Ligation-Mediated Polymerase Chain Reaction Detection of 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine and 5-Hydroxycytosine at the Codon 176 of the p53 Gene of Hepatitis C-Associated Hepatocellular Carcinoma Patients


Abstract
:1. Introduction
2. Results
2.1. Demographic Variables
2.2. Measurement of M1dG Standard
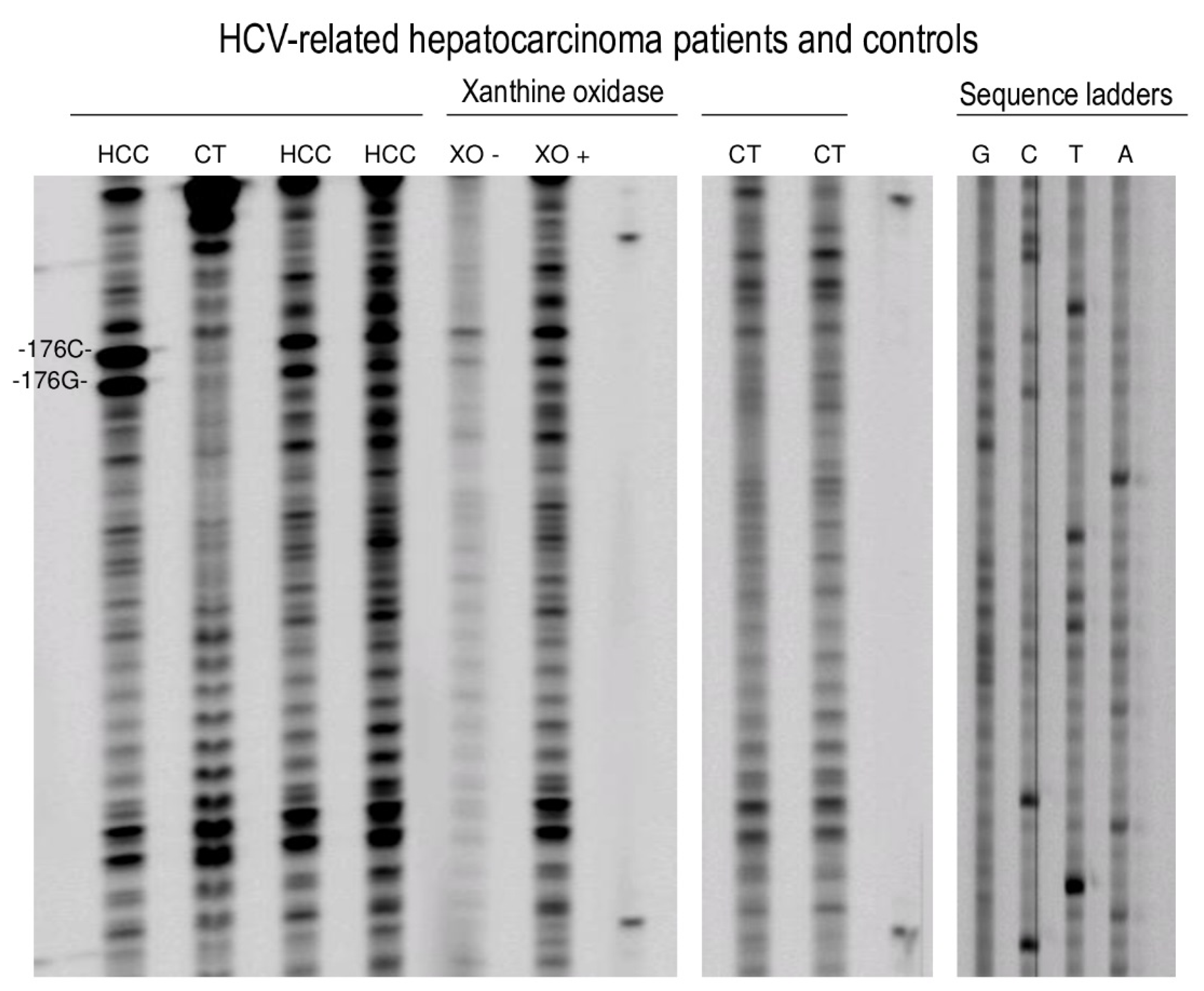
2.3. Detection of 8-oxodG, 5-OHC, and M1dG in HCV-Associated HCC Patients and HepG2 Cells
3. Discussion
4. Material and Methods
4.1. Cell Culture, ROS Treatment, and DNA Isolation
4.2. Study Participants
4.3. Ligation Mediated-PCR Assay
4.4. Preparation of Reference Adduct Standard
4.5. Mass spectrometry Assay
4.6. 32P-Postlabeling Assay
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16. [Google Scholar] [CrossRef]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.-Y.; Wong, D.K.-H.; Tsui, V.W.-M.; Seto, W.-K.; Mak, L.-Y.; Cheung, T.-T.; Lai, K.K.Y.; Yuen, M.-F. Targeted genomic profiling identifies frequent deleterious mutations in FAT4 and TP53 genes in HBV-associated hepatocellular carcinoma. BMC Cancer 2019, 19, 789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yan, K.; Zhou, J.; Zhang, N.; Wang, M.; Song, J.; Zhao, J.; Zhang, Y.; Cai, S.; Zhao, Y.; et al. Relationship of liver cancer with LRP1B or TP53 mutation and tumor mutation burden and survival. J. Clin. Oncol. 2019, 37, 1573. [Google Scholar] [CrossRef]
- Lombardo, D.; Saitta, C.; Giosa, D.; Di Tocco, F.C.; Musolino, C.; Caminiti, G.; Chines, V.; FranzÃ, M.S.; Alibrandi, A.; Navarra, G.; et al. Frequency of somatic mutations in TERT promoter, TP53 and CTNNB1 genes in patients with hepatocellular carcinoma from Southern Italy. Oncol. Lett. 2020, 19, 2368–2374. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Huang, M.-H.; Jiang, J.-D.; Peng, Z.-G. Hepatitis C: From inflammatory pathogenesis to anti-inflammatory/hepatoprotective therapy. World J. Gastroenterol. 2018, 24, 5297–5311. [Google Scholar] [CrossRef]
- Lemon, S.M.; McGivern, D.R. Is Hepatitis C Virus Carcinogenic? Gastroenterology 2012, 142, 1274–1278. [Google Scholar] [CrossRef] [Green Version]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-H.; Gelhaus, S.; Vedantam, S.; Oliva, A.L.; Batra, A.; Blair, I.A.; Troxel, A.B.; Field, J.; Penning, T.M. The pattern of p53 mutations caused by PAH o-quinones is driven by 8-oxo-dGuo formation while the spectrum of mutations is determined by biological selection for dominance. Chem. Res. Toxicol. 2008, 21, 1039–1049. [Google Scholar] [CrossRef]
- Jeong, Y.C.; Swenberg, J.A. Formation of M1G-dR from endogenous and exogenous ROS-inducing chemicals. Free Radic. Biol. Med. 2005, 39, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Taghizadeh, K.; Dedon, P.C. Chemical and biological evidence for base propenals as the major source of the endogenous M1dG adduct in cellular DNA. J. Biol. Chem. 2005, 280, 25377–25382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peluso, M.; Bollati, V.; Munnia, A.; Srivatanakul, P.; Jedpiyawongse, A.; Sangrajrang, S.; Piro, S.; Ceppi, M.; Bertazzi, P.A.; Boffetta, P.; et al. DNA methylation differences in exposed workers and nearby residents of the Ma Ta Phut industrial estate, Rayong, Thailand. Int. J. Epidemiol. 2012, 41, 1753–1760; discussion 1761–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peluso, M.E.; Munnia, A.; Bollati, V.; Srivatanakul, P.; Jedpiyawongse, A.; Sangrajrang, S.; Ceppi, M.; Giese, R.W.; Boffetta, P.; Baccarelli, A.A. Aberrant Methylation of Hypermethylated-in-Cancer-1 and Exocyclic DNA Adducts in Tobacco Smokers. Toxicol. Sci. 2014, 137, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Munnia, A.; Amasio, M.E.; Peluso, M. Exocyclic malondialdehyde and aromatic DNA adducts in larynx tissues. Free Radic. Biol. Med. 2004, 37, 850–858. [Google Scholar] [CrossRef]
- Peluso, M.; Munnia, A.; Risso, G.G.; Catarzi, S.; Piro, S.; Ceppi, M.; Giese, R.W.; Brancato, B. Breast fine-needle aspiration malondialdehyde deoxyguanosine adduct in breast cancer. Free Radic. Res. 2011, 45, 477–482. [Google Scholar] [CrossRef]
- Loft, S.; Moller, P. Oxidative DNA damage and human cancer: Need for cohort studies. Antioxid. Redox Signal. 2006, 8, 1021–1031. [Google Scholar] [CrossRef]
- Loft, S.; Olsen, A.; Moller, P.; Poulsen, H.E.; Tjonneland, A. Association between 8-oxo-7,8-dihydro-2’-deoxyguanosine excretion and risk of postmenopausal breast cancer: Nested case-control study. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1289–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brancato, B.; Munnia, A.; Cellai, F.; Ceni, E.; Mello, T.; Bianchi, S.; Catarzi, S.; Risso, G.G.; Galli, A.; Peluso, M.E. 8-Oxo-7,8-dihydro-2’-deoxyguanosine and other lesions along the coding strand of the exon 5 of the tumour suppressor gene P53 in a breast cancer case-control study. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2016, 23, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Cellai, F.; Munnia, A.; Viti, J.; Doumett, S.; Ravagli, C.; Ceni, E.; Mello, T.; Polvani, S.; Giese, W.R.; Baldi, G.; et al. Magnetic hyperthermia and oxidative damage to DNA of human hepatocarcinoma cells. Int. J. Mol. Sci. 2017, 18, 939. [Google Scholar] [CrossRef]
- Wang, P.; Gao, J.; Li, G.; Shimelis, O.; Giese, R.W. Nontargeted analysis of DNA adducts by mass-tag MS: Reaction of p-benzoquinone with DNA. Chem. Res Toxicol. 2012, 25, 2737–2743. [Google Scholar] [CrossRef] [PubMed]
- Goda, Y.; Marnett, L.J. High-performance liquid chromatography with electrochemical detection for determination of the major malondialdehyde-guanine adduct. Chem. Res. Toxicol. 1991, 4, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Grollman, A.P.; Shibutani, S. Comparison of the mutagenic properties of 8-oxo-7,8-dihydro-2’-deoxyadenosine and 8-oxo-7,8-dihydro-2’-deoxyguanosine DNA lesions in mammalian cells. Carcinogenesis 1999, 20, 2287–2292. [Google Scholar] [CrossRef] [PubMed]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Gungor, N.; Knaapen, A.M.; Munnia, A.; Peluso, M.; Haenen, G.R.; Chiu, R.K.; Godschalk, R.W.; van Schooten, F.J. Genotoxic effects of neutrophils and hypochlorous acid. Mutagenesis 2009, 25, 149–154. [Google Scholar] [CrossRef]
- Cooks, T.; Harris, C.C.; Oren, M. Caught in the cross fire: p53 in inflammation. Carcinogenesis 2014, 35, 1680–1690. [Google Scholar] [CrossRef]
- Ubertini, V.; Norelli, G.; D’Arcangelo, D.; Gurtner, A.; Cesareo, E.; Baldari, S.; Gentileschi, M.P.; Piaggio, G.; Nistico, P.; Soddu, S.; et al. Mutant p53 gains new function in promoting inflammatory signals by repression of the secreted interleukin-1 receptor antagonist. Oncogene 2015, 34, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vurusaner, B.; Poli, G.; Basaga, H. Tumor suppressor genes and ROS: Complex networks of interactions. Free Radic. Biol. Med. 2012, 52, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Pacchiana, R.; Masetto, F.; Mullappilly, N.; Riganti, C.; Donadelli, M. Mutant p53-Associated Molecular Mechanisms of ROS Regulation in Cancer Cells. Biomolecules 2020, 10, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, H.; Weng, M.W.; Chen, W.C.; Tang, M.S. Chromium (VI) induces both bulky DNA adducts and oxidative DNA damage at adenines and guanines in the p53 gene of human lung cells. Carcinogenesis 2012, 33, 1993–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, M.-W.; Lee, H.-W.; Choi, B.; Wang, H.-T.; Hu, Y.; Mehta, M.; Desai, D.; Amin, S.; Zheng, Y.; Tang, M.-S. AFB1 hepatocarcinogenesis is via lipid peroxidation that inhibits DNA repair, sensitizes mutation susceptibility and induces aldehyde-DNA adducts at p53 mutational hotspot codon 249. Oncotarget 2017, 8, 18213–18226. [Google Scholar] [CrossRef]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Sukowati, C.H.C.; El-Khobar, K.E.; Ie, S.I.; Anfuso, B.; Muljono, D.H.; Tiribelli, C. Significance of hepatitis virus infection in the oncogenic initiation of hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 1497–1512. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, F.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Frequency and geographic distribution of TERT promoter mutations in primary hepatocellular carcinoma. Infect. Agents Cancer 2017, 12, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Li, Z.; Ye, Y.; Xie, L.; Li, W. Oxidative Stress and Liver Cancer: Etiology and Therapeutic Targets. Oxid. Med. Cell. Longev. 2016, 2016, 7891574. [Google Scholar] [CrossRef] [Green Version]
- Shawki, S.M.; Meshaal, S.S.; El Dash, A.S.; Zayed, N.A.; Hanna, M.O. Increased DNA damage in hepatitis C virus-related hepatocellular carcinoma. DNA Cell Biol. 2014, 33, 884–890. [Google Scholar] [CrossRef]
- Piciocchi, M.; Cardin, R.; Cillo, U.; Vitale, A.; Cappon, A.; Mescoli, C.; Guido, M.; Rugge, M.; Burra, P.; Floreani, A.; et al. Differential timing of oxidative DNA damage and telomere shortening in hepatitis C and B virus related liver carcinogenesis. Transl. Res. 2016, 168, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Fujita, N.; Sugimoto, R.; Urawa, N.; Horiike, S.; Kobayashi, Y.; Iwasa, M.; Ma, N.; Kawanishi, S.; Watanabe, S.; et al. Hepatic oxidative DNA damage is associated with increased risk for hepatocellular carcinoma in chronic hepatitis C. Br. J. Cancer 2008, 98, 580–586. [Google Scholar] [CrossRef] [Green Version]
- Maki, A.; Kono, H.; Gupta, M.; Asakawa, M.; Suzuki, T.; Matsuda, M.; Fujii, H.; Rusyn, I. Predictive power of biomarkers of oxidative stress and inflammation in patients with hepatitis C virus-associated hepatocellular carcinoma. Ann. Surg. Oncol. 2007, 14, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Ma-On, C.; Sanpavat, A.; Whongsiri, P.; Suwannasin, S.; Hirankarn, N.; Tangkijvanich, P.; Boonla, C. Oxidative stress indicated by elevated expression of Nrf2 and 8-OHdG promotes hepatocellular carcinoma progression. Med. Oncol. 2017, 34, 57. [Google Scholar] [CrossRef]
- Rebbani, K.; Tsukiyama-Kohara, K. HCV-Induced Oxidative Stress: Battlefield-Winning Strategy. Oxid. Med. Cell. Longev. 2016, 2016, 7425628. [Google Scholar] [CrossRef] [Green Version]
- Machida, K.; Cheng, K.T.N.; Sung, V.M.H.; Shimodaira, S.; Lindsay, K.L.; Levine, A.M.; Lai, M.-Y.; Lai, M.M.C. Hepatitis C virus induces a mutator phenotype: Enhanced mutations of immunoglobulin and protooncogenes. Proc. Natl. Acad. Sci. USA 2004, 101, 4262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceppi, M.; Munnia, A.; Cellai, F.; Bruzzone, M.; Peluso, M.E.M. Linking the generation of DNA adducts to lung cancer. Toxic Vitr. 2017, 390, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Denissenko, M.F.; Chen, J.X.; Tang, M.-s.; Pfeifer, G.P. Cytosine methylation determines hot spots of DNA damage in the human gene. Proc. Natl. Acad. Sci. USA 1997, 94, 3893–3898. [Google Scholar] [CrossRef] [Green Version]
- Cleary, S.P.; Jeck, W.R.; Zhao, X.; Chen, K.; Selitsky, S.R.; Savich, G.L.; Tan, T.-X.; Wu, M.C.; Getz, G.; Lawrence, M.S.; et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology 2013, 58, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, T.B.; Hudari, F.; Munnia, A.; Peluso, M.; Godschalk, R.W.; Zanoni, M.V.; den Hartog, G.J.; Bast, A.; Barros, S.B.; Maria-Engler, S.S.; et al. The oxidation of p-phenylenediamine, an ingredient used for permanent hair dyeing purposes, leads to the formation of hydroxyl radicals: Oxidative stress and DNA damage in human immortalized keratinocytes. Toxicol. Lett. 2015, 239, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat. Res. 2005, 591, 45–59. [Google Scholar] [CrossRef]
- Ruano, G.; Kidd, K.K. Coupled amplification and sequencing of genomic DNA. Proc. Natl. Acad. Sci. USA 1991, 88, 2815–2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Oxidative Base Lesions at the Codon 176 of the p53 Gene and Generation of M1dG in Genomic DNA | |||||
|---|---|---|---|---|---|
| 8-oxodG (–TGC–) a | 5-OHC (–TGC–) a | M1dG | |||
| N | RI b ± SE | RI b ± SE | RAL c × 108 ± SE | ||
| Hospital Based Population | |||||
| Controls | 22 | 0.28 ± 0.06 | 0.29 ± 0.01 | 1.67 ± 0.18 | |
| HCV-HCC patients | 31 | 0.61 ± 0.40 | 0.95 ± 0.27 | 3.25 ± 0.41 | |
| Experimental Hepatocytes | |||||
| Untreated HepG2 cells | 6 | 0.20 ± 0.01 | 0.22 ± 0.02 | 0.35 ± 0.2 | |
| XO-treated HepG2 cells | 6 | 0.34 ± 0.03 | 0.44 ± 0.04 | 3.2 ± 0.3 | |
| Mean Ratio and 95% Confidence Interval of Biomarkers Under Study | |||
|---|---|---|---|
| 8-oxodG (–TGC–) a | 5-OHC (–TGC–) a | M1dG | |
| MR, 95% C.I. | MR, 95% C.I. | MR, 95% C.I. | |
| Controls | Reference | Reference | Reference |
| HCV-HCC patients | 2.3, 1.57–3.48 | 3.8, 0.71–20.3 | 1.87, 1.27–2.77 |
| p-value b | <0.0001 | 0.115 | 0.001 |
| Position a | Nomenclature | DNA Adducts | RI ± SE b | Mean Ratio, 95% C.I. | Mutation Type | Mutation Rate |
|---|---|---|---|---|---|---|
| (–TGC–) | c.527G | 8-oxodG | 0.61 ± 0.40 | 130%, 95% C.I. 1.57–3.48 | G:C > A:T | 41.67% |
| (–TGC–) | c.528C | 5-OHC | 0.95 ± 0.27 | 280%, 95% C.I. 0.71–20.3 | G:C > T:A | 8.33% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galli, A.; Munnia, A.; Cellai, F.; Tarocchi, M.; Ceni, E.; van Schooten, F.J.; Godschalk, R.; Giese, R.W.; Peluso, M. Ligation-Mediated Polymerase Chain Reaction Detection of 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine and 5-Hydroxycytosine at the Codon 176 of the p53 Gene of Hepatitis C-Associated Hepatocellular Carcinoma Patients. Int. J. Mol. Sci. 2020, 21, 6753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186753
Galli A, Munnia A, Cellai F, Tarocchi M, Ceni E, van Schooten FJ, Godschalk R, Giese RW, Peluso M. Ligation-Mediated Polymerase Chain Reaction Detection of 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine and 5-Hydroxycytosine at the Codon 176 of the p53 Gene of Hepatitis C-Associated Hepatocellular Carcinoma Patients. International Journal of Molecular Sciences. 2020; 21(18):6753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186753
Chicago/Turabian StyleGalli, Andrea, Armelle Munnia, Filippo Cellai, Mirko Tarocchi, Elisabetta Ceni, Frederik Jan van Schooten, Roger Godschalk, Roger W. Giese, and Marco Peluso. 2020. "Ligation-Mediated Polymerase Chain Reaction Detection of 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine and 5-Hydroxycytosine at the Codon 176 of the p53 Gene of Hepatitis C-Associated Hepatocellular Carcinoma Patients" International Journal of Molecular Sciences 21, no. 18: 6753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186753