Activate or Inhibit? Implications of Autophagy Modulation as a Therapeutic Strategy for Alzheimer’s Disease

 ,
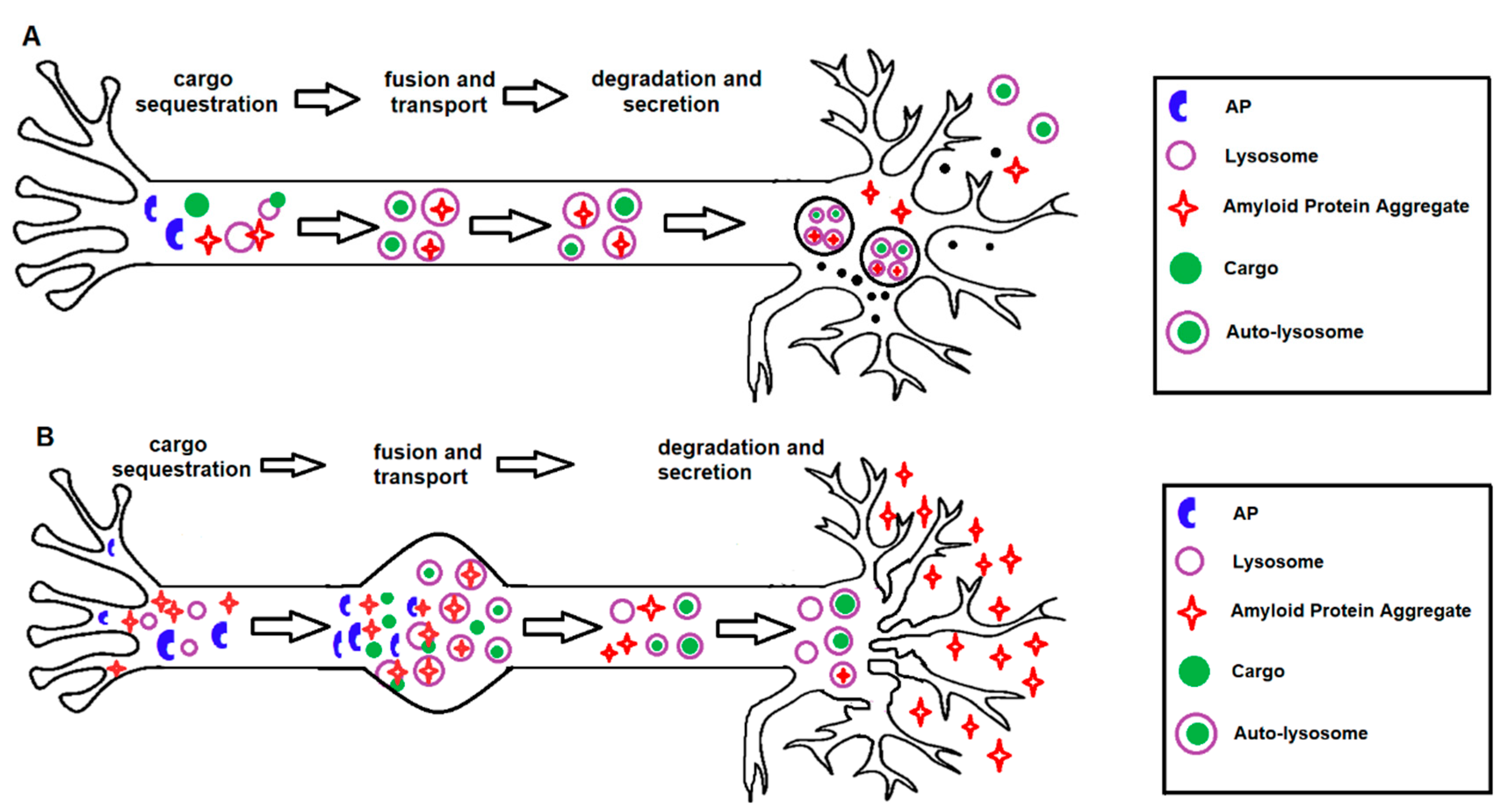
, 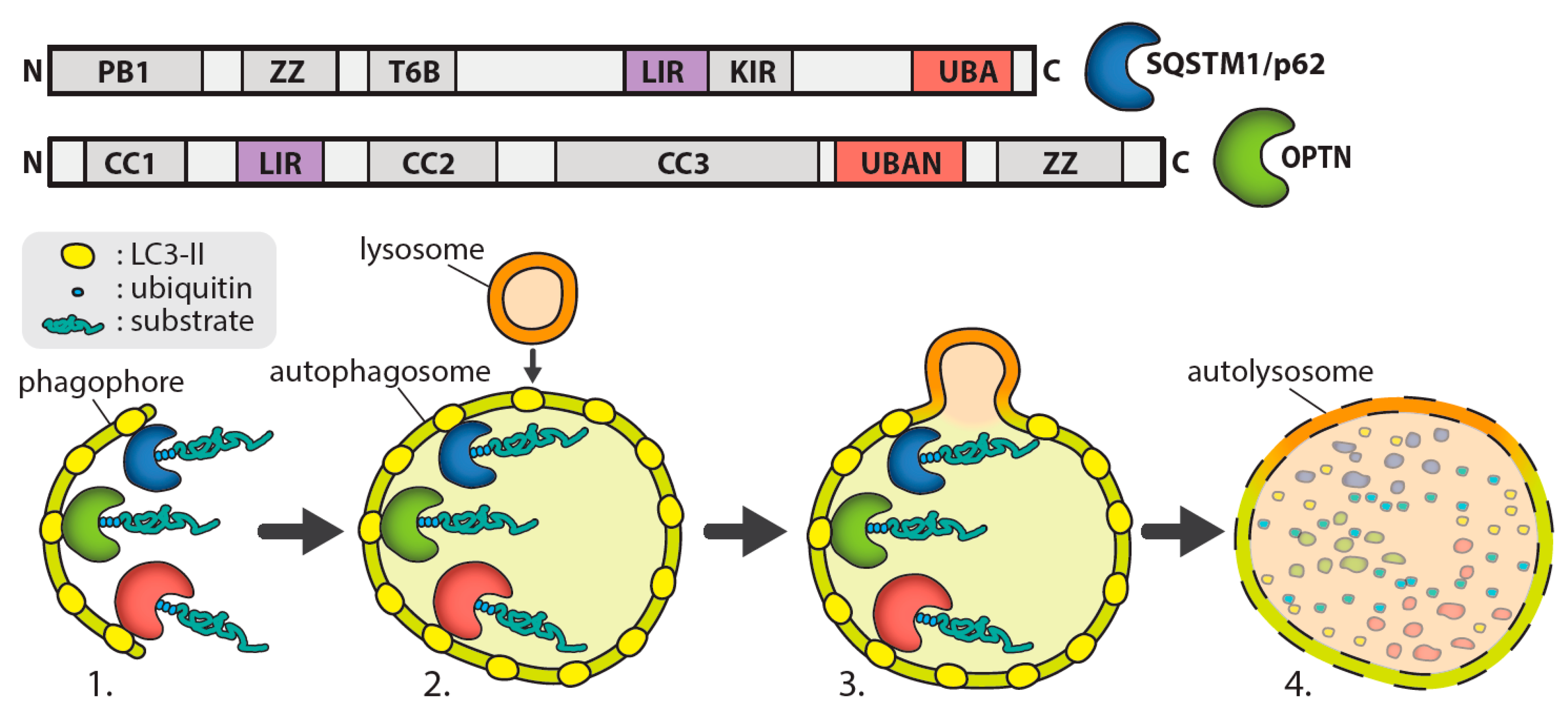{kind=link}
{kind=link}
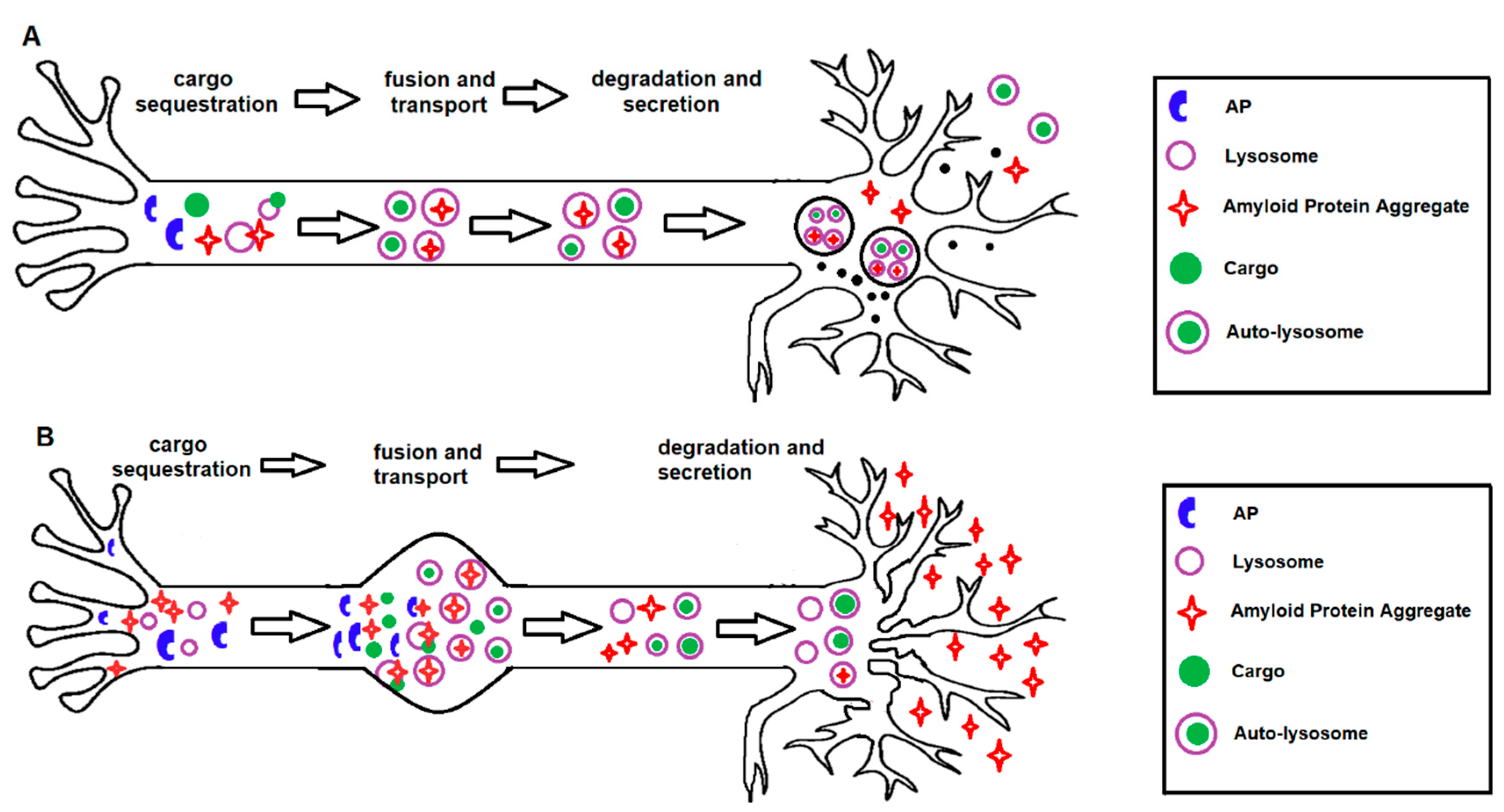{kind=link}
Abstract
:1. Introduction
2. Autophagy Process
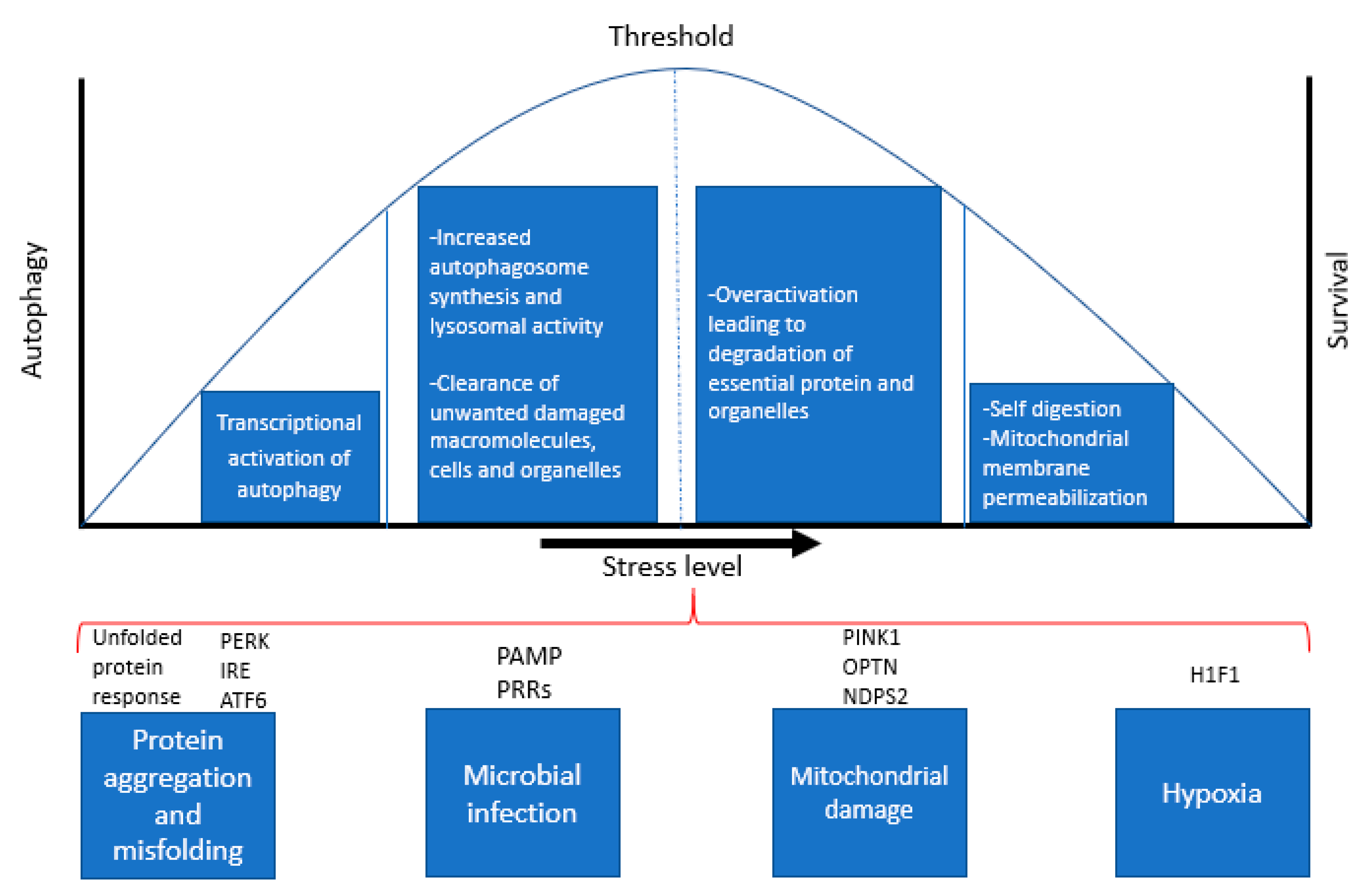
3. Role of Autophagy as a Stress Response Pathway
4. Dual Role of Autophagy: Crosstalk between Autophagy and Apoptosis
5. Autophagy and Ageing
6. Role for Autophagy Genes in AD
7. Role of Autophagy in AD Pathogenesis
8. Autophagy Therapeutics for AD
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Krisko, A.; Radman, M. Protein damage, ageing and age-related diseases. Open Biol. 2019, 9, 180249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiseman, R.L.; Powers, E.T.; Buxbaum, J.N.; Kelly, J.W.; Balch, W.E. An Adaptable Standard for Protein Export from the Endoplasmic Reticulum. Cell 2007, 131, 809–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mputhia, Z.; Hone, E.; Tripathi, T.; Sargeant, T.J.; Martins, R.N.; Bharadwaj, P. Autophagy Modulation as a Treatment of Amyloid Diseases. Molecules 2019, 24, 3372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, W.; Liu, R.; Huang, H.; Zhang, R.; Sun, L. Effect of p62 on tau hyperphosphorylation in a rat model of Alzheimer’s disease. Neural Regen. Res. 2012, 7, 1304–1311. [Google Scholar]
- Pierzynowska, K.; Gaffke, L.; Cyske, Z.; Puchalski, M.; Rintz, E.; Bartkowski, M.; Osiadły, M.; Pierzynowski, M.; Mantej, J.; Piotrowska, E.; et al. Autophagy stimulation as a promising approach in treatment of neurodegenerative diseases. Metab. Brain Dis. 2018, 33, 989–1008. [Google Scholar] [CrossRef]
- Kanazawa, T.; Taneike, I.; Akaishi, R.; Yoshizawa, F.; Furuya, N.; Fujimura, S.; Kadowaki, M. Amino Acids and Insulin Control Autophagic Proteolysis through Different Signaling Pathways in Relation to mTOR in Isolated Rat Hepatocytes. J. Biol. Chem. 2003, 279, 8452–8459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mordier, S.; Deval, C.; Bechet, D.M.; Tassa, A.; Ferrara, M. Leucine Limitation Induces Autophagy and Activation of Lysosome-dependent Proteolysis in C2C12 Myotubes through a Mammalian Target of Rapamycin-independent Signaling Pathway. J. Biol. Chem. 2000, 275, 29900–29906. [Google Scholar] [CrossRef] [Green Version]
- Høyer-Hansen, M.; Jäättelä, M. AMP-Activated Protein Kinase: A Universal Regulator of Autophagy? Autophagy 2007, 3, 381–383. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Shao, S.H.; Xu, Z.-X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1–AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zientara-Rytter, K.; Subramani, S. The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy. Cells 2019, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, B.R.; Maddison, D.C.; Smith, G.A.; Peters, O. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain 2019, 12, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Moreau, K.; Rubinsztein, D.C. Protein misfolding disorders and macroautophagy. Curr. Opin. Cell Biol. 2011, 23, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Martínez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.-M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2013, 24, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Natural 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; González-Polo, R.A.; Casares, N.; Perfettini, J.-L.; Dessen, P.; Larochette, N.; Métivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of Macroautophagy Triggers Apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B.; Yoshimorim, T. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchberger, A.; Bukau, B.; Sommer, T. Protein Quality Control in the Cytosol and the Endoplasmic Reticulum: Brothers in Arms. Mol. Cell 2010, 40, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Sagua, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic Reticulum and the Unfolded Protein Response. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef] [Green Version]
- Sumpter, R.; Levine, B. Autophagy and innate immunity: Triggering, targeting and tuning. Semin. Cell Dev. Biol. 2010, 21, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.-F.; Walker, U.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.J.; Ding, Y.; Kohtz, S.; Mizushima, N.; Cristea, I.M.; Rout, M.P.; Chait, B.T.; Zhong, Y.; Heintz, N.; Yue, Z. Induction of Autophagy in Axonal Dystrophy and Degeneration. J. Neurosci. 2006, 26, 8057–8068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puyal, J.; Clarke, P.G. Targeting autophagy to prevent neonatal stroke damage. Autophagy 2009, 5, 1060–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Wirawan, E.; Walle, L.V.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Betin, V.M.S.; Lane, J.D. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 2009, 122, 2554–2566. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.-G.; Zhang, L.; Chen, G.; Zhang, T.; Liu, J.; Jin, M.; Ma, X.; Ma, D.; Yuan, J. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy 2010, 6, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.-U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Natural 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-S.; Kumar, A.; Stavrides, P.; Peterson, J.N.; Peterhoff, C.M.; Pawlik, M.; Levy, E.; Cataldo, A.M.; Nixon, R.A. Neuronal Apoptosis and Autophagy Cross Talk in Aging PS/APP Mice, a Model of Alzheimer’s Disease. Am. J. Pathol. 2008, 173, 665–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, R.C.; Juhasz, G.; Neufeld, T.P. Direct Induction of Autophagy by Atg1 Inhibits Cell Growth and Induces Apoptotic Cell Death. Curr. Biol. 2007, 17, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginet, V.; Puyal, J.; Clarke, P.G.; Truttmann, A.C. Enhancement of Autophagic Flux after Neonatal Cerebral Hypoxia-Ischemia and Its Region-Specific Relationship to Apoptotic Mechanisms. Am. J. Pathol. 2009, 175, 1962–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.-S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.-I.; Ezaki, J.; Murata, S.; et al. Homeostatic Levels of p62 Control Cytoplasmic Inclusion Body Formation in Autophagy-Deficient Mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherra, S.J.; Chu, C.T. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008, 3, 309–323. [Google Scholar]
- Koike, M.; Shibata, M.; Tadakoshi, M.; Gotoh, K.; Komatsu, M.; Waguri, S.; Kawahara, N.; Kuida, K.; Nagata, S.; Kominami, E.; et al. Inhibition of Autophagy Prevents Hippocampal Pyramidal Neuron Death after Hypoxic-Ischemic Injury. Am. J. Pathol. 2008, 172, 454–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samara, C.; Syntichaki, P.; Tavernarakis, N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2007, 15, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Renna, M.; Jimenez-Sanchez, M.; Sarkar, S.; Rubinsztein, D.C. Chemical Inducers of Autophagy That Enhance the Clearance of Mutant Proteins in Neurodegenerative Diseases. J. Biol. Chem. 2010, 285, 11061–11067. [Google Scholar] [CrossRef] [Green Version]
- Puyal, J.; Ginet, V.; Grishchuk, Y.; Truttmann, A.C.; Clarke, P.G.H. Neuronal Autophagy as a Mediator of Life and Death. Neuroscientist 2011, 18, 224–236. [Google Scholar] [CrossRef]
- Martinet, W.; Agostinis, P.; Vanhoecke, B.; Dewaele, M.; De Meyer, G. Autophagy in disease: A double-edged sword with therapeutic potential. Clin. Sci. 2009, 116, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Fletcher, G.C.; Tolkovsky, A.M. Autophagy Is Activated by Apoptotic Signalling in Sympathetic Neurons: An Alternative Mechanism of Death Execution. Mol. Cell. Neurosci. 1999, 14, 180–198. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Vicente, M.; Sovak, G.; Cuervo, A.M. Protein degradation and aging. Exp. Gerontol. 2005, 40, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, E.; Kovacs, J. Exploring the age-related changes in hormone-regulated protein breakdown by the use of a physiologic model of stimulation of liver autophagy. In Protein Metabolism in Aging; Segal, H.L., Rothstein, M., Bergamini, E., Eds.; Wiley–Liss: New York, NY, USA, 1989; pp. 361–370. [Google Scholar]
- Roso, A.D.; Vittorini, S.; Cavallini, G.; Donati, A.; Gori, Z.; Masini, M.; Pollera, M.; Bergamini, E. Ageing-related changes in the in vivo function of rat liver macroautophagy and proteolysis. Exp. Gerontol. 2003, 38, 519–527. [Google Scholar] [CrossRef]
- Donati, A.; Cavallini, G.; Paradiso, C.; Vittorini, S.; Pollera, M.; Gori, Z.; Bergamini, E. Age-Related Changes in the Autophagic Proteolysis of Rat Isolated Liver Cells: Effects of Antiaging Dietary Restrictions. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2001, 56, B375–B383. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Dice, J.F. Age-related Decline in Chaperone-mediated Autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, N.; Athonvarangkul, D.; Singh, R. Autophagy and Aging. Adv. Exp. Med. Biol. 2015, 847, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.Q.; Kumar, A.V.; Mills, J.; Lapierre, L.R. Autophagy in aging and longevity. Qual. Life Res. 2019, 139, 277–290. [Google Scholar] [CrossRef]
- Markaki, M.; Metaxakis, A.; Tavernarakis, N. The role of autophagy in aging: Molecular mechanisms. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2017; pp. 123–138. [Google Scholar]
- Pyo, J.-O.; Yoo, S.-M.; Ahn, H.-H.; Nah, J.; Hong, S.-H.; Kam, T.-I.; Jung, S.; Jung, Y.-K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Cuervo, A.M. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 2008, 14, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkis, G.J.; Ashcom, J.D.; Hawdon, J.M.; A Jacobson, L. Decline in protease activities with age in the nematode caenorhabditis elegans. Mech. Ageing Dev. 1988, 45, 191–201. [Google Scholar] [CrossRef]
- Minnerly, J.; Zhang, J.; Parker, T.; Kaul, T.; Jia, K. The cell non-autonomous function of ATG-18 is essential for neuroendocrine regulation of Caenorhabditis elegans lifespan. PLoS Genet. 2017, 13, e1006764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Masoro, E.J.; Nelson, J.F.; Strong, R.; Mcmahan, C.A.; Richardson, A. Genetic mouse models of extended lifespan. Exp. Gerontol. 2003, 38, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and Aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuele, E.; Minoretti, P.; Sanchis-Gomar, F.; Pareja-Galeano, H.; Yilmaz, Y.; Garatachea, N.; Lucia, A. Can Enhanced Autophagy Be Associated with Human Longevity? Serum Levels of the Autophagy Biomarker Beclin-1 Are Increased in Healthy Centenarians. Rejuvenation Res. 2014, 17, 518–524. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef]
- Gupta, V.K.; Scheunemann, L.; Eisenberg, T.; Mertel, S.; Bhukel, A.; Koemans, T.S.; Kramer, J.M.; Liu, K.S.Y.; Schroeder, S.; Stunnenberg, H.G.; et al. Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat. Neurosci. 2013, 16, 1453–1460. [Google Scholar] [CrossRef]
- Gupta, V.K.; Pech, U.; Bhukel, A.; Fulterer, A.; Ender, A.; Mauermann, S.F.; Andlauer, T.F.; Antwi-Adjei, E.; Beuschel, C.B.; Thriene, K.; et al. Spermidine Suppresses Age-Associated Memory Impairment by Preventing Adverse Increase of Presynaptic Active Zone Size and Release. PLoS Biol. 2016, 14, e1002563. [Google Scholar] [CrossRef]
- Barja, G. Free radicals and aging. Trends Neurosci. 2004, 27, 595–600. [Google Scholar] [CrossRef]
- Nassif, M.; Woehlbier, U.; Manque, P.A. The delicate balance of autophagy in neurodegeneration. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2017; pp. 387–399. [Google Scholar]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 37, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Rezzani, R.; Stacchiotti, A.; Rodella, L.F. Morphological and biochemical studies on aging and autophagy. Ageing Res. Rev. 2012, 11, 10–31. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.-S.; Müllendorff, K.; Cheng, I.H.; Miranda, R.D.; Huang, Y.; Mahley, R.W. Reactivity of Apolipoprotein E4 and Amyloid β Peptide. J. Biol. Chem. 2005, 281, 2683–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, R. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.C.; Grosso, R.; Fader, C.M. Hallmarks of Aging: An Autophagic Perspective. Front. Endocrinol. 2019, 9, 9. [Google Scholar] [CrossRef]
- Majcher, V.; Goode, A.; James, V.; Layfield, R. Autophagy receptor defects and ALS-FTLD. Mol. Cell. Neurosci. 2015, 66, 43–52. [Google Scholar] [CrossRef]
- Cuyvers, E.; Van Der Zee, J.; Bettens, K.; Engelborghs, S.; Vandenbulcke, M.; Robberecht, C.; Dillen, L.; Merlin, C.; Geerts, N.; Graff, C.; et al. Genetic variability in SQSTM1 and risk of early-onset Alzheimer dementia: A European early-onset dementia consortium study. Neurobiol. Aging 2010, 36, 2005.e15–2005.e22. [Google Scholar] [CrossRef]
- Verheijen, J.; Van Der Zee, J.; Gijselinck, I.; Bossche, T.V.D.; Dillen, L.; Heeman, B.; Gomez-Tortosa, E.; Lladó, A.; Sanchez-Valle, R.; Graff, C.; et al. Common and rare TBK1 variants in early-onset Alzheimer disease in a European cohort. Neurobiol. Aging 2018, 62, 245.e1–245.e7. [Google Scholar] [CrossRef] [PubMed]
- Bonvicini, C.; Scassellati, C.; Benussi, L.; Di Maria, E.; Maj, C.; Ciani, M.; Fostinelli, S.; Mega, A.; Bocchetta, M.; Lanzi, G.; et al. Next Generation Sequencing Analysis in Early Onset Dementia Patients. J. Alzheimer’s Dis. 2019, 67, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Zou, F.; Chai, H.S.; Younkin, C.S.; Allen, M.; Crook, J.; Pankratz, V.S.; Carrasquillo, M.M.; Rowley, C.N.; Nair, A.A.; Middha, S.; et al. Brain Expression Genome-Wide Association Study (eGWAS) Identifies Human Disease-Associated Variants. PLoS Genet. 2012, 8, e1002707. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.-C. Genetics of Alzheimer’s disease: Where we are, and where we are going. Curr. Opin. Neurobiol. 2020, 61, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.-L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, N.; Tanabe, H.; Kuribayashi, K.; Tsuji, N.; Tanaka, M.; Kobayashi, D. Sesamin induces autophagy in colon cancer cells by reducing tyrosine phosphorylation of EphA1 and EphB2. Int. J. Oncol. 2011, 39, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Alnasser, H.A.; Guan, Q.; Zhang, F.; Gleave, M.E.; Nguan, C.Y.C.; Du, C. Requirement of clusterin expression for prosurvival autophagy in hypoxic kidney tubular epithelial cells. Am. J. Physiol. Physiol. 2016, 310, F160–F173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Kumano, M.; Beraldi, E.; Fazli, L.; Du, C.; Moore, S.; Sorensen, P.; Zoubeidi, A.; Gleave, M.E. Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat. Commun. 2014, 5, 5775. [Google Scholar] [CrossRef]
- Moreau, K.; Fleming, A.; Imarisio, S.; Ramirez, A.L.; Mercer, J.L.; Jimenez-Sanchez, M.; Bento, C.F.; Puri, C.; Zavodszky, E.; Siddiqi, F.; et al. PICALM modulates autophagy activity and tau accumulation. Nat. Commun. 2014, 5, 4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Füllgrabe, J.; Lynch-Day, M.A.; Heldring, N.; Li, W.; Struijk, R.B.; Ma, Q.; Hermanson, O.; Rosenfeld, M.G.; Klionsky, D.J.; Joseph, B. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 2013, 500, 468–471. [Google Scholar] [CrossRef]
- Tsai, C.-W.; Lai, F.-J.; Sheu, H.-M.; Lin, Y.-S.; Chang, T.-H.; Jan, M.-S.; Chen, S.-M.; Hsu, P.-C.; Huang, T.-T.; Sheen, M.-C.; et al. WWOX suppresses autophagy for inducing apoptosis in methotrexate-treated human squamous cell carcinoma. Cell Death Dis. 2013, 4, e792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Luo, Y.; Zhang, X.; Yang, X.; Hong, X.-Y.; Sun, D.-S.; Li, X.-C.; Hu, Y.; Li, X.-G.; Zhang, J.-F.; et al. MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: A vicious cycle in Alzheimer neurodegeneration. Autophagy 2019, 16, 641–658. [Google Scholar] [CrossRef]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Krishnaswamy, S.; Verdile, G.; Groth, D.; Kanyenda, L.; Martins, R.N. The structure and function of Alzheimer’s gamma secretase enzyme complex. Crit. Rev. Clin. Lab. Sci. 2009, 46, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Atwood, C.S.; Obrenovich, M.E.; Liu, T.; Chan, H.; Perry, G.; Smith, M.A.; Martins, R.N. Amyloid-β: A chameleon walking in two worlds: A review of the trophic and toxic properties of amyloid-β. Brain Res. Rev. 2003, 43, 1–16. [Google Scholar] [CrossRef]
- Bharadwaj, P.R.; Dubey, A.K.; Masters, C.L.; Martins, R.N.; Macreadie, I. Aβ aggregation and possible implications in Alzheimer’s disease pathogenesis. J. Cell. Mol. Med. 2009, 13, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Tipping, K.W.; Van Oosten-Hawle, P.; Hewitt, E.W.; Radford, S.E. Amyloid Fibres: Inert End-Stage Aggregates or Key Players in Disease? Trends Biochem. Sci. 2015, 40, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.H.; Paillusson, S.; Hartopp, N.; Rupawala, H.; Mórotz, G.M.; Gomez-Suaga, P.; Greig, J.; Troakes, C.; Noble, W.; Miller, C.C.J. Disruption of endoplasmic reticulum-mitochondria tethering proteins in post-mortem Alzheimer’s disease brain. Neurobiol. Dis. 2020, 143, 105020. [Google Scholar] [CrossRef]
- Haghi, M.; Masoudi, R.; Najibi, S.M. Distinctive alteration in the expression of autophagy genes in Drosophila models of amyloidopathy and tauopathy. Upsala J. Med Sci. 2020, 1–9. [Google Scholar] [CrossRef]
- Moloudizargari, M.; Asghari, M.H.; Ghobadi, E.; Fallah, M.; Rasouli, S.; Abdollahi, M. Autophagy, its mechanisms and regulation: Implications in neurodegenerative diseases. Ageing Res. Rev. 2017, 40, 64–74. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; DiFiglia, M.; Heintz, N.; Nixon, R.A.; Qin, Z.-H.; Ravikumar, B.; Stefanis, L.; Tolkovsky, A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 2005, 1, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007, 6, 304–312. [Google Scholar] [CrossRef]
- Williams, A.; Jahreiss, L.; Sarkar, S.; Saiki, S.; Menzies, F.; Ravikumar, B.; Rubinsztein, D. Aggregate-prone proteins are cleared from the cytosol by autophagy: Therapeutic implications. Curr. Top. Dev. Biol. 2006, 89–101. [Google Scholar] [CrossRef]
- Martínez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domise, M.; Sauvé, F.; Didier, S.; Caillerez, R.; Bégard, S.; Carrier, S.; Colin, M.; Marinangeli, C.; Buée, L.; Vingtdeux, V. Neuronal AMP-activated protein kinase hyper-activation induces synaptic loss by an autophagy-mediated process. Cell Death Dis. 2019, 10, 221. [Google Scholar] [CrossRef] [Green Version]
- Lippai, M.; Csikós, G.; Maròy, P.; Lukacsovich, T.; Juhasz, G.; Sass, M. SNF4Aγ, the Drosophila AMPK γ subunit is required for regulation of developmental and stress-induced autophagy. Autophagy 2008, 4, 476–486. [Google Scholar] [CrossRef]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy Induction and Autophagosome Clearance in Neurons: Relationship to Autophagic Pathology in Alzheimer’s Disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, P.; Martins, R.N. PRKAG2 Gene Expression Is Elevated and its Protein Levels Are Associated with Increased Amyloid-β Accumulation in the Alzheimer’s Disease Brain. J. Alzheimer’s Dis. 2020, 74, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, P.; Martins, R.N. Autophagy modulates Aβ accumulation and formation of aggregates in yeast. Mol. Cell. Neurosci. 2020, 104, 103466. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- A Nixon, R.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.-H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [CrossRef] [PubMed]
- A Nixon, R. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Kumar, A.; Peterhoff, C.; Kulnane, L.S.; Uchiyama, Y.; Lamb, B.; Cuervo, A.M.; A Nixon, R. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: Implications for β-amyloid peptide over-production and localization in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2004, 36, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, L.; A Tsvetanov, K.; Jones, P.; Bevan-Jones, W.; Arnold, R.; Borchert, R.; Mak, E.; Su, L.; O’Brien, J.; Rowe, J.B. Neuroinflammation and Functional Connectivity in Alzheimer’s Disease: Interactive Influences on Cognitive Performance. J. Neurosci. 2019, 39, 7218–7226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharm. 2014, 88, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Parpura, V.; Pekna, M.; Pekny, M.; Sofroniew, M. Glia in the pathogenesis of neurodegenerative diseases. Biochem. Soc. Trans. 2014, 42, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, S.; Jiang, Q.; Lee, C.Y.D.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Landreth, G.E. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. 2009, 29, 4252–4262. [Google Scholar] [CrossRef] [Green Version]
- Pihlaja, R.; Koistinaho, J.; Malm, T.; Sikkilä, H.; Vainio, S.; Koistinaho, M. Transplanted astrocytes internalize deposited β-amyloid peptides in a transgenic mouse model of Alzheimer’s disease. Glia 2007, 56, 154–163. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Witton, J.; Olabarria, M.; Noristani, H.N.; Verkhratsky, A. Increase in the density of resting microglia precedes neuritic plaque formation and microglial activation in a transgenic model of Alzheimer’s disease. Cell Death Dis. 2010, 1, e1. [Google Scholar] [CrossRef]
- Mohamed, A.; De Chaves, E.P. Aβ Internalization by Neurons and Glia. Int. J. Alzheimer’s Dis. 2011, 2011, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.L.; Zinn, R.; Hohensinn, B.; Konen, L.M.; Beynon, S.B.; Tan, R.P.; Clark, I.A.; Abdipranoto, A.; Vissel, B. Neuroinflammation and Neuronal Loss Precede Aβ Plaque Deposition in the hAPP-J20 Mouse Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e59586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomilio, C.; Pavía, P.; Gorojod, R.M.; Vinuesa, A.; Alaimo, A.; Galvan, V.; Kotler, M.L.; Beauquis, J.; Saravia, F. Glial alterations from early to late stages in a model of Alzheimer’s disease: Evidence of autophagy involvement in Aβ internalization. Hippocampus 2015, 26, 194–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, T.; Kalita, P.; Martins, R.; Bharadwaj, P. Autophagy Promotes Memory Formation. ACS Chem. Neurosci. 2019, 10, 3337–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wüllner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2005, 15, 433–442. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Vacher, C.; Berger, Z.; E Davies, J.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, U.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. α-Synuclein Is Degraded by Both Autophagy and the Proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [Green Version]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Dong, G.; Wang, L. High-frequency transcranial magnetic stimulation protects APP/PS1 mice against Alzheimer’s disease progress by reducing APOE and enhancing autophagy. Brain Behav. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, C.J.; Qin, K.; Cook, J.; Solanki, A.; Mastrianni, J.A. Rapamycin delays disease onset and prevents prp plaque deposition in a mouse model of gerstmann-straussler-scheinker disease. J. Neurosci. 2012, 32, 12396–12405. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.-T.; Zhu, X.-C.; Zhang, Q.-Q.; Cao, L.; Wang, H.-F.; Tan, M.-S.; Gao, Q.; Qin, H.; Zhang, Y.-D.; et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 2014, 85, 121–130. [Google Scholar] [CrossRef]
- Ozcelik, S.; Fraser, G.; Castets, P.; Schaeffer, V.; Skachokova, Z.; Breu, K.; Clavaguera, F.; Sinnreich, M.; Kappos, L.; Goedert, M.; et al. Rapamycin Attenuates the Progression of Tau Pathology in P301S Tau Transgenic Mice. PLoS ONE 2013, 8, e62459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by Rapamycin Abolishes Cognitive Deficits and Reduces Amyloid-β Levels in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef] [Green Version]
- Wander, S.A.; Hennessy, B.T.; Slingerland, J.M. Next-generationmTOR inhibitors in clinical oncology: How pathway complexity informs thera-peutic strategy. J. Clin. Investig. 2011, 121, 1231–1241. [Google Scholar] [CrossRef] [Green Version]
- DeBosch, B.J.; Heitmeier, M.R.; Mayer, A.L.; Higgins, C.B.; Crowley, J.R.; Kraft, T.E.; Chi, M.; Newberry, E.P.; Chen, Z.; Finck, B.N.; et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci. Signal. 2016, 9, ra21. [Google Scholar] [CrossRef] [Green Version]
- Tanji, K.; Miki, Y.; Maruyama, A.; Mimura, J.; Matsumiya, T.; Mori, F.; Imaizumi, T.; Itoh, K.; Wakabayashi, K. Trehalose intake induces chaperone molecules along with autophagy in a mouse model of Lewy body disease. Biochem. Biophys. Res. Commun. 2015, 465, 746–752. [Google Scholar] [CrossRef]
- Son, J.H.; Shim, J.H.; Kim, K.-H.; Ha, J.-Y.; Han, J.Y. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 2012, 44, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.W.; Lachenmayer, M.L.; Ju, S.; Stock, A.; Liken, J.; Kim, S.H.; Delgado, L.M.; E Alfaro, I.; Bernales, S.; Verdile, G.; et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer’s mouse model. Mol. Psychiatry 2012, 18, 889–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, P.R.; A Bates, K.; Porter, T.; Teimouri, E.; Perry, G.; Steele, J.W.; Gandy, S.; Groth, D.; Martins, R.N.; Verdile, G. Latrepirdine: Molecular mechanisms underlying potential therapeutic roles in Alzheimer’s and other neurodegenerative diseases. Transl. Psychiatry 2013, 3, e332. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Martins, R. A rapid absorbance-based growth assay to screen oligomer Aβ toxicity and protection against cell death in yeast. Neural Regen. Res. 2019. accepted. [Google Scholar]
- Bharadwaj, P.R.; Verdile, G.; Barr, R.K.; Gupta, V.; Steele, J.W.; Lachenmayer, M.L.; Yue, Z.; Ehrlich, M.E.; Petsko, G.; Ju, S.; et al. Latrepirdine (Dimebon™) enhances autophagy and reduces intracellular GFP-Aβ42 levels in yeast. J. Alzheimer’s Dis. 2012, 32, 949–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntsapi, C.; Swart, C.; Lumkwana, D.; Loos, B. Autophagic flux failure in neurodegeneration: Identifying the defect and compensating flux offset. Autophagy Current Trends Cell. Physiol. Pathol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.C.; Jensen, E.H.; Rexach, J.E.; Vinters, H.V.; Hsieh-Wilson, L.C. Loss ofO-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 15120–15125. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnan, S.; Shrestha, Y.; Jayatunga, D.P.W.; Rea, S.; Martins, R.; Bharadwaj, P. Activate or Inhibit? Implications of Autophagy Modulation as a Therapeutic Strategy for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6739. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186739
Krishnan S, Shrestha Y, Jayatunga DPW, Rea S, Martins R, Bharadwaj P. Activate or Inhibit? Implications of Autophagy Modulation as a Therapeutic Strategy for Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(18):6739. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186739
Chicago/Turabian StyleKrishnan, Sharmeelavathi, Yasaswi Shrestha, Dona P. W. Jayatunga, Sarah Rea, Ralph Martins, and Prashant Bharadwaj. 2020. "Activate or Inhibit? Implications of Autophagy Modulation as a Therapeutic Strategy for Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 18: 6739. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186739