Methods Favoring Homology-Directed Repair Choice in Response to CRISPR/Cas9 Induced-Double Strand Breaks

 ,
,
Abstract
:1. Introduction
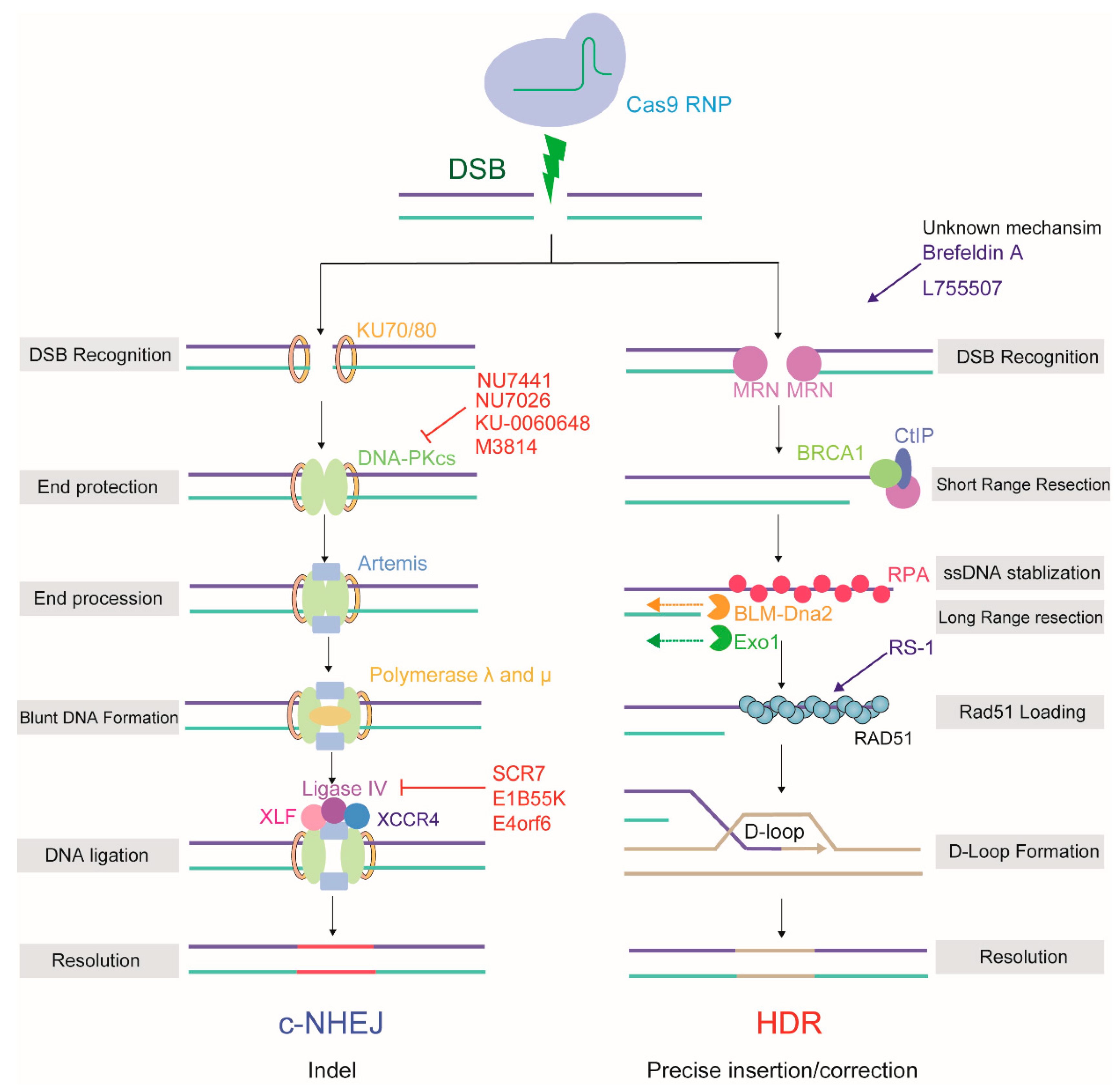
2. The Mechanism of DSB Repair Used by the CRISPR/Cas9 System
2.1. HDR
2.2. NHEJ/c-NHEJ
2.3. Two Additional Pathways
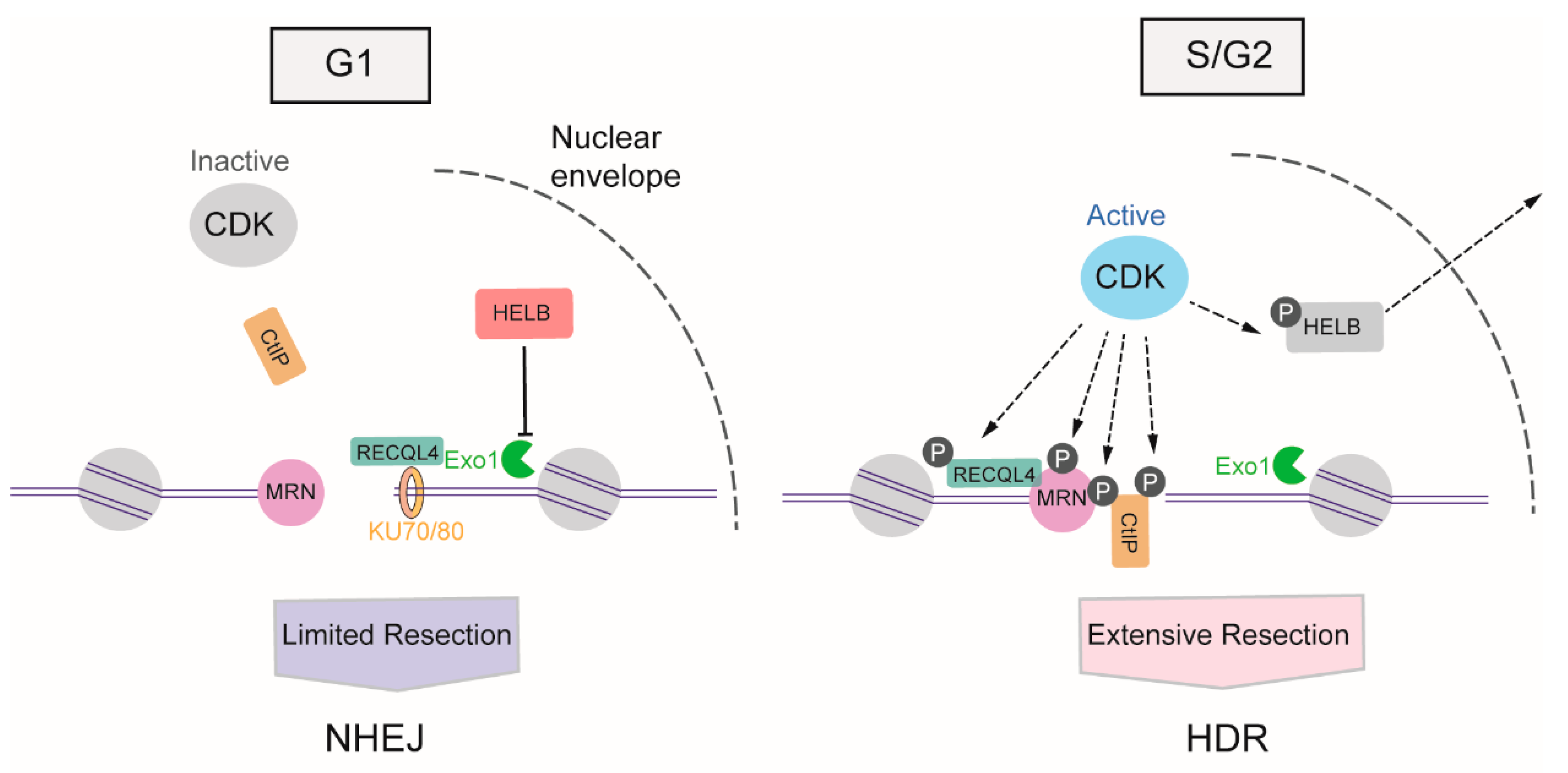
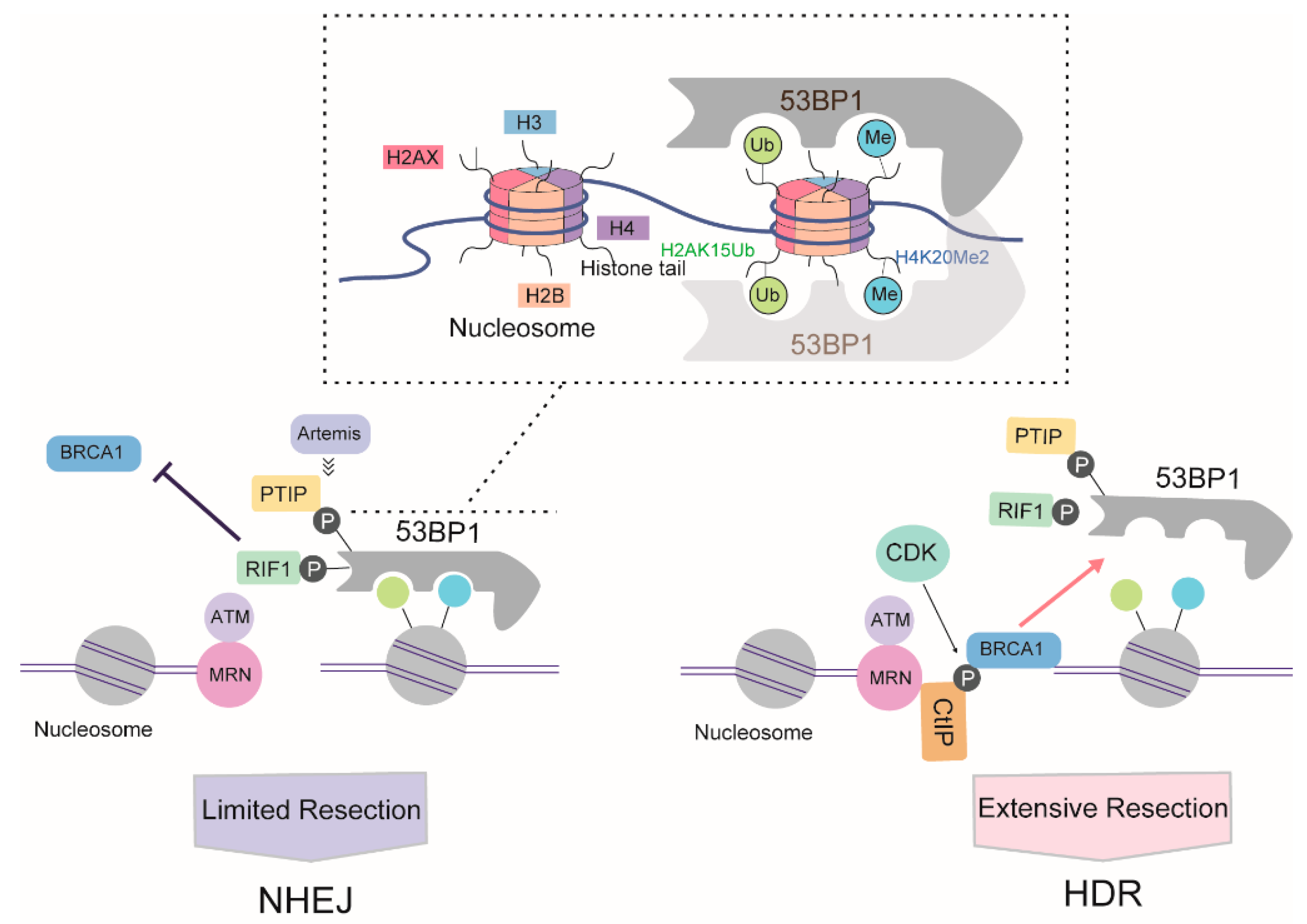
3. The Mechanism Regulating the Choice between the HDR and NHEJ Repair Pathways
3.1. The Regulation of DNA Resection by Cyclin-Dependent Kinases
3.2. The Regulation of DNA Resection by Ubiquitin
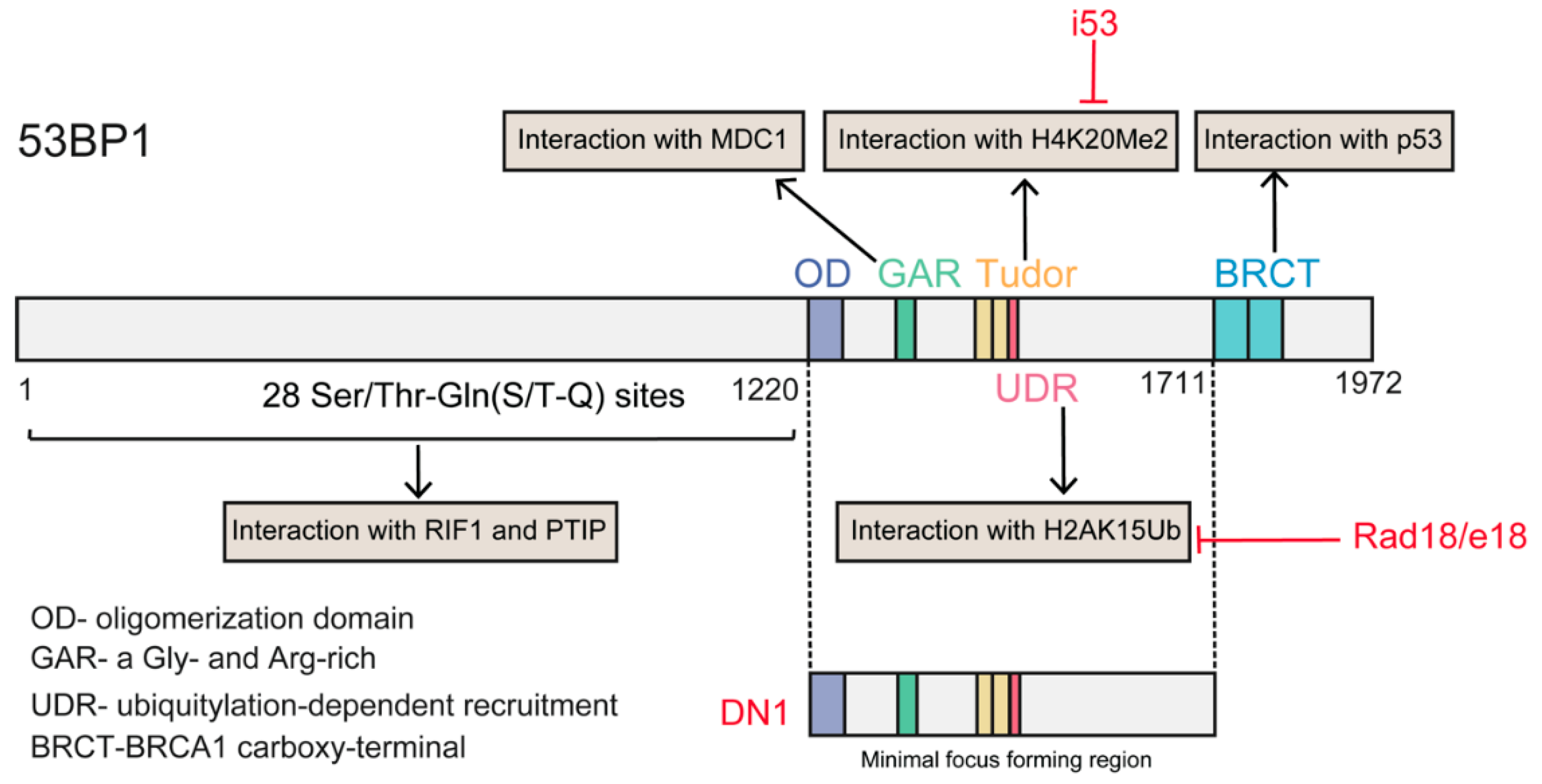
3.3. The Battle between 53BP1 and BRCA1 for DNA Resection
4. Methods to Enhance CRISPR/Cas9-Mediated HDR Choice
4.1. Regulation of the Trade-Off between c-NHEJ and HDR
4.1.1. Suppression of Key NHEJ Factors
4.1.2. In Favor of HDR Factors
4.1.3. Manipulation of the Relationship between 53BP1 and BRCA1
4.2. Cas9 Activity Paired with the HDR-Active Cell Cycle Phase
4.3. All the Components in One HDR Complex
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- WareJoncas, Z.; Campbell, J.M.; Martinez-Galvez, G.; Gendron, W.A.C.; Barry, M.A.; Harris, P.C.; Sussman, C.R.; Ekker, S.C. Precision gene editing technology and applications in nephrology. Nat. Rev. Nephrol. 2018, 14, 663–677. [Google Scholar] [CrossRef]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrangou, R.; Doudna, J.A. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016, 34, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Robledo, J.E.; Barrera, M.C.; Tobon, G.J. CRISPR/Cas: From adaptive immune system in prokaryotes to therapeutic weapon against immune-related diseases. Int. Rev. Immunol. 2020, 39, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Terns, M.P. CRISPR-based technologies: Impact of RNA-targeting systems. Mol. Cell 2018, 72, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Bhattacharjee, O.; Mandal, D.; Sen, M.K.; Dey, D.; Dasgupta, A.; Kazi, T.A.; Gupta, R.; Sinharoy, S.; Acharya, K.; et al. CRISPR-Cas9 system: A new-fangled dawn in gene editing. Life Sci. 2019, 232, 116636. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Mojica, F.J.M.; Diez-Villasenor, C.; Garcia-Martinez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155 Pt 3, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Taylor, D.W.; Chen, J.S.; Kornfeld, J.E.; Zhou, K.; Thompson, A.J.; Nogales, E.; Doudna, J.A. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 2016, 351, 867–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Clark, K.J.; Campbell, J.M.; Panetta, M.R.; Guo, Y.; Ekker, S.C. Making designer mutants in model organisms. Development 2014, 141, 4042–4054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J.; Dellaire, G. Precision genome editing in the CRISPR era. Biochem. Cell Biol. 2017, 95, 187–201. [Google Scholar] [CrossRef]
- Dumitrache, L.C.; Hu, L.; Son, M.Y.; Li, H.; Wesevich, A.; Scully, R.; Stark, J.; Hasty, P. Trex2 enables spontaneous sister chromatid exchanges without facilitating DNA double-strand break repair. Genetics 2011, 188, 787–797. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Sulli, G.; Di Micco, R.; d’Adda di Fagagna, F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 2012, 12, 709–720. [Google Scholar] [CrossRef]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef]
- Nowsheen, S.; Aziz, K.; Aziz, A.; Deng, M.; Qin, B.; Luo, K.; Jeganathan, K.B.; Zhang, H.; Liu, T.; Yu, J.; et al. L3MBTL2 orchestrates ubiquitin signalling by dictating the sequential recruitment of RNF8 and RNF168 after DNA damage. Nat. Cell Biol. 2018, 20, 455–464. [Google Scholar] [CrossRef]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Jackson, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, V.; Phelps, S.E.; Gray, S.; Neale, M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 2011, 479, 241–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley, J.M.; Jimenez-Sainz, J.; Wang, W.; Miller, A.S.; Xue, X.; Nguyen, K.A.; Jensen, R.B.; Sung, P. Enhancement of BLM-DNA2-mediated long-range DNA end resection by CtIP. Cell Rep. 2017, 21, 324–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renkawitz, J.; Lademann, C.A.; Jentsch, S. Mechanisms and principles of homology search during recombination. Nat. Rev. Mol. Cell Biol. 2014, 15, 369–383. [Google Scholar] [CrossRef]
- Bhat, K.P.; Cortez, D. RPA and RAD51: Fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 2018, 25, 446–453. [Google Scholar] [CrossRef]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [Green Version]
- West, S.C. The search for a human Holliday junction resolvase. Biochem. Soc. Trans. 2009, 37 Pt 3, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [Green Version]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Kragelund, B.B.; Weterings, E.; Hartmann-Petersen, R.; Keijzers, G. The Ku70/80 ring in Non-Homologous End-Joining: Easy to slip on, hard to remove. Front. Biosci. 2016, 21, 514–527. [Google Scholar]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Conlin, M.P.; Reid, D.A.; Small, G.W.; Chang, H.H.; Watanabe, G.; Lieber, M.R.; Ramsden, D.A.; Rothenberg, E. DNA ligase IV guides end-processing choice during nonhomologous end joining. Cell Rep. 2017, 20, 2810–2819. [Google Scholar] [CrossRef] [Green Version]
- Stinson, B.M.; Moreno, A.T.; Walter, J.C.; Loparo, J.J. A mechanism to minimize errors during non-homologous end joining. Mol. Cell 2020, 77, 1080–1091. [Google Scholar] [CrossRef]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xu, X. Microhomology-mediated end joining: New players join the team. Cell Biosci. 2017, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Barboule, N.; Frit, P.; Gomez, D.; Bombarde, O.; Couderc, B.; Ren, G.S.; Salles, B.; Calsou, P. Ku counteracts mobilization of PARP1 and MRN in chromatin damaged with DNA double-strand breaks. Nucleic Acids Res. 2011, 39, 9605–9619. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Sfeir, A.; Symington, L.S. Microhomology-mediated end joining: A back-up survival mechanism or dedicated pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Nakade, S.; Tsubota, T.; Sakane, Y.; Kume, S.; Sakamoto, N.; Obara, M.; Daimon, T.; Sezutsu, H.; Yamamoto, T.; Sakuma, T.; et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014, 5, 5560. [Google Scholar] [CrossRef]
- Sakuma, T.; Nakade, S.; Sakane, Y.; Suzuki, K.T.; Yamamoto, T. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 2016, 11, 118–133. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, S.; Hur, J.K. CRISPR and target-specific DNA endonucleases for efficient DNA Knock-in in eukaryotic genomes. Mol. Cells 2018, 41, 943–952. [Google Scholar]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of single-strand annealing and its role in genome maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi-Iwai, Y.; Sonoda, E.; Buerstedde, J.M.; Bezzubova, O.; Morrison, C.; Takata, M.; Shinohara, A.; Takeda, S. Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell Biol. 1998, 18, 6430–6435. [Google Scholar] [CrossRef] [Green Version]
- Stone, H.R.; Morris, J.R. DNA damage emergency: Cellular garbage disposal to the rescue? Oncogene 2014, 33, 805–813. [Google Scholar] [CrossRef]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef]
- Buis, J.; Stoneham, T.; Spehalski, E.; Ferguson, D.O. Mre11 regulates CtIP-dependent double-strand break repair by interaction with CDK2. Nat. Struct. Mol. Biol. 2012, 19, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wu, L.C.; Bowcock, A.M.; Aronheim, A.; Baer, R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998, 273, 25388–25392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomimatsu, N.; Mukherjee, B.; Catherine Hardebeck, M.; Ilcheva, M.; Vanessa Camacho, C.; Louise Harris, J.; Porteus, M.; Llorente, B.; Khanna, K.K.; Burma, S. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat. Commun. 2014, 5, 3561. [Google Scholar] [CrossRef]
- Tkac, J.; Xu, G.; Adhikary, H.; Young, J.T.F.; Gallo, D.; Escribano-Diaz, C.; Krietsch, J.; Orthwein, A.; Munro, M.; Sol, W.; et al. HELB Is a feedback inhibitor of DNA end resection. Mol. Cell 2016, 61, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Shamanna, R.A.; Keijzers, G.; Anand, R.; Rasmussen, L.J.; Cejka, P.; Croteau, D.L.; Bohr, V.A. RECQL4 Promotes DNA end resection in repair of DNA double-strand breaks. Cell Rep. 2016, 16, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Shamanna, R.A.; de Freitas, J.K.; Okur, M.; Khadka, P.; Kulikowicz, T.; Holland, P.P.; Tian, J.; Croteau, D.L.; Davis, A.J.; et al. Cell cycle-dependent phosphorylation regulates RECQL4 pathway choice and ubiquitination in DNA double-strand break repair. Nat. Commun. 2017, 8, 2039. [Google Scholar] [CrossRef] [Green Version]
- Uckelmann, M.; Sixma, T.K. Histone ubiquitination in the DNA damage response. DNA Repair 2017, 56, 92–101. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Ismail, I.H.; Gagne, J.P.; Genois, M.M.; Strickfaden, H.; McDonald, D.; Xu, Z.; Poirier, G.G.; Masson, J.Y.; Hendzel, M.J. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat. Cell Biol. 2015, 17, 1446–1457. [Google Scholar] [CrossRef]
- Brown, J.S.; Lukashchuk, N.; Sczaniecka-Clift, M.; Britton, S.; le Sage, C.; Calsou, P.; Beli, P.; Galanty, Y.; Jackson, S.P. Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell Rep. 2015, 11, 704–714. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Chen, J. The E3 ligase RNF8 regulates KU80 removal and NHEJ repair. Nat. Struct. Mol. Biol. 2012, 19, 201–206. [Google Scholar] [CrossRef]
- Himmels, S.F.; Sartori, A.A. Controlling DNA-end resection: An emerging task for ubiquitin and SUMO. Front. Genet. 2016, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Lafranchi, L.; de Boer, H.R.; de Vries, E.G.; Ong, S.E.; Sartori, A.A.; van Vugt, M.A. APC/C(Cdh1) controls CtIP stability during the cell cycle and in response to DNA damage. EMBO J. 2014, 33, 2860–2879. [Google Scholar] [CrossRef] [Green Version]
- Steger, M.; Murina, O.; Huhn, D.; Ferretti, L.P.; Walser, R.; Hanggi, K.; Lafranchi, L.; Neugebauer, C.; Paliwal, S.; Janscak, P.; et al. Prolyl isomerase PIN1 regulates DNA double-strand break repair by counteracting DNA end resection. Mol. Cell 2013, 50, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef]
- Mirman, Z.; de Lange, T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef] [Green Version]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef]
- Escribano-Diaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkac, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Alvarez-Quilon, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Setiaputra, D.; Durocher, D. Shieldin—The protector of DNA ends. EMBO Rep. 2019, 20, e47560. [Google Scholar] [CrossRef]
- Arnoult, N.; Correia, A.; Ma, J.; Merlo, A.; Garcia-Gomez, S.; Maric, M.; Tognetti, M.; Benner, C.W.; Boulton, S.J.; Saghatelian, A.; et al. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature 2017, 549, 548–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Aroumougame, A.; Lobrich, M.; Li, Y.; Chen, D.; Chen, J.; Gong, Z. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014, 28, 2693–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza-Aghazadeh-Attari, M.; Mohammadzadeh, A.; Yousefi, B.; Mihanfar, A.; Karimian, A.; Majidinia, M. 53BP1: A key player of DNA damage response with critical functions in cancer. DNA Repair 2019, 73, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Garcia, A.; Lopez-Saavedra, A.; Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014, 9, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, H.; Chen, Y.; Yang, X.; Wang, P.; Liu, T.; Deng, M.; Qin, B.; Correia, C.; Lee, S.; et al. A cell cycle-dependent BRCA1-UHRF1 cascade regulates DNA double-strand break repair pathway choice. Nat. Commun. 2016, 7, 10201. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isono, M.; Niimi, A.; Oike, T.; Hagiwara, Y.; Sato, H.; Sekine, R.; Yoshida, Y.; Isobe, S.Y.; Obuse, C.; Nishi, R.; et al. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017, 18, 520–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vartak, S.V.; Raghavan, S.C. Inhibition of nonhomologous end joining to increase the specificity of CRISPR/Cas9 genome editing. FEBS J. 2015, 282, 4289–4294. [Google Scholar] [CrossRef]
- Beumer, K.J.; Trautman, J.K.; Mukherjee, K.; Carroll, D. Donor DNA utilization during gene targeting with zinc-finger nucleases. G3 2013, 3, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Singh, P.; Schimenti, J.C.; Bolcun-Filas, E. A mouse geneticist’s practical guide to CRISPR applications. Genetics 2015, 199, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, Z.; Ge, W. An efficient platform for generating somatic point mutations with germline transmission in the zebrafish by CRISPR/Cas9-mediated gene editing. J. Biol. Chem. 2018, 293, 6611–6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chen, W.; Zhang, X.; Yu, L.; Dong, W.; Pan, S.; Gao, S.; Huang, X.; Zhang, L. Increasing the efficiency of CRISPR/Cas9-mediated precise genome editing in rats by inhibiting NHEJ and using Cas9 protein. RNA Biol. 2016, 13, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Shi, Z.; Guo, X.; Jiang, B.; Wang, G.; Luo, D.; Chen, Y.; Zhu, Y.S. Ligase IV inhibitor SCR7 enhances gene editing directed by CRISPR-Cas9 and ssODN in human cancer cells. Cell Biosci. 2018, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhang, X.; Zhong, C.; Mo, J.; Quan, R.; Yang, J.; Liu, D.; Li, Z.; Yang, H.; Wu, Z. Small molecules enhance CRISPR/Cas9-mediated homology-directed genome editing in primary cells. Sci. Rep. 2017, 7, 8943. [Google Scholar] [CrossRef]
- Song, J.; Yang, D.; Xu, J.; Zhu, T.; Chen, Y.E.; Zhang, J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016, 7, 10548. [Google Scholar] [CrossRef] [Green Version]
- Gutschner, T.; Haemmerle, M.; Genovese, G.; Draetta, G.F.; Chin, L. Post-translational Regulation of Cas9 during G1 enhances homology-directed repair. Cell Rep. 2016, 14, 1555–1566. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Pang, D.; Wang, K.; Li, M.; Guo, N.; Yuan, H.; Li, J.; Zou, X.; Jiao, H.; Ouyang, H.; et al. Optimization of a CRISPR/Cas9-mediated Knock-in Strategy at the Porcine Rosa26 locus in porcine foetal fibroblasts. Sci. Rep. 2017, 7, 3036. [Google Scholar] [CrossRef] [Green Version]
- Jayavaradhan, R.; Pillis, D.M.; Malik, P. A versatile tool for the quantification of CRISPR/Cas9-induced genome editing events in human hematopoietic cell lines and hematopoietic stem/progenitor cells. J. Mol. Biol. 2019, 431, 102–110. [Google Scholar] [CrossRef]
- Canny, M.D.; Moatti, N.; Wan, L.C.K.; Fradet-Turcotte, A.; Krasner, D.; Mateos-Gomez, P.A.; Zimmermann, M.; Orthwein, A.; Juang, Y.C.; Zhang, W.; et al. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol. 2018, 36, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, E.J.; Lovendahl, K.N.; St Martin, A.; Harris, R.S.; Gordon, W.R. Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun. Biol. 2018, 1, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, G.E.; Matsumoto, Y.; Brooks, R.C.; Lu, Z.; Lieber, M.R.; Tomkinson, A.E. SCR7 is neither a selective nor a potent inhibitor of human DNA ligase IV. DNA Repair. 2016, 43, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, D.; Zhang, X.; Quan, R.; Zhong, C.; Mo, J.; Huang, Y.; Wang, H.; Ruan, X.; Xu, Z.; et al. Suppressing Ku70/Ku80 expression elevates homology-directed repair efficiency in primary fibroblasts. Int. J. Biochem. Cell Biol. 2018, 99, 154–160. [Google Scholar] [CrossRef]
- Robert, F.; Barbeau, M.; Ethier, S.; Dostie, J.; Pelletier, J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015, 7, 93. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, Y.A.; Nguyen, D.T.; Chow, S.; Chung, R.S.; Guillemin, G.J.; Cole, N.J.; Hesselson, D. Chemical reprogramming enhances homology-directed genome editing in zebrafish embryos. Commun. Biol. 2019, 2, 198. [Google Scholar] [CrossRef] [Green Version]
- Riesenberg, S.; Maricic, T. Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Nat. Commun. 2018, 9, 2164. [Google Scholar] [CrossRef] [Green Version]
- Riesenberg, S.; Chintalapati, M.; Macak, D.; Kanis, P.; Maricic, T.; Paabo, S. Simultaneous precise editing of multiple genes in human cells. Nucleic Acids Res. 2019, 47, e116. [Google Scholar] [CrossRef]
- Frank, K.M.; Sekiguchi, J.M.; Seidl, K.J.; Swat, W.; Rathbun, G.A.; Cheng, H.L.; Davidson, L.; Kangaloo, L.; Alt, F.W. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 1998, 396, 173–177. [Google Scholar] [CrossRef]
- Jayathilaka, K.; Sheridan, S.D.; Bold, T.D.; Bochenska, K.; Logan, H.L.; Weichselbaum, R.R.; Bishop, D.K.; Connell, P.P. A chemical compound that stimulates the human homologous recombination protein RAD51. Proc. Natl. Acad. Sci. USA 2008, 105, 15848–15853. [Google Scholar] [CrossRef] [Green Version]
- Pinder, J.; Salsman, J.; Dellaire, G. Nuclear domain ’knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015, 43, 9379–9392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurihara, T.; Kouyama-Suzuki, E.; Satoga, M.; Li, X.; Badawi, M.; Baig, D.N.; Yanagawa, T.; Uemura, T.; Mori, T.; Tabuchi, K. DNA repair protein RAD51 enhances the CRISPR/Cas9-mediated knock-in efficiency in brain neurons. Biochem. Biophys. Res. Commun. 2020, 524, 621–628. [Google Scholar] [CrossRef]
- Haber, J.E. In vivo biochemistry: Physical monitoring of recombination induced by site-specific endonucleases. Bioessays 1995, 17, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.H.; Powell, S.N. Molecular pathways: Understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin. Cancer Res. 2012, 18, 6400–6406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Primio, C.; Galli, A.; Cervelli, T.; Zoppe, M.; Rainaldi, G. Potentiation of gene targeting in human cells by expression of Saccharomyces cerevisiae Rad52. Nucleic Acids Res. 2005, 33, 4639–4648. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Ren, C.; Liu, Z.; Bai, Y.; Chen, Z.; Wei, Z.; Wang, X.; Zhang, Z.; Xu, K. Enhancing CRISPR/Cas9-mediated homology-directed repair in mammalian cells by expressing Saccharomyces cerevisiae Rad52. Int. J. Biochem. Cell Biol. 2017, 92, 43–52. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Guo, Y.; Du, W.; Yin, Y.; Zhang, T.; Lu, H. Enhancing targeted genomic DNA editing in chicken cells using the CRISPR/Cas9 system. PLoS ONE 2017, 12, e0169768. [Google Scholar] [CrossRef]
- Paulsen, B.S.; Mandal, P.K.; Frock, R.L.; Boyraz, B.; Yadav, R.; Upadhyayula, S.; Gutierrez-Martinez, P.; Ebina, W.; Fasth, A.; Kirchhausen, T.; et al. Ectopic expression of RAD52 and dn53BP1 improves homology-directed repair during CRISPR-Cas9 genome editing. Nat. Biomed. Eng. 2017, 1, 878–888. [Google Scholar] [CrossRef]
- Charpentier, M.; Khedher, A.H.Y.; Menoret, S.; Brion, A.; Lamribet, K.; Dardillac, E.; Boix, C.; Perrouault, L.; Tesson, L.; Geny, S.; et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018, 9, 1133. [Google Scholar] [CrossRef]
- Tran, N.T.; Bashir, S.; Li, X.; Rossius, J.; Chu, V.T.; Rajewsky, K.; Kuhn, R. Enhancement of precise gene editing by the association of Cas9 With homologous recombination factors. Front. Genet. 2019, 10, 365. [Google Scholar] [CrossRef] [Green Version]
- Reuven, N.; Adler, J.; Broennimann, K.; Myers, N.; Shaul, Y. Recruitment of DNA repair MRN complex by intrinsically disordered protein domain fused to Cas9 improves efficiency of CRISPR-mediated genome editing. Biomolecules 2019, 9, 584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015, 16, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, A.; Hartlerode, A.; Stucki, M.; Odate, S.; Puget, N.; Kwok, A.; Nagaraju, G.; Yan, C.; Alt, F.W.; Chen, J.; et al. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol. Cell 2007, 28, 1045–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayavaradhan, R.; Pillis, D.M.; Goodman, M.; Zhang, F.; Zhang, Y.; Andreassen, P.R.; Malik, P. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019, 10, 2866. [Google Scholar] [CrossRef] [Green Version]
- Nambiar, T.S.; Billon, P.; Diedenhofen, G.; Hayward, S.B.; Taglialatela, A.; Cai, K.; Huang, J.W.; Leuzzi, G.; Cuella-Martin, R.; Palacios, A.; et al. Stimulation of CRISPR-mediated homology-directed repair by an engineered RAD18 variant. Nat. Commun. 2019, 10, 3395. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014, 3, e04766. [Google Scholar] [CrossRef]
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [Green Version]
- Lomova, A.; Clark, D.N.; Campo-Fernandez, B.; Flores-Bjurstrom, C.; Kaufman, M.L.; Fitz-Gibbon, S.; Wang, X.; Miyahira, E.Y.; Brown, D.; DeWitt, M.A.; et al. Improving gene editing outcomes in human hematopoietic stem and progenitor cells by temporal control of DNA repair. Stem Cells 2019, 37, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Wienert, B.; Nguyen, D.N.; Guenther, A.; Feng, S.J.; Locke, M.N.; Wyman, S.K.; Shin, J.; Kazane, K.R.; Gregory, G.L.; Carter, M.A.M.; et al. Timed inhibition of CDC7 increases CRISPR-Cas9 mediated templated repair. Nat. Commun. 2020, 11, 2109. [Google Scholar] [CrossRef]
- Gerlach, M.; Kraft, T.; Brenner, B.; Petersen, B.; Niemann, H.; Montag, J. Efficient Knock-in of a Point Mutation in Porcine Fibroblasts Using the CRISPR/Cas9-GMNN Fusion Gene. Genes 2018, 9, 296. [Google Scholar] [CrossRef] [Green Version]
- Howden, S.E.; McColl, B.; Glaser, A.; Vadolas, J.; Petrou, S.; Little, M.H.; Elefanty, A.G.; Stanley, E.G. A Cas9 variant for efficient generation of indel-free knockin or gene-corrected human pluripotent stem cells. Stem Cell Rep. 2016, 7, 508–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Wang, C.; Hong, L.; Sun, N.; Chen, D.; Chen, S.; Han, F. Programmable DNA repair with CRISPRa/i enhanced homology-directed repair efficiency with a single Cas9. Cell Discov. 2018, 4, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devkota, S. The road less traveled: Strategies to enhance the frequency of homology-directed repair (HDR) for increased efficiency of CRISPR/Cas-mediated transgenesis. BMB Rep. 2018, 51, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Carlson-Stevermer, J.; Abdeen, A.A.; Kohlenberg, L.; Goedland, M.; Molugu, K.; Lou, M.; Saha, K. Assembly of CRISPR ribonucleoproteins with biotinylated oligonucleotides via an RNA aptamer for precise gene editing. Nat. Commun. 2017, 8, 1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.; Zhuang, F.; Hu, X.; Wang, B.; Wen, X.Z.; Ji, J.F.; Xi, J.J. Efficient generation of mice carrying homozygous double-floxp alleles using the Cas9-Avidin/Biotin-donor DNA system. Cell Res. 2017, 27, 578–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savic, N.; Ringnalda, F.C.; Lindsay, H.; Berk, C.; Bargsten, K.; Li, Y.; Neri, D.; Robinson, M.D.; Ciaudo, C.; Hall, J.; et al. Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. Elife 2018, 7, e33761. [Google Scholar] [CrossRef]
- Liang, X.; Potter, J.; Kumar, S.; Ravinder, N.; Chesnut, J.D. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 2017, 241, 136–146. [Google Scholar] [CrossRef]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Song, F.; Stieger, K. Optimizing the DNA Donor Template for Homology-Directed Repair of Double-Strand Breaks. Mol. Ther Nucleic Acids 2017, 7, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Zhang, J.P.; Li, X.L.; Li, G.H.; Chen, W.; Arakaki, C.; Botimer, G.D.; Baylink, D.; Zhang, L.; Wen, W.; Fu, Y.W.; et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017, 18, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
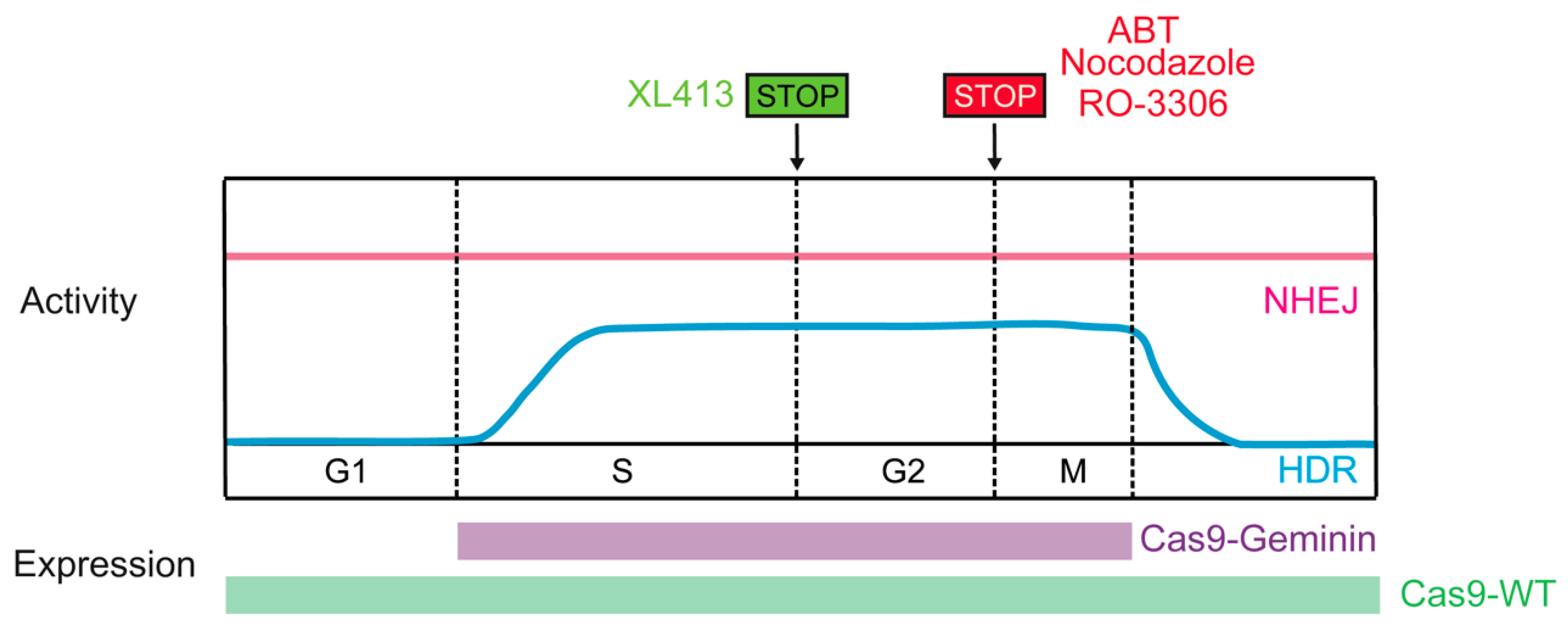{kind=link}
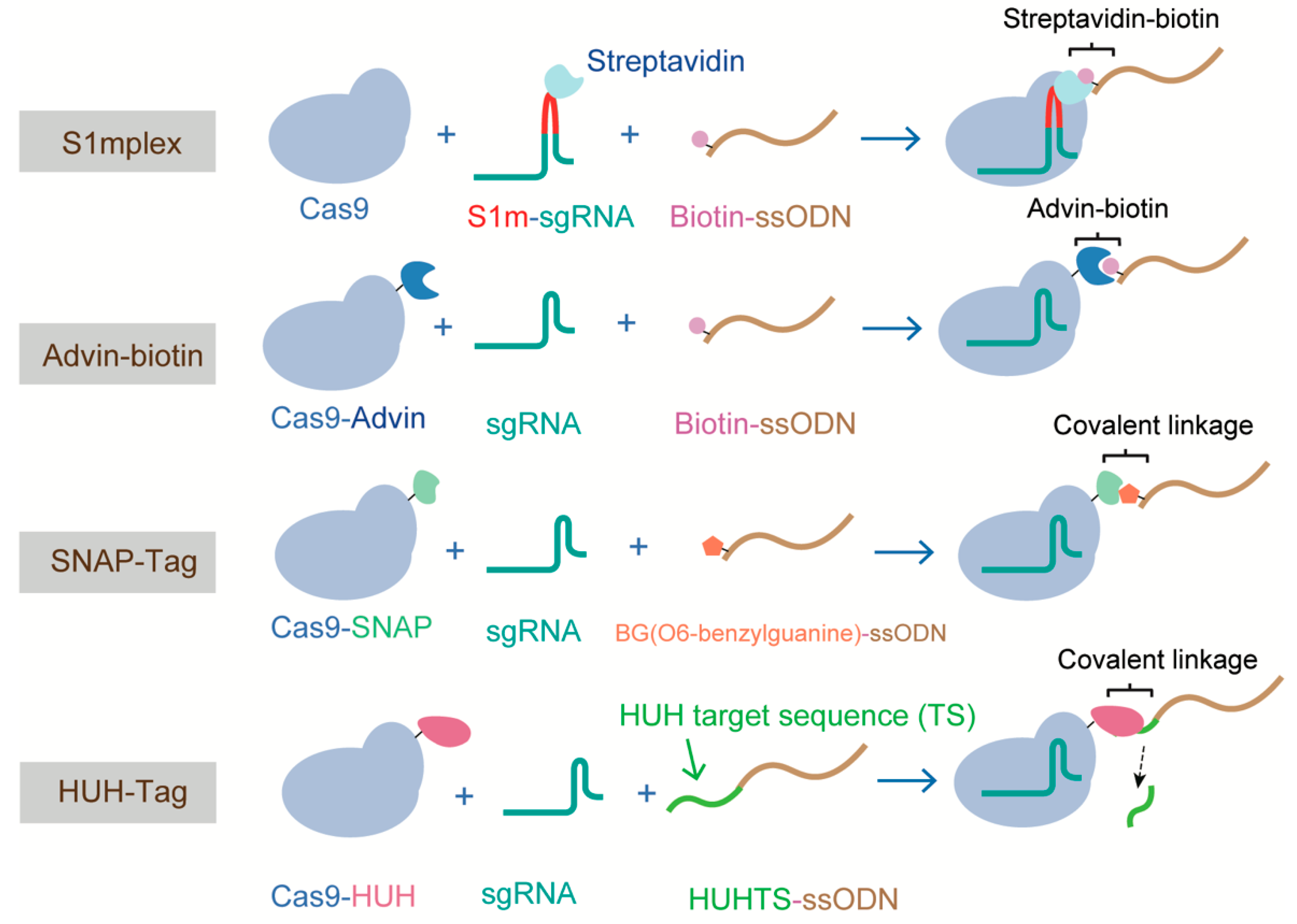{kind=link}
| Target | Treatment | HDR Increasement | Cell Type | Gene Locus | Editing Type | Ref. |
|---|---|---|---|---|---|---|
| DNA Ligase IV inhibitor | SCR7 | 3-fold | A549 | TSG101 | Stop cassette KI | [80] |
| 19-fold | Mel-JuSo | TSG101 | Stop cassette KI | [80] | ||
| 13-fold(4.58–58.3%) | DC2.4 | Tap1 | Venus KI fluorescent gene KI | [80] | ||
| 4.5-fold(5–22.7%) | Mouse zygotes | Igk | LPETG KI | [80] | ||
| 5-fold(5–25%) | HEK293 | AAVS1-TLR | GC | [82] | ||
| 10-fold(5.8–56.2%) | Mouse embryos | Tex1 | GC | [81] | ||
| 2.5-fold(2.7–6.6%) | MCF-7 | AAVS | GC | [85] | ||
| 10-fold(1.1–4.3%) | HCT-116 | AAVS | MCS KI | [85] | ||
| 4-fold (5.6–11.2%) | Porcine fetal fibroblasts | eGFP | MCS KI | [86] | ||
| 1.9-fold (26.22–49.66%) | Primary porcine cell | ROSA26 | Neomycin KI | [86] | ||
| 3.5-fold (1.9–6.6%) | MCF-7 /GFP-mut | - | GC | [86] | ||
| 1.7-fold (8.4–14.6%) | HCT-116 ΔTCT | β-catenin | GC | [86] | ||
| 3.7-fold (15–55%) | Zebrafish embryos | Ybx 1 | S82A GC | [83] | ||
| 2.2-fold (18–39%) | Rat zygotes | Fabp2 | Cre KI | [84] | ||
| 1.4-fold (46–64%) | Rat zygotes | Dbndd | Cre ER T2 KI | [84] | ||
| NS | Rabbit embryos | RLL | EGFP KI | [87] | ||
| NS | HEK-293T | rOSA26 | Neomycin KI resistance gene KI | [88] | ||
| NS | Porcine foetal Fibroblasts | Rosa26 | EGFP KI | [89] | ||
| NS | HSPC | Rosa26 | EGFP KI | [90] | ||
| NS | U2OS | LMNA-GFP | GC | [91] | ||
| NS | HEK-293T | GAPDH | HIBIT KI | [92] | ||
| PK-cs inhibitor | NU7441 | 3~9-fold | MEF | TP53 | GC | [95] |
| 2-fold (1.9–3.8%) | HEK293-TLR | - | GC | [95] | ||
| 13.5-fold (4–53%) | Zebra embryos | - | mCherry KI | [96] | ||
| 2.5-fold (8.6–21.5%) | A549 | CD45 | GFP KI | [90] | ||
| 2.4-fold (8–19.2%) | CD34+ HSPC | CD45 gene/GFP insertion | GFP KI | [90] | ||
| 1.3-fold (4.6–6%) | U2OS | LMNA | mClover KI insertion | [91] | ||
| KU-0060648 | 2~8–fold | MEF | TP53 | GC | [95] | |
| 2.2-fold (1.9–4.1%) | HEK293-TLR | - | GC | [95] | ||
| NU7026 | 1.6-fold (3.7–6.0%) | iPSCs | AAVS | KI | [97] | |
| 1.7-fold (19–32%) | hiPSCs-409B2 | FRMD7 | GC | [98] | ||
| 2.4-fold (2.5–6%) | HEK293 | GFP | KI | [130] | ||
| M3814 | 4-fold (18–81%) | K562 cells-409B2 hiPSCs | FRMD7 | GC | [98] | |
| Rad51 agonist | RS-1 | EP 3-fold(2.5–7.5%)epEP/electroporation | HEK293A | LMNA | Clover KI | [101] |
| Lipo 6-fold(3.5–21%)/lipofection | HEK293A | LMNA | Clover KI | [101] | ||
| 8.5-fold (0.8–6.8%) | Rabbit embryos | RLL | EGFP KI | [87] | ||
| 2.1-fold (8–16.8%) | CD34+ HSPC | CD45 gene/GFP insertion | GFP KI | [90] | ||
| 1.6-fold(15–24%) | Zebrafish embryos | Ybx1 | S82A GC | [83] | ||
| NS | iPSC | CTNB1/PRDM14 | mNeoGreen KI | [97] | ||
| Cell cycle Synchronization | Nocodazole | 2.2-fold(9–20%) | HEK293T | EMX1, DYRK1 | MCS KI | [116] |
| 1.7-fold (13–22%) | iPSC | PRDM14 | mCherry KI | [131] | ||
| 1.7-fold (7–12%) | iPSC | CTNNB | mCherry KI | [131] | ||
| 1.4-fold (14–19.2%) | HEK293T | MALAT1 | MCS KI | [88] | ||
| 6.7-fold (7.86–52.49%) | DiPSC | NEUROD | Puromycin KI | [117] | ||
| 3.4-fold (17.46–59.77%) | H1 | NEUROD | Puromycin KI KI | [117] | ||
| ABT | 6.8-fold (7.86–53.50%) | DiPSC | NEUROD | Puromycin KI | [117] | |
| 4.6-fold (17.46–79.91%) | H1 | NEUROD | Puromycin KI | [117] | ||
| RO-3306 | 1.3-fold(18.1–23.5%) | PBSC | β-globin | GC | [118] | |
| Golgi apparatus | Brefeldin A | 2-fold (17.7%–27.2%) | E14 mouse ESCs | Nanog | GFP KI | [112] |
| inhibitor | 1.3-fold (13%–17%) | iPSC | CTNB1 | KI | [131] | |
| β3-adrenergic receptor agonist | L755507 | 3-fold (17.7–33.3%) | E14 mouse ESCs | Nanog | GFP KI | [112] |
| 8.9-fold (0.35–3.13%) | iPSC | SOD1 | A4V GC | [112] | ||
| 2-fold (0.8–1.6%) | HUVEC | ACTA2 | Venus KI | [112] | ||
| 1.9-fold (5.6–10.9%) | Porcine fetal fibroblasts | eGFP | GC | [86] | ||
| NS | iPSC | CTNB1/PRDM14 | mNeoGreen KI | [97] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Ren, S.; Yu, S.; Pan, H.; Li, T.; Ge, S.; Zhang, J.; Xia, N. Methods Favoring Homology-Directed Repair Choice in Response to CRISPR/Cas9 Induced-Double Strand Breaks. Int. J. Mol. Sci. 2020, 21, 6461. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186461
Yang H, Ren S, Yu S, Pan H, Li T, Ge S, Zhang J, Xia N. Methods Favoring Homology-Directed Repair Choice in Response to CRISPR/Cas9 Induced-Double Strand Breaks. International Journal of Molecular Sciences. 2020; 21(18):6461. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186461
Chicago/Turabian StyleYang, Han, Shuling Ren, Siyuan Yu, Haifeng Pan, Tingdong Li, Shengxiang Ge, Jun Zhang, and Ningshao Xia. 2020. "Methods Favoring Homology-Directed Repair Choice in Response to CRISPR/Cas9 Induced-Double Strand Breaks" International Journal of Molecular Sciences 21, no. 18: 6461. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186461