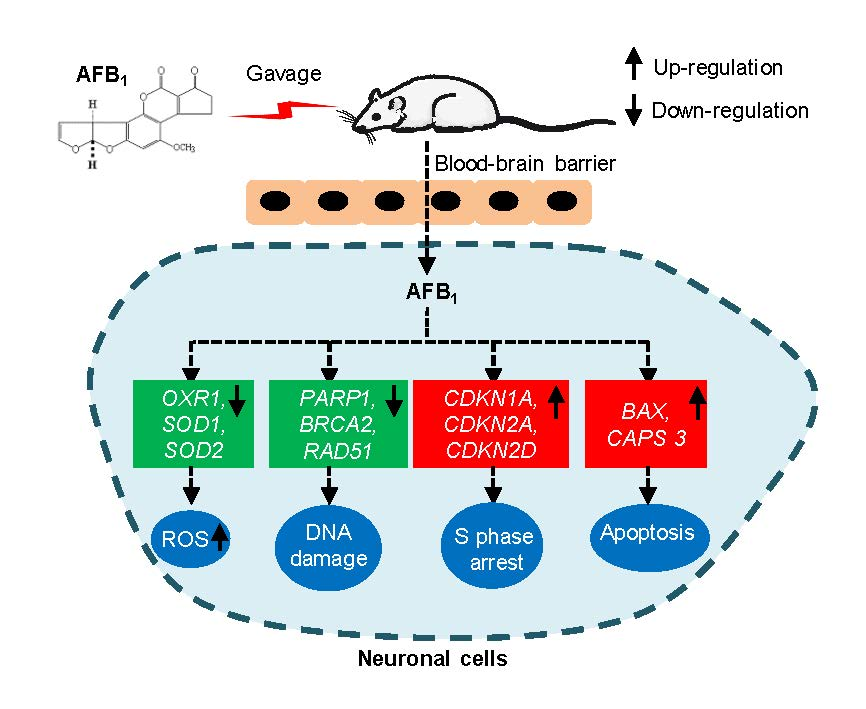
Aflatoxin B1 Induces Neurotoxicity through Reactive Oxygen Species Generation, DNA Damage, Apoptosis, and S-Phase Cell Cycle Arrest


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
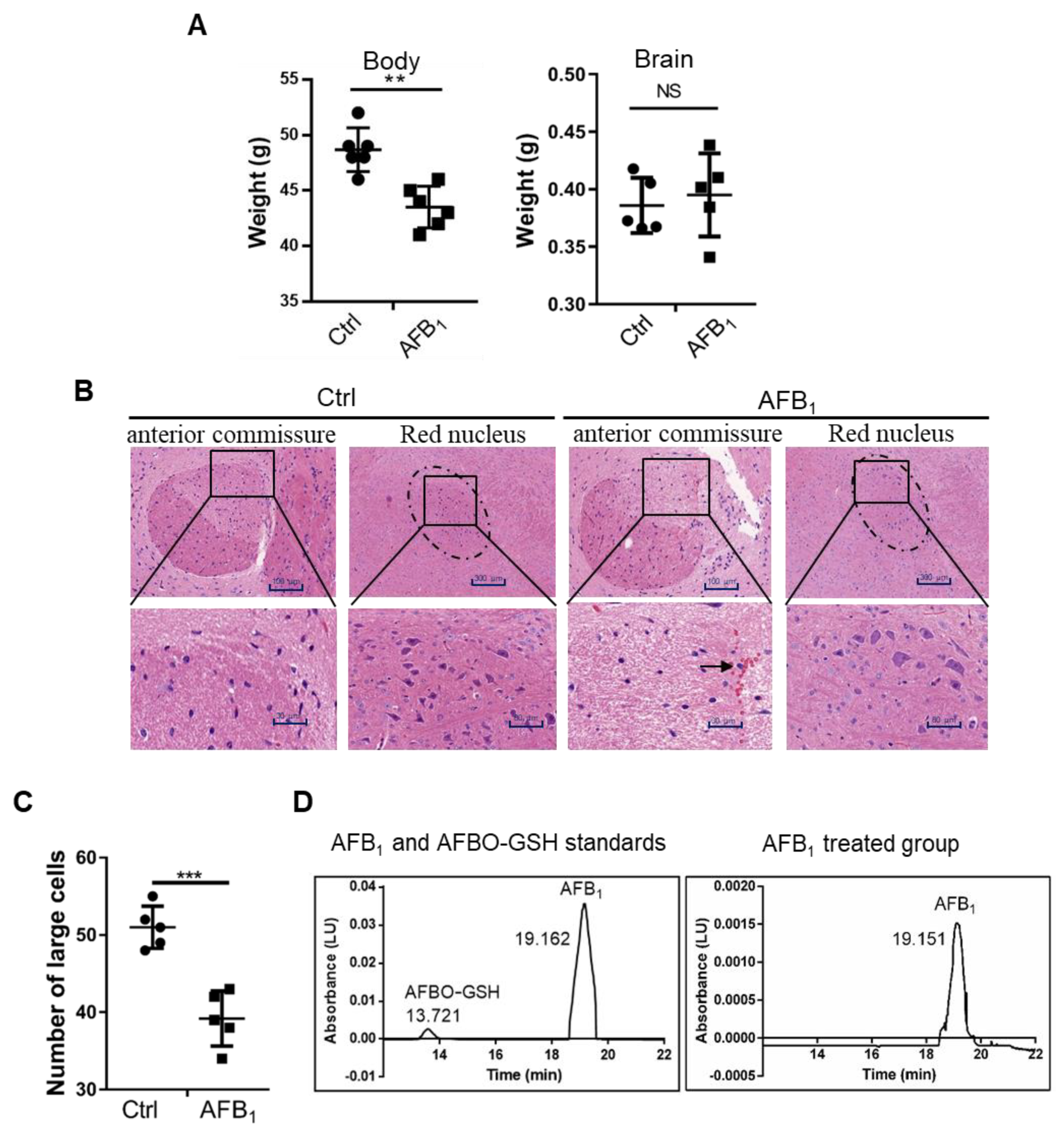
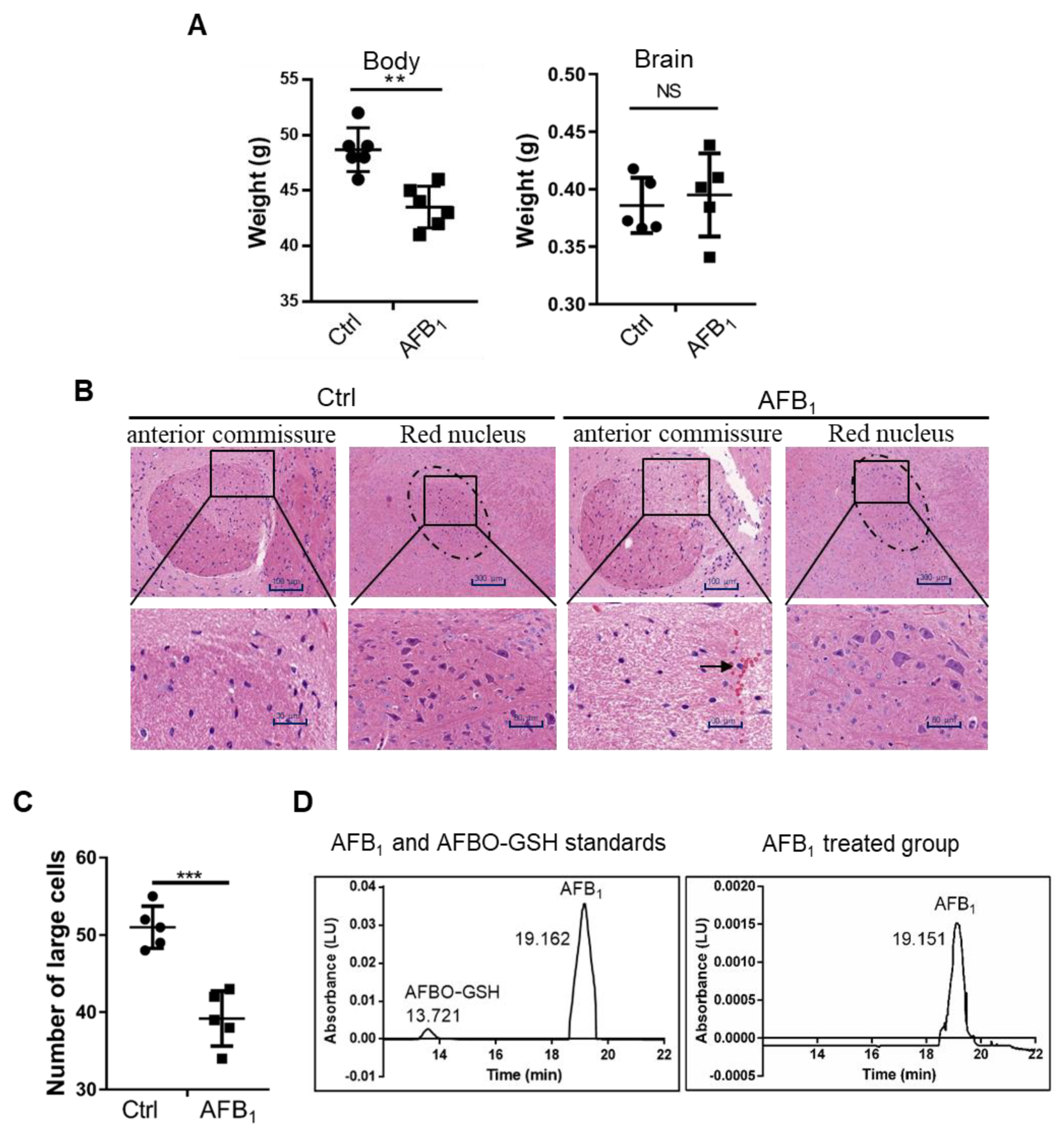
2.1. AFB1 Induces Brain Damage in Mice
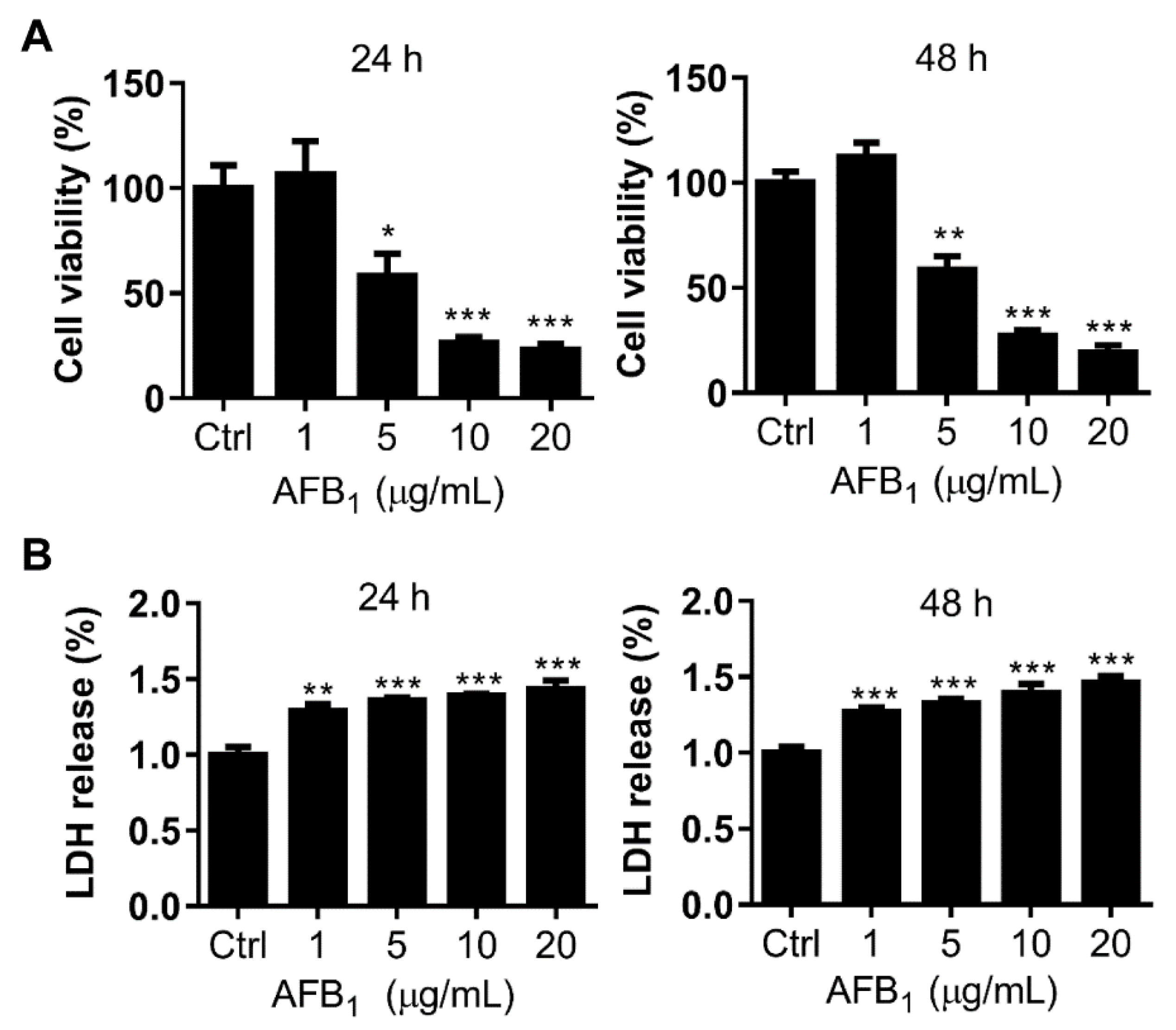
2.2. AFB1 Inhibits Cell Proliferation of IMR-32 Cells
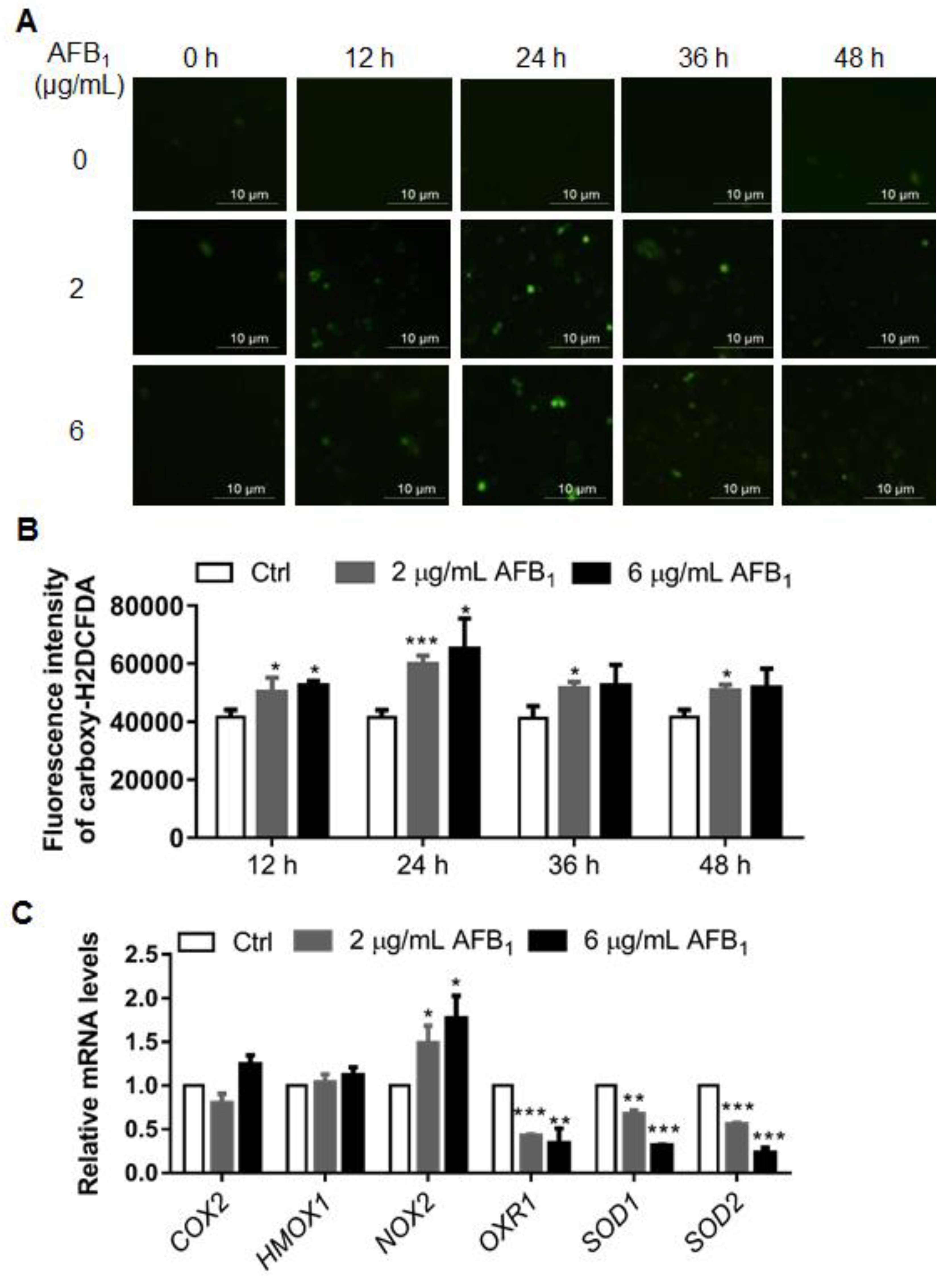
2.3. AFB1 Enhances Intracellular ROS Levels
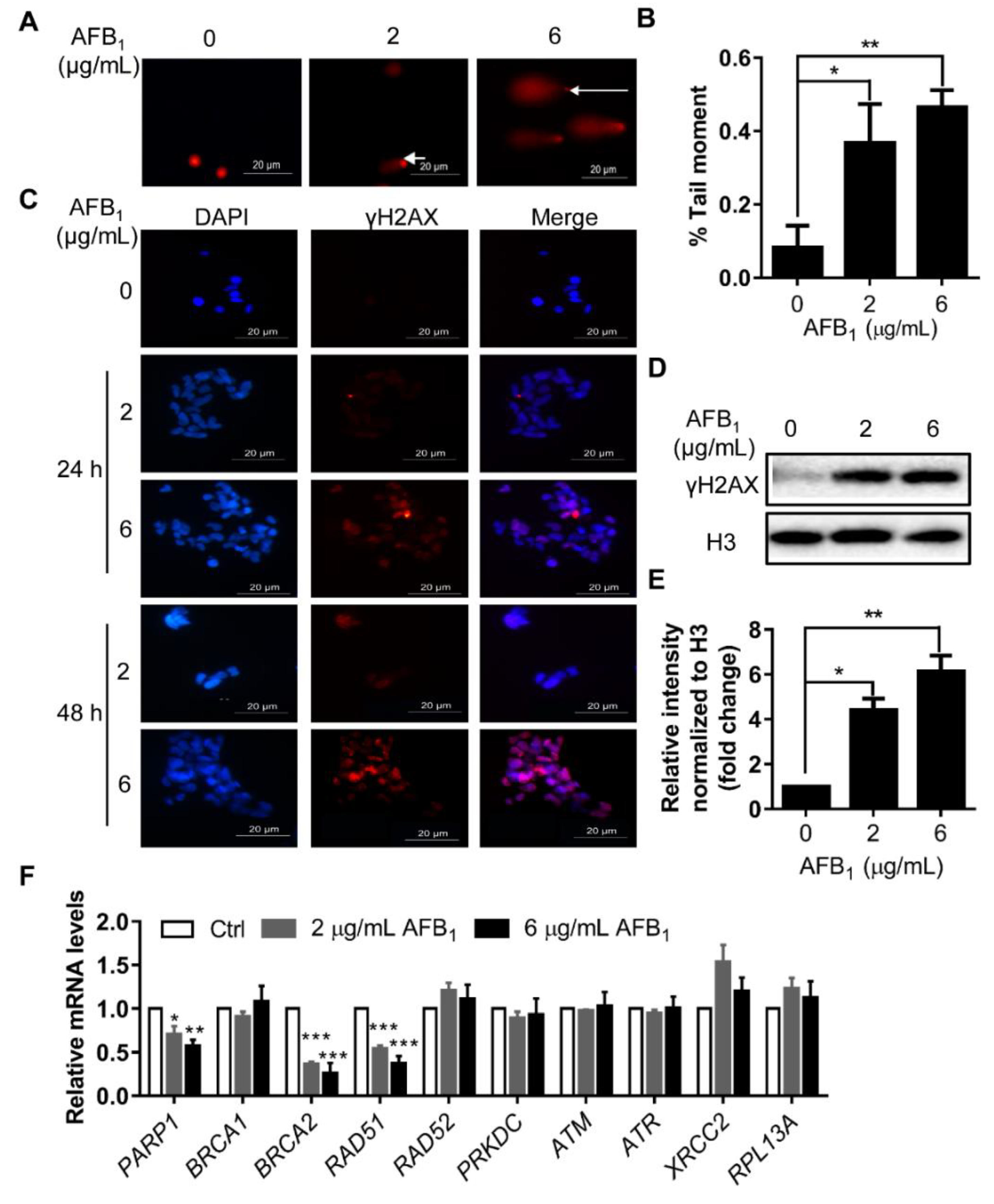
2.4. AFB1 Induces DNA Damage
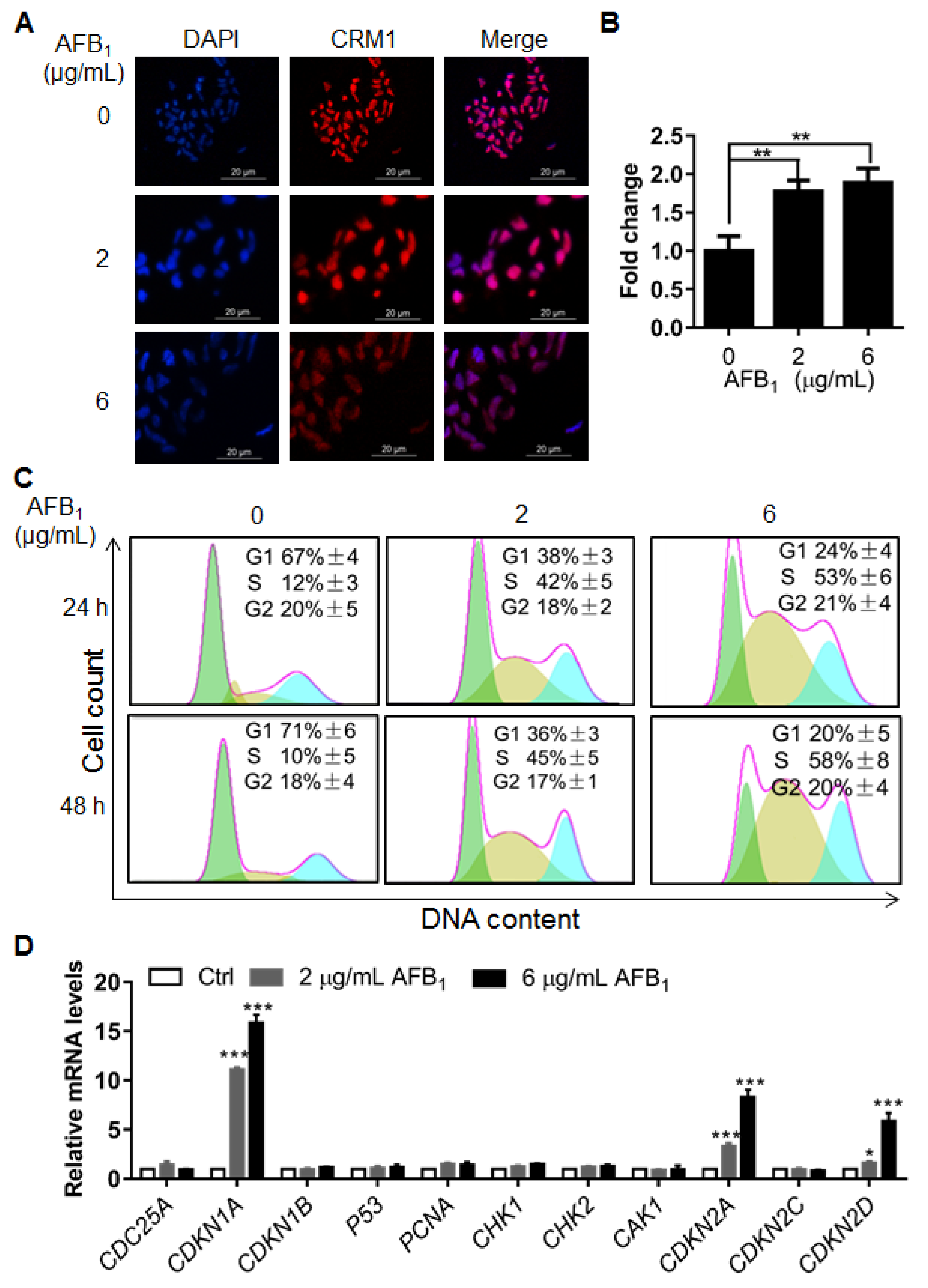
2.5. AFB1 Induces S-Phase Cell Cycle Arrest
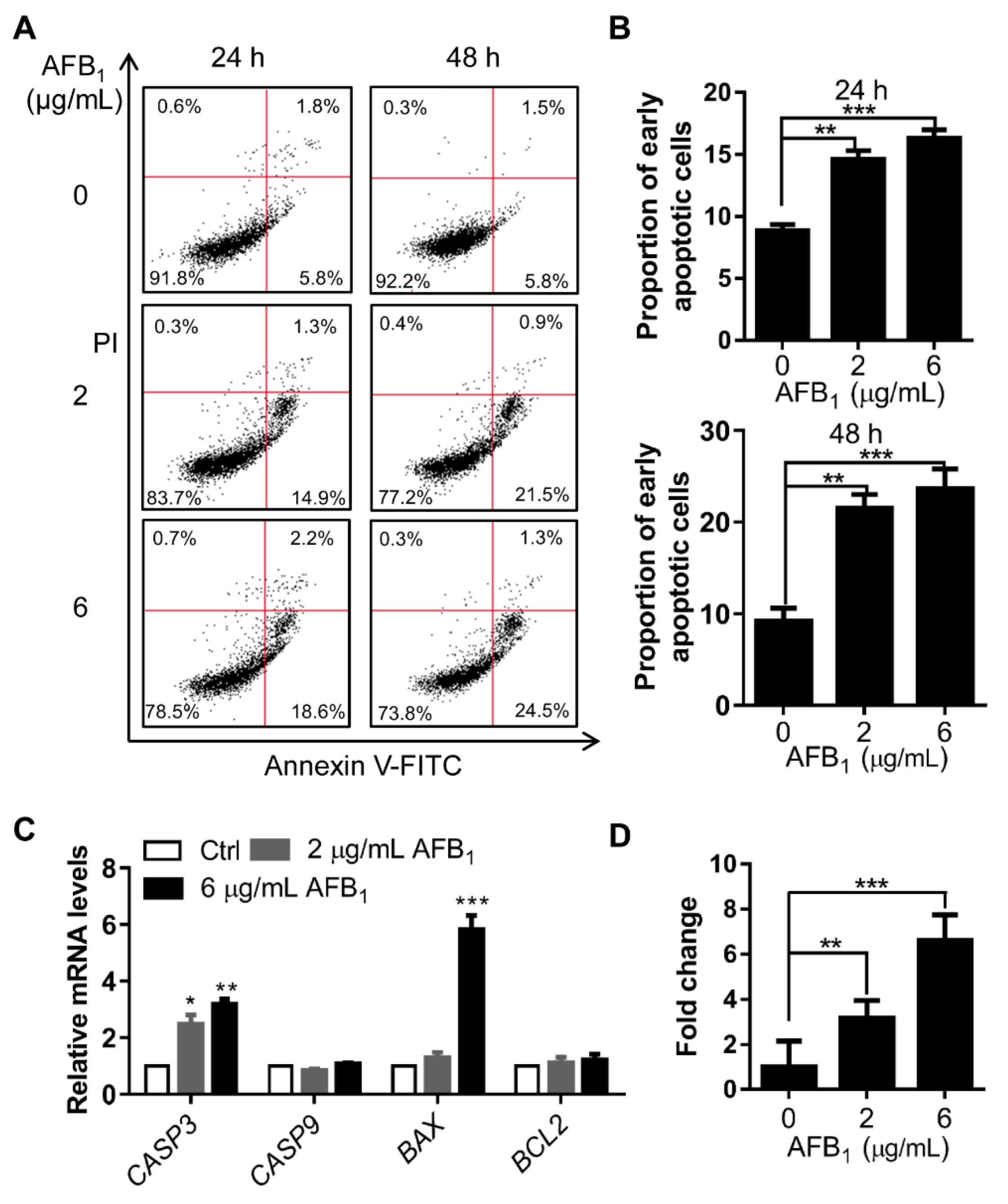
2.6. AFB1 Induces Apoptosis
3. Discussion
4. Materials and Methods
4.1. Animals and Cells
4.2. HPLC Analysis of AFB1 and AFBO
4.3. Cell Viability Assay and LDH Release Assay
4.4. RNA Extraction and qRT-PCR
4.5. Western Blot
4.6. Immunofluorescence
4.7. Comet Assay
4.8. Cell Cycle, ROS, and Apoptosis Analysis
4.9. Caspase-3 Activity Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wei, Y.K.; Zhao, X.M.; Li, M.M.; Yu, J.X.; Gurudeeban, S.; Hu, Y.F.; Ji, G.F.; Wei, D.Q. Detoxification of aflatoxins on prospective approach: Effect on structural, mechanical, and optical properties under pressures. Interdiscip. Sci. 2018, 10, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Stoev, S.D. Foodborne mycotoxicoses, risk assessment and underestimated hazard of masked mycotoxins and joint mycotoxin effects or interaction. Environ. Toxicol. Pharmacol. 2015, 39, 794–809. [Google Scholar] [CrossRef]
- Hamid, A.S.; Tesfamariam, I.G.; Zhang, Y.; Zhang, Z.G. Aflatoxin B1-induced hepatocellular carcinoma in developing countries: Geographical distribution, mechanism of action and prevention. Oncol. Lett. 2013, 5, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Bulatao-Jayme, J.; Almero, E.M.; Castro, M.C.; Jardeleza, M.T.; Salamat, L.A. A case-control dietary study of primary liver cancer risk from aflatoxin exposure. Int. J. Epidemiol. 1982, 11, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Qian, G.S.; Ross, R.K.; Yu, M.C.; Yuan, J.M.; Gao, Y.T.; Henderson, B.E.; Wogan, G.N.; Groopman, J.D. A follow-up study of urinary markers of aflatoxin exposure and liver cancer risk in Shanghai, People’s Republic of China. Cancer Epidemiol. Biomarkeres Prev. 1994, 3, 3–10. [Google Scholar]
- Fouad, A.M.; Ruan, D.; El-Senousey, H.K.; Chen, W.; Jiang, S.Q.; Zheng, C.T. Harmful Effects and Control Strategies of Aflatoxin B-1 Produced by Aspergillus flavus and Aspergillus parasiticus Strains on Poultry: Review. Toxins 2019, 11, 176. [Google Scholar] [CrossRef] [Green Version]
- Ikegwuonu, F.I. The neurotoxicity of aflatoxin B1 in the rat. Toxicology 1983, 28, 247–259. [Google Scholar] [CrossRef]
- Trebak, F.; Alaoui, A.; Alexandre, D.; El Ouezzani, S.; Anouar, Y.; Chartrel, N.; Magoul, R. Impact of aflatoxin B1 on hypothalamic neuropeptides regulating feeding behavior. Neurotoxicology 2015, 49, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Baldissera, M.D.; Souza, C.F.; Zeppenfeld, C.C.; Descovi, S.N.; Moreira, K.L.S.; da Rocha, M.; da Veiga, M.L.; da Silva, A.S.; Baldisserotto, B. Aflatoxin B1-contaminated diet disrupts the blood-brain barrier and affects fish behavior: Involvement of neurotransmitters in brain synaptosomes. Environ. Toxicol. Pharmacol. 2018, 60, 45–51. [Google Scholar] [CrossRef]
- Makhlouf, M.M.M. Histological and ultrastructural study of AflatoxinB1 induced neurotoxicity in Sciatic nerve of adult male Albino rats. Ultrastruct. Pathol. 2020, 44, 52–60. [Google Scholar] [CrossRef]
- Alsayyah, A.; ElMazoudy, R.; Al-Namshan, M.; Al-Jafary, M.; Alaqeel, N. Chronic neurodegeneration by aflatoxin B1 depends on alterations of brain enzyme activity and immunoexpression of astrocyte in male rats. Ecotoxicol. Environ. Saf. 2019, 182, 109407. [Google Scholar] [CrossRef] [PubMed]
- Benkerroum, N. Chronic and Acute Toxicities of Aflatoxins: Mechanisms of Action. Int. J. Environ. Res. Public Health 2020, 17, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vipin, A.V.; Rao, R.; Kurrey, N.K.; KA, K.K.; Venkateswaran, G. Protective effects of phenolics rich extract of ginger against Aflatoxin B1-induced oxidative stress and hepatotoxicity. Biochem. Pharmacol. 2017, 91, 415–424. [Google Scholar]
- Dey, D.K.; Kang, S.C. Aflatoxin B1 induces reactive oxygen species-dependent caspase-mediated apoptosis in normal human cells, inhibits Allium cepa root cell division, and triggers inflammatory response in zebrafish larvae. Sci. Total Environ. 2020, 737, 139704. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Q.C.; Han, J.; Xiong, B.; Sun, S.C. Aflatoxin B1 is toxic to porcine oocyte maturation. Mutagenesis 2015, 30, 527–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.J.; Xu, Z.L.; Yu, C.; Xu, X.H. Effects of aflatoxin B1 on mitochondrial respiration, ROS generation and apoptosis in broiler cardiomyocytes. Anim. Sci. J. 2017, 88, 1561–1568. [Google Scholar] [CrossRef]
- Zamir-Nasta, T.; Razi, M.; Shapour, H.; Malekinejad, H. Roles of p21, p53, cyclin D1, CDK-4, estrogen receptor alpha in aflatoxin B1-induced cytotoxicity in testicular tissue of mice. Environ. Toxicol. 2018, 33, 385–395. [Google Scholar] [CrossRef]
- Huang, B.; Mu, P.; Chen, X.; Tang, S.; Ye, W.; Zhu, W.; Deng, Y. Aflatoxin B1 induces S phase arrest by upregulating the expression of p21 via MYC, PLK1 and PLD1. Biochem. Pharmacol. 2019, 166, 108–119. [Google Scholar] [CrossRef]
- Yin, H.; Jiang, M.; Peng, X.; Cui, H.; Zhou, Y.; He, M.; Zuo, Z.; Ouyang, P.; Fan, J.; Fang, J. The molecular mechanism of G2M cell cycle arrest induced by AFB1 in the jejunum. Oncotarget 2016, 7, 35592–35606. [Google Scholar] [CrossRef] [Green Version]
- Mahato, D.K.; Lee, K.E.; Kamle, M.; Devi, S.; Dewangan, K.N.; Kumar, P.; Kang, S.G. Aflatoxins in Food and Feed: An Overview on Prevalence, Detection and Control Strategies. Front. Microbiol. 2019, 10, 2266. [Google Scholar] [CrossRef]
- Xu, Q.; Shi, W.; Lv, P.; Meng, W.; Mao, G.; Gong, C.; Chen, Y.; Wei, Y.; He, X.; Zhao, J.; et al. Critical role of caveolin-1 in aflatoxin B1-induced hepatotoxicity via the regulation of oxidation and autophagy. Cell. Death Dis. 2020, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, F. Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ. Health Perspect. 2010, 118, 818–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, Z.M.; Bush, G.; Rauch, S.L.; Cosgrove, G.R.; Eskandar, E.N. Human anterior cingulate neurons and the integration of monetary reward with motor responses. Nat. Neurosci. 2004, 7, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Ikehara, H.; Nagasawa, K.; Hishiki, H.; Uchikawa, H.; Shimojo, N. Anterior Commissure Involvement in Humanherpes Virus 6 Encephalitis. Child. Neurol. Open 2014, 1, 2329048X14544473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefranc, M.; Manto, M.; Merle, P.; Tir, M.; Montpellier, D.; Constant, J.M.; Le Gars, D.; Macron, J.M.; Krystkowiak, P. Targeting the red nucleus for cerebellar tremor. Cerebellum 2014, 13, 372–377. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, N.; Liu, J.; Li, F.D.; Li, S.L.; Wang, J.Q. Aflatoxin B1 and aflatoxin M1 induced cytotoxicity and DNA damage in differentiated and undifferentiated Caco-2 cells. Food Chem. Toxicol. 2015, 83, 54–60. [Google Scholar] [CrossRef]
- Zhou, H.; George, S.; Hay, C.; Lee, J.; Qian, H.; Sun, X. Individual and combined effects of Aflatoxin B1, Deoxynivalenol and Zearalenone on HepG2 and RAW 264.7 cell lines. Food Chem. Toxicol. 2017, 103, 18–27. [Google Scholar] [CrossRef]
- Yang, C.F.; Shen, H.M.; Ong, C.N. Protective effect of ebselen against hydrogen peroxide-induced cytotoxicity and DNA damage in HepG2 cells. Biochem. Pharmacol. 1999, 57, 273–279. [Google Scholar] [CrossRef]
- Mughal, M.J.; Xi, P.; Yi, Z.; Jing, F. Aflatoxin B1 invokes apoptosis via death receptor pathway in hepatocytes. Oncotarget 2017, 8, 8239–8249. [Google Scholar] [CrossRef]
- Zhang, Z.; Ren, Z.; Chen, S.; Guo, X.; Liu, F.; Guo, L.; Mei, N. ROS generation and JNK activation contribute to 4-methoxy-TEMPO-induced cytotoxicity, autophagy, and DNA damage in HepG2 cells. Arch. Toxicol. 2018, 92, 717–728. [Google Scholar] [CrossRef]
- Lin, Y.C.; Owen, N.; Minko, I.G.; Lange, S.S.; Tomida, J.; Li, L.; Stone, M.P.; Wood, R.D.; McCullough, A.K.; Lloyd, R.S. DNA polymerase zeta limits chromosomal damage and promotes cell survival following aflatoxin exposure. PNAS 2016, 113, 13774–13779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat. Res. 2012, 751, 158–246. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, J.; Elia, A.; Carroll, V.A.; Moumen, A. DNA damage-induced S and G2/M cell cycle arrest requires mTORC2-dependent regulation of Chk1. Oncotarget 2015, 6, 427–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.; Guo, W.; Yang, J.; Li, L.; Wang, M.; Lei, Y.; Wan, Y.; Zhao, X.; Luo, N.; Cheng, P.; et al. PNAS-4, an Early DNA Damage Response Gene, Induces S Phase Arrest and Apoptosis by Activating Checkpoint Kinases in Lung Cancer Cells. J. Biol. Chem. 2015, 290, 14927–14944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Yu, H.; Zhang, Y.; Li, D.; Xing, X.; Chen, L.; Zeng, X.; Xu, D.; Fan, Q.; Xiao, Y.; et al. Upregulation of miR-34a-5p antagonizes AFB1-induced genotoxicity in F344 rat liver. Toxicon 2015, 106, 46–56. [Google Scholar] [CrossRef]
- Zhu, L.; Gao, J.; Huang, K.; Luo, Y.; Zhang, B.; Xu, W. miR-34a screened by miRNA profiling negatively regulates Wnt/beta-catenin signaling pathway in Aflatoxin B1 induced hepatotoxicity. Sci. Rep. 2015, 5, 16732. [Google Scholar] [CrossRef] [Green Version]
- Ladouceur, A.M.; Dorn, J.F.; Maddox, P.S. Mitotic chromosome length scales in response to both cell and nuclear size. J. Cell. Biol. 2015, 209, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Barr, A.R.; Cooper, S.; Heldt, F.S. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun. 2017, 8, 14728. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, N.; Furukawa, Y.; Sutheesophon, K.; Nakamura, M.; Kishi, K.; Okuda, K.; Sato, Y.; Kano, Y. Suppression of ARG kinase activity by STI571 induces cell cycle arrest through up-regulation of CDK inhibitor p18/INK4c. Oncogene 2003, 22, 4074–4082. [Google Scholar] [CrossRef] [Green Version]
- Croglio, M.P.; Haake, J.M.; Ryan, C.P.; Wang, V.S.; Lapier, J.; Schlarbaum, J.P.; Dayani, Y.; Artuso, E.; Prandi, C.; Koltai, H.; et al. Analogs of the novel phytohormone, strigolactone, trigger apoptosis and synergize with PARP inhibitors by inducing DNA damage and inhibiting DNA repair. Oncotarget 2016, 7, 13984–14001. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. BBA Mol. Cell. Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.B.; Li, L.; Luo, X.D.; Lin, H. BMSCs protect against liver injury via suppressing hepatocyte apoptosis and activating TGF-beta1/Bax singling pathway. Biochem. Pharmacol. 2017, 96, 1395–1402. [Google Scholar]
- Mehrzad, J.; Fazel, F.; Pouyamehr, N.; Hosseinkhani, S.; Dehghani, H. Naturally Occurring Level of Aflatoxin B1 Injures Human, Canine and Bovine Leukocytes Through ATP Depletion and Caspase Activation. Int. J. Toxicol. 2020, 39, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Pena-Blanco, A.; Garcia-Saez, A.J. Bax, Bak and beyond-mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef]
- Naaz, F.; Abdin, M.Z.; Javed, S. Protective effect of esculin against prooxidant aflatoxin B1-induced nephrotoxicity in mice. Mycotoxin Res. 2014, 30, 25–32. [Google Scholar] [CrossRef]
- Kimbrough, T.D.; Llewellyn, G.C.; Weekley, L.B. The effect of aflatoxin B1 exposure on serotonin metabolism: Response to a tryptophan load. Metab. Brain Dis. 1992, 7, 175–182. [Google Scholar] [CrossRef]
- Spijker, S.; Faliagkas, L.; Rao-Ruiz, P. Dissection of Rodent Brain Regions: Guided Free-Hand Slicing and Dissection of Frozen Tissue. In Neuromethods; Li, K.W., Ed.; Humana: New York, NY, USA, 2019; pp. 7–19. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, B.; Chen, Q.; Wang, L.; Gao, X.; Zhu, W.; Mu, P.; Deng, Y. Aflatoxin B1 Induces Neurotoxicity through Reactive Oxygen Species Generation, DNA Damage, Apoptosis, and S-Phase Cell Cycle Arrest. Int. J. Mol. Sci. 2020, 21, 6517. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186517
Huang B, Chen Q, Wang L, Gao X, Zhu W, Mu P, Deng Y. Aflatoxin B1 Induces Neurotoxicity through Reactive Oxygen Species Generation, DNA Damage, Apoptosis, and S-Phase Cell Cycle Arrest. International Journal of Molecular Sciences. 2020; 21(18):6517. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186517
Chicago/Turabian StyleHuang, Boyan, Qingmei Chen, Lingling Wang, Xiaojuan Gao, Wenya Zhu, Peiqiang Mu, and Yiqun Deng. 2020. "Aflatoxin B1 Induces Neurotoxicity through Reactive Oxygen Species Generation, DNA Damage, Apoptosis, and S-Phase Cell Cycle Arrest" International Journal of Molecular Sciences 21, no. 18: 6517. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186517