Hedgehog Signaling in Skeletal Development: Roles of Indian Hedgehog and the Mode of Its Action

Department of Cell Biology, Institute of Biomedical Sciences, Nagasaki University, Nagasaki 852-8588, Japan
Int. J. Mol. Sci. 2020, 21(18), 6665; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186665
Submission received: 30 July 2020
/
Revised: 8 September 2020
/
Accepted: 9 September 2020
/
Published: 11 September 2020
(This article belongs to the Special Issue Hedgehog Signaling 2.0)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Hedgehog (Hh) signaling is highly conserved among species and plays indispensable roles in various developmental processes. There are three Hh members in mammals; one of them, Indian hedgehog (Ihh), is expressed in prehypertrophic and hypertrophic chondrocytes during endochondral ossification. Based on mouse genetic studies, three major functions of Ihh have been proposed: (1) Regulation of chondrocyte differentiation via a negative feedback loop formed together with parathyroid hormone-related protein (PTHrP), (2) promotion of chondrocyte proliferation, and (3) specification of bone-forming osteoblasts. Gli transcription factors mediate the major aspect of Hh signaling in this context. Gli3 has dominant roles in the growth plate chondrocytes, whereas Gli1, Gli2, and Gli3 collectively mediate biological functions of Hh signaling in osteoblast specification. Recent studies have also highlighted postnatal roles of the signaling in maintenance and repair of skeletal tissues.
1. Introduction
Hedgehog (Hh) signaling plays essential roles in various developmental process in vertebrates and insects, and is a highly conserved pathway among species. Hh was first identified as a segment polarity gene by extensive screening for genes responsible for the Drosophila body plan. The name Hedgehog refers to the disorganized bristles covering the hh-null embryos, which resemble hedgehog spines. Hh is secreted from a certain population in developing tissues, travels over the tissues, and regulates the activity of cells as a morphogen, which regulates the activity of recipient cells according to its concentration. The range that Hh diffuses over varies depending on the tissues: Up to 50 μm in the imaginal disc of Drosophila and 300 μm in the limb bud of vertebrates [1,2].
Mammals have three hedgehog genes: Sonic hedgehog (Shh), Indian hedgehog (Ihh), and Desert hedgehog (Dhh). Shh is involved in the L-R axis determination, the patterning of body segments, the neural tube, and limbs and morphogenesis of the hair, teeth, lungs, gut, and muscle. In particular, Shh expressed in the zone of polarizing activity in developing limbs and in the notochord and floor plate of the neural tube determines the patterning of digits and cell fates of neural progenitors, respectively. Dhh regulates spermatogenesis and the formation of the peripheral nerve sheaths. Ihh is implicated in angiogenesis and hematopoiesis as well as skeletal formation [1,2].
A large number of studies have revealed various and indispensable functions of Hh signaling for the development and maintenance of skeletal tissues, as several concise and comprehensive reviews recently overviewed [3,4,5]. This review particularly aims to organize and summarize the functions of Hh signaling and the mode of Hh action in skeletal development, based mainly on the findings of mouse genetic studies.
2. Two Ossification Processes in Mammals
Skeletal elements develop through two ossification processes in mammals: Intramembranous ossification and endochondral ossification. In intramembranous ossification, mesenchymal cells condense at the region where future bone is formed. The condensed mesenchyme directly differentiates into bone-forming osteoblasts. Intramembranous bones include the frontal bone, parietal bone, facial bone, part of the temporal bone, and part of the clavicle.
Endochondral ossification begins with the formation of the cartilage mold; the condensed mesenchyme initially differentiates into chondrocytes, which produce a variety of glycosaminoglycan and cartilage matrix proteins, including type II, type IX, and type XI collagens. Chondrocytes proliferate and maturate. Terminal differentiation of chondrocytes is characterized by its post-mitotic hypertrophy; the hypertrophic chondrocytes express type X collagen, matrix metalloproteinase 13 (MMP13), and vascular endothelial growth factor (VEGF) and induce calcification of their surrounding matrixes. Vascular invasion is facilitated by the aforementioned vascularization-promoting factors. Hypertrophic chondrocytes eventually undergo apoptosis. The cartilage matrixes are then absorbed by chondroclasts, which are recruited into the developing cartilage along with vascular invasion.
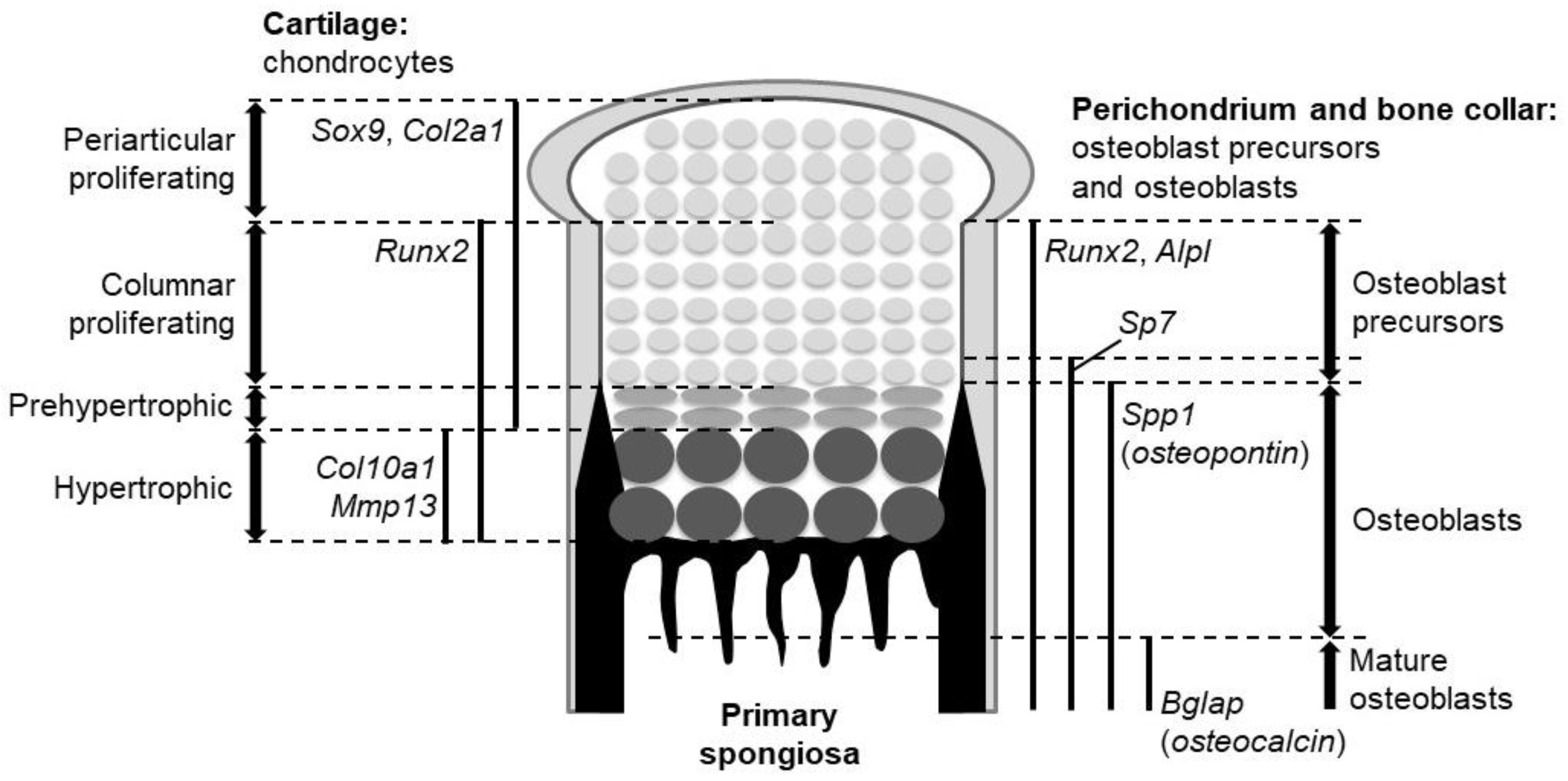
The fibrous layer that surrounds the developing cartilage is called the perichondrium, and it is known as a source of osteo-chondroprogenitors. Some cells in the perichondrium are specified into the osteoblast lineage. The perichondrial cell-derived osteoblasts form the bone collar, a predecessor of cortical bones. The specified population becomes bone-forming osteoblasts through several precursors. Upon their specification, perichondrial cells initially give rise to osteoblast precursors expressing runt-related transcription factor 2 (Runx2). The Runx2-positive precursors then express Sp7 to become Runx2-Sp7 double-positive precursors. Runx2 and Sp7 are master regulators of osteoblast development, as osteoblasts are absent in mutant mice lacking either of them [6,7]. The double positive precursors eventually differentiate into osteoblasts and mature, secreting bone matrices. Alkaline phosphatase (Alpl), secreted phosphoprotein 1 (Spp1, also known as osteopontin), and bone gla protein (Bglap, also known as osteocalcin) characterize osteoblast precursors, osteoblasts, and mature osteoblasts, respectively.
The fetal cartilage of endochondral skeletons is referred to as the growth plate. In the growth plate, chondrocytes are sequentially layered and aligned according to their differentiation stages: The proliferating stage, prehypertrophic stage, and hypertrophic stage (Figure 1). Importantly, the bone collar is formed at the perichondrium in synchronization with chondrocyte hypertrophy; it always occurs at the perichondrial region adjacent to hypertrophic chondrocytes (Figure 1). These features represent the chronological process of chondrocyte maturation and osteoblast differentiation from the epiphysis toward the diaphysis (Figure 1).
Among the three Hh ligands in mammals, Ihh is notable for its role in endochondral ossification, where it acts as a major Hh input for the biological action of Hh. Ihh is expressed strongly in prehypertrophic chondrocytes and weakly in hypertrophic chondrocytes of the growth plate. The expression of Ptch1 and Gli1, readouts of Hh signaling activation, is observed in proliferating chondrocytes, perichondrial cells, and primary spongiosa. Cells closer to the Ihh-producing cells express the readouts at higher levels.
3. Hh Signaling Transduction
Hh proteins are synthesized as <45 kDa precursor proteins in Hh-producing cells. The precursor protein is cleaved and divided into two fragments: A 19-kDa N-terminal peptide (Hh-N) and a 25-kDa C-terminal peptide (Hh-C). The Hh-N, which represents all of the Hh signaling activity, is subjected to sequentially occurring dual lipidation: Cholesterol modification to its C-terminus via covalent bonds and a subsequent palmitoylation at its N-terminus. The palmitoylation is mediated by skinny hedgehog (Ski), a membrane-bound O-acyltransferase (MBOAT). The processing and lipidation regulate not only the biological activity of Hh-N, but also affinity for the cell membrane. The affinity controls the range of Hh-N mobility over the tissue and resulting signaling gradient. Hh-C is further digested by the proteasome. The processed Hh-N is transported to the cell surface and released from the Hh-producing cells. Dispatched (Disp), a 12-pass transmembrane protein, regulates the releasing process; Disp1 and Disp2 have been identified in mice [1,2,8].
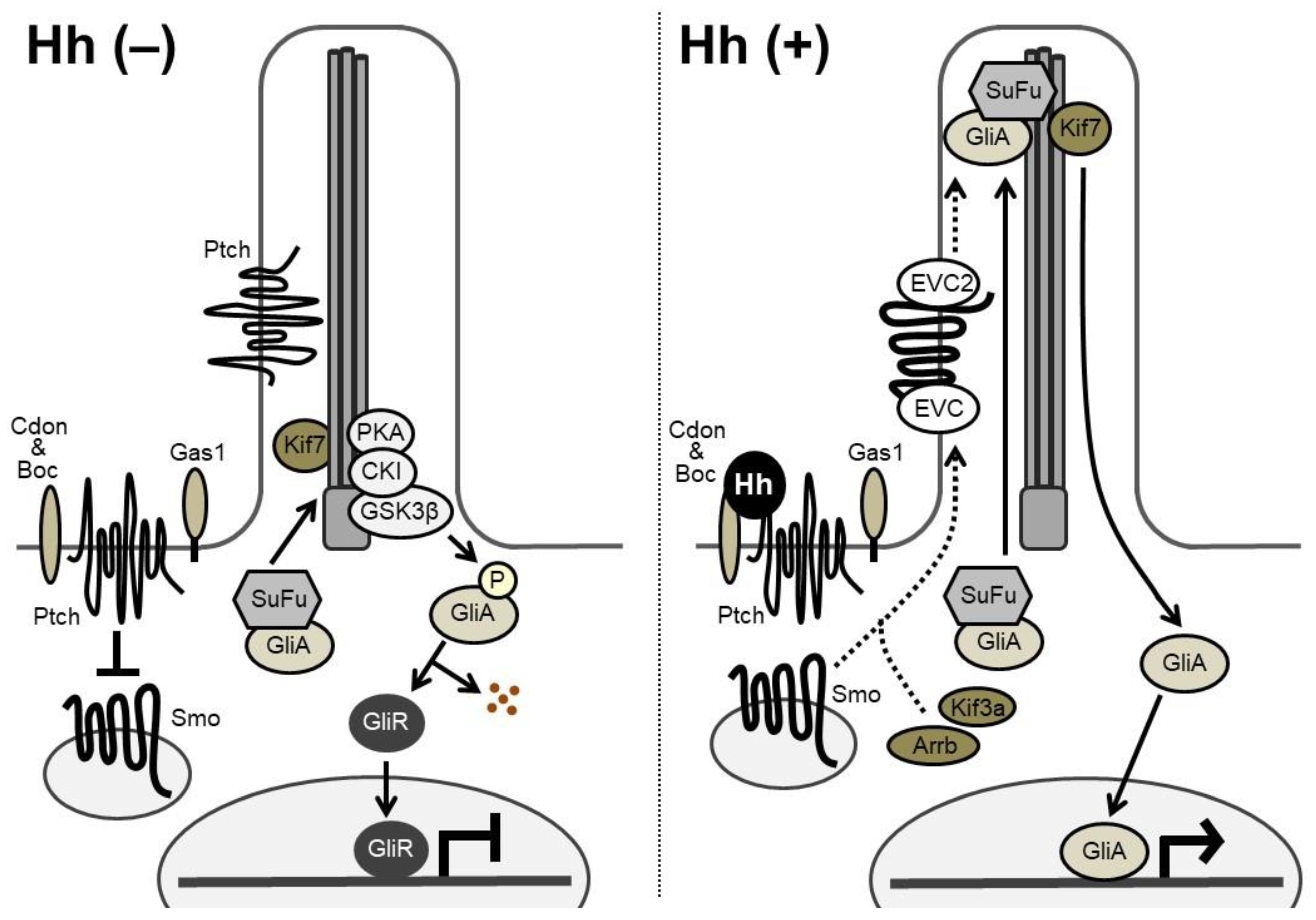
Hh signaling transduction is illustrated in Figure 2. In Hh-receiving cells, Smoothened (Smo), a seven-pass transmembrane protein, has an intrinsic intracellular signaling activity that is repressed by Patched (Ptch), a 12-pass transmembrane receptor of Hh. Two Ptch genes, Ptch1 and Ptch2, have been identified in vertebrates. Cdon (cell adhesion molecule-related/downregulated by oncogenes), Boc (brother of Cdo), and Gas1 (growth arrest specific 1) are known to act as co-receptors of Ptch, enhancing the binding affinity between Hh and Ptch. Without Hh ligands, Ptch proteins are located mainly around the primary cilia and inhibit the Smo activity. When Hh ligands bind to Ptch on the target cell, Ptch exits from the cilia; the action relieves the repressive effect of Ptch on Smo, which then shuttles through the cilia, initiating Hh signal transduction [1,2,8].
In vertebrates, Hh-responsive transcription is mediated through the zinc finger transcription factors Gli1, Gli2, and Gli3 [1]. Gli1 is one of the target genes of Hh signaling, and functions as a strong transcriptional activator. Gli2 and Gli3 are thought to act as both full-length activator forms and truncated repressor forms. Suppressor of Fused (SuFu) forms a complex with Gli factors and sequesters them in the cytoplasm [2,9]. Without Hh input, Gli2 and Gli3 are phosphorylated by protein kinase A (PKA), casein kinase 1α (CKI), and glycogen synthase kinase 3β (GSK3β) and processed into transcriptional repressor forms at the base of the cilia [2]. Speckle-type POZ protein (Spop), a substrate-binding adaptor for the cullin3-based ubiquitin E3 ligase, targets Gli2 and Gli3 for ubiquitination and proteasomal degradation [2,9,10]. Upon Hh input, released Smo prevents proteolytic processing of Gli2 and Gli3. Thus, the Gli-activator forms translocate into the nucleus to activate the transcription of target genes, while a decrease in Gli transcriptional repressors induces derepression of another set of genes. Gli2 was suggested to function primarily as a transcriptional activator, and Gli3 as a transcriptional repressor, although a few studies have shown the opposite [1].
4. Roles of Hh Signaling in Endochondral Ossification
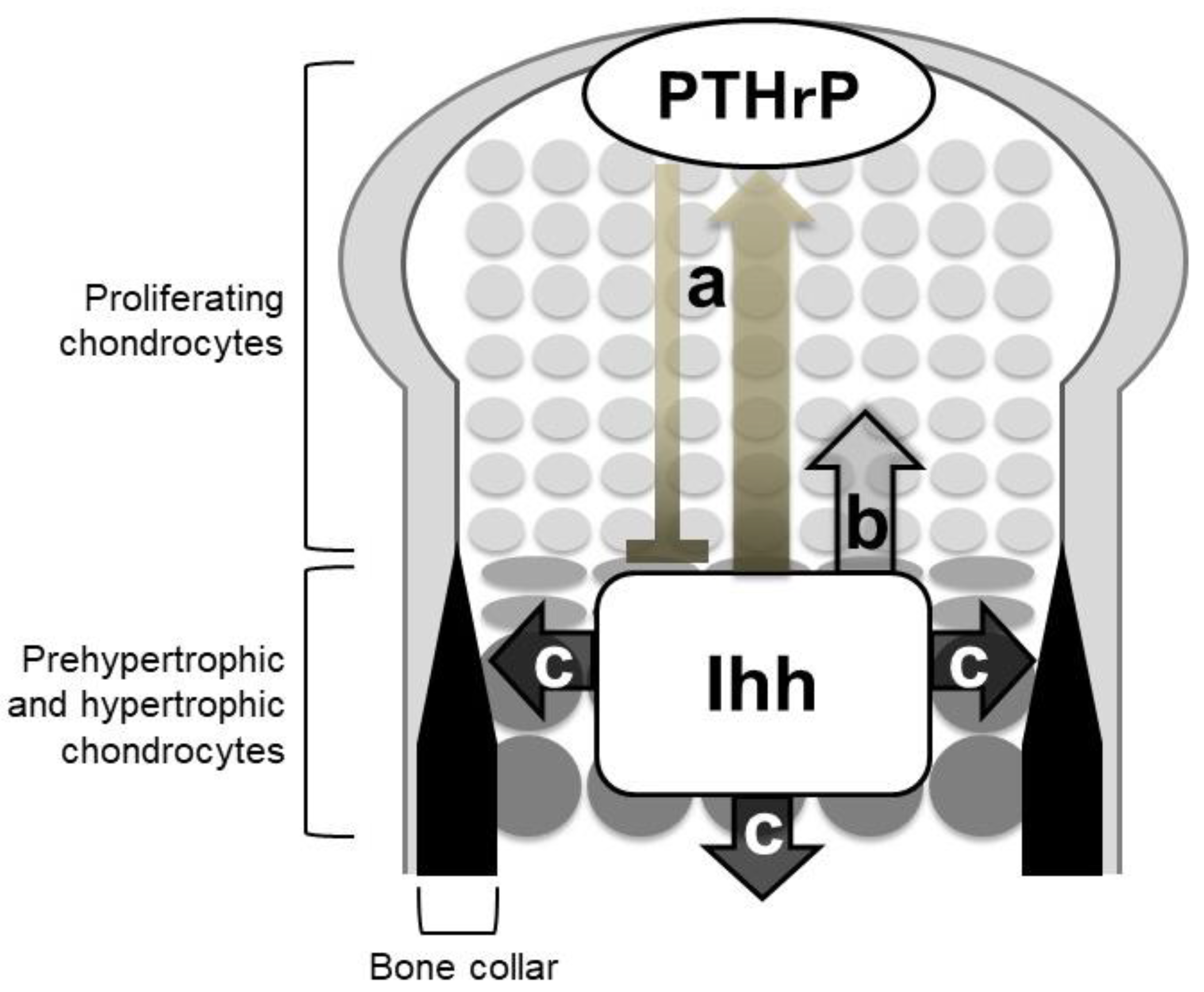
The phenotypes of Ihh-null mutant mice clearly demonstrated the three Hh signaling-dependent processes in endochondral ossification (Figure 3): (1) Chondrocyte differentiation, (2) chondrocyte proliferation, and (3) specification of bone-forming osteoblasts. Other genetic studies have supported the role of Hh signaling with some mechanistic insights. Ihh indirectly regulates the chondrocyte differentiation via a negative feedback loop with parathyroid hormone-related protein (PTHrP), whereas it acts on chondrocyte proliferation and osteoblast specification in a direct manner.
4.1. An Ihh-PTHrP Negative Feedback Loop Maintains the Growth Plate Length
Periarticular proliferating chondrocytes and the perichondrium express PTHrP (officially named as parathyroid hormone like hormone: PTHLH), whereas parathyroid hormone 1 receptor (PTH1R; also described as the PTH/PTHrP receptor or PPR) is expressed strongly in prehypertrophic chondrocytes and weakly in proliferating chondrocytes [11,12]. The phenotypes of mutant mice with the deletion of Pthrp, Ppr, or both Pth and Pthrp [13,14,15,16] and those with the chondrocyte-specific overexpression of Pthrp or the constitutively active Ppr [17,18,19] suggested that PTHrP suppressed chondrocyte hypertrophy via PPR and thereby kept chondrocytes proliferating.
The role of Ihh in association with the action of PTHrP was initially proposed by Lanske et al. and Vortkamp et al. in 1996 [15,20]. Vortkamp et al. demonstrated that Ihh overexpression in chick limbs suppressed chondrocyte hypertrophy with increased Pthrp expression in the periarticular perichondrium [20]. The addition of PTHrP rescued premature hypertrophy of Pthrp−/− mouse limbs, whereas Shh had no effect on Pthrp−/− mouse limbs. This result suggests that Hh acts upstream of PTHrP in the growth plate. In the work by Lanske et al., PTHrP and Shh treatment both induced elongation of the growth plate with the suppression of chondrocyte hypertrophy in wild-type limbs, although neither of them had effects on Ppr−/− limbs [15]. These two studies indicated that the transition from proliferating chondrocytes to hypertrophic ones was regulated by both PTHrP-PPR signaling and Ihh; they constitute a common feedback loop, where the PTHrP-PPR signaling mediated the effect of Ihh on the transition.
In 1999, St-Jacques et al. reported skeletal phenotypes of Ihh−/− mice [21]. Ihh−/− mice showed acceleration of chondrocyte hypertrophy, which resulted in shortening of the proliferating chondrocyte layer, with loss of Pthrp expression in the periarticular regions [21]. Thus, the Ihh−/− mice phenocopied the mice with the loss of PTHrP function in terms of the growth plate structure. In contrast, activation of Hh signaling in chondrocytes by the loss of Ptch1 caused a delay of chondrocyte hypertrophy and upregulation of Pthrp expression in mice [22]. These studies supported the aforementioned link between Ihh and PTHrP.
Karp et al. genetically determined the link by analyzing the phenotypes of compound mutant mice [23]. Ihh−/−; Pthrp−/− mutant limbs exhibited abnormalities similar to those of Ihh−/− mutant limbs [23]: Premature hypertrophy of chondrocytes and lack of the growth plate, trabecular bone, and bone collar. Importantly, activation of PTHrP signaling in the Ihh−/− mutant background cancelled the premature hypertrophy of Ihh−/− chondrocytes, but did not increase the number of mitotically active chondrocytes [23]. These results suggest that: (1) Ihh is required for both the differentiation and the proliferation of growth plate chondrocytes; (2) PTHrP partly mediates the Ihh function in maintaining a pool of proliferating chondrocytes; and (3) Ihh positively regulates chondrocyte proliferation in a PTHrP-independent manner.
To gain insight into the specific cellular interaction underlying the functional Ihh-PTHrP link, Chung et al. analyzed the growth plate of chimeric mice carrying Ppr−/− cells and WT cells [24]. Ppr−/− chondrocytes ectopically became hypertrophic in columnar proliferating regions and expressed Ihh. The chimeric mice also showed the upregulation of Pthrp in periarticular WT cells and an elongation of the growth plate. In chimeric mice carrying Ppr−/−; Ihh−/− cells and WT cells, Ppr−/−; Ihh−/− chondrocytes still exhibited ectopic hypertrophy, but the upregulation of Pthrp and the elongation of the growth plate were no longer observed [25].
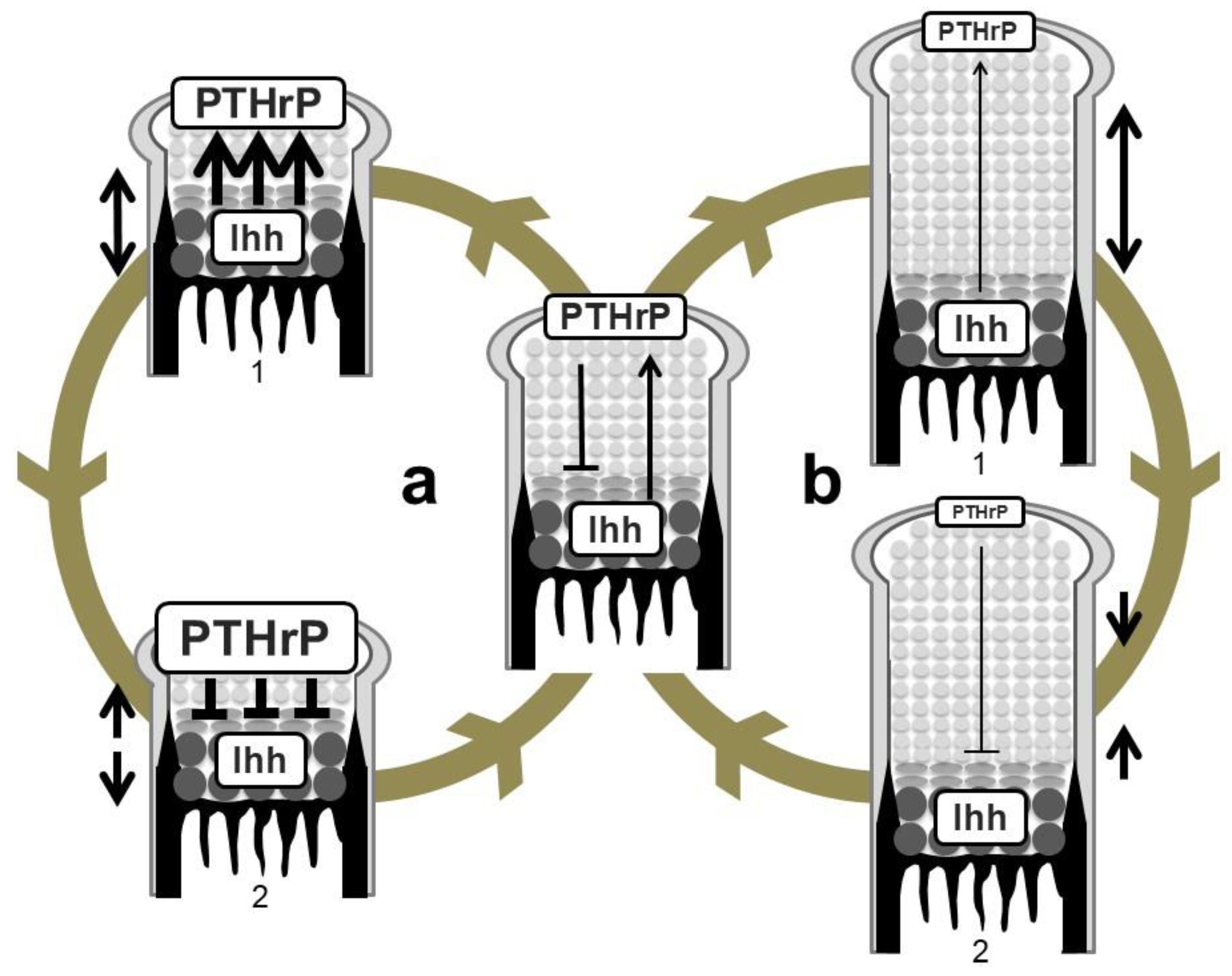
The above-described genetic studies establish the idea of an Ihh-PTHrP negative feedback loop. Ihh and PTHrP both contribute to maintenance of the growth plate length via the following functional loop (Figure 3 and Figure 4): Ihh produced by prehypertrophic chondrocytes facilitates the expression of PTHrP in cells around the periarticular regions, i.e., periarticular chondrocytes and perichondrial cells. Ihh possibly regulates the PTHrP expression in a concentration-dependent manner by acting as a morphogen. PTHrP then exerts a negative impact on chondrocyte hypertrophy by acting on PPR-expressing prehypertrophic chondrocytes. The suppressive effect of PTHrP on hypertrophy keeps chondrocytes proliferating, which leads to an increase in the distance between PTHrP- and Ihh-producing cells. This functional loop maintains a pool of proliferating chondrocytes by tightly regulating the distance; changes in the distance alter the PTHrP expression level at the periarticular region, leading to the correction of the distance (Figure 4). For example, reduction of the distance between PTHrP- and Ihh-producing cells facilitates PTHrP expression, probably due to the increased amount of Ihh acting on the periarticular region, and thereby the distance is increased, and vice versa. This notion is supported by the growth plate phenotype of Ppr−/−/WT chimeric mice, in which ectopic hypertrophy of Ppr−/− chondrocytes occurred much closer to the periarticular region than that of WT chondrocytes, and the length of the columns of WT proliferating chondrocytes was increased with enhanced Pthrp expression at the periarticular region. Thus, the Ihh-PTHrP negative feedback loop maintains a certain length of the growth plate in order to maximize skeletal growth.
Several studies support the requirement of direct Ihh input for Pthrp expression in periarticular proliferating chondrocytes and the periarticular perichondrium. When Smo was removed specifically from subsets of growth plate chondrocytes in mice, Pthrp expression became little or absent in the periarticular domain where Hh signaling was not activated [26]. Removal of the perichondrium from chicken embryonic tibiotarsi caused the expansion of the hypertrophic chondrocyte layer, and the addition of parathyroid hormone rescued the expansion [27]. As mentioned earlier, Ihh overexpression increased Pthrp expression in the periarticular perichondrium [20]. The latter two studies specifically suggest the involvement of the periarticular perichondrium as a source of PTHrP in the Ihh-PTHrP negative feedback loop.
4.2. Ihh Directly Regulates the Proliferation of Growth Plate Chondrocytes
The decreased proliferation of chondrocytes in Ihh−/− mice and chondrocyte-specific Smo−/− mice [21,28] indicates the direct and positive regulation of chondrocyte propagation by Ihh input in the growth plate (Figure 3), as Karp et al. suggested [23]. Hh signaling has also been shown to directly regulate the differentiation of growth plate chondrocytes at multiple steps independently of PTHrP. Kobayashi et al. proposed that Ihh drove the differentiation of periarticular proliferating chondrocytes into columnar proliferating chondrocytes in a PTHrP-independent manner [29,30]. In addition, chondrocyte-specific removal of Smo delayed chondrocyte hypertrophy without PTHrP in mice [31].
4.3. Ihh Is Required for Specification of Osteoblasts
Hh signaling act as a master regulator of osteoblast development during endochondral ossification. In endochondral bones, osteoblasts first emerge in a perichondrial region adjacent to pre-hypertrophic and hypertrophic chondrocytes. Mouse genetic studies demonstrate that Ihh produced by these chondrocytes acts on osteoblast progenitors to execute an osteoblast specification program in the perichondrium and the primary spongiosa. Ihh−/− mice showed no bone collar and lacked Runx2 and Bglap expressions in the perichondrium [21,32]. The analysis of Ppr−/−/WT chimeric mice and Ppr−/−; Ihh−/−/WT chimeric mice further supported the involvement of Ihh and Ihh-producing cells, i.e., pre- and hypertrophic chondrocytes, in bone collar formation [24,25]. In Ppr−/−/WT chimeras, ectopic calcification was observed in the perichondrium adjacent to ectopically hypertrophic Ppr−/− cells. The ectopic calcification disappeared in Ppr−/−; Ihh−/−/WT chimeras, although Ppr−/−; Ihh−/− cells still showed ectopic hypertrophy with robust expressions of bone morphogenetic protein (Bmp) 2 and 6. These chimeric mouse studies suggest that hypertrophic chondrocytes induce bone formation at their adjacent regions by secreting Ihh. Another important point is that BMP2 and 6 produced by hypertrophic chondrocytes may not be sufficient to induce bones at this step.
The requirement of direct Hh input for osteoblast specification is supported by the phenotypes of Smo mutant mice. Ablation of Smo from perichondrial cells caused absence of Runx2 expression and bone collar formation in the perichondrium [33]. In Smo−/−/WT chimeric mice, Smo−/− perichondrial cells, which did not differentiate into osteoblasts, expressed chondrocyte marker genes including type II collagen (Col2a1) and type X collagen (Col10a1) in the region where the bone collar is formed under physiological conditions. Smo−/− cells did not contribute to bone-forming regions in the primary spongiosa [33]. In contrast, the chondrocyte-specific overexpression of Ihh and the ablation of Ptch1 from perichondrial cells led to acceleration of bone collar formation, accompanied by the activation of Hh signaling in the perichondrium [22,33].
The above-described studies demonstrate that Ihh produced by pre- and hypertrophic chondrocytes is essential for osteoblastogenesis in the perichondrium and the primary spongiosa during endochondral ossification (Figure 3). In particular, Hh signaling is necessary for the specification of progenitors into Runx2-positive osteoblast precursors. As Smo deletion in Sp7-positive cells showed no obvious abnormality in osteoblast development, Hh signaling is dispensable for the later phase of osteoblastogenesis [34]. Given that chondrocytic phenotypes were acquired by perichondrial cells that cannot receive Hh input [33], Hh signaling may specify the cell fates of the osteo-chondroprogenitor population into the osteoblastic lineages in the perichondrium.
4.4. Factors Acting Downstream of Hh Signaling During Endochondral Ossification
Which Gli factors act downstream of Ihh in endochondral ossification? In regulation of growth plate chondrocytes, Ihh is likely to utilize Gli3 to exert its biological effects. Removal of Gli3 on an Ihh−/− background rescued abnormalities in the proliferation and maturation of chondrocytes in Ihh−/− mice [32,35]. The Gli3−/−; Ihh−/− mice also showed recovery of Pthrp expression in the periarticular regions [32,35]. Together with the fact that Gli3 primarily acts as a transcriptional repressor, Ihh-mediated suppression of the Gli3 repressor activity may derepress the Pthrp expression in an Ihh-PTHrP negative feedback loop. Because neither Gli1−/−, Gli2−/−, nor Gli1−/−; Gli2−/− mouse embryos demonstrated any obvious defects in the growth plate [16,36,37,38], Gli1 and Gli2 are unlikely to have dominant roles in maintenance of the growth plate.
In contrast, osteoblast development is mediated by all of the Gli factors, Gli1, Gli2, and Gli3, upon Hh input. The activator function of Gli2 was initially a focus in this context. Shimoyama et al. reported that Ihh induced osteoblast differentiation in a Gli2-dependent manner; they proposed that Runx2 induction and the physical interaction between Runx2 and Gli2 underlay the Ihh-Gli2 axis [39]. Joeng and Long provided further evidence supporting the importance of Gli2 in osteoblastogenesis; the skeletal phenotypes of Ihh−/− embryos were completely rescued in Ihh−/−; Gli3−/−; C2-NGli2 embryos, in which NGli2 (an N-terminally truncated, constitutively active form of Gli2) was exogenously expressed in Col2a1-positive cells under an Ihh−/−; Gli3−/− background. Based on this result, they proposed that the Gli2 activator and the Gli3 repressor collectively mediated all major aspects of the Ihh function in endochondral ossification [40].
We have proposed that Gli1 is also involved in Hh-mediated osteoblast specification cooperatively with Gli2 and Gli3, based on the following findings: First, Gli1−/− mice showed impairment of bone formation [37]. Second, Gli1−/− perichondrial cells expressed Col2a1 and Col10a1, but not Runx2 or Sp7 [37]. Third, Gli1−/−;Gli2−/− mice showed more severe skeletal phenotypes than either Gli1−/− or Gli2−/− mice [37]. Fourth, osteoblast differentiation was impaired in Gli1−/−; Gli3−/− perichondrial cells compared to that in Gli3−/− cells in vitro [37]. Lastly, Gli1 activated the transcription of early and middle marker genes for osteoblasts by directly binding to the 5′ regulatory regions of the genes [37], and it interfered with the Sox9-mediated transactivation of chondrocyte marker genes by suppressing the DNA binding of Sox9 [38]. We also found that the Gli3 repressor suppressed the Runx2-mediated transcription of osteoblastic genes by antagonizing the DNA binding of Runx2 [41].
Given that Runx2 expression is lost in the perichondrium of mice defective with Hh signaling, one might expect that Runx2 is involved in Hh-mediated osteoblast specification. However, the recovery of Runx2 expression in the Ihh−/− perichondrium did not cancel the abnormality of osteoblast differentiation in Ihh−/− mice [42]. Thus, Runx2 alone is unlikely to account for the function of Hh signaling in this context.
5. Roles of Hh Signaling in Craniofacial Development
Key features that distinguish cranial skeletal components forming the face and the rostral cranial vault from others in the head, limbs, and trunk are that the former components (1) originate from the cranial neural crest cells (CNCCs) and (2) generate osteoblasts via the intramembranous ossification.
Neural crest cells are multipotent and self-renewing. Notably, they have a greater capacity for differentiation than the cells from which they originate: They give rise to not only ectodermal lineages, but also mesodermal lineages. In vertebrates, the neural crest progenitors are derived from the ectodermal germ layer, arising in the neural plate border, a region between the neural plate and non-neural ectoderm that forms the future epidermis. The induction of the neural plate border, i.e., the induction of neural crest progenitors, is mediated by a dynamic interplay of patterning signaling pathways: BMP, FGF, Wnt, and Notch (extensively reviewed in [43]).
Hh signaling plays indispensable roles in the formation of CNCC-derived cranial skeletons by acting on postmigratory CNCCs. Jeong et al. found that the removal of Smo in CNCCs using the Wnt1-Cre driver resulted in extensive loss of CNCC-derived skeletal elements in mice, although the generation and migration of CNCCs were not affected and the mesoderm-derived skeletons remained intact [44]. They further found that five Forkhead box (Fox) family transcription factors (Foxc2, Foxd1, Foxd2, Foxf1, and Foxf2), which were selected through a transcriptional profiling of Shh mutant and WT heads, were specifically downregulated in craniofacial regions of the CNCC-specific Smo mutants, which suggested that the Fox genes were direct and major mediators of the function of Hh in craniofacial development [44]. Importantly, Smo deletion affected not only intramembranous bones, but also endochondral bones including rostral half of the basisphenoid. Thus, the involvement of Fox genes in craniofacial development may be supported by a recent paper, which demonstrates that Fox genes are necessary for Sox9-mediated induction of chondrogenic genes in zebrafish [45].
Shh is thought to be responsible for this action, since Shh, but not Ihh, is expressed in epithelial populations in the developing face from E9.5 to E12.5; Ptch1 is expressed not only in the epithelium, but also in the mesenchyme, which contains a high density of CNCCs [44], and this fact supports the functional importance of the signaling in this population. Although Ihh is unlikely to play major roles in this context, Ihh mutants have mild craniofacial defects [21].
The expressions of Shh and Ihh in murine cranial sutures have been debated (reviewed in [8]). Ihh has been shown to be expressed in ossifying bones and osteogenic fronts, whereas Shh is expressed in the suture mesenchyme [8,46]. Based on findings in Ihh−/− mouse cranial skeletons and the Ihh misexpression in chick embryonic heads, Abzhanov et al. speculated that in cranial intramembranous bones, the Ihh produced by mature osteoblasts acts as a feedback-inhibitor for the transition of pre-osteoblasts to the chondrocyte-like osteoblasts, which they proposed, and mature osteoblasts [47]. Amano et al. recently reported phenotypes of mutant mice in which Ihh was deleted in CNCCs by a Wnt1-Cre driver line [48]. The mutant showed disruption of the midface structure, where intersphenoid synchondrosis and nasal cartilage were greatly impaired, and a shortened mandible. This study also supports the idea that Ihh plays roles in the craniofacial skeleton.
Mutations in cilium-related factors caused abnormalities in cranial skeletons, in line with the fact that Hh signaling transduction takes place at the primary cilia. In particular, mutations of several intraflagellar transport proteins (IFTs), which are involved in Hh signaling transduction at primary cilia, have been implicated in craniofacial development. These IFTs include IFT144 [49] and Kif3a [50,51].
6. Postnatal Roles of Hh in Skeleton
Several genetic studies have highlighted the roles of Hh signaling in postnatal cartilage and the growth plate. When Ihh was deleted from Col2a1-expressing cells at the postnatal stages in mice, ectopic chondrocyte hypertrophy, decreased proliferation of chondrocytes, and a disorganized growth plate was observed [52]. Postnatal deletion of Ihh in Prrx1-expressing skeletal progenitors caused lack of the growth plate and a secondary ossification center in mice [53]. Thus, Ihh is necessary for postnatal maintenance of the growth plate and progression of ossification in endochondral bones. Ihh can function in both PTHrP-dependent and -independent manners in this context; the forced expression of constitutively active Ppr temporally cancelled the ectopic chondrocyte hypertrophy led by the deletion of Ihh from Col2a1-expressing cells, but did not correct the decreased proliferation of chondrocytes [54]. These data suggest the following. (1) Ihh and PTHrP functionally interact to control hypertrophy of growth plate chondrocytes at postnatal stages, as they do at embryonic stages. (2) Ihh promotes chondrocyte proliferation at the postnatal stage independently of PTHrP-PPR signaling. In line with the first notion, chondrocyte-specific ablation of Ppr in postnatal mice led to the acceleration of hypertrophy, followed by premature closure of the growth plate, in association with increased chondrocyte apoptosis [55]. However, the work by Maeda et al. also suggests that the contribution of the Ihh-PTHrP interaction to chondrocyte hypertrophy may occur less at postnatal stages than at embryonic stages [52].
We and others have demonstrated that Hh signaling also regulates postnatal bone mass. Ptch1+/− mice showed high bone mass with a high bone turnover phenotype in adults; both bone formation and bone resorption were accelerated in the mutant mice [41]. The mouse phenotype was partly recapitulated in patients with Gorlin syndrome caused by inactivating mutations of one of the PTCH1 alleles [41]. Ptch1 deletion in Bglap-expressing mature osteoblasts also led to high bone turnover in mice [56]. However, unlike Ptch1+/− mice, the mutant mice showed fragile long bones with low bone mass [3,56]. These results indicate that indirect effects of Hh signaling on osteoclasts may affect bone metabolism more than its direct effects on osteoblasts, when Hh signaling was activated specifically in mature osteoblasts. Indeed, the expression of Receptor activator of nuclear factor kappa-B ligand (RANKL) was increased in osteoblasts of both mutants. Mak et al. further showed the involvement of the PTHrP-PKA-CREB (cAMP responsive element binding protein) axis in upregulation of the RANKL expression upon Hh signaling activation in mature osteoblasts [56]. We also reported the effect of attenuation of Hh signaling on adult bone mass; Gli1 haploinsufficiency resulted in decreased bone mass with reduced bone formation and accelerated bone resorption, and impairment of fracture healing in adult mice [57]. The osteoblast phenotype of Gli1+/− mice supports the positive roles of Hh signaling in osteoblast differentiation. However, it is still debated how Hh signaling regulates osteoclastogenesis. Studies so far suggest not only its indirect actions via osteoblasts, but also direct actions on osteoclast precursors [58], and the signaling is likely to exert both positive and negative effects on osteoclastogenesis in context- or stage-dependent manners. In addition, regarding the involvement of Hh signaling in human adult bones, we need to consider that the growth plate, a major source of Ihh in the skeleton, is closed after puberty in humans. The alternative source of Hh ligands and its contribution to adult bone metabolism remain to be fully elucidated.
The postnatal expression of Ihh in the articular cartilage has been implicated in osteoarthritis (OA). Ihh expression is increased in human OA cartilages, whereas the expression is at the low level in healthy cartilages [59]. Postnatal deletion of Ihh attenuated OA progression in a surgically induced OA model compared to the WT mice [60]. Activation of Hh signaling in turn developed OA in mice [61]. Thus, Ihh can be a therapeutic target for the treatment of OA; suppression of the Ihh activity or its downstream signaling in the articular cartilage may modify OA progression or even inhibit its development in adults.
Recently, postnatally existing skeletal stem cell-like populations have drawn attention [62,63,64]. Shi et al. reported that Gli-positive cells worked as metaphyseal mesenchymal progenitors (MMPs) at the postnatal stage [65]. The Gli1-positive MMPs, residing immediately below the growth plate, expressed a set of mesenchymal stem cell markers and produced cancellous bone osteoblasts in postnatal mice. They also found that the Gli1-positive MMPs contribute to fracture healing by giving rise to both osteoblasts and chondrocytes. Haraguchi et al. also investigated the cell fate of Gli1-expressing cells present in hypertrophic chondrocytes and the chondro-osseous junction at postnatal stages in mice [66]. In this context, Gli1-expressing cells are supposed to be Ihh-responding cells in and around the growth plate. Descendants of the Gli1-expressing cells contributed to osteoblastic cells in the periosteum, trabecular bones, and cortical bones [66]. These data suggest that Hh-responding cells are involved in physiological bone formation at postnatal stages as well as embryonic ones.
Lastly, we recently demonstrated the positive role of Hh signaling in human osteoblastogenesis and its therapeutic potential by utilizing disease-specific induced pluripotent stem cells (iPSCs) [67]. We focused on two genetic diseases, Gorlin syndrome (see also the next Section 7.) and McCune Albright syndrome (MAS). Gorlin syndrome-derived iPSCs showed augmentation of osteoblastogenesis with Hh signaling activation, whereas MAS-specific iPSCs showed impaired osteoblastogenesis with low Hh signaling activity. Importantly, impaired osteoblastogenesis of MAS-specific iPSCs was restored by treatment with a Smo agonist.
7. Skeletal Diseases Caused by Abnormalities of Hh Signaling
Consistent with its critical roles revealed by mouse genetic studies, disruption of normal Hh signaling is known to cause human skeletal diseases. GWAS for genetic variants influencing human height identified IHH and Hh signaling components, PTCH1 and HHIP (hedgehog interacting protein), as height-associated loci [68,69]. This result supports the prerequisite role of Ihh in maintenance of the growth plate via the negative feedback loop formed together with PTHrP, in order to maximize skeletal growth.
Most of other Hh-related skeletal diseases are characterized by patterning defects. Heterozygous mutations of IHH also cause brachydactyly type AI (BDA1; OMIM number: 112500), in which the middle phalanges of all the digits are rudimentary or fused with the terminal phalanges [70,71,72]. Mice carrying one of the BDA1 mutations, E95K, recapitulated the phenotype of human patients with BDA1 [72]. Phalanges of the heterozygous BDA1 mutant mice were mildly affected, whereas homozygous mutants showed a classic BDA1 phenotype [72]. Altered expression pattern of Ptch1, a readout of Hh signaling, in the mutants’ growth plate indicates a change in the Ihh activity gradient, probably due to decreased binding capacity of the E95K-Ihh mutant to the receptor Ptch1 [72]. Greig cephalopolysyndactyly syndrome (GCPS; OMIM number: 175700), Pallister-Hall syndrome (PHS; OMIM number: 146510), and postaxial polydactyly type A1 and type B3 (PAPA1 and PAPB; OMIM number: 174200) are resulted from heterozygous mutations of GLI3 [73,74,75]. These diseases are characterized by various bone-related anomalies, including abnormalities of skull or limbs, syndactyly, and postaxial polydactyly. Gorlin syndrome, also known as basal cell nevus syndrome (BCNS; OMIM number: 109400), is a rare autosomal-dominant disorder caused by heterozygous inactivating mutations of PTCH1. Patients with Gorlin syndrome show skeletal abnormalities, craniofacial abnormalities, large body size, and tumors, including basal cell carcinomas of the skin and cerebellar medulloblastomas [76].
Impaired Hh signaling is thought to underlie Smith-Lemli-Opitz syndrome (SLOS; OMIM number: 270400), an autosomal recessive multiple congenital malformation and mental retardation syndrome. SLOS is caused by mutations of the gene encoding the cholesterol biosynthetic enzyme 7-dehydrocholesterol reductase (DHCR7) [77], and its clinical features include polydactyly and syndactyly as seen in the aforementioned diseases with aberrant Hh signaling. Indeed, the DHCR7 mutations perturb its ability to catalyze the conversion of 7-dehydrocholesterol (7DHC) to cholesterol; the reduced cholesterol level affects Hh signaling due to reduced Smo activities [78].
Aberrant Hh signaling was recently associated with ectopic ossification. Progressive osseous heteroplasia (POH; OMIM number: 166350) is known to show progressive ankylosis and growth retardation caused by ectopic ossification due to mutations in GNAS, which encodes Gαs [79]. Regard et al. found that patients with POH showed upregulation of Hh signaling; they also demonstrated that GNAS inhibited Hh signaling through cAMP-mediated PKA activation, and suppression of the Hh signaling activity partially rescued the POH phenotypes [80]. Thus, Hh signaling activation by GNAS mutation is likely to underlie heterotopic ossification in POH.
8. Conclusions
As reviewed in this paper, understanding of the Hh signaling function in skeletal development has greatly advanced in the last two decades. Hh signaling not only couples chondrogenesis with osteogenesis during endochondral ossification, but also regulates intramembranous ossification in the cranial skeleton. In addition, the molecular mechanisms underlying the function have been revealed with particular focuses on Gli transcription factors and other signaling components. However, two central questions remain to be addressed. First, the gene regulatory networks underlying these actions are unclear. What genes act downstream of the Hh-Gli axis in the skeleton? What transcription factors engage the Gli-mediated network during skeletal development? The recent advancement in next generation sequencer (NGS)-based genome-wide analyses and single-cell analysis will provide insight into these questions. Second, the regulatory landscape underlying Ihh transcription is still poorly understood. How do the enhancer-promoter interaction and distal enhancer networks regulate the Ihh transcription? Is the disruption of the network relevant to human disease? Recently, Will et al. identified five enhancer regions upstream of the Ihh gene; they have overlapping activities in vivo, and a cluster of the redundant enhancers regulates the Ihh expression in skeletal development [81]. The core enhancer elements and transcription factors engaging the elements remain to be identified. Thus, further studies are expected to elucidate the gene regulatory networks of Ihh transcription itself and those downstream of the Hh-Gli axis. This knowledge will enable us to understand the fate specification and subsequent maturation of skeletal cells, and the potential link between disruption of the process with human diseases when combined with GWAS and/or disease-specific iPSCs.
Author Contributions
S.O. conceived, designed, and wrote the paper. The author has read and agreed to the published version of the manuscript.
Funding
This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS: 17H04403 and 18K19636), Rising Star Award from American Society for Bone and Mineral Research, Mochida Memorial Foundation Research Grants, The Naito Grant for the Advancement of Natural Science, and Terumo Life Science Foundation R&D Subsidy.
Acknowledgments
The author thanks the members of Department of Cell Biology, Nagasaki University, Hironori Hojo (University of Tokyo), and Ung-il Chung (University of Tokyo) for their helpful discussions.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| Hh | Hedgehog |
| Ihh | Indian hedgehog |
| PTHrP | parathyroid hormone-related protein |
| Shh | Sonic hedgehog |
| Dhh | Desert hedgehog |
| MMP13 | matrix metalloproteinase 13 |
| VEGF | vascular endothelial growth factor |
| Runx2 | runt-related transcription factor 2 |
| Alpl | alkaline phosphatase |
| Spp1 | secreted phosphoprotein 1 |
| Bglap | bone gla protein |
| Ski | skinny hedgehog |
| MBOAT | membrane-bound O-acyltransferase |
| Disp | Dispatched |
| Smo | Smoothened |
| Ptch | Patched |
| Cdon | cell adhesion molecule-related/downregulated by oncogenes |
| Boc | brother of Cdo |
| Gas1 | growth arrest specific 1 |
| SuFu | Suppressor of Fused |
| PKA | protein kinase A |
| CKI | casein kinase 1α |
| GSK3β | glycogen synthase kinase 3β |
| Spop | Speckle-type POZ protein |
| Gpcrk2 | G protein-coupled receptor kinase 2 |
| Arrb | β-arrestin |
| EVC | Ellis-van Creveld syndrome protein |
| PTH1R | parathyroid hormone 1 receptor |
| PPR | PTH/PTHrP receptor |
| WT | wild-type |
| BMP | bone morphogenetic protein |
| CNCC | cranial neural crest cell |
| FGF | Fibroblast growth factor |
| Fox | Forkhead box |
| IFT | intraflagellar transport proteins |
| RANKL | Receptor activator of nuclear factor kappa-B ligand |
| CREB | cAMP responsive element binding protein |
| OA | osteoarthritis |
| MMP | metaphyseal mesenchymal progenitor |
| iPSC | induced pluripotent stem cell |
| MAS | McCune Albright syndrome |
| GWAS | genome-wide association study |
| HHIP | hedgehog interacting protein |
| BDA1 | brachydactyly type AI |
| GCPS | Greig cephalopolysyndactyly syndrome |
| PHS | Pallister-Hall syndrome |
| PAPA1 | postaxial polydactyly type A1 |
| PAPB | postaxial polydactyly type B3 |
| BCNS | basal cell nevus syndrome |
| SLOS | Smith-Lemli-Opitz syndrome |
| DHCR7 | 7-dehydrocholesterol reductase |
| 7DHC | 7-dehydrocholesterol |
| POH | progressive osseous heteroplasia |
| NGS | next-generation sequencers |
References
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Andre, P.; Ye, L.; Yang, Y.Z. The Hedgehog signalling pathway in bone formation. Int. J. Oral Sci. 2015, 7, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S. Hedgehog signaling in endochondral ossification. J. Dev. Biol. 2016, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, R.; Kitazawa, R.; Kohara, Y.; Ikedo, A.; Imai, Y.; Kitazawa, S. Recent insights into long bone development: Central role of hedgehog signaling pathway in regulating growth plate. Int. J. Mol. Sci. 2019, 20, 5840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Pan, A.; Chang, L.; Nguyen, A.; James, A.W. A review of hedgehog signaling in cranial bone development. Front. Physiol. 2013, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Pan, Y.; Wang, B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 2010, 137, 2001–2009. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Liu, A. Spop promotes skeletal development and homeostasis by positively regulating Ihh signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 14751–14756. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Deeds, J.D.; Segre, G.V. Expression of parathyroid hormone-related peptide and its receptor messenger ribonucleic acids during fetal development of rats. Endocrinology 1995, 136, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Lanske, B.; Karaplis, A.C.; Deeds, J.D.; Kohno, H.; Nissenson, R.A.; Kronenberg, H.M.; Segre, G.V. Parathyroid hormone-related peptide delays terminal differentiation of chondrocytes during endochondral bone development. Endocrinology 1996, 137, 5109–5118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amizuka, N.; Warshawsky, H.; Henderson, J.E.; Goltzman, D.; Karaplis, A.C. Parathyroid hormone-related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J. Cell Biol. 1994, 126, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Karaplis, A.C.; Luz, A.; Glowacki, J.; Bronson, R.T.; Tybulewicz, V.L.; Kronenberg, H.M.; Mulligan, R.C. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994, 8, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef]
- Miao, D.; Liu, H.; Plut, P.; Niu, M.; Huo, R.; Goltzman, D.; Henderson, J.E. Impaired endochondral bone development and osteopenia in Gli2-deficient mice. Exp. Cell Res. 2004, 294, 210–222. [Google Scholar] [CrossRef]
- Weir, E.C.; Philbrick, W.M.; Amling, M.; Neff, L.A.; Baron, R.; Broadus, A.E. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc. Natl. Acad. Sci. USA 1996, 93, 10240–10245. [Google Scholar] [CrossRef] [Green Version]
- Schipani, E.; Lanske, B.; Hunzelman, J.; Luz, A.; Kovacs, C.S.; Lee, K.; Pirro, A.; Kronenberg, H.M.; Juppner, H. Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc. Natl. Acad. Sci. USA 1997, 94, 13689–13694. [Google Scholar] [CrossRef] [Green Version]
- Philbrick, W.M.; Dreyer, B.E.; Nakchbandi, I.A.; Karaplis, A.C. Parathyroid hormone-related protein is required for tooth eruption. Proc. Natl. Acad. Sci. USA 1998, 95, 11846–11851. [Google Scholar] [CrossRef] [Green Version]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [Green Version]
- Mak, K.K.; Chen, M.H.; Day, T.F.; Chuang, P.T.; Yang, Y. Wnt/β-catenin signaling interacts differentially with Ihh signaling in controlling endochondral bone and synovial joint formation. Development 2006, 133, 3695–3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, S.J.; Schipani, E.; St-Jacques, B.; Hunzelman, J.; Kronenberg, H.; McMahon, A.P. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein-dependent and -independent pathways. Development 2000, 127, 543–548. [Google Scholar] [PubMed]
- Chung, U.I.; Lanske, B.; Lee, K.; Li, E.; Kronenberg, H. The parathyroid hormone/parathyroid hormone-related peptide receptor coordinates endochondral bone development by directly controlling chondrocyte differentiation. Proc. Natl. Acad. Sci. USA 1998, 95, 13030–13035. [Google Scholar] [CrossRef] [Green Version]
- Chung, U.I.; Schipani, E.; McMahon, A.P.; Kronenberg, H.M. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J. Clin. Investig. 2001, 107, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Hilton, M.J.; Tu, X.; Long, F. Tamoxifen-inducible gene deletion reveals a distinct cell type associated with trabecular bone, and direct regulation of PTHrP expression and chondrocyte morphology by Ihh in growth region cartilage. Dev. Biol. 2007, 308, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Long, F.; Linsenmayer, T.F. Regulation of growth region cartilage proliferation and differentiation by perichondrium. Development 1998, 125, 1067–1073. [Google Scholar] [PubMed]
- Long, F.; Zhang, X.M.; Karp, S.; Yang, Y.; McMahon, A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 2001, 128, 5099–5108. [Google Scholar]
- Kobayashi, T.; Chung, U.I.; Schipani, E.; Starbuck, M.; Karsenty, G.; Katagiri, T.; Goad, D.L.; Lanske, B.; Kronenberg, H.M. PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development 2002, 129, 2977–2986. [Google Scholar]
- Kobayashi, T.; Soegiarto, D.W.; Yang, Y.; Lanske, B.; Schipani, E.; McMahon, A.P.; Kronenberg, H.M. Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J. Clin. Investig. 2005, 115, 1734–1742. [Google Scholar] [CrossRef]
- Mak, K.K.; Kronenberg, H.M.; Chuang, P.T.; Mackem, S.; Yang, Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 2008, 135, 1947–1956. [Google Scholar] [CrossRef] [Green Version]
- Hilton, M.J.; Tu, X.; Cook, J.; Hu, H.; Long, F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development 2005, 132, 4339–4351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, F.; Chung, U.I.; Ohba, S.; McMahon, J.; Kronenberg, H.M.; McMahon, A.P. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 2004, 131, 1309–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodda, S.J.; McMahon, A.P. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 2006, 133, 3231–3244. [Google Scholar] [CrossRef] [Green Version]
- Koziel, L.; Wuelling, M.; Schneider, S.; Vortkamp, A. Gli3 acts as a repressor downstream of Ihh in regulating two distinct steps of chondrocyte differentiation. Development 2005, 132, 5249–5260. [Google Scholar] [CrossRef] [Green Version]
- Park, H.L.; Bai, C.; Platt, K.A.; Matise, M.P.; Beeghly, A.; Hui, C.C.; Nakashima, M.; Joyner, A.L. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 2000, 127, 1593–1605. [Google Scholar] [PubMed]
- Hojo, H.; Ohba, S.; Yano, F.; Saito, T.; Ikeda, T.; Nakajima, K.; Komiyama, Y.; Nakagata, N.; Suzuki, K.; Takato, T.; et al. Gli1 Protein Participates in Hedgehog-mediated Specification of Osteoblast Lineage during Endochondral Ossification. J. Biol. Chem. 2012, 287, 17860–17869. [Google Scholar] [CrossRef] [Green Version]
- Hojo, H.; Ohba, S.; Taniguchi, K.; Shirai, M.; Yano, F.; Saito, T.; Ikeda, T.; Nakajima, K.; Komiyama, Y.; Nakagata, N.; et al. Hedgehog-Gli Activators Direct Osteo-chondrogenic Function of Bone Morphogenetic Protein toward Osteogenesis in the Perichondrium. J. Biol. Chem. 2013, 288, 9924–9932. [Google Scholar] [CrossRef] [Green Version]
- Shimoyama, A.; Wada, M.; Ikeda, F.; Hata, K.; Matsubara, T.; Nifuji, A.; Noda, M.; Amano, K.; Yamaguchi, A.; Nishimura, R.; et al. Ihh/Gli2 signaling promotes osteoblast differentiation by regulating Runx2 expression and function. Mol. Biol. Cell. 2007, 18, 2411–2418. [Google Scholar] [CrossRef] [Green Version]
- Joeng, K.S.; Long, F.X. The Gli2 transcriptional activator is a crucial effector for Ihh signaling in osteoblast development and cartilage vascularization. Development 2009, 136, 4177–4185. [Google Scholar] [CrossRef] [Green Version]
- Ohba, S.; Kawaguchi, H.; Kugimiya, F.; Ogasawara, T.; Kawamura, N.; Saito, T.; Ikeda, T.; Fujii, K.; Miyajima, T.; Kuramochi, A.; et al. Patched1 haploinsufficiency increases adult bone mass and modulates Gli3 repressor activity. Dev. Cell. 2008, 14, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, X.; Joeng, K.S.; Long, F. Indian hedgehog requires additional effectors besides Runx2 to induce osteoblast differentiation. Dev. Biol. 2012, 362, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milet, C.; Monsoro-Burq, A.H. Neural crest induction at the neural plate border in vertebrates. Dev. Biol. 2012, 366, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Mao, J.; Tenzen, T.; Kottmann, A.H.; McMahon, A.P. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004, 18, 937–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Balczerski, B.; Ciozda, A.; Louie, K.; Oralova, V.; Huysseune, A.; Crump, J.G. Fox proteins are modular competency factors for facial cartilage and tooth specification. Development 2018, 145, dev165498. [Google Scholar] [CrossRef] [Green Version]
- Lenton, K.; James, A.W.; Manu, A.; Brugmann, S.A.; Birker, D.; Nelson, E.R.; Leucht, P.; Helms, J.A.; Longaker, M.T. Indian hedgehog positively regulates calvarial ossification and modulates bone morphogenetic protein signaling. Genesis 2011, 49, 784–796. [Google Scholar] [CrossRef]
- Abzhanov, A.; Rodda, S.J.; McMahon, A.P.; Tabin, C.J. Regulation of skeletogenic differentiation in cranial dermal bone. Development 2007, 134, 3133–3144. [Google Scholar] [CrossRef] [Green Version]
- Amano, K.; Okuzaki, D.; Aikawa, T.; Kogo, M. Indian hedgehog in craniofacial neural crest cells links to skeletal malocclusion by regulating associated cartilage formation and gene expression. FASEB J. 2020, 34, 6791–6807. [Google Scholar] [CrossRef] [Green Version]
- Ashe, A.; Butterfield, N.C.; Town, L.; Courtney, A.D.; Cooper, A.N.; Ferguson, C.; Barry, R.; Olsson, F.; Liem, K.F., Jr.; Parton, R.G.; et al. Mutations in mouse Ift144 model the craniofacial, limb and rib defects in skeletal ciliopathies. Hum. Mol. Genet. 2012, 21, 1808–1823. [Google Scholar] [CrossRef] [Green Version]
- Koyama, E.; Young, B.; Nagayama, M.; Shibukawa, Y.; Enomoto-Iwamoto, M.; Iwamoto, M.; Maeda, Y.; Lanske, B.; Song, B.; Serra, R.; et al. Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development 2007, 134, 2159–2169. [Google Scholar] [CrossRef] [Green Version]
- Brugmann, S.A.; Allen, N.C.; James, A.W.; Mekonnen, Z.; Madan, E.; Helms, J.A. A primary cilia-dependent etiology for midline facial disorders. Hum. Mol. Genet. 2010, 19, 1577–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, Y.; Nakamura, E.; Nguyen, M.T.; Suva, L.J.; Swain, F.L.; Razzaque, M.S.; Mackem, S.; Lanske, B. Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc. Natl. Acad. Sci. USA 2007, 104, 6382–6387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, K.; Densmore, M.J.; Lanske, B. Conditional deletion of Indian Hedgehog in limb mesenchyme results in complete loss of growth plate formation but allows mature osteoblast differentiation. J. Bone Miner. Res. 2015, 30, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Schipani, E.; Densmore, M.J.; Lanske, B. Partial rescue of postnatal growth plate abnormalities in Ihh mutants by expression of a constitutively active PTH/PTHrP receptor. Bone 2010, 46, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Hirai, T.; Chagin, A.S.; Kobayashi, T.; Mackem, S.; Kronenberg, H.M. Parathyroid hormone/parathyroid hormone-related protein receptor signaling is required for maintenance of the growth plate in postnatal life. Proc. Natl. Acad. Sci. USA 2011, 108, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Mak, K.K.; Bi, Y.; Wan, C.; Chuang, P.T.; Clemens, T.; Young, M.; Yang, Y. Hedgehog signaling in mature osteoblasts regulates bone formation and resorption by controlling PTHrP and RANKL expression. Dev. Cell. 2008, 14, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Kitaura, Y.; Hojo, H.; Komiyama, Y.; Takato, T.; Chung, U.I.; Ohba, S. Gli1 haploinsufficiency leads to decreased bone mass with an uncoupling of bone metabolism in adult mice. PLoS ONE 2014, 9, e109597. [Google Scholar] [CrossRef]
- Kohara, Y.; Haraguchi, R.; Kitazawa, R.; Imai, Y.; Kitazawa, S. Hedgehog Inhibitors suppress osteoclastogenesis in in vitro cultures, and deletion of smo in macrophage/osteoclast lineage prevents age-related bone loss. Int. J. Mol. Sci. 2020, 21, 2745. [Google Scholar] [CrossRef]
- Wei, F.; Zhou, J.; Wei, X.; Zhang, J.; Fleming, B.C.; Terek, R.; Pei, M.; Chen, Q.; Liu, T.; Wei, L.; et al. Activation of Indian hedgehog promotes chondrocyte hypertrophy and upregulation of MMP-13 in human osteoarthritic cartilage. Osteoarthr. Cartil. 2012, 20, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Chen, Q.; Lanske, B.; Fleming, B.C.; Terek, R.; Wei, X.; Zhang, G.; Wang, S.; Li, K.; Wei, L.; et al. Disrupting the Indian hedgehog signaling pathway in vivo attenuates surgically induced osteoarthritis progression in Col2a1-CreERT2; Ihhfl/fl mice. Arthritis Res. Ther. 2014, 16, R11. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.C.; Seeto, B.L.; Bartoszko, J.M.; Khoury, M.A.; Whetstone, H.; Ho, L.; Hsu, C.; Ali, S.A.; Alman, B.A. Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat. Med. 2009, 15, 1421–1425. [Google Scholar] [CrossRef]
- Ransom, R.C.; Carter, A.C.; Salhotra, A.; Leavitt, T.; Marecic, O.; Murphy, M.P.; Lopez, M.L.; Wei, Y.; Marshall, C.D.; Shen, E.Z.; et al. Mechanoresponsive stem cells acquire neural crest fate in jaw regeneration. Nature 2018, 563, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.K.F.; Gulati, G.S.; Sinha, R.; Tompkins, J.V.; Lopez, M.; Carter, A.C.; Ransom, R.C.; Reinisch, A.; Wearda, T.; Murphy, M.; et al. Identification of the human skeletal stem cell. Cell 2018, 175, 43–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.K.; Seo, E.Y.; Chen, J.Y.; Lo, D.; McArdle, A.; Sinha, R.; Tevlin, R.; Seita, J.; Vincent-Tompkins, J.; Wearda, T.; et al. Identification and specification of the mouse skeletal stem cell. Cell 2015, 160, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; He, G.; Lee, W.C.; McKenzie, J.A.; Silva, M.J.; Long, F. Gli1 identifies osteogenic progenitors for bone formation and fracture repair. Nat. Commun. 2017, 8, 2043. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, R.; Kitazawa, R.; Imai, Y.; Kitazawa, S. Growth plate-derived hedgehog-signal-responsive cells provide skeletal tissue components in growing bone. Histochem. Cell Biol. 2018, 149, 365–373. [Google Scholar] [CrossRef]
- Onodera, S.; Saito, A.; Hojo, H.; Nakamura, T.; Zujur, D.; Watanabe, K.; Morita, N.; Hasegawa, D.; Masaki, H.; Nakauchi, H.; et al. Hedgehog activation regulates human osteoblastogenesis. Stem. Cell Rep. 2020, 15, 125–139. [Google Scholar] [CrossRef]
- Lettre, G.; Jackson, A.U.; Gieger, C.; Schumacher, F.R.; Berndt, S.I.; Sanna, S.; Eyheramendy, S.; Voight, B.F.; Butler, J.L.; Guiducci, C.; et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 2008, 40, 584–591. [Google Scholar] [CrossRef]
- Weedon, M.N.; Lango, H.; Lindgren, C.M.; Wallace, C.; Evans, D.M.; Mangino, M.; Freathy, R.M.; Perry, J.R.; Stevens, S.; Hall, A.S.; et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 2008, 40, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Guo, J.; She, C.; Shu, A.; Yang, M.; Tan, Z.; Yang, X.; Guo, S.; Feng, G.; He, L. Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat. Genet. 2001, 28, 386–388. [Google Scholar] [CrossRef]
- Gao, B.; He, L. Answering a century old riddle: Brachydactyly type A1. Cell Res. 2004, 14, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Hu, J.; Stricker, S.; Cheung, M.; Ma, G.; Law, K.F.; Witte, F.; Briscoe, J.; Mundlos, S.; He, L.; et al. A mutation in Ihh that causes digit abnormalities alters its signalling capacity and range. Nature 2009, 458, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A.; Gessler, M.; Grzeschik, K.H. GLI3 zinc-finger gene interrupted by translocations in Greig syndrome families. Nature 1991, 352, 539–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Graham, J.M., Jr.; Olney, A.H.; Biesecker, L.G. GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nat. Genet. 1997, 15, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishna, U.; Wild, A.; Grzeschik, K.H.; Antonarakis, S.E. Mutation in GLI3 in postaxial polydactyly type A. Nat. Genet. 1997, 17, 269–271. [Google Scholar] [CrossRef]
- Gorlin, R.J. Nevoid basal-cell carcinoma syndrome. Medicine 1987, 66, 98–113. [Google Scholar] [CrossRef]
- Shefer, S.; Salen, G.; Batta, A.K.; Honda, A.; Tint, G.S.; Irons, M.; Elias, E.R.; Chen, T.C.; Holick, M.F. Markedly inhibited 7-dehydrocholesterol-delta 7-reductase activity in liver microsomes from Smith-Lemli-Opitz homozygotes. J. Clin. Investig. 1995, 96, 1779–1785. [Google Scholar] [CrossRef]
- Blassberg, R.; Macrae, J.I.; Briscoe, J.; Jacob, J. Reduced cholesterol levels impair Smoothened activation in Smith-Lemli-Opitz syndrome. Hum. Mol. Genet. 2016, 25, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, F.S.; Hahn, G.V.; Zasloff, M.A. Heterotopic ossification: Two rare forms and what they can teach us. J. Am. Acad. Orthop. Surg. 1994, 2, 288–296. [Google Scholar] [CrossRef]
- Regard, J.B.; Malhotra, D.; Gvozdenovic-Jeremic, J.; Josey, M.; Chen, M.; Weinstein, L.S.; Lu, J.; Shore, E.M.; Kaplan, F.S.; Yang, Y.; et al. Activation of Hedgehog signaling by loss of GNAS causes heterotopic ossification. Nat. Med. 2013, 19, 1505–1512. [Google Scholar] [CrossRef] [Green Version]
- Will, A.J.; Cova, G.; Osterwalder, M.; Chan, W.L.; Wittler, L.; Brieske, N.; Heinrich, V.; de Villartay, J.P.; Vingron, M.; Klopocki, E.; et al. Composition and dosage of a multipartite enhancer cluster control developmental expression of Ihh (Indian hedgehog). Nat. Genet. 2017, 49, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The expression domains of marker genes and the location of the different cell types in the growth plate of endochondral bones.
Figure 1.
The expression domains of marker genes and the location of the different cell types in the growth plate of endochondral bones.
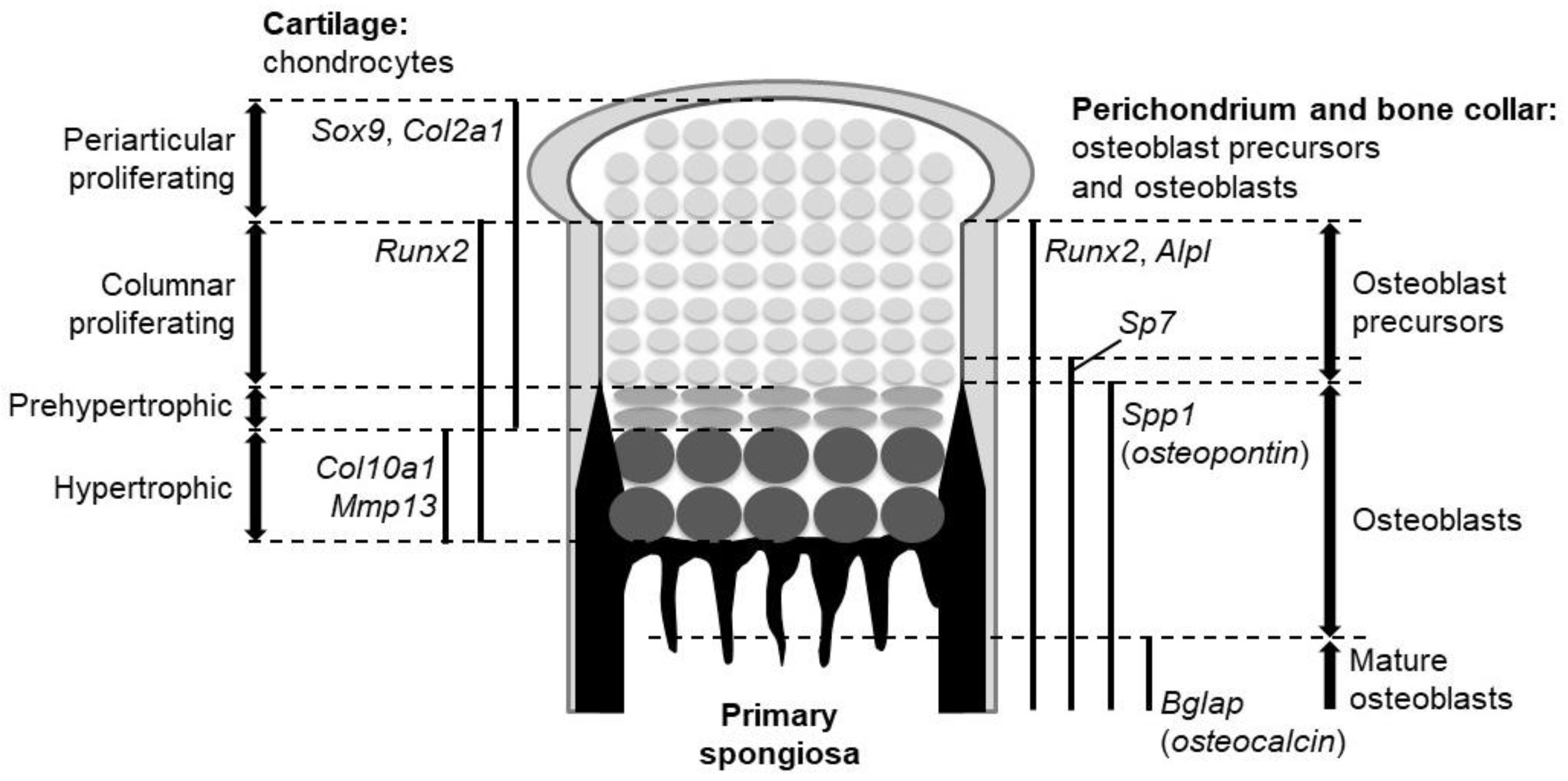
Figure 2.
Hh signaling transduction. (Left) In the absence of Hh ligands, Ptch inhibits intrinsic Smo activities. The full-length Gli factor (Gli activators: GliA), exists in complex with Suppressor of Fused (SuFu). The full-length GliA is phosphorylated by PKA, CKI, and GSK3β at the base of the cilia. The phosphorylation causes proteolytic cleavage of GliA to generate the transcriptional repressor form of Gli (Gli repressors: GliR). GliR represses transcription of target genes in the nucleus, since it possesses the transcriptional repressor domain and the DNA-binding domain, but lacks the transactivation domain. (Right) Binding of Hh ligands relieves the repressive effect of Ptch on Smo. Smo is phosphorylated by G protein-coupled receptor kinase 2 (Gpcrk2) and CKI. Smo also interacts with Kif3a and β-arrestin (Arrb) and accumulates in the cilia in association with Ellis-van Creveld syndrome protein (EVC) and EVC2 (dashed arrows). The levels of GliA and SuFu are increased in the cilia, leading to the dissociation of the GliA-SuFu complex in the cilia. GliA then escapes from proteolytic cleavage and activates transcription of target genes in the nucleus.
Figure 2.
Hh signaling transduction. (Left) In the absence of Hh ligands, Ptch inhibits intrinsic Smo activities. The full-length Gli factor (Gli activators: GliA), exists in complex with Suppressor of Fused (SuFu). The full-length GliA is phosphorylated by PKA, CKI, and GSK3β at the base of the cilia. The phosphorylation causes proteolytic cleavage of GliA to generate the transcriptional repressor form of Gli (Gli repressors: GliR). GliR represses transcription of target genes in the nucleus, since it possesses the transcriptional repressor domain and the DNA-binding domain, but lacks the transactivation domain. (Right) Binding of Hh ligands relieves the repressive effect of Ptch on Smo. Smo is phosphorylated by G protein-coupled receptor kinase 2 (Gpcrk2) and CKI. Smo also interacts with Kif3a and β-arrestin (Arrb) and accumulates in the cilia in association with Ellis-van Creveld syndrome protein (EVC) and EVC2 (dashed arrows). The levels of GliA and SuFu are increased in the cilia, leading to the dissociation of the GliA-SuFu complex in the cilia. GliA then escapes from proteolytic cleavage and activates transcription of target genes in the nucleus.
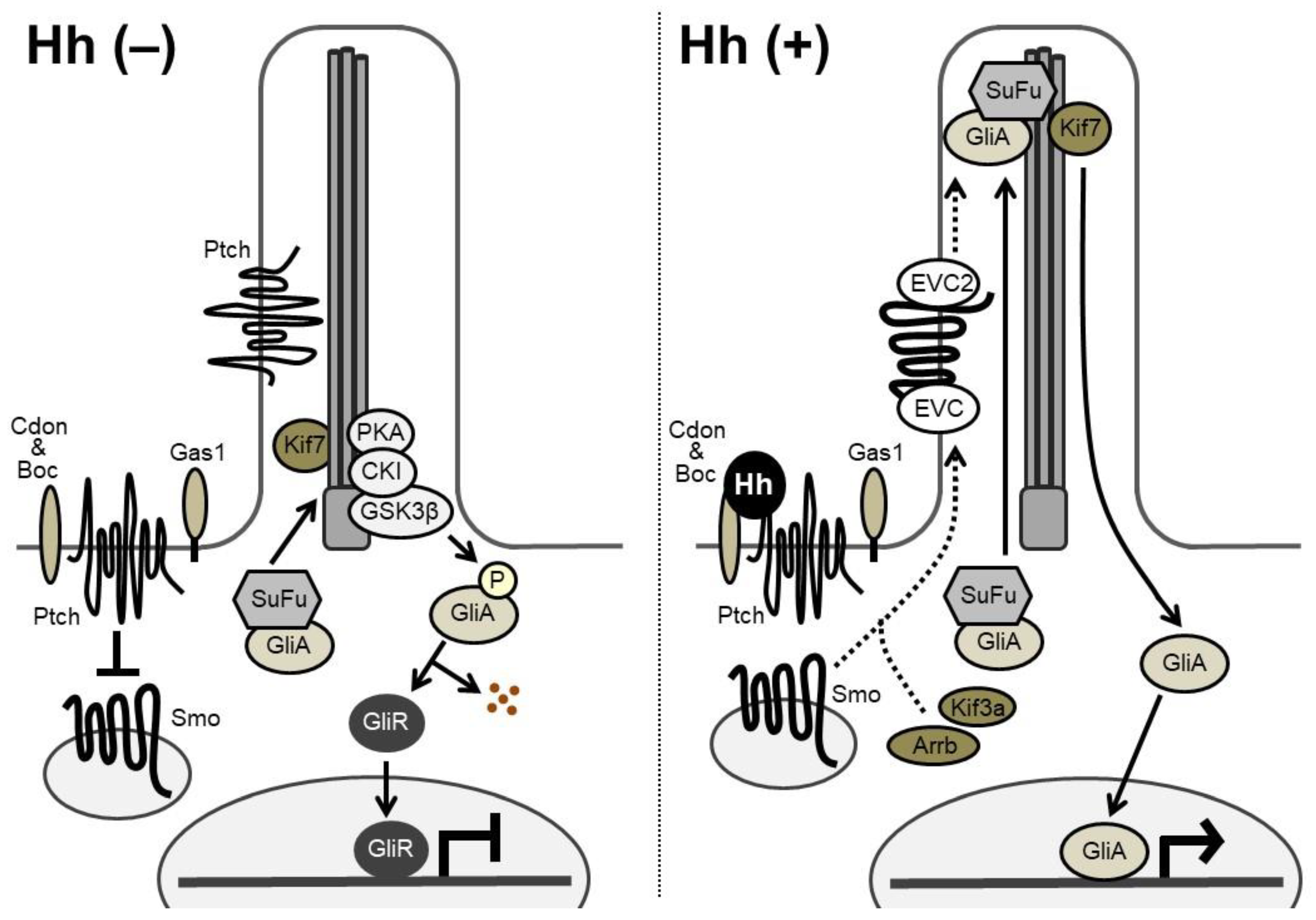
Figure 3.
Roles of Indian hedgehog (Ihh) in endochondral ossification. Mouse genetic studies support three major roles of Ihh in endochondral ossification: (a) Chondrocyte differentiation via the negative feedback loop with parathyroid hormone-related protein (PTHrP), (b) chondrocyte proliferation, and (c) specification of bone-forming osteoblasts. Ihh acts on chondrocyte proliferation and osteoblast specification in a direct manner.
Figure 3.
Roles of Indian hedgehog (Ihh) in endochondral ossification. Mouse genetic studies support three major roles of Ihh in endochondral ossification: (a) Chondrocyte differentiation via the negative feedback loop with parathyroid hormone-related protein (PTHrP), (b) chondrocyte proliferation, and (c) specification of bone-forming osteoblasts. Ihh acts on chondrocyte proliferation and osteoblast specification in a direct manner.
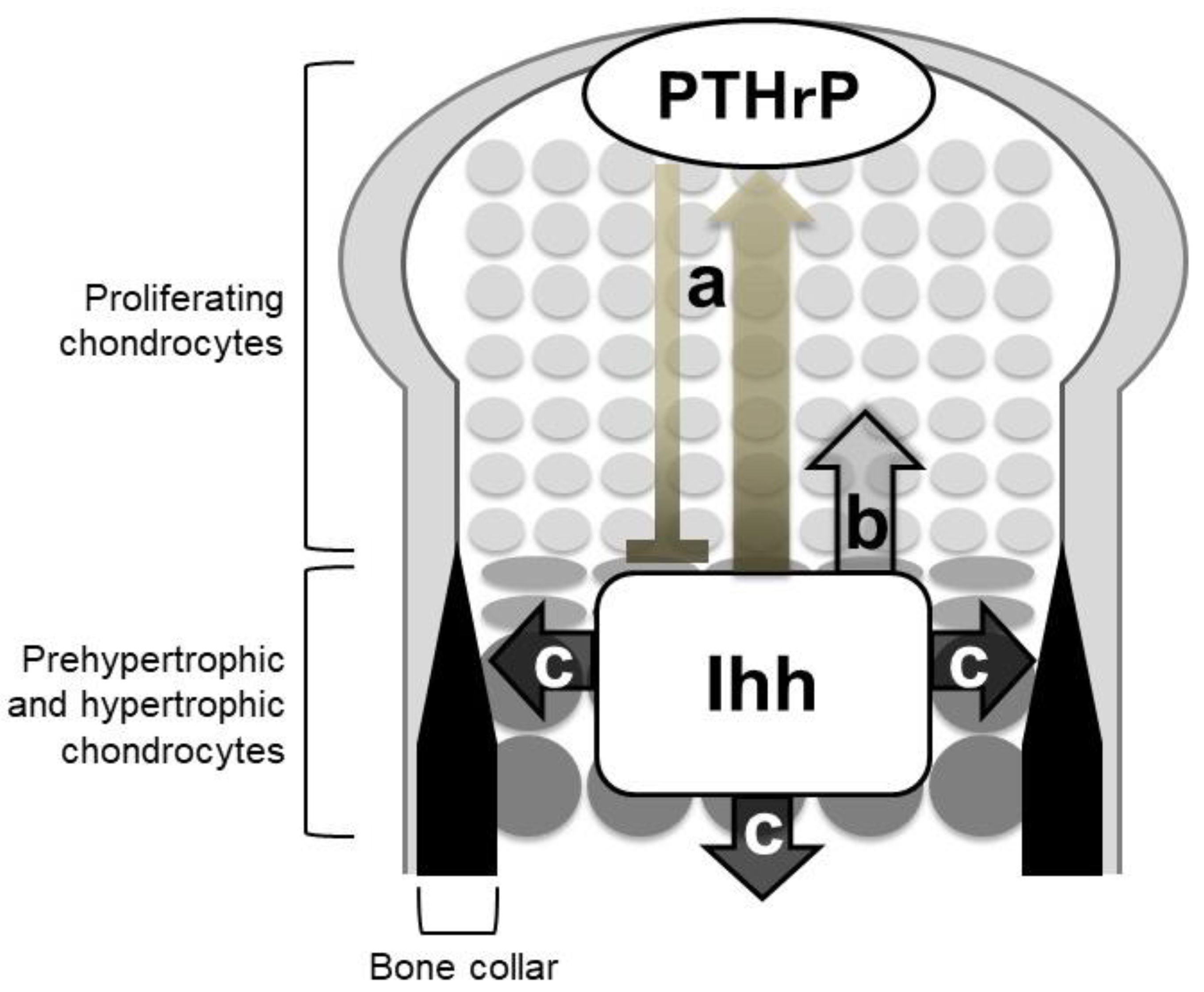
Figure 4.
Regulation of growth-plate length by the Ihh-PTHrP negative feedback loop. (a) Shortening of the length of the growth plate (double-headed arrow on the left of 1) increases PTHrP expression in periarticular proliferating chondrocytes possibly due to the increase in the amount of Ihh reaching the PTHrP-producing population (wide arrows in 1). This causes greater suppression of hypertrophy (wide lines in 2) compared to an equilibrium state. The growth plate thereby elongates to restore the equilibrium state (head-to-head arrows on the left of 2). (b) Elongation of the growth plate (double-headed arrow on the right of 1) in turn decreases PTHrP expression due to the decrease in the amount of Ihh reaching the PTHrP-producing population (thin arrow in 1). PTHrP activity on hypertrophy is attenuated (thin line in 2), leading to shortening of the length of the growth plate (head-to-head arrows on the right of 2).
Figure 4.
Regulation of growth-plate length by the Ihh-PTHrP negative feedback loop. (a) Shortening of the length of the growth plate (double-headed arrow on the left of 1) increases PTHrP expression in periarticular proliferating chondrocytes possibly due to the increase in the amount of Ihh reaching the PTHrP-producing population (wide arrows in 1). This causes greater suppression of hypertrophy (wide lines in 2) compared to an equilibrium state. The growth plate thereby elongates to restore the equilibrium state (head-to-head arrows on the left of 2). (b) Elongation of the growth plate (double-headed arrow on the right of 1) in turn decreases PTHrP expression due to the decrease in the amount of Ihh reaching the PTHrP-producing population (thin arrow in 1). PTHrP activity on hypertrophy is attenuated (thin line in 2), leading to shortening of the length of the growth plate (head-to-head arrows on the right of 2).

© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ohba, S. Hedgehog Signaling in Skeletal Development: Roles of Indian Hedgehog and the Mode of Its Action. Int. J. Mol. Sci. 2020, 21, 6665. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186665
AMA Style
Ohba S. Hedgehog Signaling in Skeletal Development: Roles of Indian Hedgehog and the Mode of Its Action. International Journal of Molecular Sciences. 2020; 21(18):6665. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186665
Chicago/Turabian StyleOhba, Shinsuke. 2020. "Hedgehog Signaling in Skeletal Development: Roles of Indian Hedgehog and the Mode of Its Action" International Journal of Molecular Sciences 21, no. 18: 6665. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186665
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.