Assessing Lysosomal Disorders in the NGS Era: Identification of Novel Rare Variants

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. NGS Results
2.2. Novel Variants
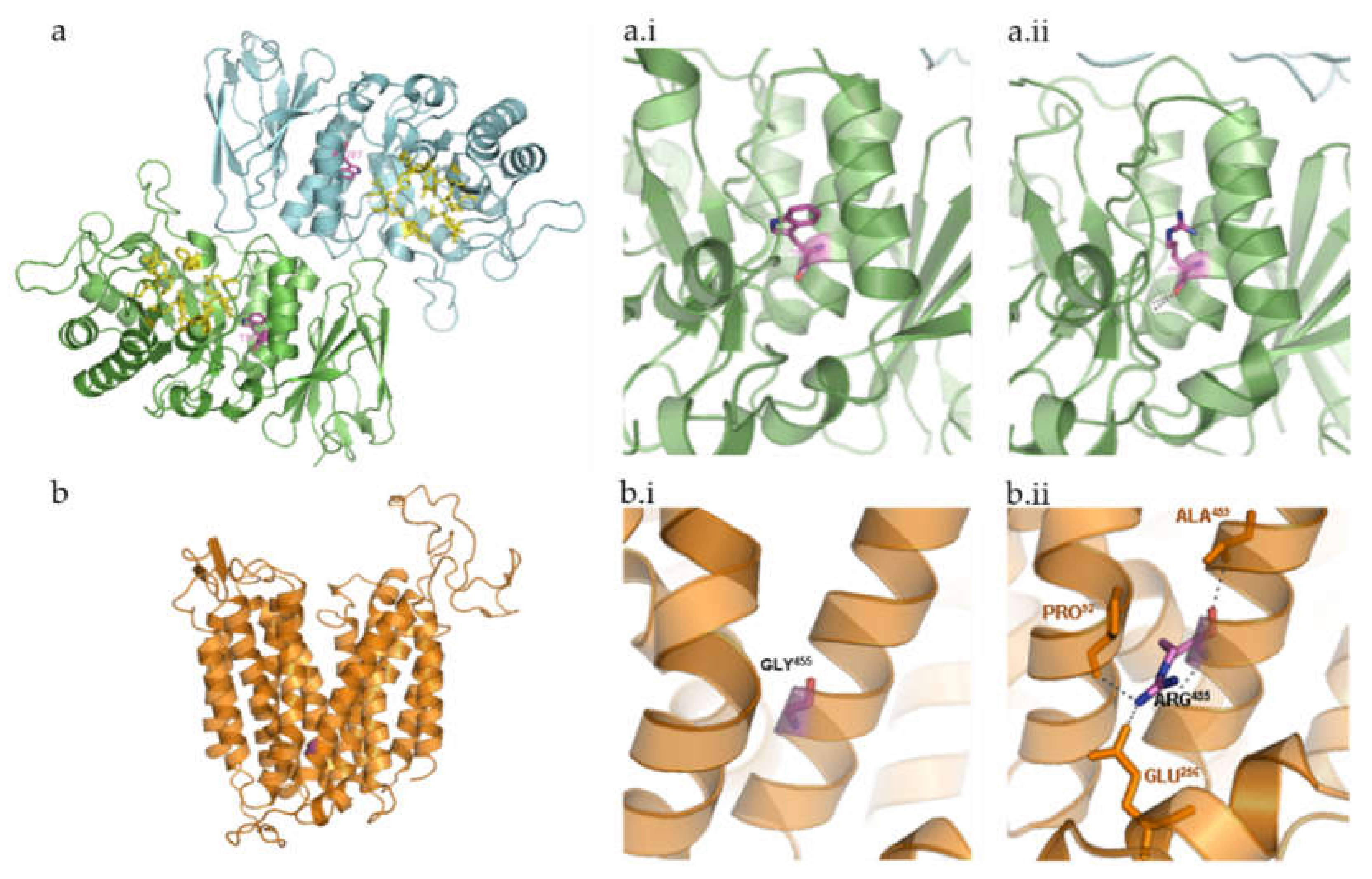
2.2.1. Shingolipidoses: p.Trp287Arg (GLA Gene), p.Gly104GlyfsTer14 (GM2A Gene) and p.Tyr205Ter (GALC Gene)
2.2.2. Neuronal Ceroid Lipofuscinoses: p.Gly455Arg (MFSD8 Gene)
2.3. Previously Reported Pathogenic Variants
2.3.1. Shingolipidoses: p.Leu483Pro (GBA Gene)
2.3.2. Mucopolysaccharidoses: p.Asp312Asn (NAGLU Gene)
2.3.3. Glycoproteinoses: c.2402dup (MAN2B1 Gene)
2.3.4. Other Enzyme Defects: p.Gly146Arg (CTSK Gene)
2.3.5. Post-Translational Modification Defects: p.Val191Ile (GNPTAB Gene)
3. Discussion
4. Materials and Methods
4.1. Sample/Subjects
4.2. Next Generation Sequencing
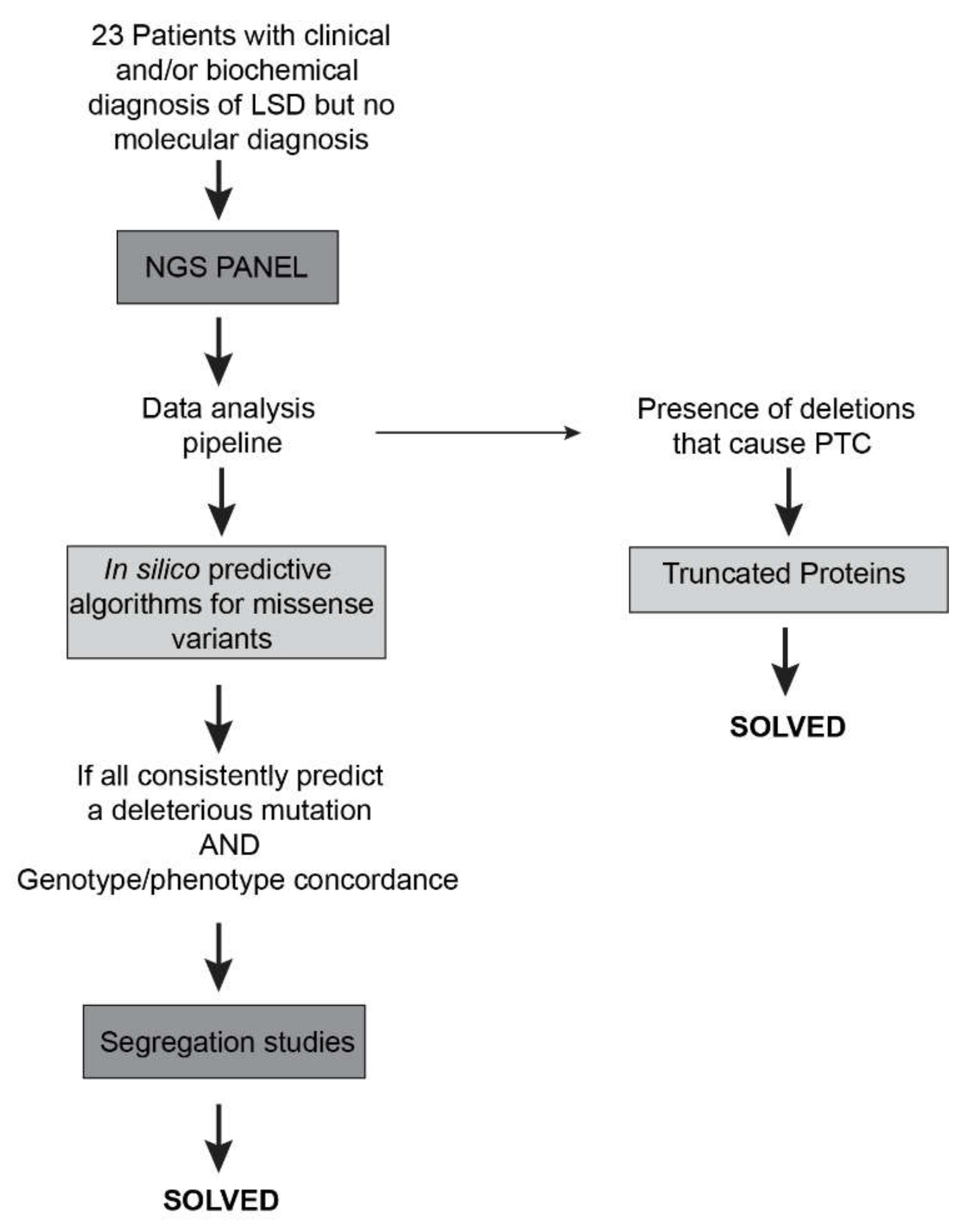
4.3. NGS Data Analysis
4.4. Sanger Sequencing Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CTSK | cathepsin K |
| GCase | β-glucocerebrosidase |
| GD | Gaucher disease |
| GLA | α-galactosidase |
| GM2AP | GM2 activator protein |
| ID | intellectual disability |
| LAMAN | α-mannosidase (also known as MAN2B1) |
| LSDs | Lysosomal Storage Diseases |
| MAF | minor allele frequency |
| MFSD8 | Major Facilitator Superfamily Domain Containing 8 |
| MGT | molecular genetic testing |
| MPS | Mucopolysaccharidosis |
| NAGLU | α-N-acetylglucosaminidase |
| NCL | neuronal ceroid lipofuscinosis |
| NGS | Next-generation sequencing |
| NMD | nonsense mediated mRNA decay |
| PTC | premature termination codon |
| SNP | single-nucleotide polymorphism |
| VUS | variants of unknown significance |
| WES | whole exome sequencing |
| WGS | whole genome sequencing |
References
- Schultz, M.L.; Tecedor, L.; Chang, M.; Davidson, B.L. Clarifying lysosomal storage diseases. Trends Neurosci. 2011, 34, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futerman, A.H.; Van Meer, G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 2004, 5, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018. [Google Scholar] [CrossRef]
- Winchester, B. Lysosomal diseases: Diagnostic update. J. Inherit. Metab. Dis. 2014, 37, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Filocamo, M.; Morrone, A. Lysosomal storage disorders: Molecular basis and laboratory testing. Hum. Genom. 2011. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wang, J.-L.; Tang, B.-S.; Sun, Z.-F.; Shi, Y.-T.; Shen, L.; Lei, L.-F.; Wei, X.-M.; Xiao, J.-J.; Hu, Z.-M.; et al. Using next-generation sequencing as a genetic diagnostic tool in rare autosomal recessive neurologic Mendelian disorders. Neurobiol. Aging 2013, 34, 2442.e11–2442.e17. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological chaperone therapy: Preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol. Ther. 2015. [Google Scholar] [CrossRef] [Green Version]
- Valenzano, K.J.; Khanna, R.; Powe, A.C.; Boyd, R.; Lee, G.; Flanagan, J.J.; Benjamin, E.R. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay Drug Dev. Technol. 2011. [Google Scholar] [CrossRef] [Green Version]
- Hechtman, P.; Gordon, B.A.; Ying, N.M.K.N. Deficiency of the hexosaminidase a activator protein in a case of GM2 gangliosidosis; variant, AB. Pediatr. Res. 1982, 16, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.; Kohlschütter, A.; Mink, J.; Simonati, A.; Williams, R. NCL diseases—Clinical perspectives. Biochim. Biophys. Acta Mol. Basis Dis. 2013. [Google Scholar] [CrossRef] [Green Version]
- Kousi, M.; Lehesjoki, A.E.; Mole, S.E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [Google Scholar] [CrossRef] [PubMed]
- Jalanko, A.; Braulke, T. Neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta Mol. Cell Res. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.R.; Dahl, H.H.M.; Canafoglia, L.; Andermann, E.; Damiano, J.; Morbin, M.; Bruni, A.C.; Giaccone, G.; Cossette, P.; Saftig, P.; et al. Cathepsin F mutations cause Type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Choudary, P.V.; Martin, B.M.; Stubblefield, B.K.; Mayor, J.A.; Barranger, J.A.; Ginns, E.I. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N. Engl. J. Med. 1987, 316, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Gelb, B.D.; Shi, G.-P.; Chapman, H.A.; Desnick, R.J. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996. [Google Scholar] [CrossRef]
- Velho, R.V.; Harms, F.L.; Danyukova, T.; Ludwig, N.F.; Friez, M.J.; Cathey, S.S.; Filocamo, M.; Tappino, B.; Güneş, N.; Tüysüz, B.; et al. The lysosomal storage disorders mucolipidosis type II, type III alpha/beta, and type III gamma: Update on GNPTAB and GNPTG mutations. Hum. Mutat. 2019, 40, 842–864. [Google Scholar] [CrossRef]
- Héron, B.; Mikaeloff, Y.; Froissart, R.; Caridade, G.; Maire, I.; Caillaud, C.; Levade, T.; Chabrol, B.; Feillet, F.; Ogier, H.; et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am. J. Med. Genet. Part A 2011. [Google Scholar] [CrossRef]
- Riise Stensland, H.M.F.; Klenow, H.B.; Van Nguyen, L.; Hansen, G.M.; Malm, D.; Nilssen, Ø. Identification of 83 novel alpha-mannosidosis-associated sequence variants: Functional analysis of MAN2B1 missense mutations. Hum. Mutat. 2012. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden markov models. Hum. Mutat. 2013. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. Mutationtaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef] [Green Version]
- Cimmaruta, C.; Citro, V.; Andreotti, G.; Liguori, L.; Cubellis, M.V.; Hay Mele, B. Challenging popular tools for the annotation of genetic variations with a real case, pathogenic mutations of lysosomal alpha-galactosidase. BMC Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garman, S.C.; Garboczi, D.N. Structural basis of Fabry disease. Mol. Genet. Metab. 2002, 77, 3–11. [Google Scholar] [CrossRef]
- Garman, S.C.; Garboczi, D.N. The molecular defect leading to fabry disease: Structure of human α-galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef]
- Davies, J.P.; Winchester, B.G.; Malcolm, S. Mutation analysis in patients with the typical form of anderson—Fabry disease. Hum. Mol. Genet. 1993, 2, 1051–1053. [Google Scholar] [CrossRef]
- Davies, J.P. Fabry disease: Fourteen α-gaiactosidase a mutations in unrelated families from the United Kingdom and other european countries. Eur. J. Hum. Genet. 1996, 4, 219–224. [Google Scholar] [CrossRef]
- Eng, C.M.; Ashley, G.A.; Burgert, T.S.; Enriquez, A.L.; D’Souza, M.; Desnick, R.J. Fabry disease: Thirty-five mutations in the α-galactosidase a gene in patients with classic and variant phenotypes. Mol. Med. 1997, 3, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Sandhoff, K.; Harzer, K. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. 2013. [Google Scholar] [CrossRef]
- Kolter, T.; Sandhoff, K. Sphingolipid metabolism diseases. Biochim. Biophys. Acta Biomembr. 2006. [Google Scholar] [CrossRef] [Green Version]
- Siintola, E.; Topcu, M.; Aula, N.; Lohi, H.; Minassian, B.A.; Paterson, A.D.; Liu, X.Q.; Wilson, C.; Lahtinen, U.; Anttonen, A.K.; et al. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am. J. Hum. Genet. 2007, 81, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kousi, M.; Siintola, E.; Dvorakova, L.; Vlaskova, H.; Turnbull, J.; Topcu, M.; Yuksel, D.; Gokben, S.; Minassian, B.A.; Elleder, M.; et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain 2009, 132, 810–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, A.J.; Ribeiro, D.; Moreira, L.; Amaral, O. In silico analysis of missense mutations as a first step in functional studies: Examples from two sphingolipidoses. Int. J. Mol. Sci. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufeld E., F.; Muenzer, J. The Mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease, 9th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Childs, B., Kinzler, K.W., Vogelstein, B., Eds.; McGrawHill: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Darvish, H.; Azcona, L.J.; Tafakhori, A.; Mesias, R.; Ahmadifard, A.; Sanchez, E.; Habibi, A.; Alehabib, E.; Johari, A.H.; Emamalizadeh, B.; et al. Phenotypic and genotypic characterization of families with complex intellectual disability identified pathogenic genetic variations in known and novel disease genes. Sci. Rep. 2020. [Google Scholar] [CrossRef] [Green Version]
- Bunge, S.; Knigge, A.; Steglich, C.; Kleijer, W.J.; van Diggelen, O.P.; Beck, M.; Gal, A. Mucopolysaccharidosis type IIIB (Sanfilippo B): Identification of 18 novel alpha-N-acetylglucosaminidase gene mutations. J. Med. Genet. 1999, 36, 28–31. [Google Scholar]
- Selmer, K.K.; Gilfillan, G.D.; Strømme, P.; Lyle, R.; Hughes, T.; Hjorthaug, H.S.; Brandal, K.; Nakken, S.; Misceo, D.; Egeland, T.; et al. A mild form of Mucopolysaccharidosis IIIB diagnosed with targeted next-generation sequencing of linked genomic regions. Eur. J. Hum. Genet. 2012. [Google Scholar] [CrossRef]
- Wood, T.C.; Harvey, K.; Beck, M.; Burin, M.G.; Chien, Y.H.; Church, H.J.; D’Almeida, V.; Van Diggelen, O.P.; Fietz, M.; Giugliani, R.; et al. Diagnosing mucopolysaccharidosis IVA. J. Inherit. Metab. Dis. 2013. [Google Scholar] [CrossRef] [Green Version]
- Kollmann, K.; Pohl, S.; Marschner, K.; Encarnação, M.; Sakwa, I.; Tiede, S.; Poorthuis, B.J.; Lübke, T.; Müller-Loennies, S.; Storch, S.; et al. Mannose phosphorylation in health and disease. Eur. J. Cell Biol. 2010. [Google Scholar] [CrossRef]
- Human Splicing Finder. Available online: www.umd.be/HSF3/technicaltips.html (accessed on 15 December 2017).
- Zampieri, S.; Cattarossi, S.; Bembi, B.; Dardis, A. GBA analysis in next-generation era: Pitfalls, challenges, and possible solutions. J. Mol. Diagn. 2017. [Google Scholar] [CrossRef] [Green Version]
- Málaga, D.R.; Brusius-Facchin, A.C.; Siebert, M.; Pasqualim, G.; Pereira, M.L.S.; De Souza, C.F.M.; Schwartz, I.V.D.; Matte, U.; Giugliani, R. Sensitivity, advantages, limitations, and clinical utility of targeted next-generation sequencing panels for the diagnosis of selected lysosomal storage disorders. Genet. Mol. Biol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Marmiesse, A.; Morey, M.; Pineda, M.; Eiris, J.; Couce, M.L.; Castro-Gago, M.; Fraga, J.M.; Lacerda, L.; Gouveia, S.; Pérez-Poyato, M.S.; et al. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet J. Rare Dis. 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Sperb-Ludwig, F.; Alegra, T.; Velho, R.V.; Ludwig, N.; Kim, C.A.; Kok, F.; Kitajima, J.P.; Van Meel, E.; Kornfeld, S.; Burin, M.G.; et al. Exome sequencing for mucolipidosis III: Detection of a novel GNPTAB gene mutation in a patient with a very mild phenotype. Mol. Genet. Metab. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Di Fruscio, G.; Schulz, A.; De Cegli, R.; Savarese, M.; Mutarelli, M.; Parenti, G.; Banfi, S.; Braulke, T.; Nigro, V.; Ballabio, A. Lysoplex: An efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy 2015, 11, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Di Fruscio, G.; Banfi, S.; Nigro, V.; Ballabio, A. Next-generation sequencing approaches to define the role of the autophagy lysosomal pathway in human disease: The example of lysoplex. Methods Mol. Biol. 2017. [Google Scholar] [CrossRef]
- Yoshida, S.; Kido, J.; Matsumoto, S.; Momosaki, K.; Mitsubuchi, H.; Shimazu, T.; Sugawara, K.; Endo, F.; Nakamura, K. Prenatal diagnosis of Gaucher disease using next-generation sequencing. Pediatr. Int. 2016. [Google Scholar] [CrossRef]
- Mori, M.; Haskell, G.; Kazi, Z.; Zhu, X.; DeArmey, S.M.; Goldstein, J.L.; Bali, D.; Rehder, C.; Cirulli, E.T.; Kishnani, P.S. Sensitivity of whole exome sequencing in detecting infantile- and late-onset Pompe disease. Mol. Genet. Metab. 2017. [Google Scholar] [CrossRef]
- Tsai, A.C.H.; Hung, Y.W.; Harding, C.; Koeller, D.M.; Wang, J.; Wong, L.J.C. Next generation deep sequencing corrects diagnostic pitfalls of traditional molecular approach in a patient with prenatal onset of Pompe disease. Am. J. Med. Genet. Part A 2017. [Google Scholar] [CrossRef]
- Song, H.K.; Sohn, Y.B.; Choi, Y.J.; Chung, Y.S.; Jang, J.H. A case report of pycnodysostosis with atypical femur fracture diagnosed by next-generation sequencing of candidate genes. Medicine 2017. [Google Scholar] [CrossRef]
- Ben Halim, N.; Ben Alaya Bouafif, N.; Romdhane, L.; Kefi Ben Atig, R.; Chouchane, I.; Bouyacoub, Y.; Arfa, I.; Cherif, W.; Nouira, S.; Talmoudi, F.; et al. Consanguinity, endogamy, and genetic disorders in Tunisia. J. Community Genet. 2013. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, M.F.; Encarnacao, M.; Matos, L.; Silva, L.; Ribeiro, D.; Santos, J.I.; Prata, M.J.; Vilarinho, L.; Alves, S. Molecular characterization of a novel splicing mutation underlying mucopolysaccharidosis (MPS) type VI-indirect proof of principle on its pathogenicity. Diagnostics 2020, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Zhao, Y.; Wang, X.; Fan, J.; Heng, J.; Liu, X.; Feng, W.; Kang, X.; Huang, B.; Liu, J.; et al. Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. USA 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef]
- Vockley, J.; Dobrowolski, S.F.; Arnold, G.L.; Guerrero, R.B.; Derks, T.G.J.; Weinstein, D.A. Complex patterns of inheritance, including synergistic heterozygosity, in inborn errors of metabolism: Implications for precision medicine driven diagnosis and treatment. Mol. Genet. Metab. 2019. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Molecular Diagnosis (#MIM) | Gene (RefSeq) | cDNA | Protein | Variant Type | In Silico Predictors # | Reference | Clinical Data (When Available)/Clinical Suspicion | Biochemical Phenotype | Origin |
|---|---|---|---|---|---|---|---|---|---|---|
| P1 | Gaucher type I (#230800) | GBA NM_001005741.2 | c. 1448T>C/c. 1448T>C | p.Leu483Pro/p.Leu483Pro | Missense | Pathogenic | [15] | At nine months: mild hepatomegaly; exuberant splenomegaly; feeding difficulties and dysphagia; bilateral convergent strabismus; marked axial hypotonia; poor facial mimic; global psychomotor development delay; cardiomegaly with dilatation of the left cavities; interstitial lung disease with multiple recurrent infections including aspiration pneumonia; three cardiac arrest events. Cerebral MRI showed supratentorial periventricular white matter alterations of tegmentum pontis and dentate nucleus, all aspects which are compatible with central nervous involvement in the context of Gaucher disease | Low GCase levels in skin fibroblasts | Cape Verdean |
| P2 | Fabry disease (#301500) | GLA NM_000169 | c.859T>C | p.Trp287Arg | Missense | Pathogenic | Novel | Fabry disease | - | Tunisian |
| P3 | Pycnodysostosis (265800) | CTSK NM_000396.3 | c.436G>C/c.436G>C | p.Gly146Arg/p.Gly146Arg | Missense | Pathogenic | [16] | Pyknodysostosis | - | Tunisian |
| P4 | Mucolipidosis type II (#252500) | GNPTAB NM_024312.4 | c.571G>A/c.571G>A | p.Val191Ile/p.Val191Ile | Missense | Mostly pathogenic | [17] | Mucolipidosis type II | - | Indian |
| P5 | CLN7 (#610951) | MFSD8 NM_152778.2 | c.1363G>C/c.1363G>C | p.Gly455Arg/p.Gly455Arg | Missense | Pathogenic | Novel | Neuronal ceroid lipofuscinosis | - | Tunisian |
| P6 | Krabbe disease (#245200) | GALC NM_000153.3 | c.613_617del/c.613_617del | p.Tyr205Ter/p.Tyr205Ter | Deletion | NA | Novel | Krabbe disease | - | Tunisian |
| P7 | GM2A-gangliosidosis AB-variant (#272750) | GM2A NM_000405.4 | c.312del/c.312del | p.Gly104Gly fsTer14/p.Gly104Gly fsTer14 | Deletion | NA | Novel | Neurodegenerative LSD (not specified) | - | Tunisian |
| P10 | MPSIIIB (#252920) | NAGLU NM_000263.3 | c.934G>A/c.934G>A | p.Asp312Asn/p.Asp312Asn | Missense | Pathogenic | [18] | Mucopolysaccharidosis | - | Tunisian |
| P13 | Alpha-mannosidosis (#248500) | MAN2B1 NM_000528.3 | c.2402dup/c.2402dup | p.Ser802Gln fsTer21/p.Ser802Gln fsTer21 | Duplication | Pathogenic | [19] | Alpha-mannosidosis | - | Tunisian |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Encarnação, M.; Coutinho, M.F.; Silva, L.; Ribeiro, D.; Ouesleti, S.; Campos, T.; Santos, H.; Martins, E.; Cardoso, M.T.; Vilarinho, L.; et al. Assessing Lysosomal Disorders in the NGS Era: Identification of Novel Rare Variants. Int. J. Mol. Sci. 2020, 21, 6355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176355
Encarnação M, Coutinho MF, Silva L, Ribeiro D, Ouesleti S, Campos T, Santos H, Martins E, Cardoso MT, Vilarinho L, et al. Assessing Lysosomal Disorders in the NGS Era: Identification of Novel Rare Variants. International Journal of Molecular Sciences. 2020; 21(17):6355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176355
Chicago/Turabian StyleEncarnação, Marisa, Maria Francisca Coutinho, Lisbeth Silva, Diogo Ribeiro, Souad Ouesleti, Teresa Campos, Helena Santos, Esmeralda Martins, Maria Teresa Cardoso, Laura Vilarinho, and et al. 2020. "Assessing Lysosomal Disorders in the NGS Era: Identification of Novel Rare Variants" International Journal of Molecular Sciences 21, no. 17: 6355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176355