Connexin Hemichannel Mimetic Peptide Attenuates Cortical Interneuron Loss and Perineuronal Net Disruption Following Cerebral Ischemia in Near-Term Fetal Sheep

 , and
, and
Abstract
:1. Introduction
2. Results
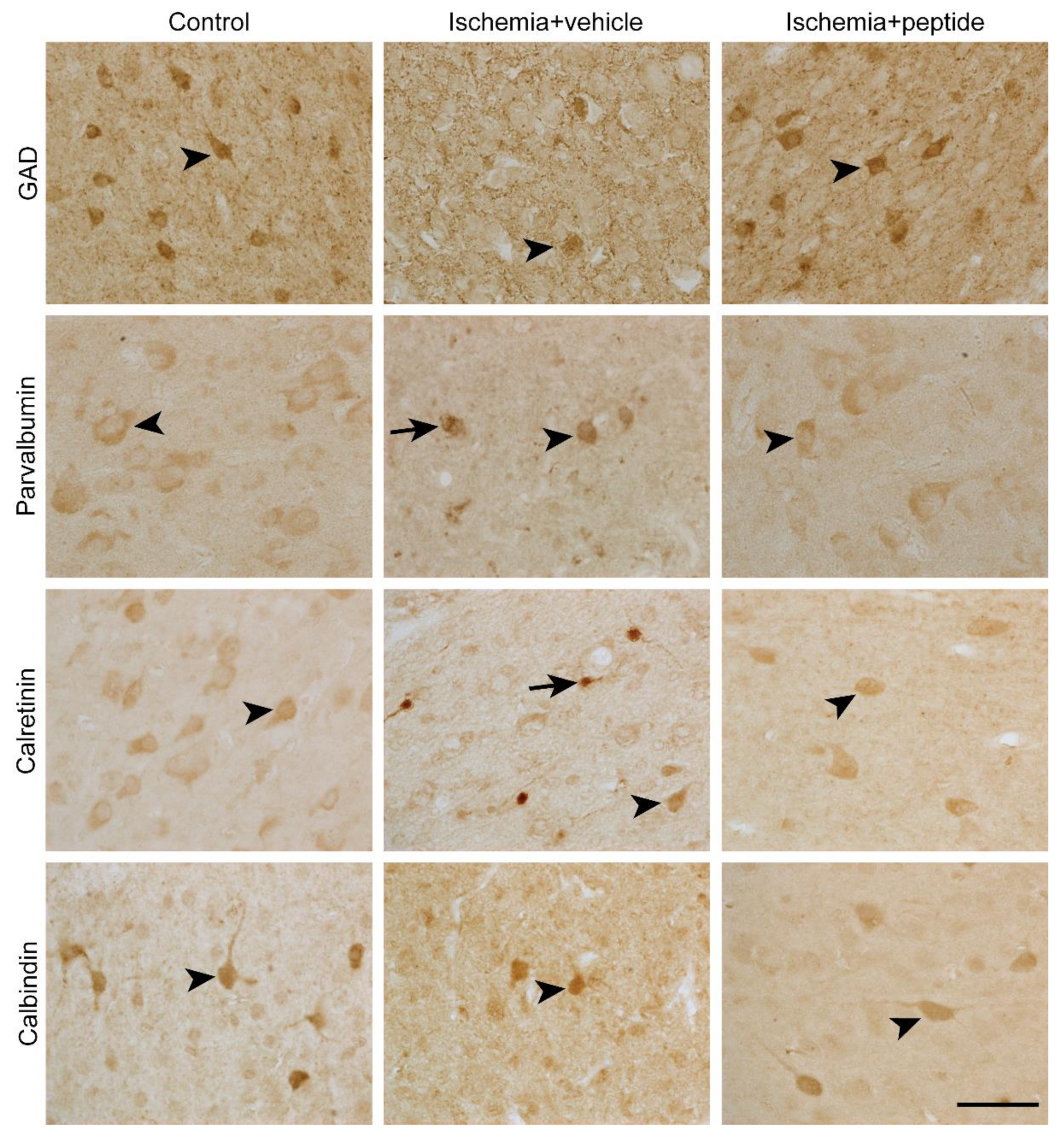
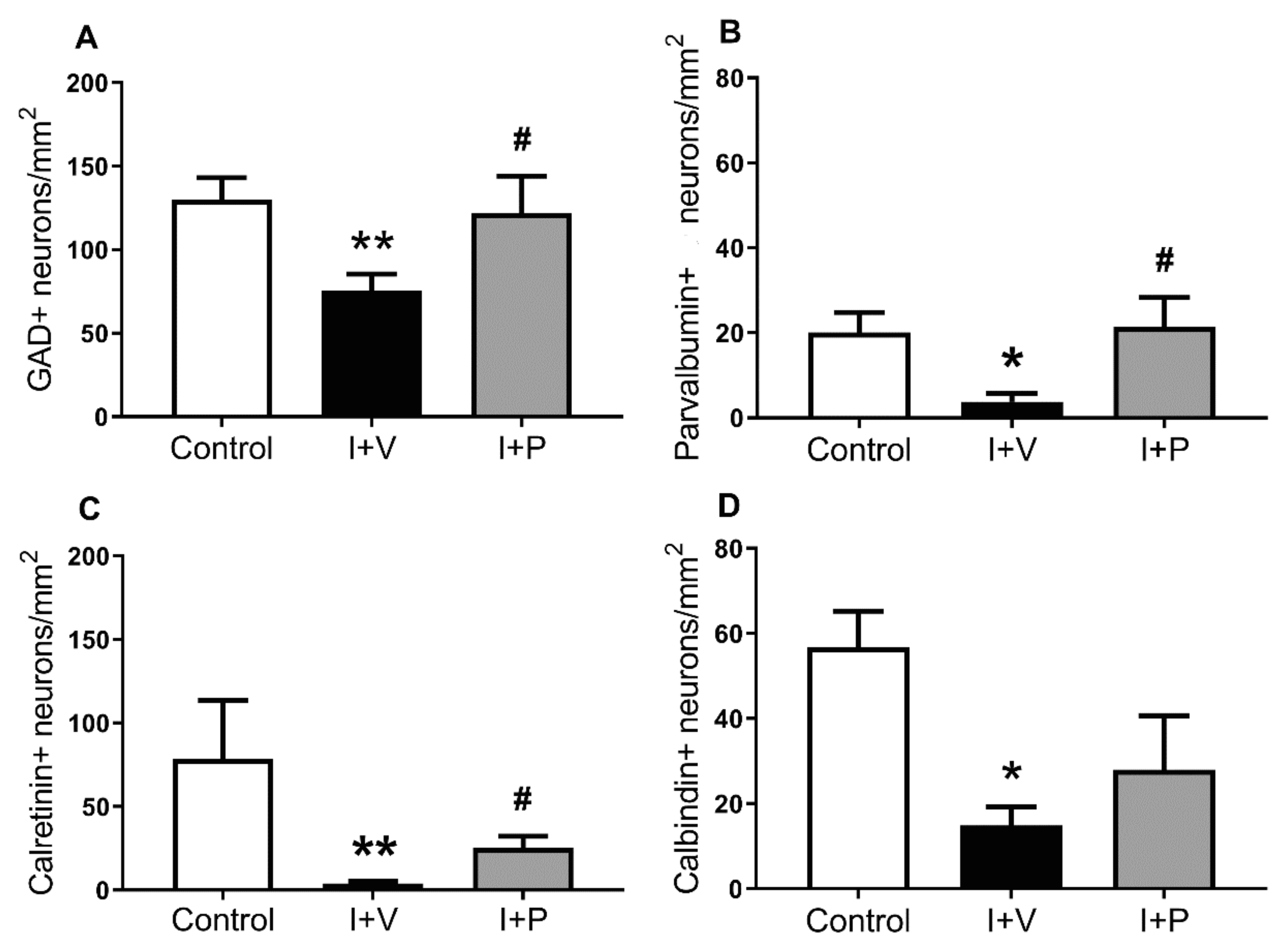
2.1. Survival of GABAergic Interneurons in the Parasagittal Cortex after Cerebral Ischemia
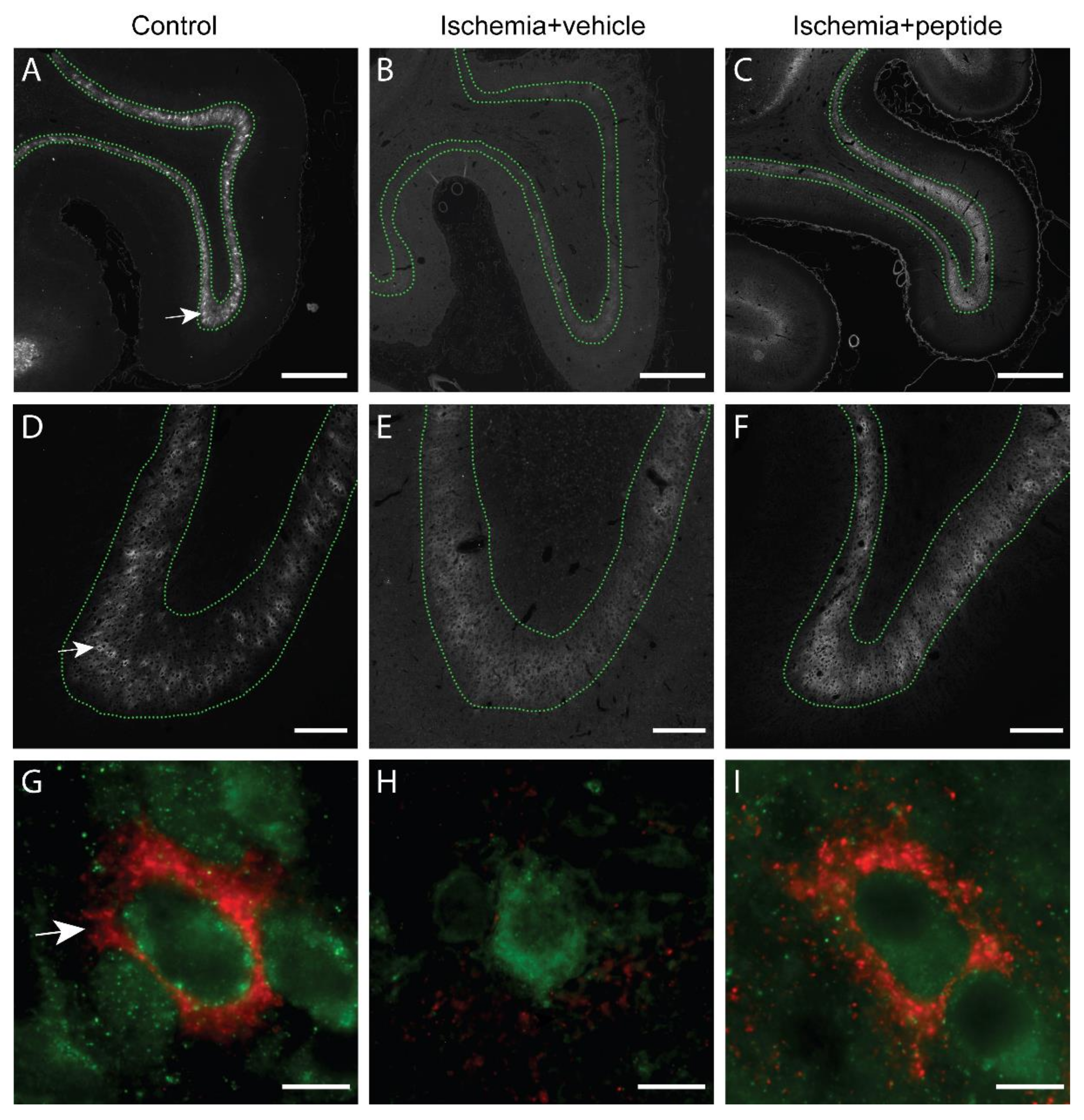
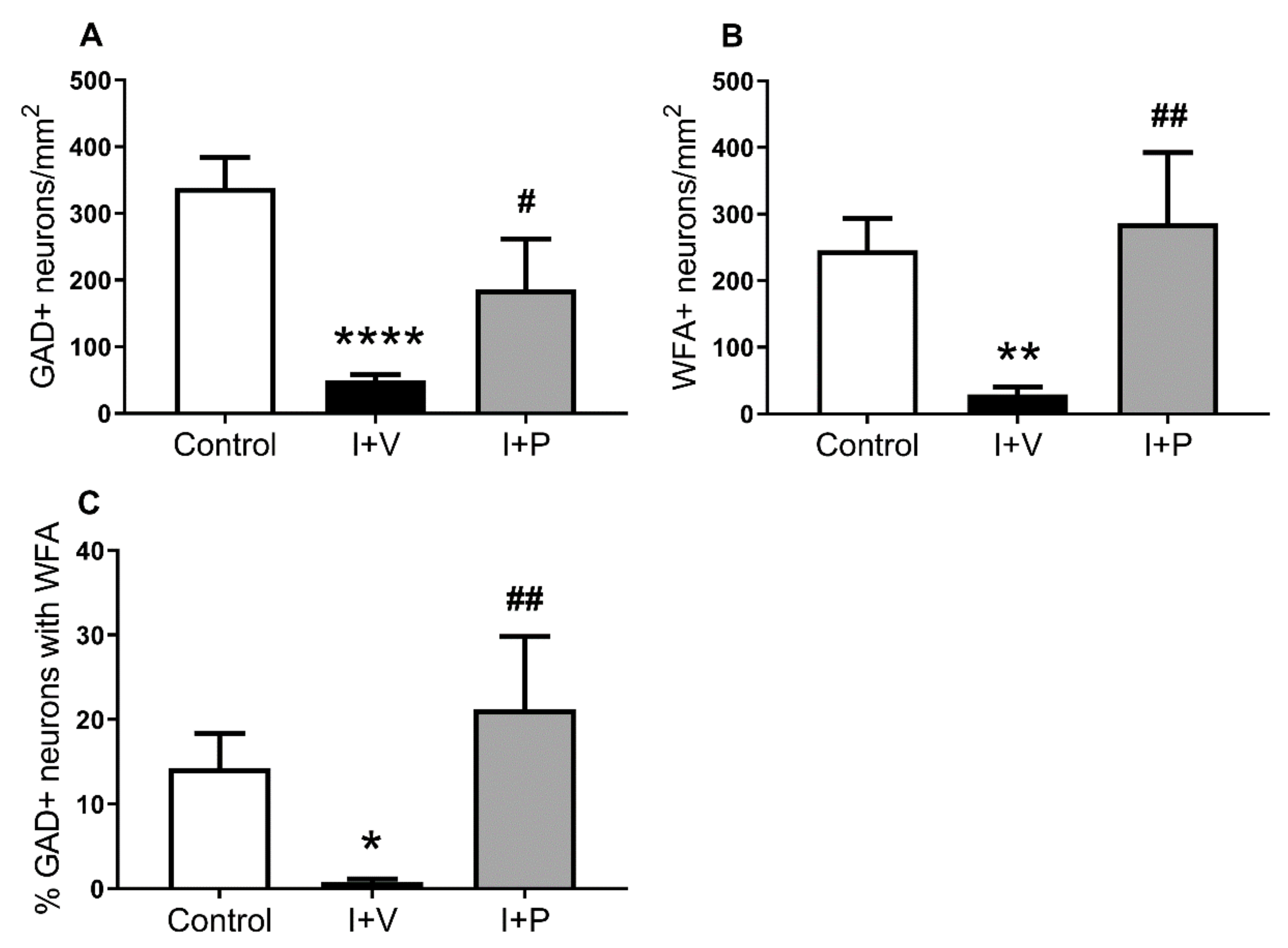
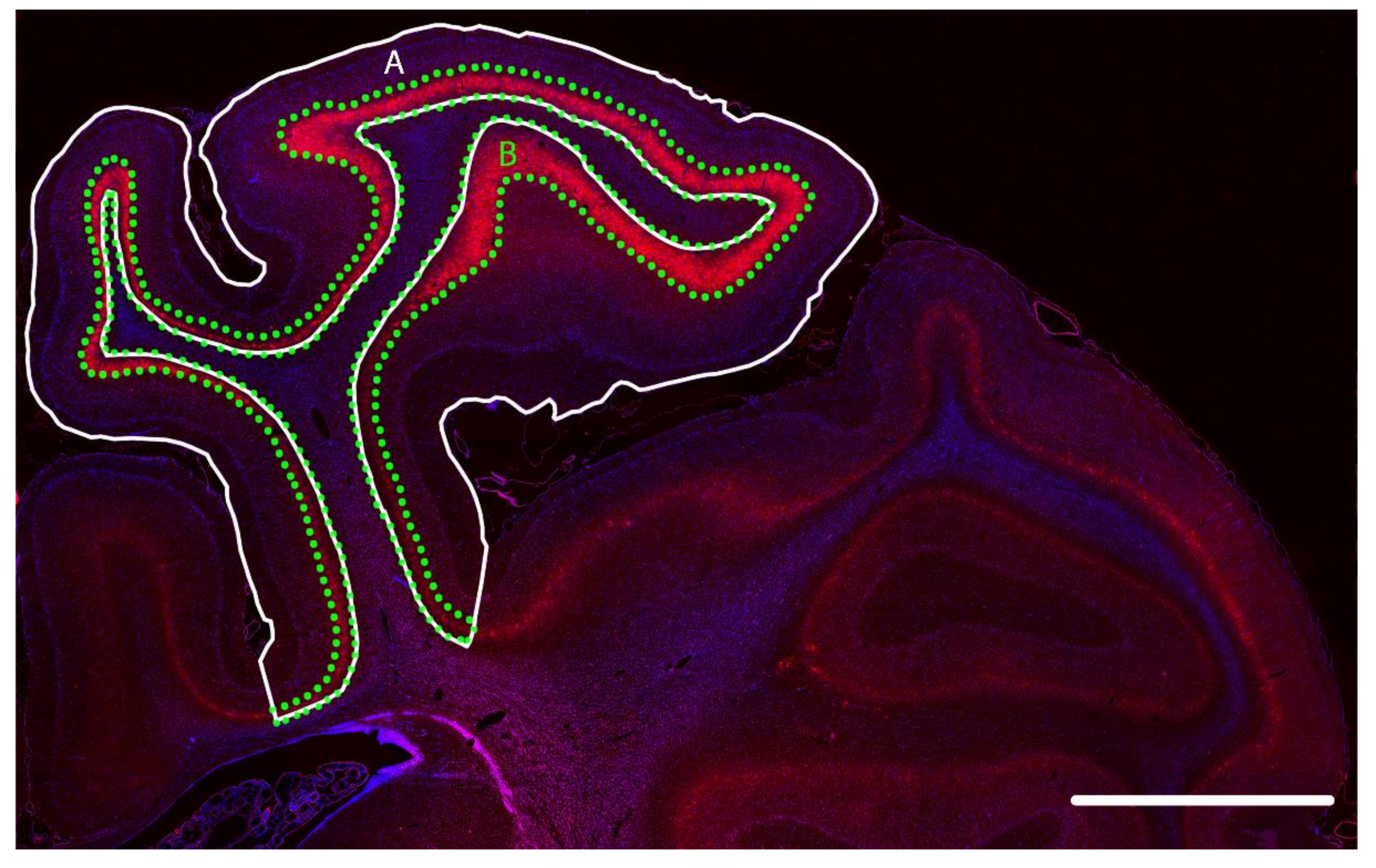
2.2. Changes in PNNs in Layer 6 of the Parasagittal Cortex Following Cerebral Ischemia
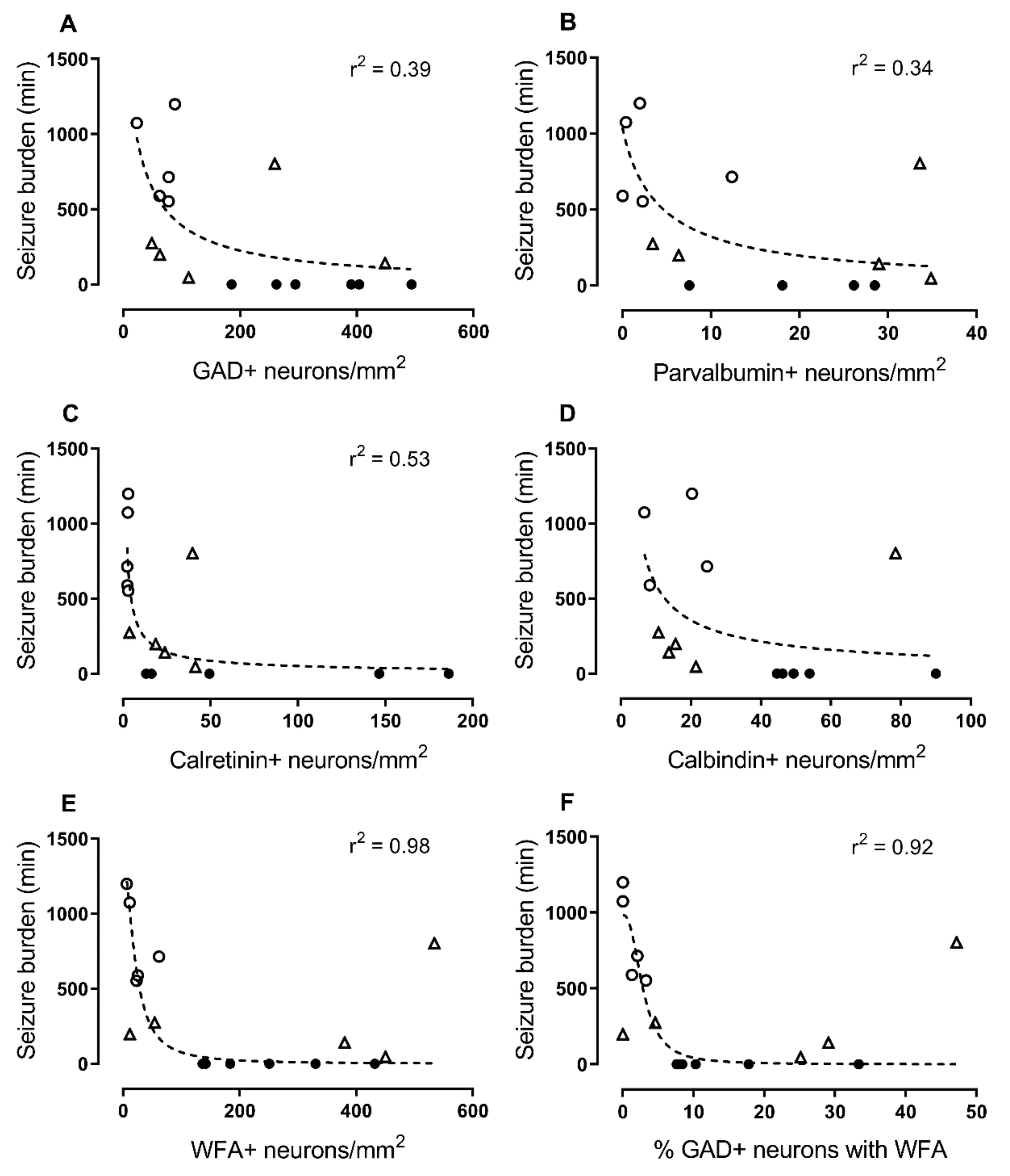
2.3. Correlations with Seizure Burden
3. Discussion
4. Materials and Methods
4.1. Animals and Surgery
4.2. Postoperative Care
4.3. Experimental Protocols
4.4. Tissue Preparation
4.5. Immunohistochemical Staining
4.6. Image Analysis
4.7. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vannucci, R.C. Hypoxic-ischemic encephalopathy. Am. J. Perinatol. 2000, 17, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Le Magueresse, C.; Monyer, H. GABAergic interneurons shape the functional maturation of the cortex. Neuron 2013, 77, 388–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tau, G.Z.; Peterson, B.S. Normal development of brain circuits. Neuropsychopharmacology 2010, 35, 147–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.H.; Scheinost, D.; Lacadie, C.; Benjamin, J.; Myers, E.H.; Qiu, M.; Schneider, K.C.; Rothman, D.L.; Constable, R.T.; Ment, L.R. GABA, resting-state connectivity and the developing brain. Neonatology 2014, 106, 149–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, D.A. Inhibitory neurons in human cortical circuits: Substrate for cognitive dysfunction in schizophrenia. Curr. Opin. Neurobiol. 2014, 26, 22–26. [Google Scholar] [CrossRef] [Green Version]
- Jacob, J. Cortical interneuron dysfunction in epilepsy associated with autism spectrum disorders. Epilepsia 2016, 57, 182–193. [Google Scholar] [CrossRef] [Green Version]
- Robinson, S.; Li, Q.; Dechant, A.; Cohen, M.L. Neonatal loss of gamma-aminobutyric acid pathway expression after human perinatal brain injury. J. Neurosurg. 2006, 104 (Suppl. 6), 396–408. [Google Scholar]
- Smith-Hicks, C.L. GABAergic dysfunction in pediatric neuro-developmental disorders. Front. Cell. Neurosci. 2013, 7, 269. [Google Scholar] [CrossRef] [Green Version]
- Celio, M.R.; Blumcke, I. Perineuronal nets—A specialized form of extracellular matrix in the adult nervous system. Brain Res. Rev. 1994, 19, 128–145. [Google Scholar] [CrossRef]
- Bozzelli, P.L.; Alaiyed, S.; Kim, E.; Villapol, S.; Conant, K. Proteolytic remodeling of perineuronal nets: Effects on synaptic plasticity and neuronal population dynamics. Neural. Plast. 2018, 2018, 5735789. [Google Scholar]
- Wen, T.H.; Binder, D.K.; Ethell, I.M.; Razak, K.A. The perineuronal ‘safety’ net? Perineuronal net abnormalities in neurological disorders. Front. Mol. Neurosci. 2018, 11, 270. [Google Scholar] [CrossRef] [PubMed]
- Hockfield, S.; Kalb, R.G.; Zaremba, S.; Fryer, H. Expression of neural proteoglycans correlates with the acquisition of mature neuronal properties in the mammalian brain. Cold Spring Harb. Symp. Quant. Biol. 1990, 55, 505–514. [Google Scholar] [CrossRef]
- Testa, D.; Prochiantz, A.; Di Nardo, A.A. Perineuronal nets in brain physiology and disease. Semin. Cell Dev. Biol. 2019, 89, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Bitanihirwe, B.K.Y.; Mauney, S.A.; Woo, T.U.W. Weaving a net of neurobiological mechanisms in schizophrenia and unraveling the underlying pathophysiology. Biol. Psychiatry 2016, 80, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Morawski, M.; Bruckner, G.; Jager, C.; Seeger, G.; Matthews, R.T.; Arendt, T. Involvement of perineuronal and perisynaptic extracellular matrix in Alzheimer’s disease neuropathology. Brain Pathol. 2012, 22, 547–561. [Google Scholar] [CrossRef] [Green Version]
- McRae, P.A.; Baranov, E.; Rogers, S.L.; Porter, B.E. Persistent decrease in multiple components of the perineuronal net following status epilepticus. Eur. J. Neurosci. 2012, 36, 3471–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowke, T.M.; Galinsky, R.; Davidson, J.O.; Wassink, G.; Karunasinghe, R.N.; Prasad, J.D.; Bennet, L.; Gunn, A.J.; Dean, J.M. Loss of interneurons and disruption of perineuronal nets in the cerebral cortex following hypoxia-ischaemia in near-term fetal sheep. Sci. Rep. 2018, 8, 17686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decrock, E.; De Vuyst, E.; Vinken, M.; Van Moorhem, M.; Vranckx, K.; Wang, N.; Van Laeken, L.; De Bock, M.; D’Herde, K.; Lai, C.P.; et al. Connexin 43 hemichannels contribute to the propagation of apoptotic cell death in a rat C6 glioma cell model. Cell Death Differ. 2009, 16, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Nadarajah, B.; Thomaidou, D.; Evans, W.H.; Parnavelas, J.G. Gap junctions in the adult cerebral cortex: Regional differences in their distribution and cellular expression of connexins. J. Comp. Neurol. 1996, 376, 326–342. [Google Scholar] [CrossRef]
- Nakase, T.; Yoshida, Y.; Nagata, K. Enhanced connexin 43 immunoreactivity in penumbral areas in the human brain following ischemia. Glia 2006, 54, 369–375. [Google Scholar] [CrossRef]
- Davidson, J.O.; Green, C.R.; Nicholson, L.F.; O’Carroll, S.J.; Fraser, M.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann. Neurol. 2012, 71, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, R.; Davidson, J.O.; Lear, C.A.; Bennet, L.; Green, C.R.; Gunn, A.J. Connexin hemichannel blockade improves survival of striatal GABA-ergic neurons after global cerebral ischaemia in term-equivalent fetal sheep. Sci. Rep. 2017, 7, 6304. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, G.H.; Baghurst, K.I.; Potter, B.J.; Hetzel, B.S. Foetal brain development in the sheep. Neuropathol. Appl. Neurobiol. 1979, 5, 103–114. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Timing of neuroblast multiplication in developing human brain. Nature 1970, 226, 639–640. [Google Scholar] [CrossRef] [PubMed]
- Stolp, H.B.; Fleiss, B.; Arai, Y.; Supramaniam, V.; Vontell, R.; Birtles, S.; Yates, A.G.; Baburamani, A.A.; Thornton, C.; Rutherford, M.; et al. Interneuron development is disrupted in preterm brains with diffuse white matter injury: Observations in mouse and human. Front. Physiol. 2019, 10, 955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, M.; Smiley, J.F.; Hui, M.; Masiello, K.; Betz, J.; Ilina, M.; Saito, M.; Wilson, D.A. Neonatal ethanol disturbs the normal maturation of parvalbumin interneurons surrounded by subsets of perineuronal nets in the cerebral cortex: Partial reversal by lithium. Cereb. Cortex 2019, 29, 1383–1397. [Google Scholar] [CrossRef]
- O’Carroll, S.J.; Alkadhi, M.; Nicholson, L.F.; Green, C.R. Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun. Adhes. 2008, 15, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Chacko, A.; Andronikou, S.; Mian, A.; Goncalves, F.G.; Vedajallam, S.; Thai, N.J. Cortical ischaemic patterns in term partial-prolonged hypoxic-ischaemic injury-the inter-arterial watershed demonstrated through atrophy, ulegyria and signal change on delayed MRI scans in children with cerebral palsy. Insights Imaging 2020, 11, 53. [Google Scholar] [CrossRef]
- Ahearne, C.E.; Boylan, G.B.; Murray, D.M. Short and long term prognosis in perinatal asphyxia: An update. World J. Clin. Pediatr. 2016, 5, 67–74. [Google Scholar] [CrossRef]
- Villani, F.; D’Incerti, L.; Granata, T.; Battaglia, G.; Vitali, P.; Chiapparini, L.; Avanzini, G. Epileptic and imaging findings in perinatal hypoxic-ischemic encephalopathy with ulegyria. Epilepsy Res. 2003, 55, 235–243. [Google Scholar] [CrossRef]
- Ouwehand, S.; Smidt, L.C.A.; Dudink, J.; Benders, M.; de Vries, L.S.; Groenendaal, F.; van der Aa, N.E. Predictors of outcomes in hypoxic-ischemic encephalopathy following hypothermia: A meta-analysis. Neonatology 2020, 1–17. [Google Scholar] [CrossRef]
- Davidson, J.O.; Green, C.R.; Nicholson, L.F.; Bennet, L.; Gunn, A.J. Deleterious effects of high dose connexin 43 mimetic peptide infusion after cerebral ischaemia in near-term fetal sheep. Int. J. Mol. Sci. 2012, 13, 6303–6319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drury, P.P.; Davidson, J.O.; Mathai, S.; van den Heuij, L.G.; Ji, H.; Bennet, L.; Tan, S.; Silverman, R.B.; Gunn, A.J. nNOS inhibition during profound asphyxia reduces seizure burden and improves survival of striatal phenotypic neurons in preterm fetal sheep. Neuropharmacology 2014, 83, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClendon, E.; Chen, K.; Gong, X.; Sharifnia, E.; Hagen, M.; Cai, V.; Shaver, D.C.; Riddle, A.; Dean, J.M.; Gunn, A.J.; et al. Prenatal cerebral ischemia triggers dysmaturation of caudate projection neurons. Ann. Neurol. 2014, 75, 508–524. [Google Scholar] [CrossRef] [PubMed]
- Ardalan, M.; Svedin, P.; Baburamani, A.A.; Supramaniam, V.G.; Ek, J.; Hagberg, H.; Mallard, C. Dysmaturation of somatostatin interneurons following umbilical cord occlusion in preterm fetal sheep. Front. Physiol. 2019, 10, 563. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.H.; Alwakeel, A.J.; Goddard, L.; Hobbs, C.E.; Gowing, E.K.; Barnett, E.R.; Kohe, S.E.; Sizemore, R.J.; Oorschot, D.E. Delayed post-treatment with bone marrow-derived mesenchymal stem cells is neurorestorative of striatal medium-spiny projection neurons and improves motor function after neonatal rat hypoxia-ischemia. Mol. Cell. Neurosci. 2015, 68, 56–72. [Google Scholar] [CrossRef]
- Lacaille, H.; Vacher, C.M.; Bakalar, D.; O’Reilly, J.J.; Salzbank, J.; Penn, A.A. Impaired interneuron development in a novel model of neonatal brain injury. eNeuro 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Louzoun-Kaplan, V.; Zuckerman, M.; Perez-Polo, J.R.; Golan, H.M. Prenatal hypoxia down regulates the GABA pathway in newborn mice cerebral cortex; partial protection by MgSO4. Int. J. Dev. Neurosci. 2008, 26, 77–85. [Google Scholar] [CrossRef]
- Van de Berg, W.D.; Kwaijtaal, M.; de Louw, A.J.; Lissone, N.P.; Schmitz, C.; Faull, R.L.; Blokland, A.; Blanco, C.E.; Steinbusch, H.W. Impact of perinatal asphyxia on the GABAergic and locomotor system. Neuroscience 2003, 117, 83–96. [Google Scholar] [CrossRef]
- Nisimov, H.; Orenbuch, A.; Pleasure, S.J.; Golan, H.M. Impaired organization of GABAergic neurons following prenatal hypoxia. Neuroscience 2018, 384, 300–313. [Google Scholar] [CrossRef]
- Failor, S.; Nguyen, V.; Darcy, D.P.; Cang, J.; Wendland, M.F.; Stryker, M.P.; McQuillen, P.S. Neonatal cerebral hypoxia-ischemia impairs plasticity in rat visual cortex. J. Neurosci. 2010, 30, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Valdez, R.; Emerson, P.; Goffigan-Holmes, J.; Kirkwood, A.; Martin, L.J.; Northington, F.J. Delayed injury of hippocampal interneurons after neonatal hypoxia-ischemia and therapeutic hypothermia in a murine model. Hippocampus 2018, 28, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Komitova, M.; Xenos, D.; Salmaso, N.; Tran, K.M.; Brand, T.; Schwartz, M.L.; Ment, L.; Vaccarino, F.M. Hypoxia-induced developmental delays of inhibitory interneurons are reversed by environmental enrichment in the postnatal mouse forebrain. J. Neurosci. 2013, 33, 13375–13387. [Google Scholar] [CrossRef] [PubMed]
- Verney, C.; Rees, S.; Biran, V.; Thompson, M.; Inder, T.; Gressens, P. Neuronal damage in the preterm baboon: Impact of the mode of ventilatory support. J. Neuropathol. Exp. Neurol. 2010, 69, 473–482. [Google Scholar] [CrossRef]
- Panda, S.; Dohare, P.; Jain, S.; Parikh, N.; Singla, P.; Mehdizadeh, R.; Klebe, D.W.; Kleinman, G.M.; Cheng, B.; Ballabh, P. Estrogen treatment reverses prematurity-induced disruption in cortical interneuron population. J. Neurosci. 2018, 38, 7378–7391. [Google Scholar] [CrossRef] [Green Version]
- Tibrewal, M.; Cheng, B.; Dohare, P.; Hu, F.; Mehdizadeh, R.; Wang, P.; Zheng, D.; Ungvari, Z.; Ballabh, P. Disruption of interneuron neurogenesis in premature newborns and reversal with estrogen treatment. J. Neurosci. 2018, 38, 1100–1113. [Google Scholar] [CrossRef]
- Katsarou, A.M.; Moshe, S.L.; Galanopoulou, A.S. Interneuronopathies and their role in early life epilepsies and neurodevelopmental disorders. Epilepsia Open 2017, 2, 284–306. [Google Scholar] [CrossRef] [Green Version]
- Galanopoulou, A.S. GABA(A) receptors in normal development and seizures: Friends or foes? Curr. Neuropharmacol. 2008, 6, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D.A.; Moghaddam, B. Cognitive dysfunction in schizophrenia: Convergence of gamma-aminobutyric acid and glutamate alterations. Arch. Neurol. 2006, 63, 1372–1376. [Google Scholar] [CrossRef]
- Morishita, H.; Kundakovic, M.; Bicks, L.; Mitchell, A.; Akbarian, S. Interneuron epigenomes during the critical period of cortical plasticity: Implications for schizophrenia. Neurobiol. Learn. Mem. 2015, 124, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Powell, E.M.; Campbell, D.B.; Stanwood, G.D.; Davis, C.; Noebels, J.L.; Levitt, P. Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J. Neurosci. 2003, 23, 622–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botting, N.; Powls, A.; Cooke, R.W.; Marlow, N. Attention deficit hyperactivity disorders and other psychiatric outcomes in very low birthweight children at 12 years. J. Child. Psychol. Psychiatry 1997, 38, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Indredavik, M.S.; Vik, T.; Evensen, K.A.; Skranes, J.; Taraldsen, G.; Brubakk, A.M. Perinatal risk and psychiatric outcome in adolescents born preterm with very low birth weight or term small for gestational age. J. Dev. Behav. Pediatr. 2010, 31, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Barnes, S.A.; Pinto-Duarte, A.; Kappe, A.; Zembrzycki, A.; Metzler, A.; Mukamel, E.A.; Lucero, J.; Wang, X.; Sejnowski, T.J.; Markou, A.; et al. Disruption of mGluR5 in parvalbumin-positive interneurons induces core features of neurodevelopmental disorders. Mol. Psychiatry 2015, 20, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Wohr, M.; Orduz, D.; Gregory, P.; Moreno, H.; Khan, U.; Vorckel, K.J.; Wolfer, D.P.; Welzl, H.; Gall, D.; Schiffmann, S.N.; et al. Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl. Psychiatry 2015, 5, e525. [Google Scholar] [CrossRef]
- Lim, L.; Mi, D.; Llorca, A.; Marin, O. Development and functional diversification of cortical interneurons. Neuron 2018, 100, 294–313. [Google Scholar] [CrossRef] [Green Version]
- Letinic, K.; Zoncu, R.; Rakic, P. Origin of GABAergic neurons in the human neocortex. Nature 2002, 417, 645–649. [Google Scholar] [CrossRef]
- Luhmann, H.J.; Kirischuk, S.; Sinning, A.; Kilb, W. Early GABAergic circuitry in the cerebral cortex. Curr. Opin. Neurobiol. 2014, 26, 72–78. [Google Scholar] [CrossRef]
- Pyka, M.; Wetzel, C.; Aguado, A.; Geissler, M.; Hatt, H.; Faissner, A. Chondroitin sulfate proteoglycans regulate astrocyte-dependent synaptogenesis and modulate synaptic activity in primary embryonic hippocampal neurons. Eur. J. Neurosci. 2011, 33, 2187–2202. [Google Scholar] [CrossRef]
- Zaremba, S.; Guimaraes, A.; Kalb, R.G.; Hockfield, S. Characterization of an activity-dependent, neuronal surface proteoglycan identified with monoclonal antibody Cat-301. Neuron 1989, 2, 1207–1219. [Google Scholar] [CrossRef]
- Frischknecht, R.; Chang, K.J.; Rasband, M.N.; Seidenbecher, C.I. Neural ECM molecules in axonal and synaptic homeostatic plasticity. Prog. Brain Res. 2014, 214, 81–100. [Google Scholar] [PubMed]
- Srivastava, T.; Diba, P.; Dean, J.M.; Banine, F.; Shaver, D.; Hagen, M.; Gong, X.; Su, W.; Emery, B.; Marks, D.L.; et al. A TLR/AKT/FoxO3 immune tolerance-like pathway disrupts the repair capacity of oligodendrocyte progenitors. J. Clin. Investig. 2018, 128, 2025–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, F.; Kakizawa, H.; Nishizuka, M.; Hirano, K.; Shuo, T.; Ida, M.; Tokita, Y.; Aono, S.; Keino, H.; Oohira, A. Changes in the amounts of chondroitin sulfate proteoglycans in rat brain after neonatal hypoxia-ischemia. J. Neurosci. Res. 2005, 81, 837–845. [Google Scholar] [CrossRef]
- Aya-ay, J.; Mayer, J.; Eakin, A.K.; Muffly, B.G.; Anello, M.; Sandy, J.D.; Gottschall, P.E. The effect of hypoxic-ischemic brain injury in perinatal rats on the abundance and proteolysis of brevican and NG2. Exp. Neurol. 2005, 193, 149–162. [Google Scholar] [CrossRef]
- Leonardo, C.C.; Eakin, A.K.; Ajmo, J.M.; Gottschall, P.E. Versican and brevican are expressed with distinct pathology in neonatal hypoxic-ischemic injury. J. Neurosci. Res. 2008, 86, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Karetko-Sysa, M.; Skangiel-Kramska, J.; Nowicka, D. Disturbance of perineuronal nets in the perilesional area after photothrombosis is not associated with neuronal death. Exp. Neurol. 2011, 231, 113–126. [Google Scholar] [CrossRef]
- Yutsudo, N.; Kitagawa, H. Involvement of chondroitin 6-sulfation in temporal lobe epilepsy. Exp. Neurol. 2015, 274, 126–133. [Google Scholar] [CrossRef]
- Pollock, E.; Everest, M.; Brown, A.; Poulter, M.O. Metalloproteinase inhibition prevents inhibitory synapse reorganization and seizure genesis. Neurobiol. Dis. 2014, 70, 21–31. [Google Scholar] [CrossRef]
- Hobohm, C.; Gunther, A.; Grosche, J.; Rossner, S.; Schneider, D.; Bruckner, G. Decomposition and long-lasting downregulation of extracellular matrix in perineuronal nets induced by focal cerebral ischemia in rats. J. Neurosci. Res. 2005, 80, 539–548. [Google Scholar] [CrossRef]
- Hartig, W.; Mages, B.; Aleithe, S.; Nitzsche, B.; Altmann, S.; Barthel, H.; Krueger, M.; Michalski, D. Damaged neocortical perineuronal nets due to experimental focal cerebral ischemia in mice, rats and sheep. Front. Integr. Neurosci. 2017, 11, 15. [Google Scholar] [CrossRef] [Green Version]
- Dityatev, A.; Bruckner, G.; Dityateva, G.; Grosche, J.; Kleene, R.; Schachner, M. Activity-dependent formation and functions of chondroitin sulfate-rich extracellular matrix of perineuronal nets. Dev. Neurobiol. 2007, 67, 570–588. [Google Scholar] [CrossRef]
- Vedunova, M.; Sakharnova, T.; Mitroshina, E.; Perminova, M.; Pimashkin, A.; Zakharov, Y.; Dityatev, A.; Mukhina, I. Seizure-like activity in hyaluronidase-treated dissociated hippocampal cultures. Front. Cell. Neurosci. 2013, 7, 149. [Google Scholar] [CrossRef] [Green Version]
- Balashova, A.; Pershin, V.; Zaborskaya, O.; Tkachenko, N.; Mironov, A.; Guryev, E.; Kurbatov, L.; Gainullin, M.; Mukhina, I. Enzymatic digestion of hyaluronan-based brain extracellular matrix in vivo can induce seizures in neonatal mice. Front. Neurosci. 2019, 13, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arranz, A.M.; Perkins, K.L.; Irie, F.; Lewis, D.P.; Hrabe, J.; Xiao, F.; Itano, N.; Kimata, K.; Hrabetova, S.; Yamaguchi, Y. Hyaluronan deficiency due to Has3 knock-out causes altered neuronal activity and seizures via reduction in brain extracellular space. J. Neurosci. 2014, 34, 6164–6176. [Google Scholar] [CrossRef] [Green Version]
- Suttkus, A.; Rohn, S.; Jager, C.; Arendt, T.; Morawski, M. Neuroprotection against iron-induced cell death by perineuronal nets—An in vivo analysis of oxidative stress. Am. J. Neurodegener. Dis. 2012, 1, 122–129. [Google Scholar] [PubMed]
- Cabungcal, J.H.; Steullet, P.; Morishita, H.; Kraftsik, R.; Cuenod, M.; Hensch, T.K.; Do, K.Q. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 9130–9135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placha, K.; Luptakova, D.; Baciak, L.; Ujhazy, E.; Juranek, I. Neonatal brain injury as a consequence of insufficient cerebral oxygenation. Neuro. Endocrinol. Lett. 2016, 37, 79–96. [Google Scholar] [PubMed]
- Davidson, J.O.; Green, C.R.; Bennet, L.; Nicholson, L.F.; Danesh-Meyer, H.; O’Carroll, S.J.; Gunn, A.J. A key role for connexin hemichannels in spreading ischemic brain injury. Curr. Drug Targets 2013, 14, 36–46. [Google Scholar] [CrossRef]
- Schulz, R.; Gorge, P.M.; Gorbe, A.; Ferdinandy, P.; Lampe, P.D.; Leybaert, L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol. Ther. 2015, 153, 90–106. [Google Scholar] [CrossRef] [Green Version]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in cardiovascular and neurovascular health and disease: Pharmacological implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef]
- Kang, J.; Kang, N.; Lovatt, D.; Torres, A.; Zhao, Z.; Lin, J.; Nedergaard, M. Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 2008, 28, 4702–4711. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belousov, A.B.; Fontes, J.D.; Freitas-Andrade, M.; Naus, C.C. Gap junctions and hemichannels: Communicating cell death in neurodevelopment and disease. BMC Cell Biol. 2017, 18 (Suppl. 1), 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, M.; Culot, M.; Wang, N.; Bol, M.; Decrock, E.; De Vuyst, E.; da Costa, A.; Dauwe, I.; Vinken, M.; Simon, A.M.; et al. Connexin channels provide a target to manipulate brain endothelial calcium dynamics and blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 2011, 31, 1942–1957. [Google Scholar] [CrossRef] [Green Version]
- Danesh-Meyer, H.V.; Kerr, N.M.; Zhang, J.; Eady, E.K.; O’Carroll, S.J.; Nicholson, L.F.; Johnson, C.S.; Green, C.R. Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischaemia. Brain 2012, 135, 506–520. [Google Scholar] [CrossRef] [Green Version]
- Mugisho, O.O.; Green, C.R.; Kho, D.T.; Zhang, J.; Graham, E.S.; Acosta, M.L.; Rupenthal, I.D. The inflammasome pathway is amplified and perpetuated in an autocrine manner through connexin43 hemichannel mediated ATP release. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 385–393. [Google Scholar] [CrossRef]
- Maatouk, L.; Yi, C.; Carrillo-de Sauvage, M.A.; Compagnion, A.C.; Hunot, S.; Ezan, P.; Hirsch, E.C.; Koulakoff, A.; Pfrieger, F.W.; Tronche, F.; et al. Glucocorticoid receptor in astrocytes regulates midbrain dopamine neurodegeneration through connexin hemichannel activity. Cell Death Differ. 2019, 26, 580–596. [Google Scholar] [CrossRef]
- Cea, L.A.; Cisterna, B.A.; Puebla, C.; Frank, M.; Figueroa, X.F.; Cardozo, C.; Willecke, K.; Latorre, R.; Saez, J.C. De novo expression of connexin hemichannels in denervated fast skeletal muscles leads to atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 16229–16234. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.Q.; Green, C.R.; Bennet, L.; Gunn, A.J.; Davidson, J.O. The role of connexin and pannexin channels in perinatal brain injury and inflammation. Front. Physiol. 2019, 10, 141. [Google Scholar] [CrossRef] [Green Version]
- Galinsky, R.; Davidson, J.O.; Dean, J.M.; Green, C.R.; Bennet, L.; Gunn, A.J. Glia and hemichannels: Key mediators of perinatal encephalopathy. Neural Regen. Res. 2018, 13, 181–189. [Google Scholar]
- Froger, N.; Orellana, J.A.; Calvo, C.F.; Amigou, E.; Kozoriz, M.G.; Naus, C.C.; Saez, J.C.; Giaume, C. Inhibition of cytokine-induced connexin43 hemichannel activity in astrocytes is neuroprotective. Mol. Cell. Neurosci. 2010, 45, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Buser, J.R.; Maire, J.; Riddle, A.; Gong, X.; Nguyen, T.; Nelson, K.; Luo, N.L.; Ren, J.; Struve, J.; Sherman, L.S.; et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann. Neurol. 2012, 71, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.H.; Romero, R.; Kim, C.J.; Koo, J.N.; Choe, G.; Syn, H.C.; Chi, J.G. High expression of tumor necrosis factor-alpha and interleukin-6 in periventricular leukomalacia. Am. J. Obstet. Gynecol. 1997, 177, 406–411. [Google Scholar] [CrossRef]
- Mallard, C.; Davidson, J.O.; Tan, S.; Green, C.R.; Bennet, L.; Robertson, N.J.; Gunn, A.J. Astrocytes and microglia in acute cerebral injury underlying cerebral palsy associated with preterm birth. Pediatr. Res. 2014, 75, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Fleiss, B.; Gressens, P. Tertiary mechanisms of brain damage: A new hope for treatment of cerebral palsy? Lancet Neurol. 2012, 11, 556–566. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Reactive gliosis in the pathogenesis of CNS diseases. Biochim. Biophys. Acta 2016, 1862, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J. Dysmaturation of premature brain: Importance, cellular mechanisms, and potential interventions. Pediatr. Neurol. 2019, 95, 42–66. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A. White matter injury in the preterm infant: Pathology and mechanisms. Acta Neuropathol. 2017, 134, 331–349. [Google Scholar] [CrossRef]
- Xing, L.; Yang, T.; Cui, S.; Chen, G. Connexin hemichannels in astrocytes: Role in CNS disorders. Front. Mol. Neurosci. 2019, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Rash, J.E.; Yasumura, T.; Dudek, F.E.; Nagy, J.I. Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J. Neurosci. 2001, 21, 1983–2000. [Google Scholar] [CrossRef] [Green Version]
- John, S.A.; Kondo, R.; Wang, S.Y.; Goldhaber, J.I.; Weiss, J.N. Connexin-43 hemichannels opened by metabolic inhibition. J. Biol. Chem. 1999, 274, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Sinovas, A.; Cabestrero, A.; Lopez, D.; Torre, I.; Morente, M.; Abellan, A.; Miro, E.; Ruiz-Meana, M.; Garcia-Dorado, D. The modulatory effects of connexin 43 on cell death/survival beyond cell coupling. Prog. Biophys. Mol. Biol. 2007, 94, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Quist, A.P.; Rhee, S.K.; Lin, H.; Lal, R. Physiological role of gap-junctional hemichannels. Extracellular calcium-dependent isosmotic volume regulation. J. Cell Biol. 2000, 148, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Hernandez, D.E.; Ezan, P.; Velarde, V.; Bennett, M.V.; Giaume, C.; Saez, J.C. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia 2010, 58, 329–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, J.O.; Drury, P.P.; Green, C.R.; Nicholson, L.F.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade is neuroprotective after asphyxia in preterm fetal sheep. PLoS ONE 2014, 9, e96558. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Green, C.R.; Nicholson, L.F.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade is neuroprotective after, but not during, global cerebral ischemia in near-term fetal sheep. Exp. Neurol. 2013, 248, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Rout, A.L.; Wassink, G.; Yuill, C.A.; Zhang, F.G.; Green, C.R.; Bennet, L.; Gunn, A.J. Non-additive effects of delayed connexin hemichannel blockade and hypothermia after cerebral ischemia in near-term fetal sheep. J. Cereb. Blood Flow Metab. 2015, 35, 2052–2061. [Google Scholar] [CrossRef] [Green Version]
- Song, I.; Dityatev, A. Crosstalk between glia, extracellular matrix and neurons. Brain Res. Bull. 2018, 136, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Wiese, S.; Karus, M.; Faissner, A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front. Pharmacol. 2012, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.V.; Bouvier, D.S. Astrocyte-secreted matricellular proteins in CNS remodelling during development and disease. Neural. Plast. 2014, 2014, 321209. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Ferrer, M.; Dityatev, A. Shaping synapses by the neural extracellular matrix. Front. Neuroanat. 2018, 12, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralay Ranaivo, H.; Hodge, J.N.; Choi, N.; Wainwright, M.S. Albumin induces upregulation of matrix metalloproteinase-9 in astrocytes via MAPK and reactive oxygen species-dependent pathways. J. Neuroinflamm. 2012, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, G.A.; Cunningham, L.A.; Wallace, J.; Alexander, S.; Estrada, E.Y.; Grossetete, M.; Razhagi, A.; Miller, K.; Gearing, A. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: Activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. 2001, 893, 104–112. [Google Scholar] [CrossRef]
- Krishnaswamy, V.R.; Benbenishty, A.; Blinder, P.; Sagi, I. Demystifying the extracellular matrix and its proteolytic remodeling in the brain: Structural and functional insights. Cell. Mol. Life Sci. 2019, 76, 3229–3248. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Nasu-Tada, K.; Fujishita, K.; Sato, K.; Koizumi, S. Secretion of matrix metalloproteinase-9 from astrocytes by inhibition of tonic P2Y14-receptor-mediated signal(s). Cell. Mol. Neurobiol. 2013, 33, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Hagen, M.W.; Riddle, A.; McClendon, E.; Gong, X.; Shaver, D.; Srivastava, T.; Dean, J.M.; Bai, J.Z.; Fowke, T.M.; Gunn, A.J.; et al. Role of recurrent hypoxia-ischemia in preterm white matter injury severity. PLoS ONE 2014, 9, e112800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Happel, M.F.K.; Frischknecht, R. Neuronal plasticity in the juvenile and adult brain regulated by the extracellular matrix. In Composition and Function of the Extracellular Matrix in the Human Body; IntechOpen: Rijeka, Croatia, 2016. [Google Scholar]
- Wen, T.H.; Afroz, S.; Reinhard, S.M.; Palacios, A.R.; Tapia, K.; Binder, D.K.; Razak, K.A.; Ethell, I.M. Genetic reduction of matrix metalloproteinase-9 promotes formation of perineuronal nets around parvalbumin-expressing interneurons and normalizes auditory cortex responses in developing fmr1 knock-out mice. Cereb. Cortex 2018, 28, 3951–3964. [Google Scholar] [CrossRef] [Green Version]
- Lemarchant, S.; Pruvost, M.; Montaner, J.; Emery, E.; Vivien, D.; Kanninen, K.; Koistinaho, J. ADAMTS proteoglycanases in the physiological and pathological central nervous system. J. Neuroinflamm. 2013, 10, 133. [Google Scholar] [CrossRef] [Green Version]
- Leonardo, C.C.; Eakin, A.K.; Ajmo, J.M.; Collier, L.A.; Pennypacker, K.R.; Strongin, A.Y.; Gottschall, P.E. Delayed administration of a matrix metalloproteinase inhibitor limits progressive brain injury after hypoxia-ischemia in the neonatal rat. J. Neuroinflamm. 2008, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Bednarek, N.; Svedin, P.; Garnotel, R.; Favrais, G.; Loron, G.; Schwendiman, L.; Hagberg, H.; Morville, P.; Mallard, C.; Gressens, P. Increased MMP-9 and TIMP-1 in mouse neonatal brain and plasma and in human neonatal plasma after hypoxia-ischemia: A potential marker of neonatal encephalopathy. Pediatr. Res. 2012, 71, 63–70. [Google Scholar] [CrossRef]
- Planas, A.M.; Sole, S.; Justicia, C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol. Dis. 2001, 8, 834–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amantea, D.; Russo, R.; Gliozzi, M.; Fratto, V.; Berliocchi, L.; Bagetta, G.; Bernardi, G.; Corasaniti, M.T. Early upregulation of matrix metalloproteinases following reperfusion triggers neuroinflammatory mediators in brain ischemia in rat. Int. Rev. Neurobiol. 2007, 82, 149–169. [Google Scholar] [PubMed]
- Cross, A.K.; Haddock, G.; Stock, C.J.; Allan, S.; Surr, J.; Bunning, R.A.; Buttle, D.J.; Woodroofe, M.N. ADAMTS-1 and -4 are up-regulated following transient middle cerebral artery occlusion in the rat and their expression is modulated by TNF in cultured astrocytes. Brain Res. 2006, 1088, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Qteishat, A.; Gaffney, J.J.; Krupinski, J.; Slevin, M. Hyaluronan expression following middle cerebral artery occlusion in the rat. NeuroReport 2006, 17, 1111–1114. [Google Scholar] [CrossRef] [PubMed]
- Romanic, A.M.; White, R.F.; Arleth, A.J.; Ohlstein, E.H.; Barone, F.C. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: Inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 1998, 29, 1020–1030. [Google Scholar] [CrossRef] [Green Version]
- Sunagawa, S.; Ichiyama, T.; Honda, R.; Fukunaga, S.; Maeba, S.; Furukawa, S. Matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in perinatal asphyxia. Brain Dev. 2009, 31, 588–593. [Google Scholar] [CrossRef]
- Al’Qteishat, A.; Gaffney, J.; Krupinski, J.; Rubio, F.; West, D.; Kumar, S.; Kumar, P.; Mitsios, N.; Slevin, M. Changes in hyaluronan production and metabolism following ischaemic stroke in man. Brain 2006, 129 Pt 8, 2158–2176. [Google Scholar] [CrossRef]
- Gu, Z.; Cui, J.; Brown, S.; Fridman, R.; Mobashery, S.; Strongin, A.Y.; Lipton, S.A. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J. Neurosci. 2005, 25, 6401–6408. [Google Scholar] [CrossRef] [Green Version]
- Cocozzelli, A.G.; White, T.W. Connexin 43 mutations lead to increased hemichannel functionality in skin disease. Int. J. Mol. Sci. 2019, 20, 6168. [Google Scholar] [CrossRef] [Green Version]
- Deva, N.C.; Zhang, J.; Green, C.R.; Danesh-Meyer, H.V. Connexin43 modulation inhibits scarring in a rabbit eye glaucoma trabeculectomy model. Inflammation 2012, 35, 1276–1286. [Google Scholar] [CrossRef]
- O’Carroll, S.J.; Gorrie, C.A.; Velamoor, S.; Green, C.R.; Nicholson, L.F. Connexin43 mimetic peptide is neuroprotective and improves function following spinal cord injury. Neurosci. Res. 2013, 75, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Gangoso, E.; Talaveron, R.; Jaraiz-Rodriguez, M.; Dominguez-Prieto, M.; Ezan, P.; Koulakoff, A.; Medina, J.M.; Giaume, C.; Tabernero, A. A c-Src inhibitor peptide based on connexin43 exerts neuroprotective effects through the inhibition of glial hemichannel activity. Front. Mol. Neurosci. 2017, 10, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonkin, R.S.; Bowles, C.; Perera, C.J.; Keating, B.A.; Makker, P.G.S.; Duffy, S.S.; Lees, J.G.; Tran, C.; Don, A.S.; Fath, T.; et al. Attenuation of mechanical pain hypersensitivity by treatment with Peptide5, a connexin-43 mimetic peptide, involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp. Neurol. 2018, 300, 1–12. [Google Scholar] [CrossRef]
- Chen, M.J.; Kress, B.; Han, X.; Moll, K.; Peng, W.; Ji, R.R.; Nedergaard, M. Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. Glia 2012, 60, 1660–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schock, S.C.; Leblanc, D.; Hakim, A.M.; Thompson, C.S. ATP release by way of connexin 36 hemichannels mediates ischemic tolerance in vitro. Biochem. Biophys. Res. Commun. 2008, 368, 138–144. [Google Scholar] [CrossRef]
- Thompson, R.J.; Jackson, M.F.; Olah, M.E.; Rungta, R.L.; Hines, D.J.; Beazely, M.A.; MacDonald, J.F.; MacVicar, B.A. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science 2008, 322, 1555–1559. [Google Scholar] [CrossRef] [Green Version]
- Weilinger, N.L.; Tang, P.L.; Thompson, R.J. Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J. Neurosci. 2012, 32, 12579–12588. [Google Scholar] [CrossRef]
- Freitas-Andrade, M.; Bechberger, J.F.; MacVicar, B.A.; Viau, V.; Naus, C.C. Pannexin1 knockout and blockade reduces ischemic stroke injury in female, but not in male mice. Oncotarget 2017, 8, 36973–36983. [Google Scholar] [CrossRef] [Green Version]
- Cisneros-Mejorado, A.; Gottlieb, M.; Cavaliere, F.; Magnus, T.; Koch-Nolte, F.; Scemes, E.; Perez-Samartin, A.; Matute, C. Blockade of P2X7 receptors or pannexin-1 channels similarly attenuates postischemic damage. J. Cereb. Blood Flow Metab. 2015, 35, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Griffin, J.M.; Harris, P.W.; Chan, S.H.; Nicholson, L.F.; Brimble, M.A.; O’Carroll, S.J.; Green, C.R. Characterizing the mode of action of extracellular connexin43 channel blocking mimetic peptides in an in vitro ischemia injury model. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 68–78. [Google Scholar] [CrossRef]
- Mazur, M.; Miller, R.H.; Robinson, S. Postnatal erythropoietin treatment mitigates neural cell loss after systemic prenatal hypoxic-ischemic injury. J. Neurosurg. Pediatr. 2010, 6, 206–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, S.; Scotter, J.; Dean, J.M.; Bennet, L.; Waldvogel, H.J.; Guan, J.; Faull, R.L.; Gunn, A.J. Induced cerebral hypothermia reduces post-hypoxic loss of phenotypic striatal neurons in preterm fetal sheep. Exp. Neurol. 2007, 203, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. Osteoarthr. Cartil. 2012, 20, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunn, A.J.; Gunn, T.R.; de Haan, H.H.; Williams, C.E.; Gluckman, P.D. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J. Clin. Investig. 1997, 99, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.E.; Gunn, A.J.; Mallard, C.; Gluckman, P.D. Outcome after ischemia in the developing sheep brain: An electroencephalographic and histological study. Ann. Neurol. 1992, 31, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Parsons, Y. Stereotaxic method and atlas for the ovine fetal forebrain. J. Dev. Physiol. 1983, 5, 101–128. [Google Scholar]
- Hartig, W.; Brauer, K.; Bruckner, G. Wisteria floribunda agglutinin-labelled nets surround parvalbumin-containing neurons. NeuroReport 1992, 3, 869–872. [Google Scholar] [CrossRef]
- Koppe, G.; Bruckner, G.; Brauer, K.; Hartig, W.; Bigl, V. Developmental patterns of proteoglycan-containing extracellular matrix in perineuronal nets and neuropil of the postnatal rat brain. Cell Tissue Res. 1997, 288, 33–41. [Google Scholar] [CrossRef]
- Scher, M.S.; Aso, K.; Beggarly, M.E.; Hamid, M.Y.; Steppe, D.A.; Painter, M.J. Electrographic seizures in preterm and full-term neonates: Clinical correlates, associated brain lesions, and risk for neurologic sequelae. Pediatrics 1993, 91, 128–134. [Google Scholar]
- Galinsky, R.; Davidson, J.O.; Bennet, L.; Green, C.R.; Gunn, A.J. Connexin hemichannel blockade improves survival of striatal neurons after perinatal cerebral ischaemia. J. Paediatr. Child Health 2015, 51, 60. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Dilution | Specificity | Source |
|---|---|---|---|
| Primary Antibodies | |||
| Gamma-aminobutyric acid 65/67 | 1:200 | GABAergic interneurons | Abcam, Melbourne, Australia |
| Parvalbumin | 1:200 | Parvalbumin interneurons | Swant Ltd., Marly, Switzerland |
| Calretinin | 1:200 | Calretinin interneurons | Swant Ltd. |
| Calbindin | 1:200 | Calbindin interneurons | Swant Ltd. |
| Biotinylated Wisteria floribunda agglutinin | 1:400 | Perineuronal nets | Sigma-Aldrich Co., Saint Louis, MO, USA |
| NeuN | 1:20 | Post-mitotic neurons | Merck Millipore, Billerica, MA, USA |
| Secondary Antibodies | |||
| Biotinylated goat anti-rabbit IgG | 1:200 | Vector Laboratories, Burlingame, CA, USA | |
| Biotinylated goat anti-mouse IgG | 1:200 | Vector Laboratories | |
| Streptavidin-conjugated IgG Alexa Fluor 594 | 1:200 | Thermo Fisher Scientific, Waltham, MA, USA | |
| Goat anti-rabbit IgG Alexa Fluor 488 | 1:200 | Thermo Fisher Scientific | |
| Goat anti-mouse IgG1 Alexa Fluor 647 | 1:100 | Thermo Fisher Scientific |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, P.; Davidson, J.O.; Fowke, T.M.; Galinsky, R.; Wassink, G.; Karunasinghe, R.N.; Prasad, J.D.; Ranasinghe, S.; Green, C.R.; Bennet, L.; et al. Connexin Hemichannel Mimetic Peptide Attenuates Cortical Interneuron Loss and Perineuronal Net Disruption Following Cerebral Ischemia in Near-Term Fetal Sheep. Int. J. Mol. Sci. 2020, 21, 6475. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186475
Yang P, Davidson JO, Fowke TM, Galinsky R, Wassink G, Karunasinghe RN, Prasad JD, Ranasinghe S, Green CR, Bennet L, et al. Connexin Hemichannel Mimetic Peptide Attenuates Cortical Interneuron Loss and Perineuronal Net Disruption Following Cerebral Ischemia in Near-Term Fetal Sheep. International Journal of Molecular Sciences. 2020; 21(18):6475. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186475
Chicago/Turabian StyleYang, Panzao, Joanne O. Davidson, Tania M. Fowke, Robert Galinsky, Guido Wassink, Rashika N. Karunasinghe, Jaya D. Prasad, Sumudu Ranasinghe, Colin R. Green, Laura Bennet, and et al. 2020. "Connexin Hemichannel Mimetic Peptide Attenuates Cortical Interneuron Loss and Perineuronal Net Disruption Following Cerebral Ischemia in Near-Term Fetal Sheep" International Journal of Molecular Sciences 21, no. 18: 6475. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186475