MicroRNAs as Biomarkers and Therapeutic Targets in Inflammation- and Ischemia-Reperfusion-Related Acute Renal Injury


Abstract
:1. Introduction
1.1. Definitions of AKI
1.2. Epidemiology of AKI
1.3. Causes of AKI
1.3.1. AKI Induced by Ischemia/Reperfusion Injury
1.3.2. AKI Induced by Sepsis
1.3.3. Contrast-Induced Nephropathy
2. MicroRNA Biogenesis and Function
2.1. The Canonical Pathway of miRNA Biogenesis
2.2. The Noncanonical Pathways of miRNA Biogenesis
2.2.1. Drosha/DGCR8-Independent Pathway
2.2.2. Dicer-Independent Pathway
3. Extracellular microRNAs in Biological Fluids
3.1. Extracellular microRNA Transport via Extracellular Vesicles
3.2. Extracellular microRNA Transport via RNA-Binding Proteins
4. miRNAs in Acute Kidney Injury
4.1. miR-687
4.2. miR-24
4.3. miR-494
4.4. miR-21
5. miRNAs as Novel Biomarkers for AKI
5.1. Circulating miRNAs
5.2. Urinary miRNAs
6. miRNAs as a Potential Therapeutic Strategy in AKI
6.1. miRNA Targeted Therapy
6.2. Stem Cell-Derived miRNAs (Stem Cell Secretome)
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AGO | argonaute |
| AGO2 | protein argonaute-2 |
| AKI | acute kidney injury |
| ARF | acute renal failure |
| ASO | antisense oligonucleotide |
| ATF3 | activating transcription factor 3 |
| CIN | Contrast-induced nephropathy |
| CKD | chronic kidney disease |
| DGCR8 | DiGeorge syndrome critical region 8 |
| ECFC | endothelial colony-forming cell |
| EPC | endothelial progenitor cell |
| ESCRT | endosomal sorting complex required for transport |
| EVs | extracellular vesicles |
| H/R | hypoxia/reoxygenation |
| HDL | high-density lipoprotein |
| HIF-1 | hypoxia-inducible factor-1 |
| hnRNPA2B1 | heterogeneous nuclear ribonucleoprotein A2B1 |
| HO-1 | heme oxygenase-1 |
| I/R | ischemia/reperfusion |
| ICU | intensive care unit |
| IL-1 | interleukin-1 |
| IL-6 | interleukin-6 |
| Kim-1 | kidney injury molecule-1 |
| m7G | 7-methylguanosine |
| MAPK | mitogen-activated protein kinase |
| MBs | multivesicular bodies |
| MCP-1 | monocyte chemoattractant protein-1 |
| miRNAs | microRNAs |
| miSFITs | miRNA silencing-mediated fine-tuners |
| MSCs | mesenchymal stem cells |
| MTP18 | mitochondrial protein 18 kDa |
| NGAL | neutrophil gelatinase-associated lipocalin |
| NPM1 | nucleophosmin 1 |
| nSMase2 | neutral sphingomyelinase 2 |
| PDCD4 | programmed cell death protein 4 |
| PT | proximal tubule |
| PTEN | phosphatase and tensin homolog |
| RanGTP | GTP-binding RAs-related nuclear protein |
| RISC | RNA-induced silencing complex |
| S1PR1 | sphingosine-1-phosphate receptor 1 |
| shRNA | short hairpin RNA |
| TNF-α | tumor necrosis factor-alpha |
| UTR | untranslated region |
References
- Lewington, A.J.; Cerda, J.; Mehta, R.L. Raising awareness of acute kidney injury: A global perspective of a silent killer. Kidney Int. 2013, 84, 457–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Dong, G.; Liang, X.; Dong, Z. Epigenetic regulation in AKI and kidney repair: Mechanisms and therapeutic implications. Nat. Rev. Nephrol. 2019, 15, 220–239. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [PubMed] [Green Version]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L.; Acute Kidney Injury Advisory Group of the American Society of Nephrology. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [Green Version]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Dong, Z.; Harris, R.; Murray, P.; Parikh, S.M.; Rosner, M.H.; Kellum, J.A.; Ronco, C.; Acute Dialysis Quality Initiative XIII Working Group. Cellular and Molecular Mechanisms of AKI. J. Am. Soc. Nephrol. 2016, 27, 1288–1299. [Google Scholar] [CrossRef]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Zarbock, A.; Gomez, H.; Kellum, J.A. Sepsis-induced acute kidney injury revisited: Pathophysiology, prevention and future therapies. Curr. Opin. Crit. Care 2014, 20, 588–595. [Google Scholar] [CrossRef]
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gomez, H.; Kellum, J.A. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019, 96, 1083–1099. [Google Scholar] [CrossRef]
- Heyman, S.N.; Rosen, S.; Rosenberger, C. Renal parenchymal hypoxia, hypoxia adaptation, and the pathogenesis of radiocontrast nephropathy. Clin. J. Am. Soc. Nephrol. 2008, 3, 288–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geenen, R.W.; Kingma, H.J.; van der Molen, A.J. Contrast-induced nephropathy: Pharmacology, pathophysiology and prevention. Insights Imaging 2013, 4, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.U.; Oner, M.G.; Meuwissen, R.L.; Genc, S. The role of microRNAs in human diseases. Methods Mol. Biol. 2014, 1107, 33–50. [Google Scholar]
- Denli, A.M.; Tops, B.B.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef]
- Forman, J.J.; Legesse-Miller, A.; Coller, H.A. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc. Natl. Acad. Sci. USA 2008, 105, 14879–14884. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [Green Version]
- Okada, C.; Yamashita, E.; Lee, S.J.; Shibata, S.; Katahira, J.; Nakagawa, A.; Yoneda, Y.; Tsukihara, T. A high-resolution structure of the pre-microRNA nuclear export machinery. Science 2009, 326, 1275–1279. [Google Scholar] [CrossRef]
- Zhang, H.; Kolb, F.A.; Jaskiewicz, L.; Westhof, E.; Filipowicz, W. Single processing center models for human Dicer and bacterial RNase III. Cell 2004, 118, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ipsaro, J.J.; Joshua-Tor, L. From guide to target: Molecular insights into eukaryotic RNA-interference machinery. Nat. Struct. Mol. Biol. 2015, 22, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, S.L.; Li, S.; Abadal, G.X.; Pauley, B.A.; Fritzler, M.J.; Chan, E.K. The C-terminal half of human Ago2 binds to multiple GW-rich regions of GW182 and requires GW182 to mediate silencing. RNA 2009, 15, 804–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Li, M.; Vilborg, A.; Lee, N.; Shu, M.D.; Yartseva, V.; Sestan, N.; Steitz, J.A. Mammalian 5′-capped microRNA precursors that generate a single microRNA. Cell 2013, 155, 1568–1580. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.S.; Maurin, T.; Robine, N.; Rasmussen, K.D.; Jeffrey, K.L.; Chandwani, R.; Papapetrou, E.P.; Sadelain, M.; O’Carroll, D.; Lai, E.C. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 15163–15168. [Google Scholar] [CrossRef] [Green Version]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.; Hannon, G.J. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 2010, 465, 584–589. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Hanke, M.; Hoefig, K.; Merz, H.; Feller, A.C.; Kausch, I.; Jocham, D.; Warnecke, J.M.; Sczakiel, G. A robust methodology to study urine microRNA as tumor marker: microRNA-126 and microRNA-182 are related to urinary bladder cancer. Urol. Oncol. 2010, 28, 655–661. [Google Scholar] [CrossRef]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Park, N.J.; Zhou, H.; Elashoff, D.; Henson, B.S.; Kastratovic, D.A.; Abemayor, E.; Wong, D.T. Salivary microRNA: Discovery, characterization, and clinical utility for oral cancer detection. Clin. Cancer Res. 2009, 15, 5473–5477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Li, M.; Wang, X.; Li, Q.; Wang, T.; Zhu, Q.; Zhou, X.; Wang, X.; Gao, X.; Li, X. Immune-related microRNAs are abundant in breast milk exosomes. Int. J. Biol. Sci. 2012, 8, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.J.; Jeong, P.; Kim, W.T.; Kim, T.H.; Lee, Y.S.; Song, P.H.; Choi, Y.H.; Kim, I.Y.; Moon, S.K.; Kim, W.J. Cell-free microRNAs in urine as diagnostic and prognostic biomarkers of bladder cancer. Int. J. Oncol. 2012, 41, 1871–1878. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H. ESCRT complexes and the biogenesis of multivesicular bodies. Curr. Opin. Cell Biol. 2008, 20, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Mittelbrunn, M.; Gutierrez-Vazquez, C.; Villarroya-Beltri, C.; Gonzalez, S.; Sanchez-Cabo, F.; Gonzalez, M.A.; Bernad, A.; Sanchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, A.J.; Hoshino, D.; Hong, N.H.; Cha, D.J.; Franklin, J.L.; Coffey, R.J.; Patton, J.G.; Weaver, A.M. KRAS-MEK Signaling Controls Ago2 Sorting into Exosomes. Cell Rep. 2016, 15, 978–987. [Google Scholar] [CrossRef] [Green Version]
- Villarroya-Beltri, C.; Gutierrez-Vazquez, C.; Sanchez-Cabo, F.; Perez-Hernandez, D.; Vazquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sanchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shurtleff, M.J.; Temoche-Diaz, M.M.; Karfilis, K.V.; Ri, S.; Schekman, R. Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. eLife 2016, 5, e19276. [Google Scholar] [CrossRef] [PubMed]
- Noferesti, S.S.; Sohel, M.M.; Hoelker, M.; Salilew-Wondim, D.; Tholen, E.; Looft, C.; Rings, F.; Neuhoff, C.; Schellander, K.; Tesfaye, D. Controlled ovarian hyperstimulation induced changes in the expression of circulatory miRNA in bovine follicular fluid and blood plasma. J. Ovarian Res. 2015, 8, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohel, M.M.; Hoelker, M.; Noferesti, S.S.; Salilew-Wondim, D.; Tholen, E.; Looft, C.; Rings, F.; Uddin, M.J.; Spencer, T.E.; Schellander, K.; et al. Exosomal and Non-Exosomal Transport of Extra-Cellular microRNAs in Follicular Fluid: Implications for Bovine Oocyte Developmental Competence. PLoS ONE 2013, 8, e78505. [Google Scholar] [CrossRef] [Green Version]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhang, S.; Weber, J.; Baxter, D.; Galas, D.J. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010, 38, 7248–7259. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Bhatt, K.; He, H.Z.; Mi, Q.S.; Haase, V.H.; Dong, Z. Targeted deletion of Dicer from proximal tubules protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2010, 21, 756–761. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, K.; Wei, Q.; Pabla, N.; Dong, G.; Mi, Q.S.; Liang, M.; Mei, C.; Dong, Z. MicroRNA-687 Induced by Hypoxia-Inducible Factor-1 Targets Phosphatase and Tensin Homolog in Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2015, 26, 1588–1596. [Google Scholar] [CrossRef]
- Lorenzen, J.M.; Kaucsar, T.; Schauerte, C.; Schmitt, R.; Rong, S.; Hubner, A.; Scherf, K.; Fiedler, J.; Martino, F.; Kumarswamy, R.; et al. MicroRNA-24 antagonism prevents renal ischemia reperfusion injury. J. Am. Soc. Nephrol. 2014, 25, 2717–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Olszanecki, R.; Rezzani, R.; Omura, S.; Stec, D.E.; Rodella, L.; Botros, F.T.; Goodman, A.I.; Drummond, G.; Abraham, N.G. Genetic suppression of HO-1 exacerbates renal damage: Reversed by an increase in the antiapoptotic signaling pathway. Am. J. Physiol. Renal. Physiol. 2007, 292, F148–F157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajwa, A.; Jo, S.K.; Ye, H.; Huang, L.; Dondeti, K.R.; Rosin, D.L.; Haase, V.H.; Macdonald, T.L.; Lynch, K.R.; Okusa, M.D. Activation of sphingosine-1-phosphate 1 receptor in the proximal tubule protects against ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2010, 21, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.F.; Chen, H.H.; Lai, P.F.; Cheng, C.F.; Huang, Y.T.; Lee, Y.C.; Chen, T.W.; Lin, H. MicroRNA-494 reduces ATF3 expression and promotes AKI. J. Am. Soc. Nephrol. 2012, 23, 2012–2023. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.H.; Lai, P.F.; Lan, Y.F.; Cheng, C.F.; Zhong, W.B.; Lin, Y.F.; Chen, T.W.; Lin, H. Exosomal ATF3 RNA attenuates pro-inflammatory gene MCP-1 transcription in renal ischemia-reperfusion. J. Cell Physiol. 2014, 229, 1202–1211. [Google Scholar] [CrossRef]
- Yoshida, T.; Sugiura, H.; Mitobe, M.; Tsuchiya, K.; Shirota, S.; Nishimura, S.; Shiohira, S.; Ito, H.; Nobori, K.; Gullans, S.R.; et al. ATF3 protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2008, 19, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Li, H.F.; Cheng, C.F.; Liao, W.J.; Lin, H.; Yang, R.B. ATF3-mediated epigenetic regulation protects against acute kidney injury. J. Am. Soc. Nephrol. 2010, 21, 1003–1013. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.F.; Jing, Y.; Hao, J.; Frankfort, N.C.; Zhou, X.; Shen, B.; Liu, X.; Wang, L.; Li, R. MicroRNA-21 in the pathogenesis of acute kidney injury. Protein Cell 2013, 4, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Godwin, J.G.; Ge, X.; Stephan, K.; Jurisch, A.; Tullius, S.G.; Iacomini, J. Identification of a microRNA signature of renal ischemia reperfusion injury. Proc. Natl. Acad. Sci. USA 2010, 107, 14339–14344. [Google Scholar] [CrossRef] [Green Version]
- Jia, P.; Teng, J.; Zou, J.; Fang, Y.; Wu, X.; Liang, M.; Ding, X. Xenon Protects Against Septic Acute Kidney Injury via miR-21 Target Signaling Pathway. Crit. Care Med. 2015, 43, e250–e259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, P.; Pan, T.; Xu, S.; Fang, Y.; Song, N.; Guo, M.; Liang, Y.; Xu, X.; Ding, X. Depletion of miR-21 in dendritic cells aggravates renal ischemia-reperfusion injury. FASEB J. 2020, 34, 11729–11740. [Google Scholar] [CrossRef]
- Du, J.; Cao, X.; Zou, L.; Chen, Y.; Guo, J.; Chen, Z.; Hu, S.; Zheng, Z. MicroRNA-21 and risk of severe acute kidney injury and poor outcomes after adult cardiac surgery. PLoS ONE 2013, 8, e63390. [Google Scholar] [CrossRef] [PubMed]
- Arvin, P.; Samimagham, H.R.; Montazerghaem, H.; Khayatian, M.; Mahboobi, H.; Ghadiri Soufi, F. Early detection of cardiac surgeryassociated acute kidney injury by microRNA-21. Bratisl. Lek. Listy 2017, 118, 626–631. [Google Scholar] [PubMed]
- Kang, Z.; Li, Z.; Huang, P.; Luo, J.; Liu, P.; Wang, Y.; Xia, T.; Zhou, Y. Remote ischemic preconditioning upregulates microRNA-21 to protect the kidney in children with congenital heart disease undergoing cardiopulmonary bypass. Pediatr. Nephrol. 2018, 33, 911–919. [Google Scholar] [CrossRef]
- Lorenzen, J.M.; Kielstein, J.T.; Hafer, C.; Gupta, S.K.; Kumpers, P.; Faulhaber-Walter, R.; Haller, H.; Fliser, D.; Thum, T. Circulating miR-210 predicts survival in critically ill patients with acute kidney injury. Clin. J. Am. Soc. Nephrol. 2011, 6, 1540–1546. [Google Scholar] [CrossRef] [Green Version]
- Aguado-Fraile, E.; Ramos, E.; Conde, E.; Rodriguez, M.; Martin-Gomez, L.; Lietor, A.; Candela, A.; Ponte, B.; Liano, F.; Garcia-Bermejo, M.L. A Pilot Study Identifying a Set of microRNAs As Precise Diagnostic Biomarkers of Acute Kidney Injury. PLoS ONE 2015, 10, e0127175. [Google Scholar] [CrossRef]
- Wu, R.; Wu, Y.; Yang, L.; Deng, Y.; Chen, D. Value of serum level of microRNA-494 in predicting prognosis of acute renal injury after cardiac surgery in children. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2019, 31, 1469–1473. [Google Scholar]
- Lin, Y.; Ding, Y.; Song, S.; Li, M.; Wang, T.; Guo, F. Expression patterns and prognostic value of miR-210, miR-494, and miR-205 in middle-aged and old patients with sepsis-induced acute kidney injury. Bosn. J. Basic Med. Sci. 2019, 19, 249–256. [Google Scholar] [CrossRef]
- Gutierrez-Escolano, A.; Santacruz-Vazquez, E.; Gomez-Perez, F. Dysregulated microRNAs involved in contrast-induced acute kidney injury in rat and human. Ren. Fail 2015, 37, 1498–1506. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.Q.; Zhang, T.; Ding, D.; Zhang, W.F.; Wang, X.L.; Sun, Z.; Hu, L.H.; Qin, S.Y.; Shen, L.H.; He, B. Circulating MicroRNA-188, -30a, and -30e as Early Biomarkers for Contrast-Induced Acute Kidney Injury. J. Am. Heart Assoc. 2016, 5, e004138. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, K.; Saikumar, J.; Bijol, V.; Koyner, J.L.; Qian, J.; Betensky, R.A.; Waikar, S.S.; Vaidya, V.S. Human miRNome profiling identifies microRNAs differentially present in the urine after kidney injury. Clin. Chem. 2013, 59, 1742–1752. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.F.; Wen, D.; Zhao, Q.; Shen, P.Y.; Shi, H.; Zhao, Q.; Chen, Y.X.; Zhang, W. Urinary MicroRNA-30c-5p and MicroRNA-192-5p as potential biomarkers of ischemia-reperfusion-induced kidney injury. Exp. Biol. Med. 2017, 242, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saikumar, J.; Hoffmann, D.; Kim, T.M.; Gonzalez, V.R.; Zhang, Q.; Goering, P.L.; Brown, R.P.; Bijol, V.; Park, P.J.; Waikar, S.S.; et al. Expression, circulation, and excretion profile of microRNA-21, -155, and -18a following acute kidney injury. Toxicol. Sci. 2012, 129, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.Y.; Zhang, H.; Wu, W.; Wu, Y.Y. Value of serum miR-21-3p in predicting acute kidney injury in children with sepsis. Zhongguo Dang Dai Er Ke Za Zhi 2020, 22, 269–273. [Google Scholar]
- Zhang, L.; Xu, Y.; Xue, S.; Wang, X.; Dai, H.; Qian, J.; Ni, Z.; Yan, Y. Implications of dynamic changes in miR-192 expression in ischemic acute kidney injury. Int. Urol. Nephrol. 2017, 49, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.H.; Lan, Y.F.; Li, H.F.; Cheng, C.F.; Lai, P.F.; Li, W.H.; Lin, H. Urinary miR-16 transactivated by C/EBPbeta reduces kidney function after ischemia/reperfusion-induced injury. Sci. Rep. 2016, 6, 27945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilflingseder, J.; Jelencsics, K.; Bergmeister, H.; Sunzenauer, J.; Regele, H.; Eskandary, F.; Reindl-Schwaighofer, R.; Kainz, A.; Oberbauer, R. miR-182-5p Inhibition Ameliorates Ischemic Acute Kidney Injury. Am. J. Pathol. 2017, 187, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Sun, H.; Song, S.; Liu, Y.; Liu, P.; Livingston, M.J.; Wang, J.; Liang, M.; Mi, Q.S.; Huo, Y.; et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J. Clin. Investig. 2018, 128, 5448–5464. [Google Scholar] [CrossRef] [Green Version]
- Gatti, S.; Bruno, S.; Deregibus, M.C.; Sordi, A.; Cantaluppi, V.; Tetta, C.; Camussi, G. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol. Dial. Transplant. 2011, 26, 1474–1483. [Google Scholar] [CrossRef] [Green Version]
- Cantaluppi, V.; Gatti, S.; Medica, D.; Figliolini, F.; Bruno, S.; Deregibus, M.C.; Sordi, A.; Biancone, L.; Tetta, C.; Camussi, G. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int. 2012, 82, 412–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinas, J.L.; Burger, D.; Zimpelmann, J.; Haneef, R.; Knoll, W.; Campbell, P.; Gutsol, A.; Carter, A.; Allan, D.S.; Burns, K.D. Transfer of microRNA-486-5p from human endothelial colony forming cell-derived exosomes reduces ischemic kidney injury. Kidney Int. 2016, 90, 1238–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Pei, L.; Lin, F.; Yin, H.; Li, X.; He, W.; Liu, N.; Gou, X. Exosomes from human-bone-marrow-derived mesenchymal stem cells protect against renal ischemia/reperfusion injury via transferring miR-199a-3p. J. Cell Physiol. 2019, 234, 23736–23749. [Google Scholar] [CrossRef] [PubMed]
- Mall, C.; Rocke, D.M.; Durbin-Johnson, B.; Weiss, R.H. Stability of miRNA in human urine supports its biomarker potential. Biomark. Med. 2013, 7, 623–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltrami, C.; Clayton, A.; Newbury, L.J.; Corish, P.; Jenkins, R.H.; Phillips, A.O.; Fraser, D.J.; Bowen, T. Stabilization of Urinary MicroRNAs by Association with Exosomes and Argonaute 2 Protein. Noncoding RNA 2015, 1, 151–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaels, Y.S.; Barnkob, M.B.; Barbosa, H.; Baeumler, T.A.; Thompson, M.K.; Andre, V.; Colin-York, H.; Fritzsche, M.; Gileadi, U.; Sheppard, H.M.; et al. Precise tuning of gene expression levels in mammalian cells. Nat. Commun. 2019, 10, 818. [Google Scholar] [CrossRef] [Green Version]
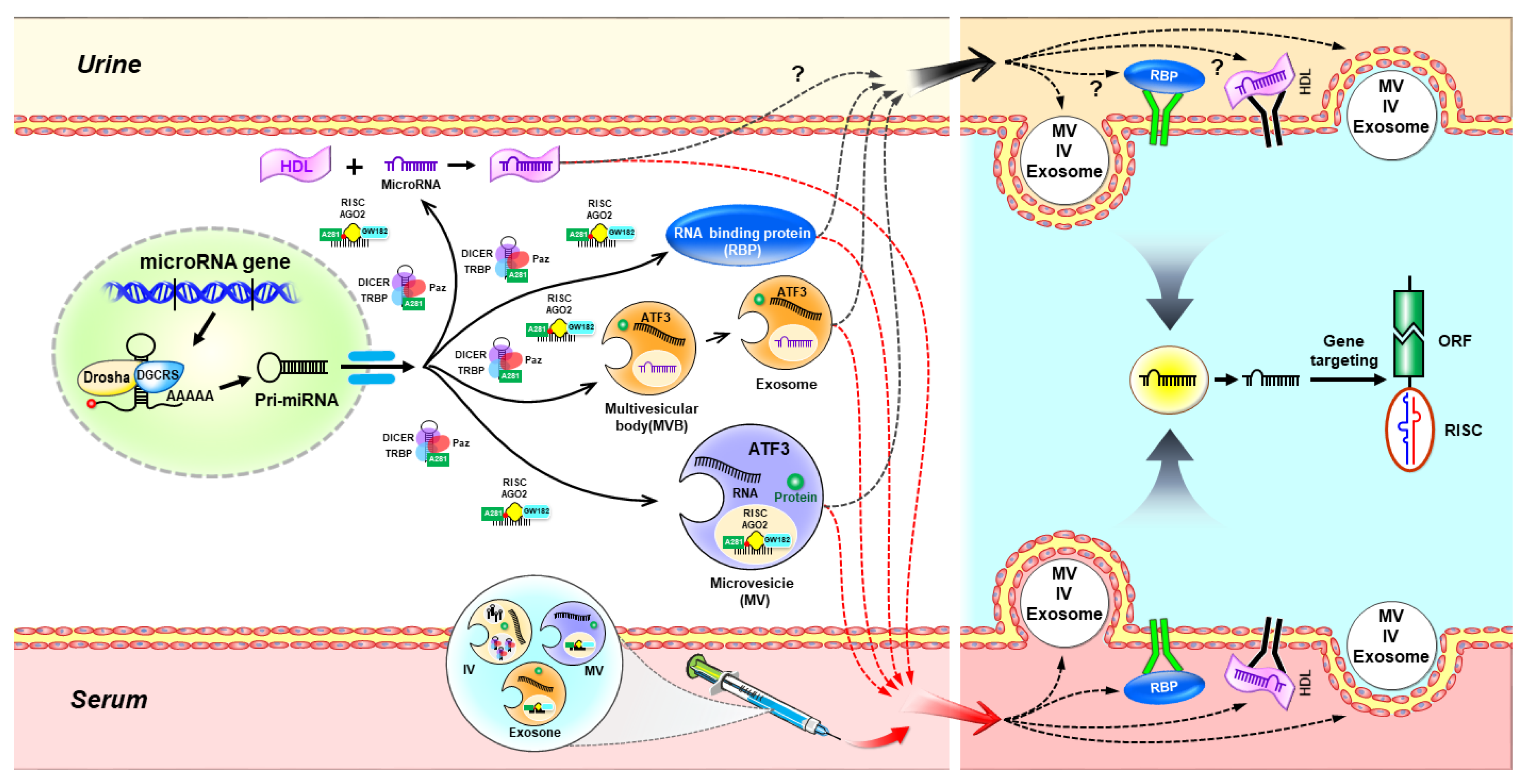

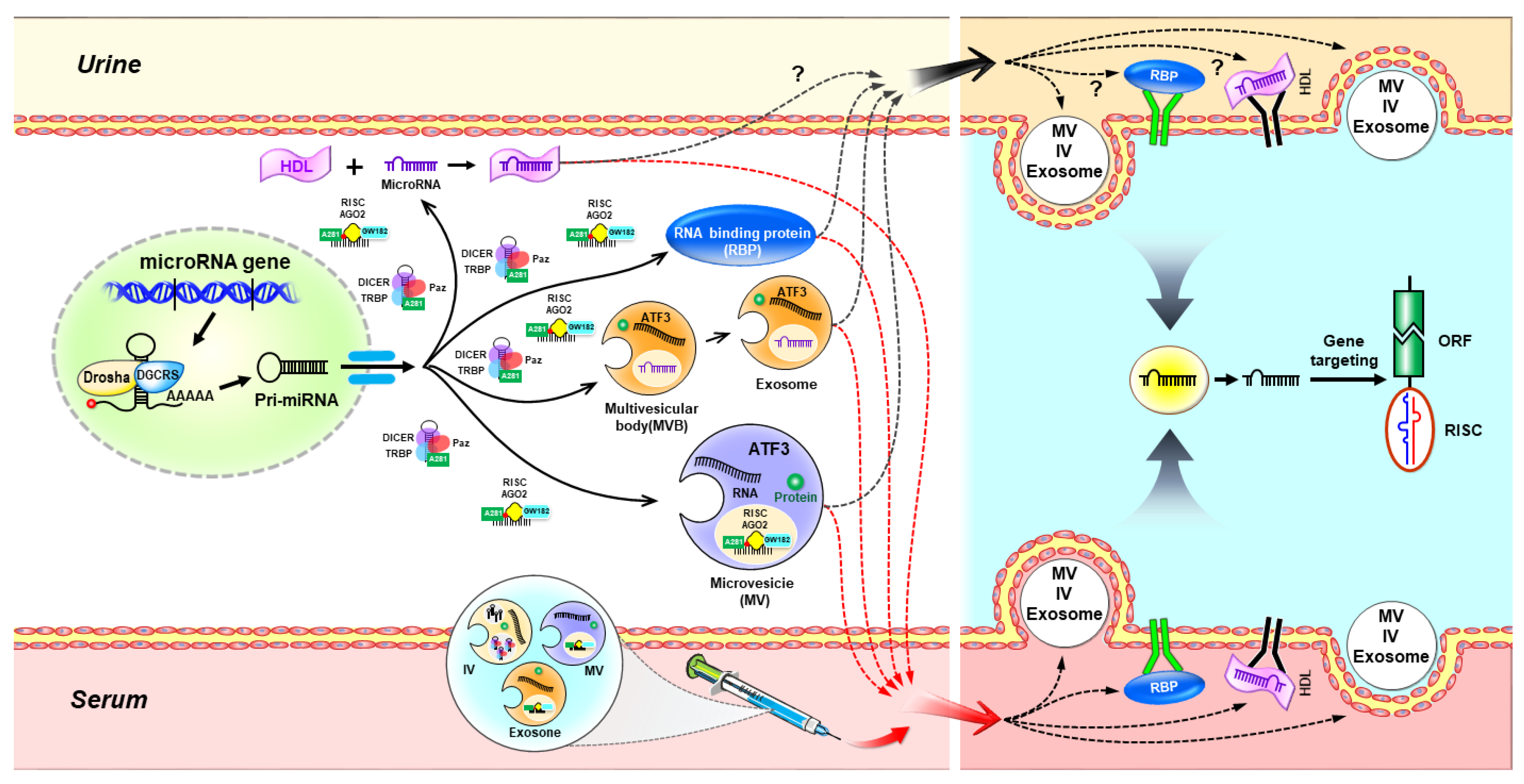{kind=link}
| Possible Pathophysiology | Species | Serum | Urine | Ref. |
|---|---|---|---|---|
| I/R | Human, mice | - | ↑ miR-494 | [55] |
| I/R | Human | ↑ miR-494 | - | [68] |
| I/R, inflammation | Human | ↓ miR-16, miR320 ↑ miR 210 | - | [66] |
| I/R, inflammation | Human | ↓ miR-101-3p, miR-127-3p, miR-210-3p, miR-126-3p, miR-26b-5p, miR-29a-3p, miR-146a-5p, miR-27a-3p, miR-93-3p and miR-10a-5p | - | [67] |
| I/R | Human | ↑ miR-21 | ↑ miR-21 | [63,65] |
| I/R | Human | ↓ miR-21 | ↓ miR-21 | [64] |
| I/R, inflammation | Human | - | ↑ miR-21 | [74] |
| I/R, inflammation | Human | ↑ miR-21-3p | - | [75] |
| I/R | Human | ↑ miR-192 | - | [76] |
| I/R, inflammation | Human | - | ↑ miR-21, miR-200c, and miR-423 ↓ miR-4640 | [72] |
| I/R | Human | - | ↑ miR-30c-5p, miR-192-5p | [73] |
| I/R, inflammation | Human, mice | - | ↑ miR-16 | [77] |
| CIN | Human | ↑ miR-30a, miR-30c, and miR-30e | - | [70] |
| CIN | Human | ↑ miR-30a, miR-30e, and miR-188 | - | [71] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.-L.; Li, H.-F.; Chen, H.-H.; Lin, H. MicroRNAs as Biomarkers and Therapeutic Targets in Inflammation- and Ischemia-Reperfusion-Related Acute Renal Injury. Int. J. Mol. Sci. 2020, 21, 6738. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186738
Wu Y-L, Li H-F, Chen H-H, Lin H. MicroRNAs as Biomarkers and Therapeutic Targets in Inflammation- and Ischemia-Reperfusion-Related Acute Renal Injury. International Journal of Molecular Sciences. 2020; 21(18):6738. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186738
Chicago/Turabian StyleWu, Yueh-Lin, Hsiao-Fen Li, Hsi-Hsien Chen, and Heng Lin. 2020. "MicroRNAs as Biomarkers and Therapeutic Targets in Inflammation- and Ischemia-Reperfusion-Related Acute Renal Injury" International Journal of Molecular Sciences 21, no. 18: 6738. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186738