P2 × 7 Receptor Inhibits Astroglial Autophagy via Regulating FAK- and PHLPP1/2-Mediated AKT-S473 Phosphorylation Following Kainic Acid-Induced Seizures

Abstract
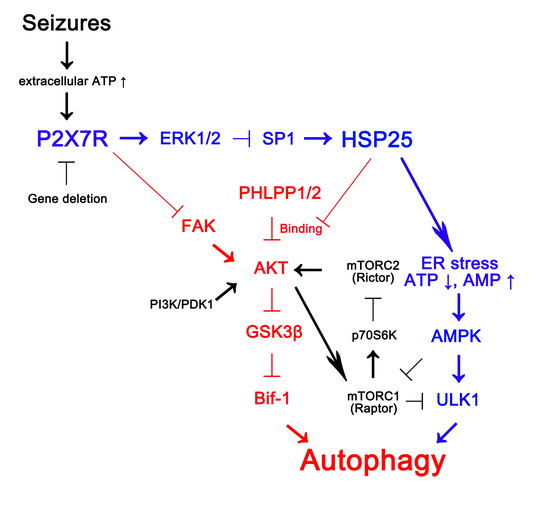
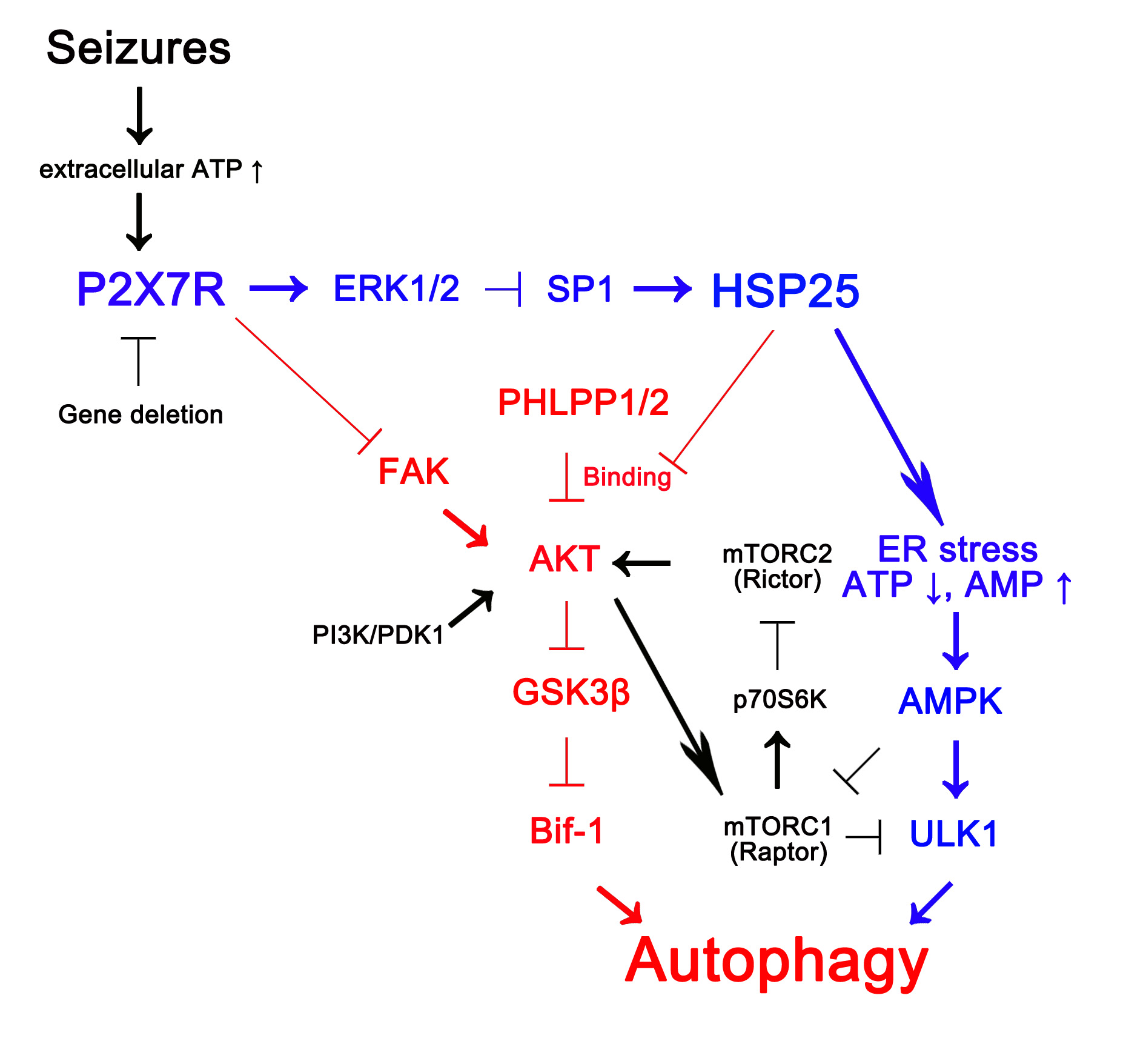
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
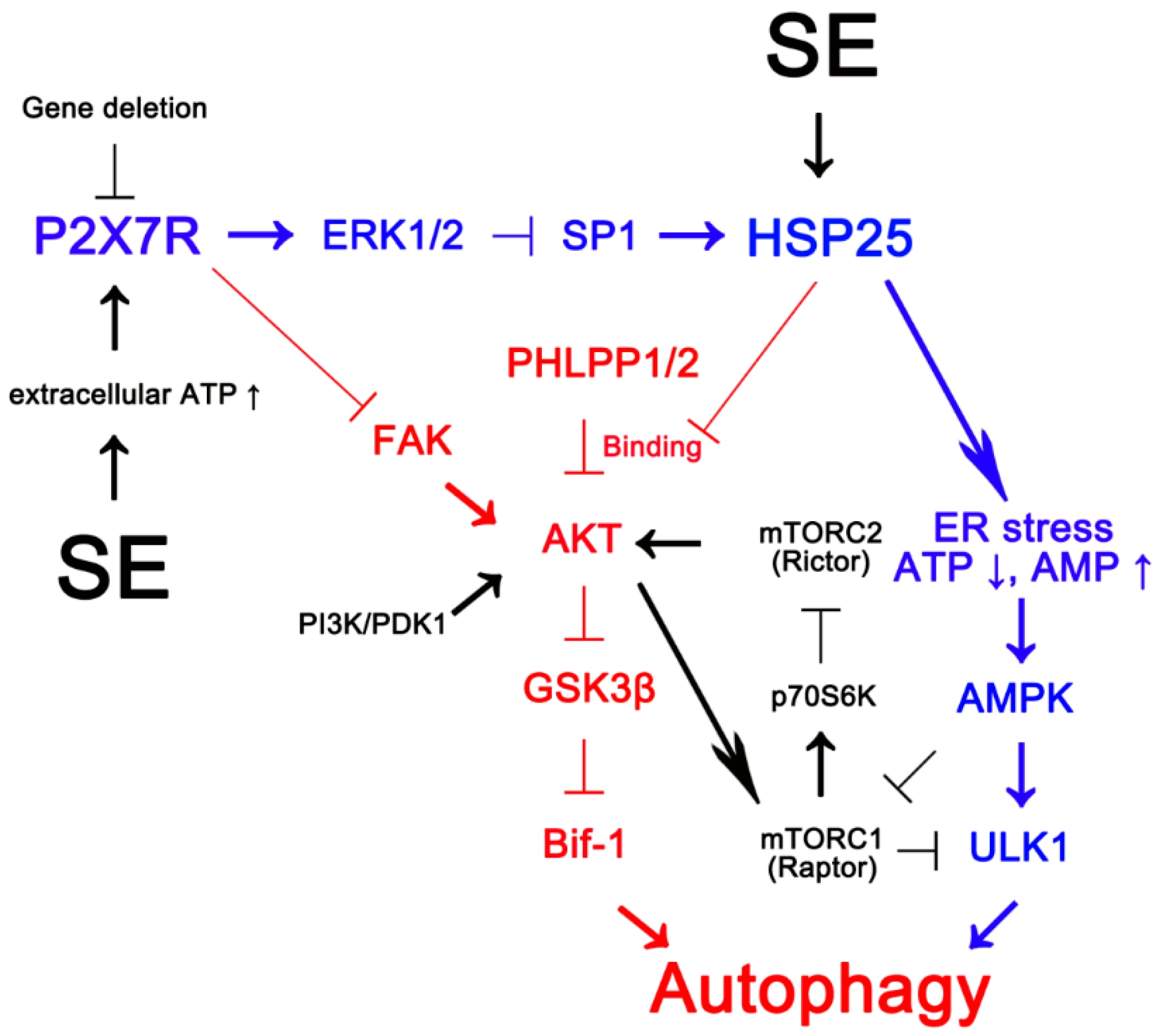
1. Introduction
2. Results
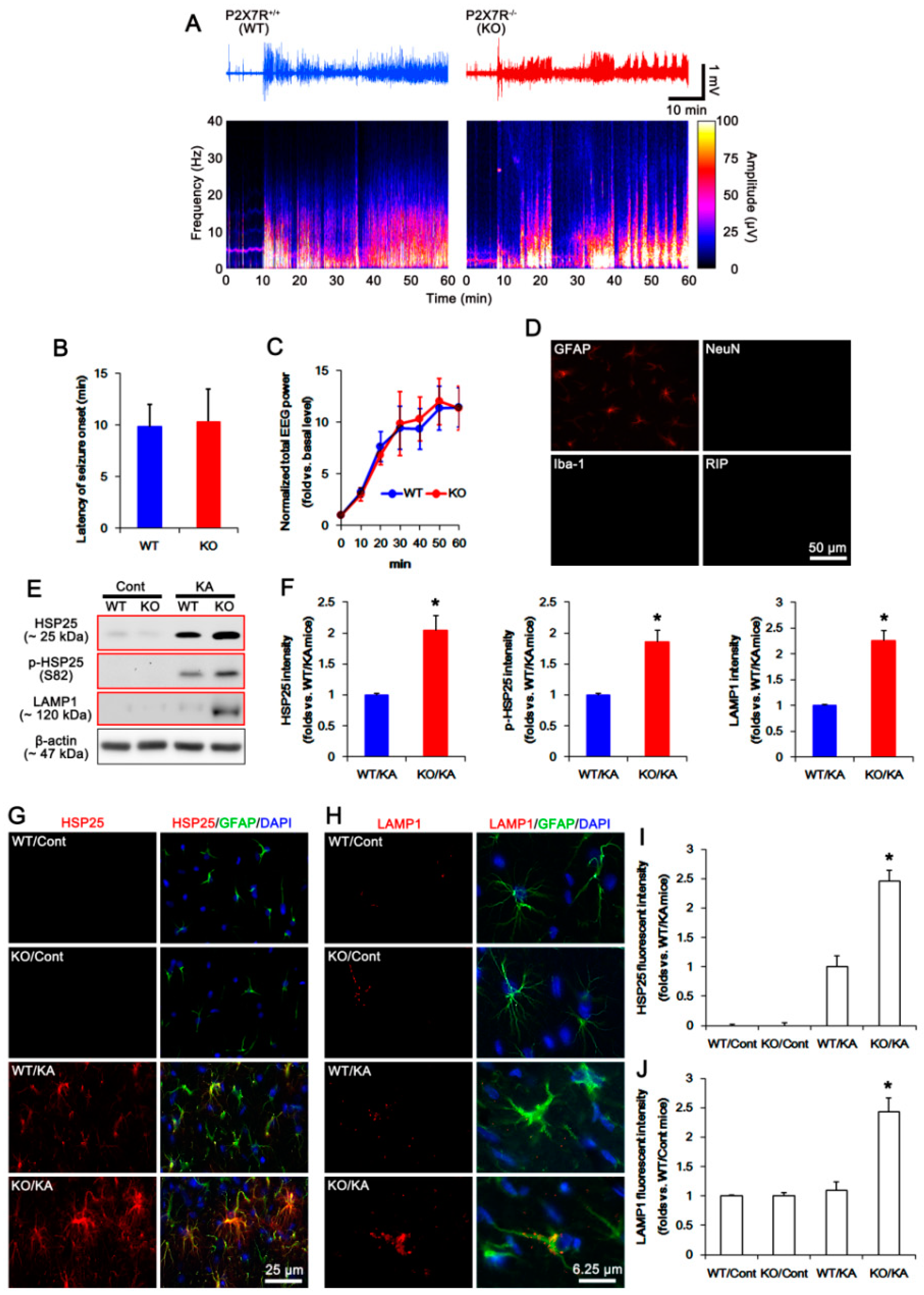
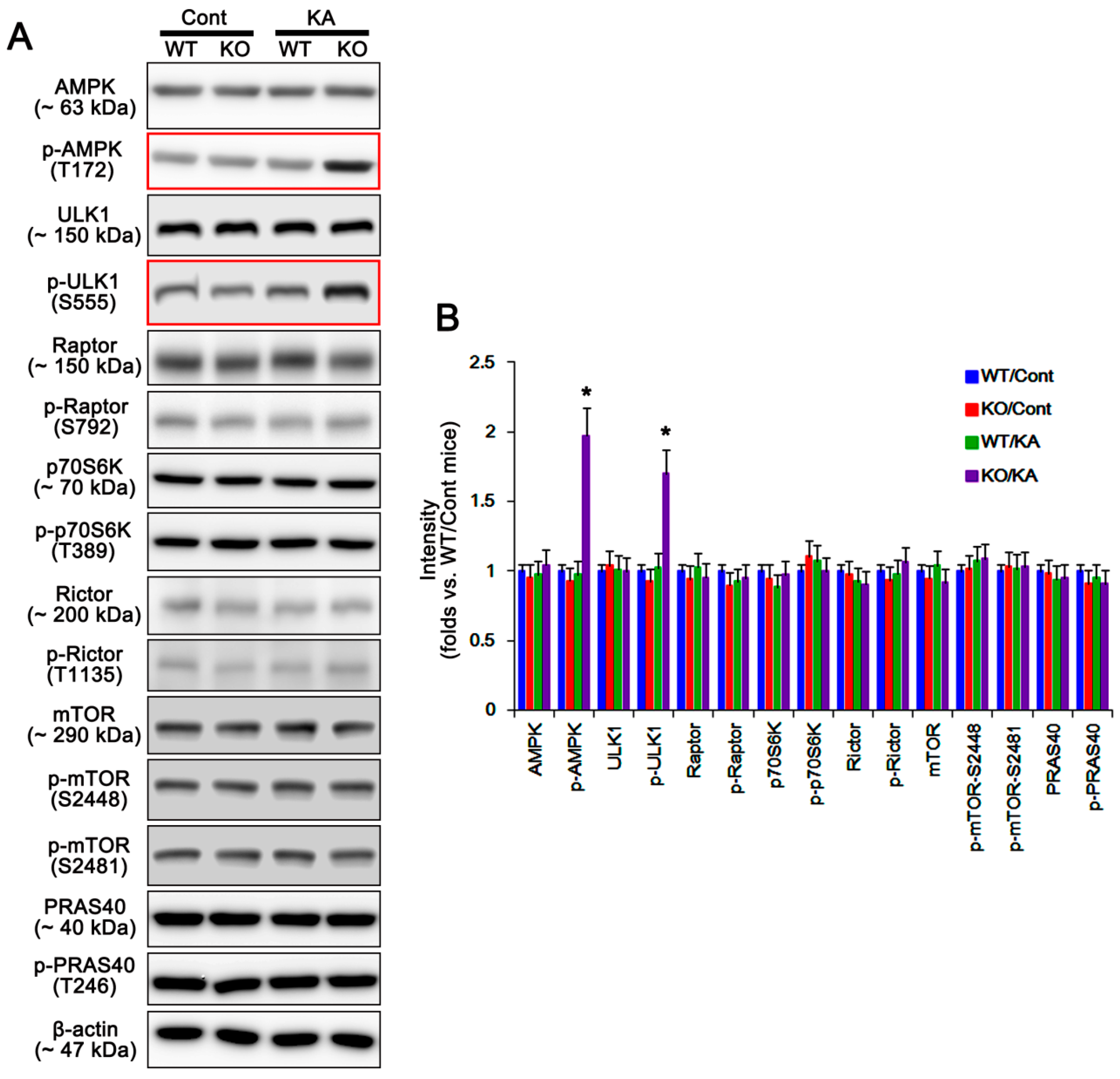
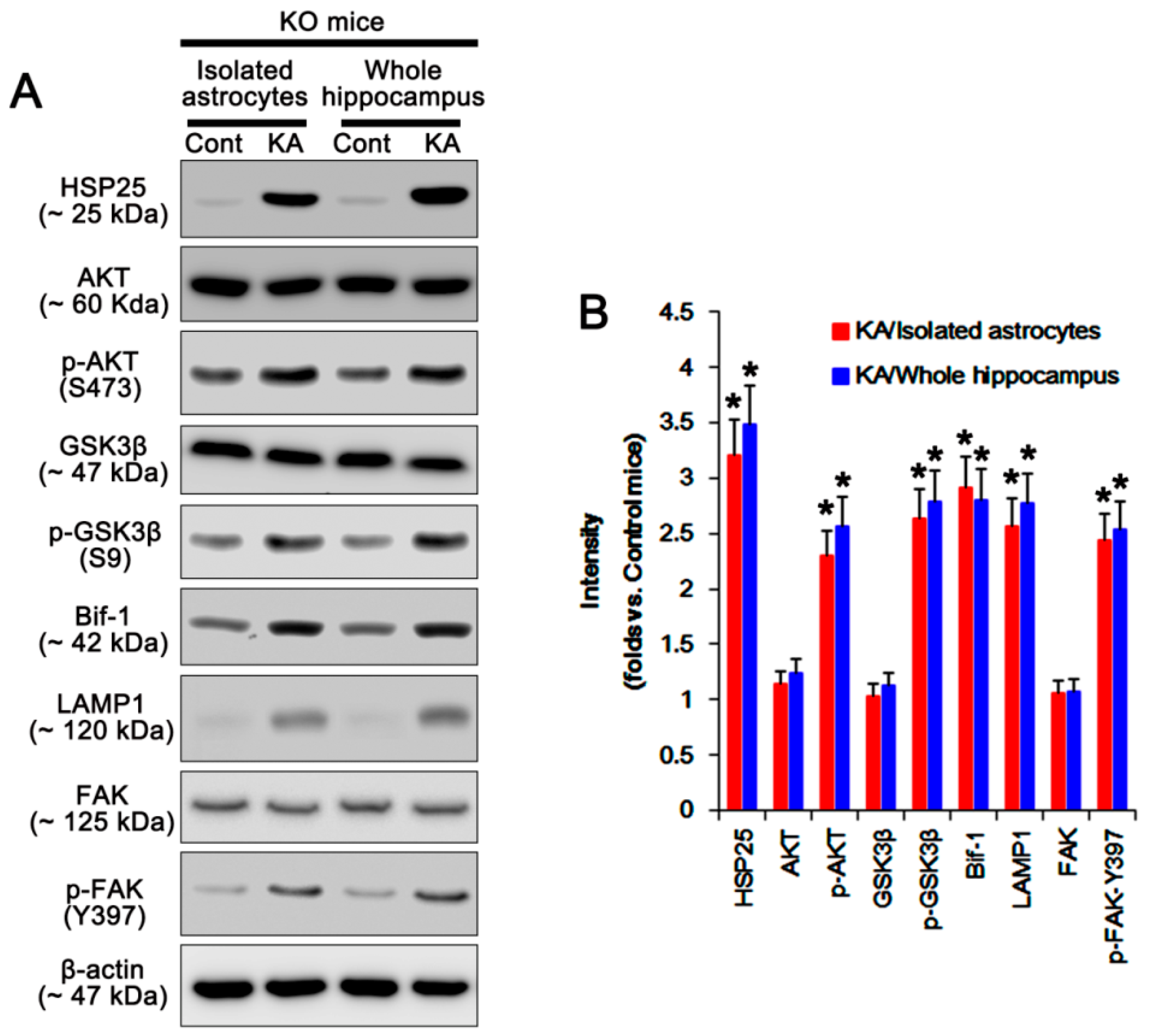
2.1. P2X7R Deletion Facilitates Astroglial Autophagy Following KA Injection
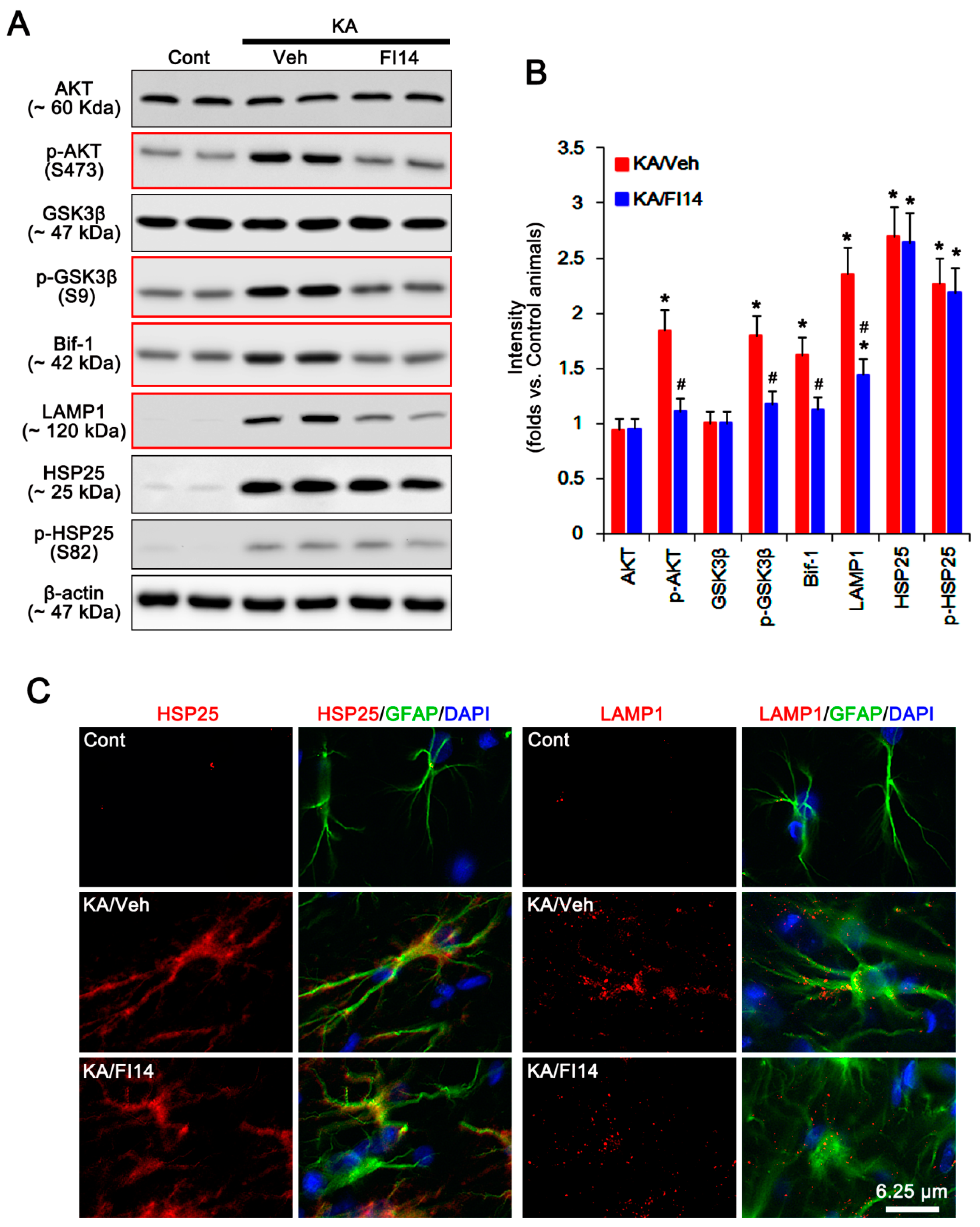
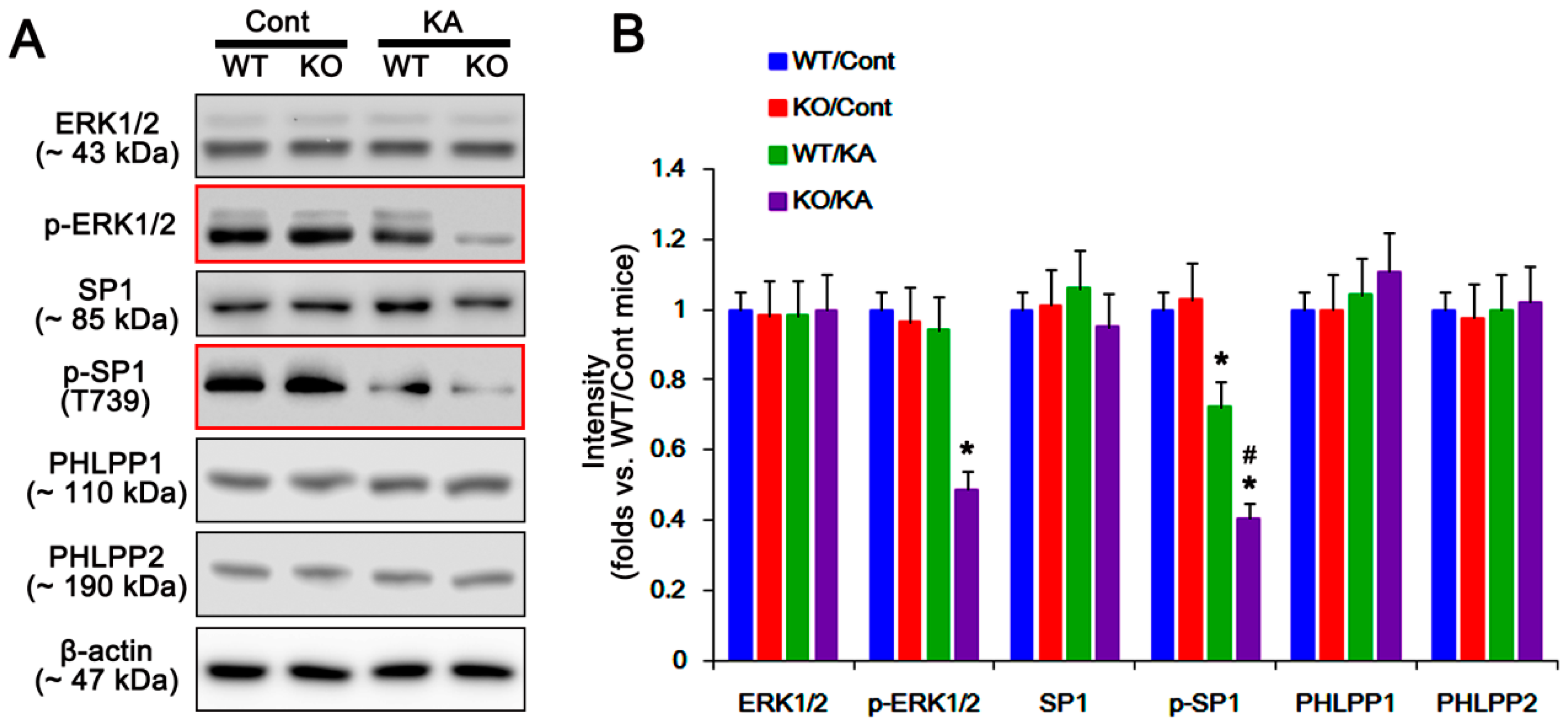
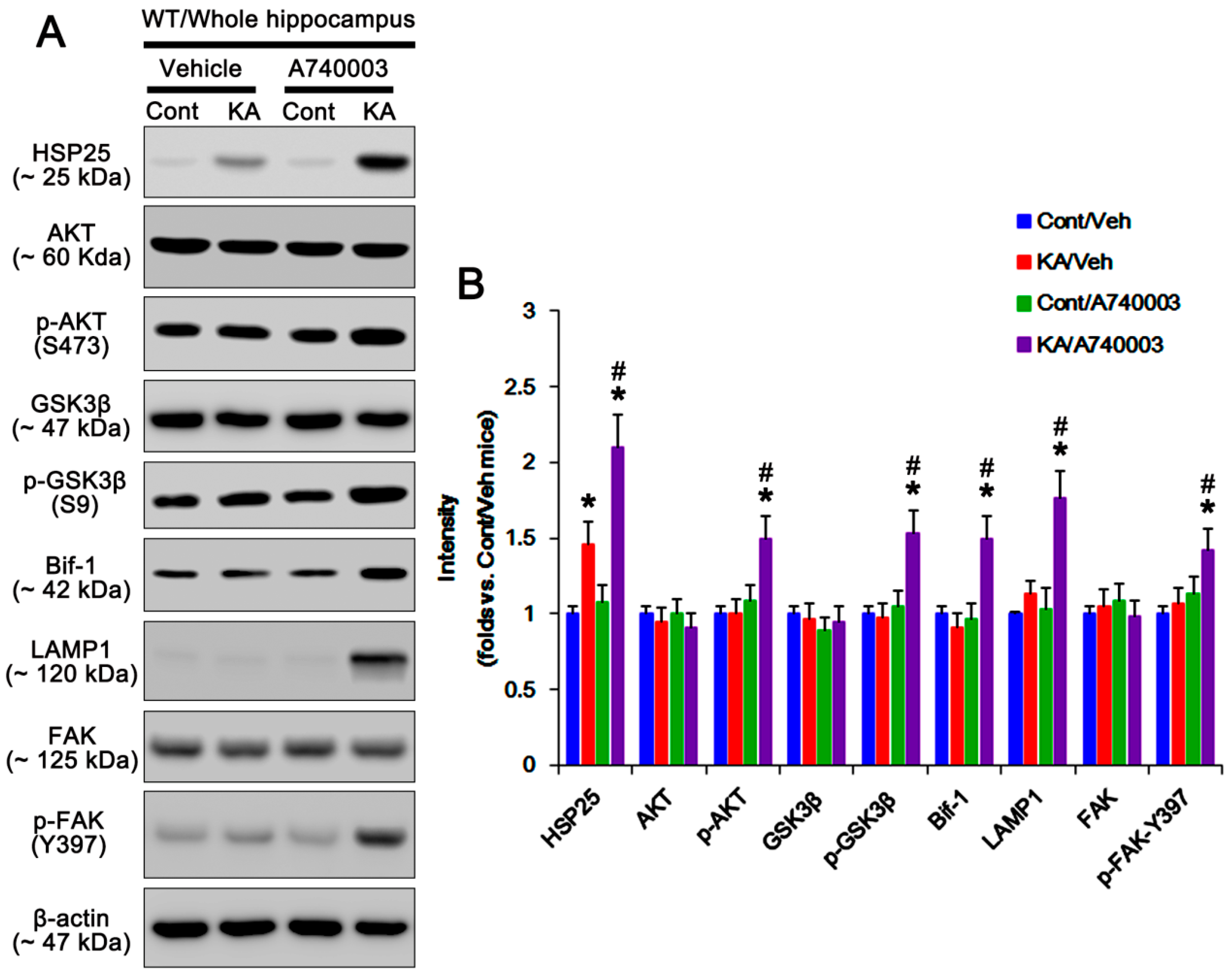
2.2. P2X7R Deletion Enhances FAK-Mediated AKT-S473 Phosphorylation in Astrocytes Independent of Pi3k/Pdk1 Signaling Pathway
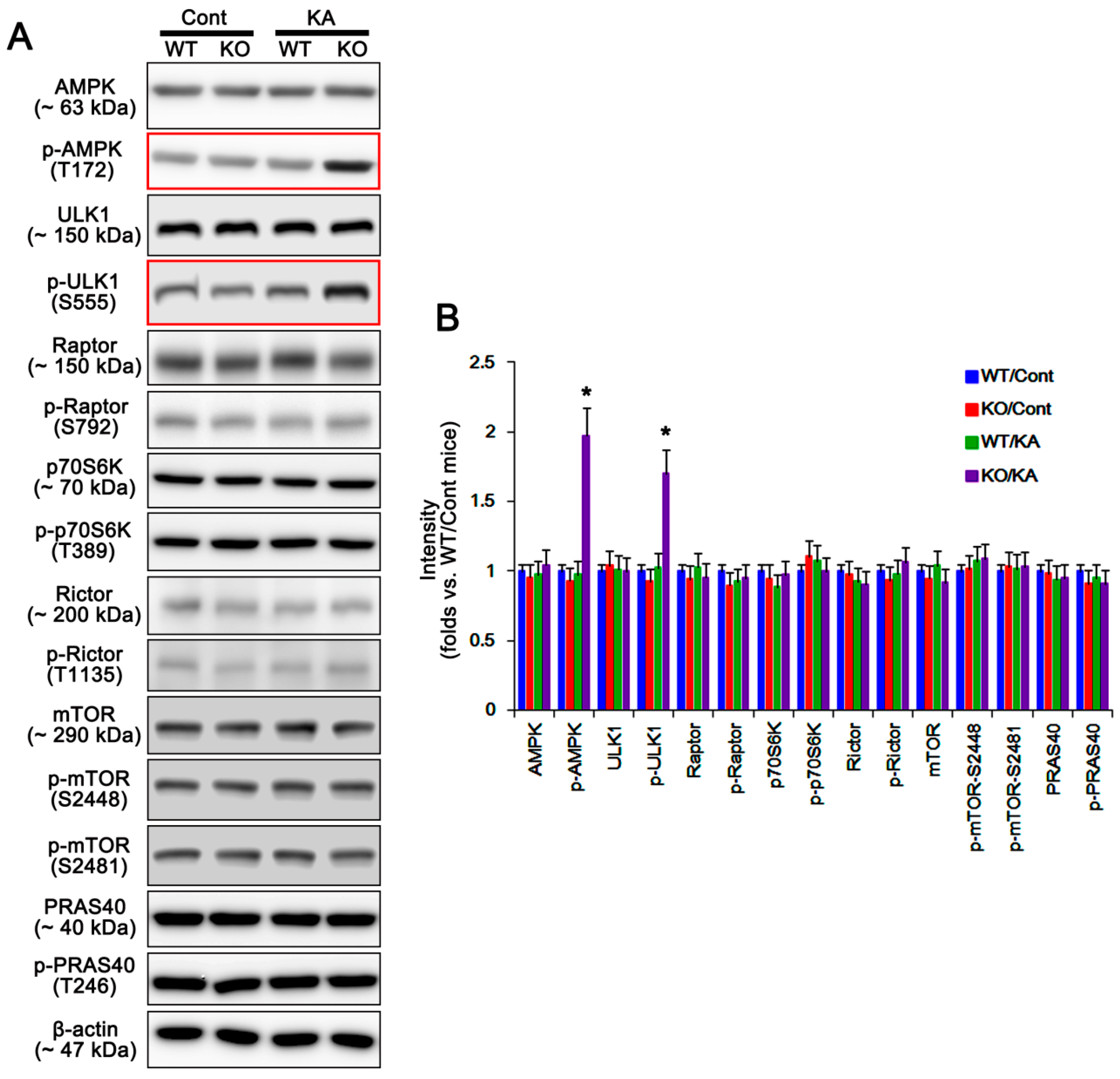
2.3. P2X7R Does Not Affect mTORC-Mediated AKT Phosphorylation Following KA Injection
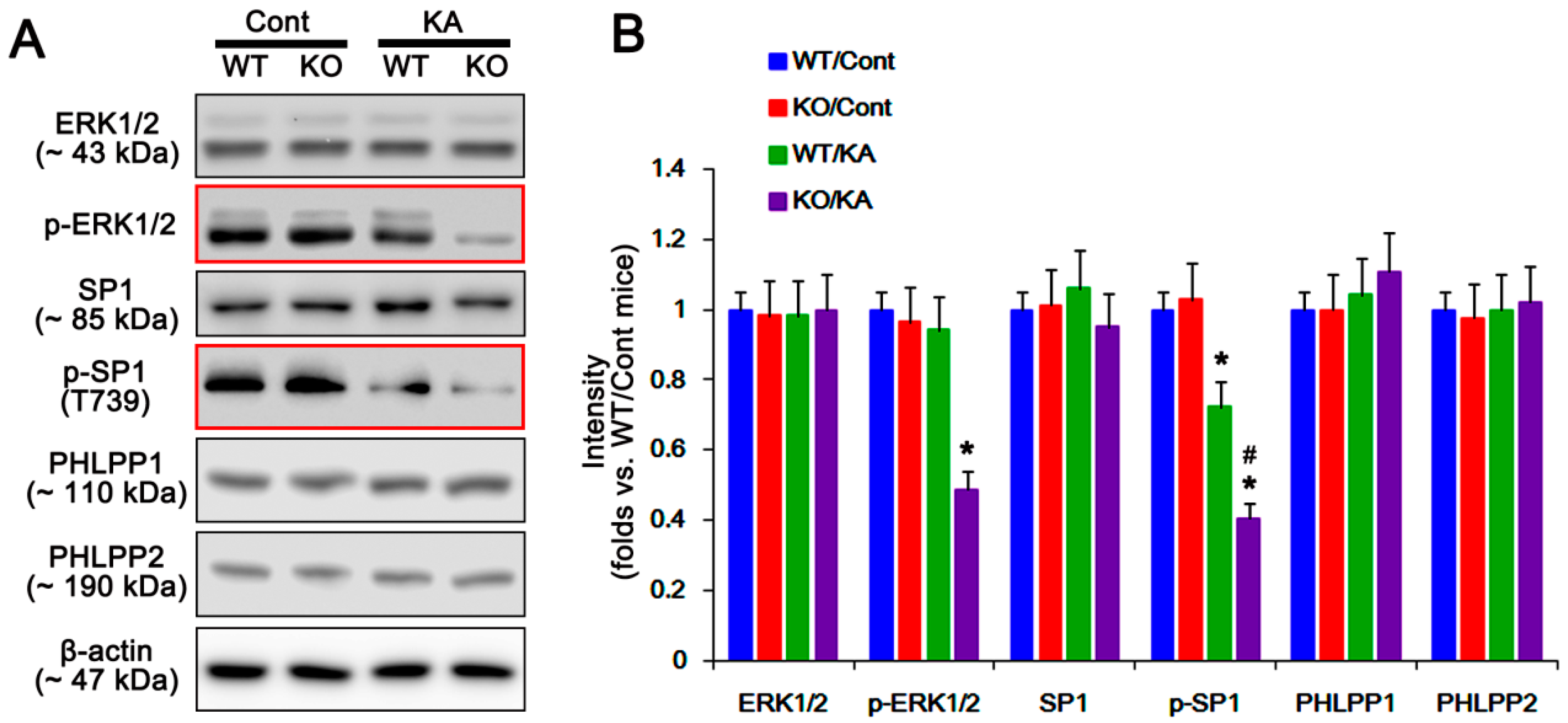
2.4. P2X7R Deletion Cannot Influence PHLPP1/2 Expression in Astrocytes Following KA Injection
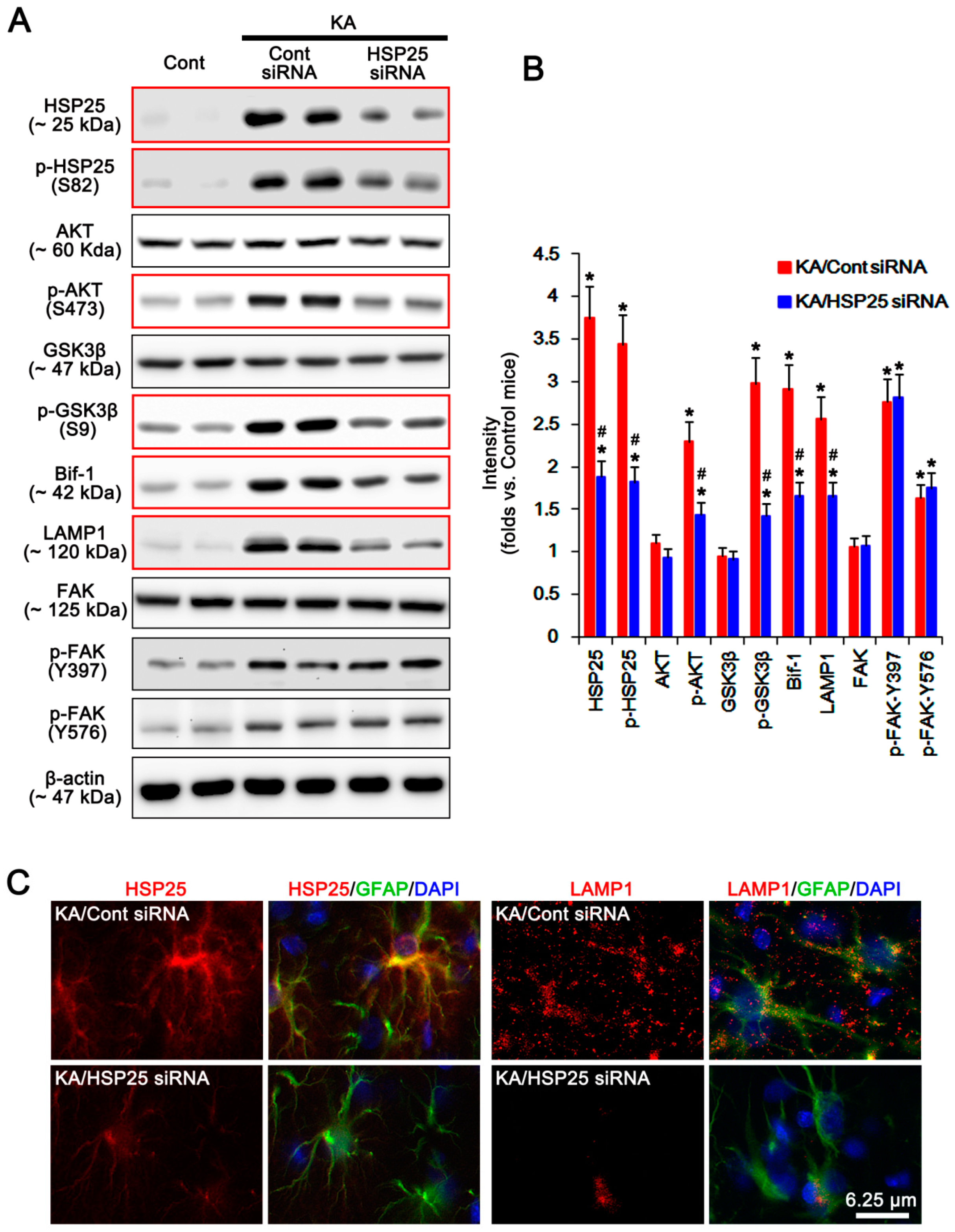
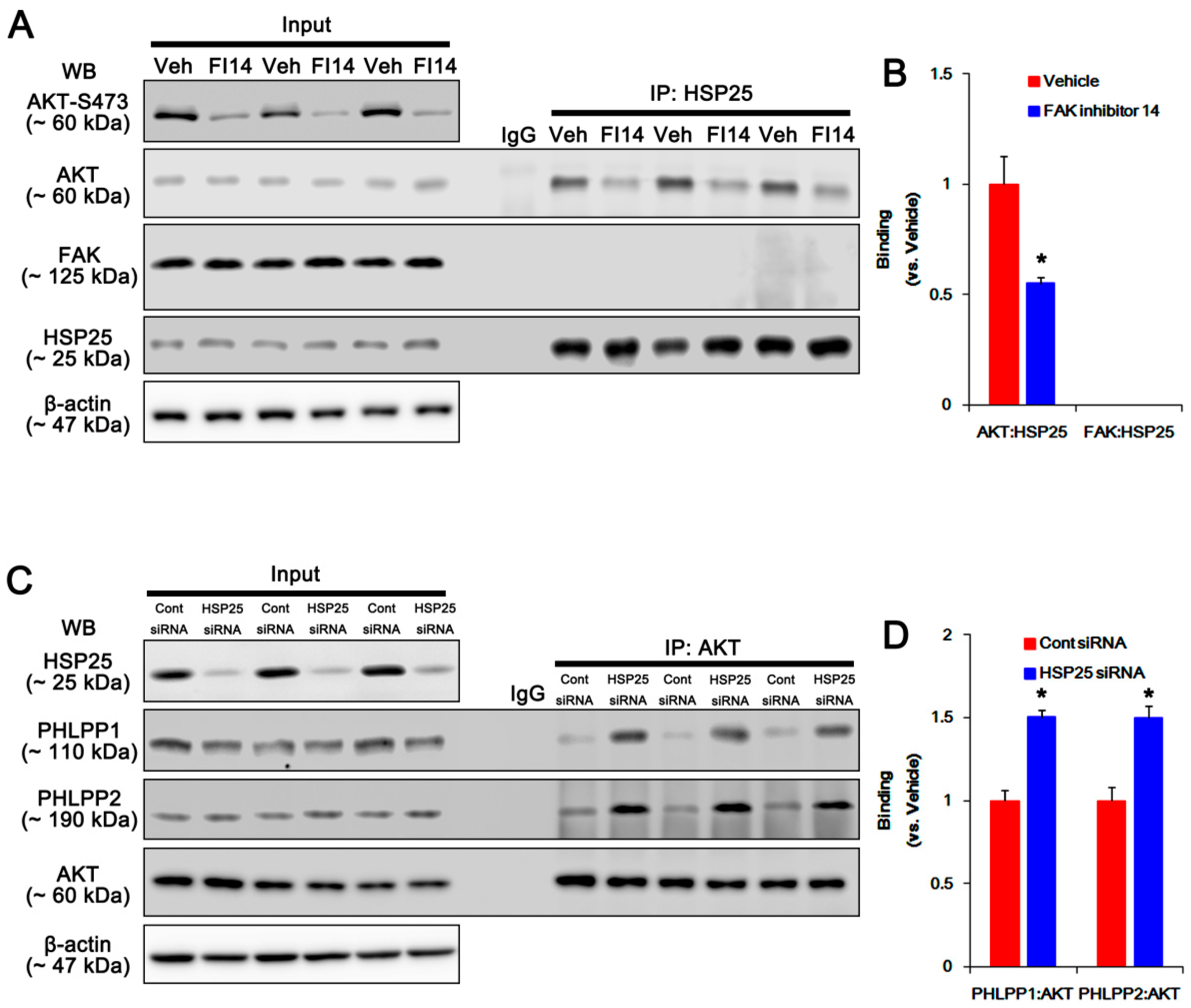
2.5. HSP25 Binding to AKT Abrogates AKT-S473 Dephosphorylation
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Chemicals
4.2. Seizure Induction and Infusions of Drug and siRNA Oligonucleotide
4.3. Electrophysiology
4.4. Astroglial Isolation
4.5. Coimmunoprecipitation and Western Blot
4.6. Double Immunofluorescence Study
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jäkel, S.; Dimou, L. Glial cells and their function in the adult brain: A journey through the history of their ablation. Front. Cell Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.C.; Kim, D.S.; Kwak, S.E.; Kim, J.E.; Won, M.H.; Kim, D.W.; Choi, S.Y.; Kwon, O.S. Epileptogenic roles of astroglial death and regeneration in the dentate gyrus of experimental temporal lobe epilepsy. Glia 2006, 54, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Lewén, A.; Noshita, N.; Gasche, Y.; Chan, P.H. Effects of global ischemia duration on neuronal, astroglial, oligodendroglial, and microglial reactions in the vulnerable hippocampal CA1 subregion in rats. J. Neurotrauma 2002, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 2010, 58, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Penfield, W. Neuroglia and microglia—The interstitial tissue of the central nervous system. In Special Cytology, the Form and Function of the Cell in Health and Disease; Cowdry, E.V., Ed.; Hoeber: New York, NY, USA, 1928; pp. 1033–1068. [Google Scholar]
- Kim, J.E.; Ko, A.R.; Hyun, H.W.; Min, S.J.; Kang, T.C. P2RX7-MAPK1/2-SP1 axis inhibits MTOR independent HSPB1-mediated astroglial autophagy. Cell Death Dis. 2018, 9, 546. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kang, T.C. p65/RelA-Ser529 NF-κB subunit phosphorylation induces autophagic astroglial death (Clasmatodendrosis) following status epilepticus. Cell Mol. Neurobiol. 2011, 31, 1071–1078. [Google Scholar] [CrossRef]
- Kim, J.E.; Ryu, H.J.; Yeo, S.I.; Kang, T.C. P2X7 receptor differentially modulates astroglial apoptosis and clasmatodendrosis in the rat brain following status epilepticus. Hippocampus 2011, 21, 1318–1333. [Google Scholar] [CrossRef]
- Chan, E.Y.; Longatti, A.; McKnight, N.C.; Tooze, S.A. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol. Cell Biol. 2009, 29, 157–171. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Ross, F.A.; Gowans, G.J.; Tibarewal, P.; Leslie, N.R.; Hardie, D.G. Phosphorylation by Akt within the ST loop of AMPK-alpha1 down-regulates its activation in tumour cells. Biochem. J. 2014, 459, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.; Lai, D.; Zhang, L.; Xu, H. Induction of autophagy and apoptosis via PI3K/AKT/TOR pathways by Azadirachtin A in Spodoptera litura cells. Sci. Rep. 2016, 6, 35482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Takahashi, Y.; Cheng, E.; Liu, J.; Terranova, P.F.; Zhao, B.; Thrasher, J.B.; Wang, H.G.; Li, B. GSK-3beta promotes cell survival by modulating Bif-1-dependent autophagy and cell death. J. Cell Sci. 2010, 123, 861–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, K.; Mahajan, N.P. PI3K-independent AKT activation in cancers: A treasure trove for novel therapeutics. J. Cell Physiol. 2012, 227, 3178–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschstein, T.; Mikkat, S.; Mikkat, U.; Bender, R.; Kreutzer, M.; Schulz, R.; Köhling, R.; Glocker, M.O. The 27-kDa heat shock protein (HSP27) is a reliable hippocampal marker of full development of pilocarpine-induced status epilepticus. Epilepsy Res. 2012, 98, 35–43. [Google Scholar] [CrossRef]
- Huynh, K.K.; Eskelinen, E.L.; Scott, C.C.; Malevanets, A.; Saftig, P.; Grinstein, S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007, 26, 313–324. [Google Scholar] [CrossRef]
- Sandilands, E.; Schoenherr, C.; Frame, M.C. p70S6K is regulated by focal adhesion kinase and is required for Src-selective autophagy. Cell Signal. 2015, 27, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell. Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Díaz-Hernandez, M.; del Puerto, A.; Díaz-Hernandez, J.I.; Diez-Zaera, M.; Lucas, J.J.; Garrido, J.J.; Miras-Portugal, M.T. Inhibition of the ATP-gated P2X7 receptor promotes axonal growth and branching in cultured hippocampal neurons. J. Cell Sci. 2008, 121, 3717–3728. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of Raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, L.A.; Carriere, A.; Moreau, J.; Roux, P.P. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol. Cell Biol. 2010, 30, 908–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the mTORC1 Protein Kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Brognard, J.; Sierecki, E.; Gao, T.; Newton, A.C. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell 2007, 25, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Gaestel, M.; Gotthardt, R.; Müller, T. Structure and organisation of a murine gene encoding small heat-shock protein Hsp25. Gene 1993, 128, 279–283. [Google Scholar] [CrossRef]
- Chuang, J.Y.; Wang, S.A.; Yang, W.B.; Yang, H.C.; Hung, C.Y.; Su, T.P.; Chang, W.C.; Hung, J.J. Sp1 phosphorylation by cyclin-dependent kinase 1/cyclin B1 represses its DNA-binding activity during mitosis in cancer cells. Oncogene 2012, 31, 4946–4959. [Google Scholar] [CrossRef] [Green Version]
- Alamuru-Yellapragada, N.P.; Vundyala, S.; Behera, S.; Parsa, K.V. LPS depletes PHLPP levels in macrophages through the inhibition of SP1 dependent transcriptional regulation. Biochem. Biophys. Res. Commun. 2017, 486, 533–538. [Google Scholar] [CrossRef]
- Konishi, H.; Matsuzaki, H.; Tanaka, M.; Takemura, Y.; Kuroda, S.; Ono, Y.; Kikkawa, U. Activation of protein kinase B (Akt/RAC-protein kinase) by cellular stress and its association with heat shock protein Hsp27. FEBS Lett. 1997, 410, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Murashov, A.K.; Haq, I.U.; Hill, C.; Park, E.; Smith, M.; Wang, X.; Wang, X.; Goldberg, D.J.; Wolgemuth, D.J. Crosstalk between p38, Hsp25 and Akt in spinal motor neurons after sciatic nerve injury. Brain Res. Mol. Brain Res. 2001, 93, 199–208. [Google Scholar] [CrossRef]
- Wu, R.; Kausar, H.; Johnson, P.; Montoya-Durango, D.E.; Merchant, M.; Rane, M.J. Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex. J. Biol. Chem. 2007, 282, 21598–21608. [Google Scholar] [CrossRef] [Green Version]
- de Graauw, M.; Tijdens, I.; Cramer, R.; Corless, S.; Timms, J.F.; van de Water, B. Heat shock protein 27 is the major differentially phosphorylated protein involved in renal epithelial cellular stress response and controls focal adhesion organization and apoptosis. J. Biol. Chem. 2005, 280, 29885–29898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Lee, S.; Shrestha, P.; Kim, J.; Park, J.A.; Ko, Y.; Ban, Y.H.; Park, D.Y.; Ha, S.J.; Koh, G.Y.; et al. AMIGO2, a novel membrane anchor of PDK1, controls cell survival and angiogenesis via Akt activation. J. Cell Biol. 2015, 211, 619–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, M.J.; Ramos, F.J.; Chen, H.; Quon, M.J.; Dong, L.Q.; Liu, F. Mouse 3-phosphoinositide-dependent protein kinase-1 undergoes dimerization and trans-phosphorylation in the activation loop. J. Biol. Chem. 2003, 278, 42913–42919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Liang, J.; Li, J.; Lu, Y.; Ariyaratna, V.; Lu, Z.; Davies, M.A.; Westwick, J.K.; Mills, G.B. Physical association of PDK1 with AKT1 is sufficient for pathway activation independent of membrane localization and phosphatidylinositol 3 kinase. PLoS ONE 2010, 5, e9910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Zhang, X.; Li, M.; Chen, P.; Zhang, B.; Guo, H.; Cao, W.; Wei, X.; Cao, X.; Hao, X.; et al. mTOR complex component Rictor interacts with PKCzeta and regulates cancer cell metastasis. Cancer Res. 2010, 70, 9360–9370. [Google Scholar] [CrossRef] [Green Version]
- McDonald, P.C.; Oloumi, A.; Mills, J.; Dobreva, I.; Maidan, M.; Gray, V.; Wederell, E.D.; Bally, M.B.; Foster, L.J.; Dedhar, S. Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Res. 2008, 68, 1618–1624. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef]
- Xia, H.; Nho, R.S.; Kahm, J.; Kleidon, J.; Henke, C.A. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J. Biol. Chem. 2004, 279, 33024–33034. [Google Scholar] [CrossRef] [Green Version]
- Eliceiri, B.P.; Puente, X.S.; Hood, J.D.; Stupack, D.G.; Schlaepfer, D.D.; Huang, X.Z.; Sheppard, D.; Cheresh, D.A. Src-mediated coupling of focal adhesion kinase to integrin α v β 5 in vascular endothelial growth factor signaling. J. Cell Biol. 2002, 157, 149–160. [Google Scholar] [CrossRef]
- Nho, R.S.; Xia, H.; Kahm, J.; Kleidon, J.; Diebold, D.; Henke, C.A. Role of integrin-linked kinase in regulating phosphorylation of Akt and fibroblast survival in type I collagen matrices through a beta1 integrin viability signaling pathway. J. Biol. Chem. 2005, 280, 26630–26639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.O.; Shin, S.; Karreth, F.A.; Buel, G.R.; Jedrychowski, M.P.; Plas, D.R.; Dedhar, S.; Gygi, S.P.; Roux, P.P.; Dephoure, N.; et al. Focal Adhesion- and IGF1R-Dependent Survival and Migratory Pathways Mediate Tumor Resistance to mTORC1/2 Inhibition. Mol. Cell 2017, 67, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Basson, M.D. AKT directly regulates focal adhesion kinase through association and serine phosphorylation: Implication for pressure-induced colon cancer metastasis. Am. J. Physiol. Cell Physiol. 2011, 300, C657–C670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Dey-Guha, I.; Alves, C.P.; Yeh, A.C.; Salony; Sole, X.; Darp, R.; Ramaswamy, S. A mechanism for asymmetric cell division resulting in proliferative asynchronicity. Mol. Cancer Res. 2015, 13, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Sandilands, E.; Serrels, B.; Wilkinson, S.; Frame, M.C. Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells. EMBO Rep. 2015, 13, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Son, T.W.; Yun, S.P.; Yong, M.S.; Seo, B.N.; Ryu, J.M.; Youn, H.Y.; Oh, Y.M.; Han, H.J. Netrin-1 protects hypoxia-induced mitochondrial apoptosis through HSP25 expression via DCC- and integrin α6β4-dependent AKT, GSK-3β, and HSF-1 in mesenchymal stem cells. Cell Death Dis. 2013, 4, e563. [Google Scholar] [CrossRef] [Green Version]
- Havasi, A.; Li, Z.; Wang, Z.; Martin, J.L.; Botla, V.; Ruchalski, K.; Schwartz, J.H.; Borkan, S.C. Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J. Biol. Chem. 2008, 283, 12305–12313. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Kwak, H.J.; Lee, J.J.; Kim, Y.N.; Lee, J.W.; Park, M.J.; Jung, S.E.; Hong, S.I.; Lee, J.H.; Lee, J.S. HSP27 regulates cell adhesion and invasion via modulation of focal adhesion kinase and MMP-2 expression. Eur. J. Cell Biol. 2008, 87, 377–387. [Google Scholar] [CrossRef]
- Liu, T.; Guevara, O.E.; Warburton, R.R.; Hill, N.S.; Gaestel, M.; Kayyali, U.S. Modulation of HSP25 alters hypoxia-induced endothelial permeability and related signaling pathways. J. Cell Physiol. 2009, 220, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, R.; Laroia, G.; Schneider, R.J. Chaperone hsp27 inhibits translation during heat shock by binding eIF4G and facilitating dissociation of cap-initiation complexes. Genes Dev. 2000, 14, 1460–1470. [Google Scholar] [PubMed]
- Bidmon, H.J.; Görg, B.; Palomero-Gallagher, N.; Behne, F.; Lahl, R.; Pannek, H.W.; Speckmann, E.J.; Zilles, K. Heat shock protein-27 is upregulated in the temporal cortex of patients with epilepsy. Epilepsia 2004, 45, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Jakubowicz-Gil, J.; Langner, E.; Bądziul, D.; Wertel, I.; Rzeski, W. Quercetin and sorafenib as a novel and effective couple in programmed cell death induction in human gliomas. Neurotox. Res. 2014, 26, 64–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Tang, C.; Li, L.; Li, R.; Fan, Y. Quercetin blocks t-AUCB-induced autophagy by Hsp27 and Atg7 inhibition in glioblastoma cells in vitro. J. Neurooncol. 2016, 129, 39–45. [Google Scholar] [CrossRef]
- Tang, G.; Yue, Z.; Talloczy, Z.; Hagemann, T.; Cho, W.; Messing, A.; Sulzer, D.L.; Goldman, J.E. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum. Mol. Genet. 2008, 17, 1540–1555. [Google Scholar] [CrossRef] [Green Version]
- Bachetti, T.; Di Zanni, E.; Balbi, P.; Ravazzolo, R.; Sechi, G.; Ceccherini, I. Beneficial effects of curcumin on GFAP filament organization and down-regulation of GFAP expression in an in vitro model of Alexander disease. Exp. Cell Res. 2012, 318, 1844–1854. [Google Scholar] [CrossRef]
- Bachetti, T.; Di Zanni, E.; Balbi, P.; Bocca, P.; Prigione, I.; Deiana, G.A.; Rezzani, A.; Ceccherini, I.; Sechi, G. In vitro treatments with ceftriaxone promote elimination of mutant glial fibrillary acidic protein and transcription down-regulation. Exp. Cell Res. 2010, 316, 2152–2165. [Google Scholar] [CrossRef]
- Bursch, W.; Ellinger, A.; Kienzl, H.; Török, L.; Pandey, S.; Sikorska, M.; Walker, R.; Hermann, R.S. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: The role of autophagy. Carcinogenesis 1996, 17, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12, 1528–1534. [Google Scholar] [CrossRef] [Green Version]
- Gozuacik, D.; Kimchi, A. Autophagy and cell death. Curr. Top. Dev. Biol. 2007, 78, 217–245. [Google Scholar]
- Jänen, S.B.; Chaachouay, H.; Richter-Landsberg, C. Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia 2010, 58, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Batiuk, M.Y.; de Vin, F.; Duqué, S.I.; Li, C.; Saito, T.; Saido, T.; Fiers, M.; Belgard, T.G.; Holt, M.G. An immunoaffinity-based method for isolating ultrapure adult astrocytes based on ATP1B2 targeting by the ACSA-2 antibody. J. Biol. Chem. 2017, 292, 8874–8891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Lee, D.S.; Kim, M.J.; Kang, T.C. PLPP/CIN-mediated NEDD4-2 S448 dephosphorylation regulates neuronal excitability via GluA1 ubiquitination. Cell Death Dis. 2019, 10, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.-S.; Kim, J.-E. P2 × 7 Receptor Inhibits Astroglial Autophagy via Regulating FAK- and PHLPP1/2-Mediated AKT-S473 Phosphorylation Following Kainic Acid-Induced Seizures. Int. J. Mol. Sci. 2020, 21, 6476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186476
Lee D-S, Kim J-E. P2 × 7 Receptor Inhibits Astroglial Autophagy via Regulating FAK- and PHLPP1/2-Mediated AKT-S473 Phosphorylation Following Kainic Acid-Induced Seizures. International Journal of Molecular Sciences. 2020; 21(18):6476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186476
Chicago/Turabian StyleLee, Duk-Shin, and Ji-Eun Kim. 2020. "P2 × 7 Receptor Inhibits Astroglial Autophagy via Regulating FAK- and PHLPP1/2-Mediated AKT-S473 Phosphorylation Following Kainic Acid-Induced Seizures" International Journal of Molecular Sciences 21, no. 18: 6476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186476