, D.; Luckow, B.; Krombach, F. Chemokine receptors Ccr1, Ccr2, and Ccr5 mediate neutrophil migration to postischemic tissue. J. Leukoc. Biol. 2006, 79, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Suhett, C.P.; Li, Z.; Marini, A.M.; Braga, M.F.; Eiden, L.E. Temporal course of changes in gene expression suggests a cytokine-related mechanism for long-term hippocampal alteration after controlled cortical impact. J. Neurotrauma 2014, 31, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.L.; Reeves, T.M.; Phillips, L.L. Osteopontin expression in acute immune response mediates hippocampal synaptogenesis and adaptive outcome following cortical brain injury. Exp. Neurol. 2014, 261, 757–771. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.; Xi, G.; Keep, R.F.; Hua, Y. Role of iron in brain lipocalin 2 upregulation after intracerebral hemorrhage in rats. Brain Res. 2013, 1505, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Egashira, Y.; Hua, Y.; Keep, R.F.; Xi, G. Acute white matter injury after experimental subarachnoid hemorrhage: Potential role of lipocalin 2. Stroke 2014, 45, 2141–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, W.; Zheng, M.; Xi, G.; Keep, R.F.; Hua, Y. Role of lipocalin-2 in brain injury after intracerebral hemorrhage. J. Cereb. Blood Flow. Metab. 2015, 35, 1454–1461. [Google Scholar] [CrossRef] [Green Version]
- Rathore, K.I.; Berard, J.L.; Redensek, A.; Chierzi, S.; Lopez-Vales, R.; Santos, M.; Akira, S.; David, S. Lipocalin 2 plays an immunomodulatory role and has detrimental effects after spinal cord injury. J. Neurosci. 2011, 31, 13412–13419. [Google Scholar] [CrossRef] [Green Version]
- Nam, Y.; Kim, J.H.; Seo, M.; Kim, J.H.; Jin, M.; Jeon, S.; Seo, J.W.; Lee, W.H.; Bing, S.J.; Jee, Y.; et al. Lipocalin-2 protein deficiency ameliorates experimental autoimmune encephalomyelitis: The pathogenic role of lipocalin-2 in the central nervous system and peripheral lymphoid tissues. J. Biol. Chem. 2014, 289, 16773–16789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowland, J.B.; Muta, T.; Borregaard, N. IL-1beta-specific up-regulation of neutrophil gelatinase-associated lipocalin is controlled by IkappaB-zeta. J. Immunol. 2006, 176, 5559–5566. [Google Scholar] [CrossRef] [Green Version]
- Devireddy, L.R.; Gazin, C.; Zhu, X.; Green, M.R. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 2005, 123, 1293–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manwani, B.; McCullough, L.D. Sexual dimorphism in ischemic stroke: Lessons from the laboratory. Womens Health 2011, 7, 319–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Yuan, R.; Benashski, S.E.; McCullough, L.D. Changes in experimental stroke outcome across the life span. J. Cereb. Blood Flow. Metab. 2009, 29, 792–802. [Google Scholar] [CrossRef] [Green Version]
- Chiang, T.; Messing, R.O.; Chou, W.H. Mouse model of middle cerebral artery occlusion. J. Vis. Exp. JOVE 2011, 48, 2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Weng, Y.C.; Wu, Y.C.; Huang, Y.T.; Chou, W.H. PKCepsilon phosphorylation regulates the mitochondrial translocation of ATF2 in ischemia-induced neurodegeneration. BMC Neurosci. 2018, 19, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, W.H.; Wang, D.; McMahon, T.; Qi, Z.H.; Song, M.; Zhang, C.; Shokat, K.M.; Messing, R.O. GABAA receptor trafficking is regulated by protein kinase C(epsilon) and the N-ethylmaleimide-sensitive factor. J. Neurosci. 2010, 30, 13955–13965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2^(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 2013, 3, 71–85. [Google Scholar]
- Rose, L.; Bakal, D.A.; Fung, T.S.; Farn, P.; Weaver, L.E. Tactile extinction and functional status after stroke. A preliminary investigation. Stroke 1994, 25, 1973–1976. [Google Scholar] [CrossRef] [Green Version]
- Dobkin, B.H. The rehabilitation of elderly stroke patients. Clin. Geriatr. Med. 1991, 7, 507–523. [Google Scholar] [CrossRef]
- Zhang, L.; Schallert, T.; Zhang, Z.G.; Jiang, Q.; Arniego, P.; Li, Q.; Lu, M.; Chopp, M. A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J. Neurosci. Methods 2002, 117, 207–214. [Google Scholar] [CrossRef]
- Manaenko, A.; Chen, H.; Kammer, J.; Zhang, J.H.; Tang, J. Comparison Evans Blue injection routes: Intravenous versus intraperitoneal, for measurement of blood-brain barrier in a mice hemorrhage model. J. Neurosci. Methods 2011, 195, 206–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahar, J.E.; Donker, N.C.; Bok, K.; Talbo, G.H.; Green, K.Y.; Kirkwood, C.D. Identification and characterization of antibody-binding epitopes on the norovirus GII.3 capsid. J. Virol. 2014, 88, 1942–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Number of Mice Used for Experiments | Mortality Rate | Number of Mice Used in the Figures |
|---|---|---|---|
| Figure 1 and Figure 2 | naive LCN2+/+: 3 | 0 | 3 |
| (staining) | LCN2+/+ tMCAo: 3 | 0 | 3 |
| Figure 3B,C | naive LCN2+/+: 3 | 0 | 3 |
| (Immunoprecipitation–Western) | naive LCN2−/−: 3 | 0 | 3 |
| LCN2+/+ tMCAo: 3 | 0 | 3 | |
| LCN2−/− tMCAo: 3 | 0 | 3 | |
| Figure 3E,F (ELISA) | LCN2+/+ tMCAo: 10 | 0 | 10 |
| LCN2+/+ tMCAo-Ab: 10 | 1/10 (10.0%) | 9 | |
| Figure 3D and Figure 6 | naive LCN2+/+: 6 | 0 | 6 |
| (real-time RT-PCR) | naive LCN2−/−: 6 | 0 | 6 |
| LCN2+/+ tMCAo: 8 | 2/8 (25.0%) | 6 | |
| LCN2−/− tMCAo: 7 | 1/7 (14.3%) | 6 | |
| LCN2+/+ tMCAo-Ab: 7 | 1/7 (14.3%) | 6 | |
| Figure 4A–E (TTC staining) | LCN2+/+ tMCAo: 8 | 1/8 (12.5%) | 7 |
| LCN2+/+ tMCAo-Ab: 8 | 1/8 (12.5%) | 7 | |
| Figure 5A,B (blood–brain barrier (BBB)) | LCN2+/+ tMCAo: 6 | 1/6 (16.6%) | 5 |
| LCN2+/+ tMCAo-Ab: 6 | 1/6 (16.6%) | 5 | |
| Figure 5C–F | LCN2+/+ tMCAo: 4 | 0 | 4 |
| (BBB–Western) | LCN2+/+ tMCAo-Ab: 4 | 0 | 4 |
| Total | 108 | 9/108 (8.3%) | 99 |
| Mouse cDNA | Primer Sequences |
|---|---|
| LCN2 | F, 5′-ATG TCA CCT CCA TCC TGG TC-3′ |
| R, 5′-CAC ACT CAC CAC CCA TTC AG-3′ | |
| iNOS | F, 5′-GCC ACC AAC AAT GGC AAC A-3′ |
| R, 5′-CGT ACC GGA TGA GCT GTG AAT T-3′ | |
| IL6 | F, 5′-AGT TGC CTT CTT GGG ACT GA-3′ |
| R, 5′-TCC ACG ATT TCC CAG AGA AC-3′ | |
| CCL2 | F, 5′-TCA GCC AGA TGC AGT TAA CG-3′ |
| R, 5′-GAT CCT CTT GTA GCT CTC CAG C-3′ | |
| CCL9 | F, 5′-CAA CAG AGA CAA AAG AAG TCC AGA G-3′ |
| R, 5′-CTT GCT GAT AAA GAT GAT GCC C-3′ | |
| GAPDH | F, 5′-ACC ACA GTC CAT GCC ATC AC-3′ |
| R, 5′-CAC CAC CCT GTT GCT GTA GCC-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, G.; Weng, Y.-C.; Chiang, I.-C.; Huang, Y.-T.; Liao, Y.-C.; Chen, Y.-C.; Kao, C.-Y.; Liu, Y.-L.; Lee, T.-H.; Chou, W.-H. Neutralization of Lipocalin-2 Diminishes Stroke-Reperfusion Injury. Int. J. Mol. Sci. 2020, 21, 6253. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176253
Wang G, Weng Y-C, Chiang I-C, Huang Y-T, Liao Y-C, Chen Y-C, Kao C-Y, Liu Y-L, Lee T-H, Chou W-H. Neutralization of Lipocalin-2 Diminishes Stroke-Reperfusion Injury. International Journal of Molecular Sciences. 2020; 21(17):6253. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176253
Chicago/Turabian StyleWang, Guona, Yi-Chinn Weng, I-Chen Chiang, Yu-Ting Huang, Yi-Chu Liao, Yi-Chun Chen, Cheng-Yuan Kao, Yu-Li Liu, Tsong-Hai Lee, and Wen-Hai Chou. 2020. "Neutralization of Lipocalin-2 Diminishes Stroke-Reperfusion Injury" International Journal of Molecular Sciences 21, no. 17: 6253. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176253