Inhaled H2 or CO2 Do Not Augment the Neuroprotective Effect of Therapeutic Hypothermia in a Severe Neonatal Hypoxic-Ischemic Encephalopathy Piglet Model

 , ,
, ,
Abstract
:1. Introduction
2. Results
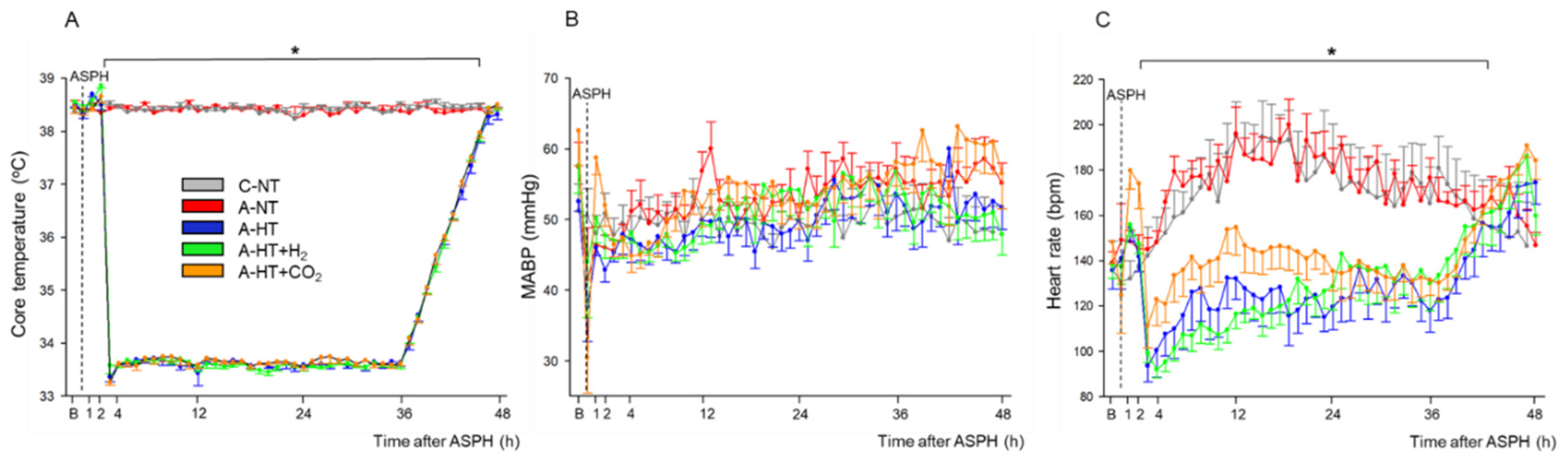
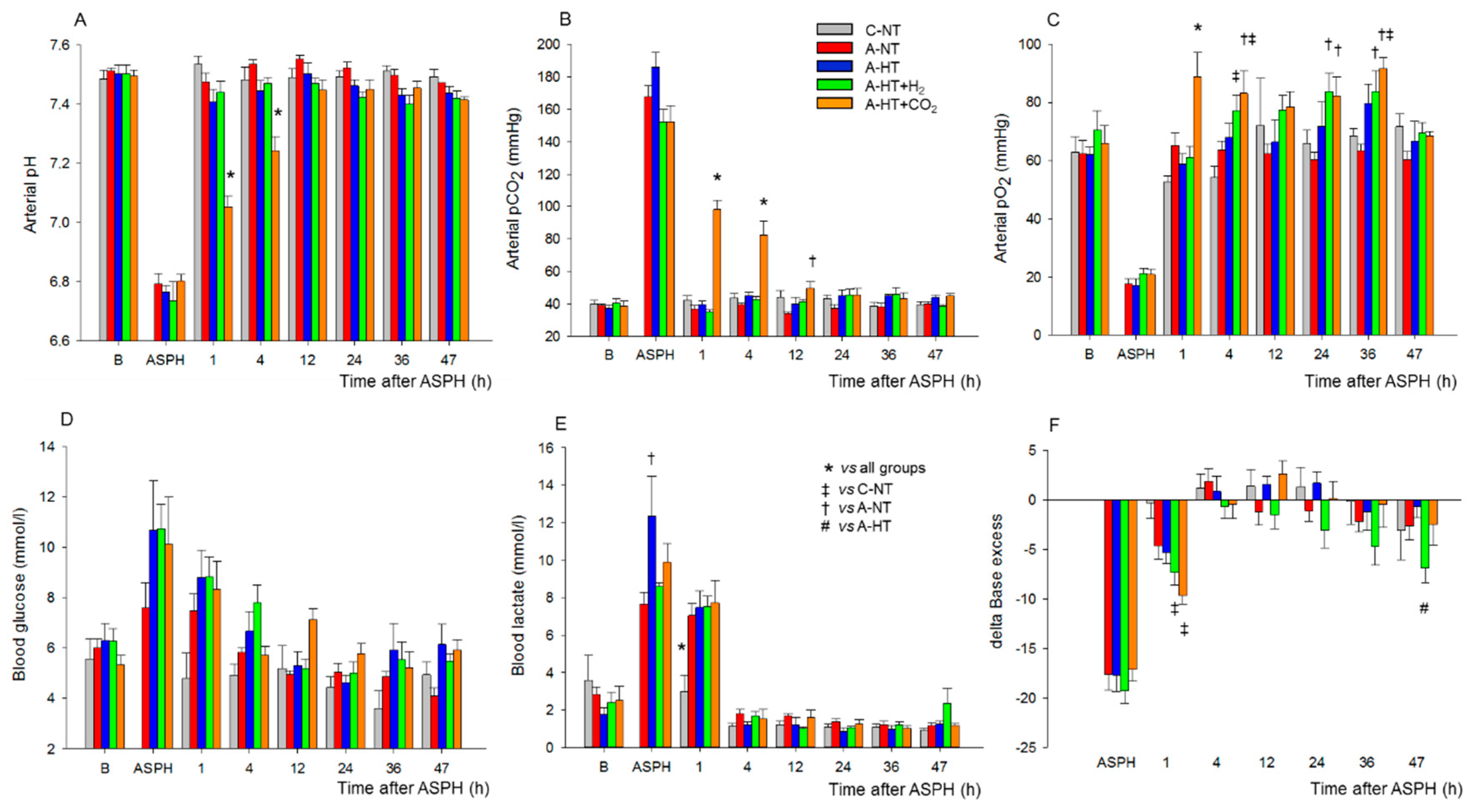
2.1. Physiological Parameters during Asphyxia and HIE Development
Brain Interstitial H2 Concentration Measurements
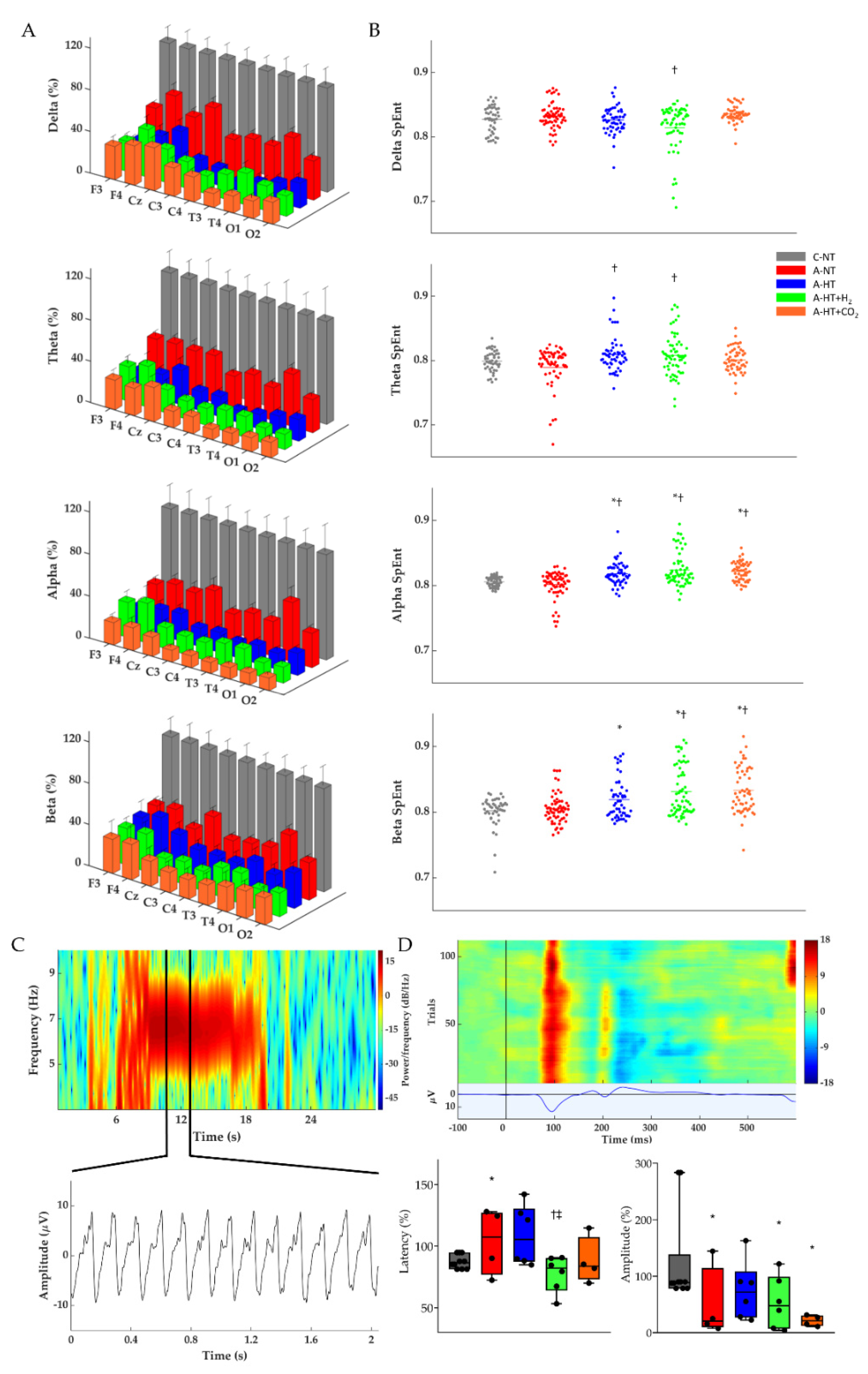
2.2. Electroencephalography (EEG) Analysis
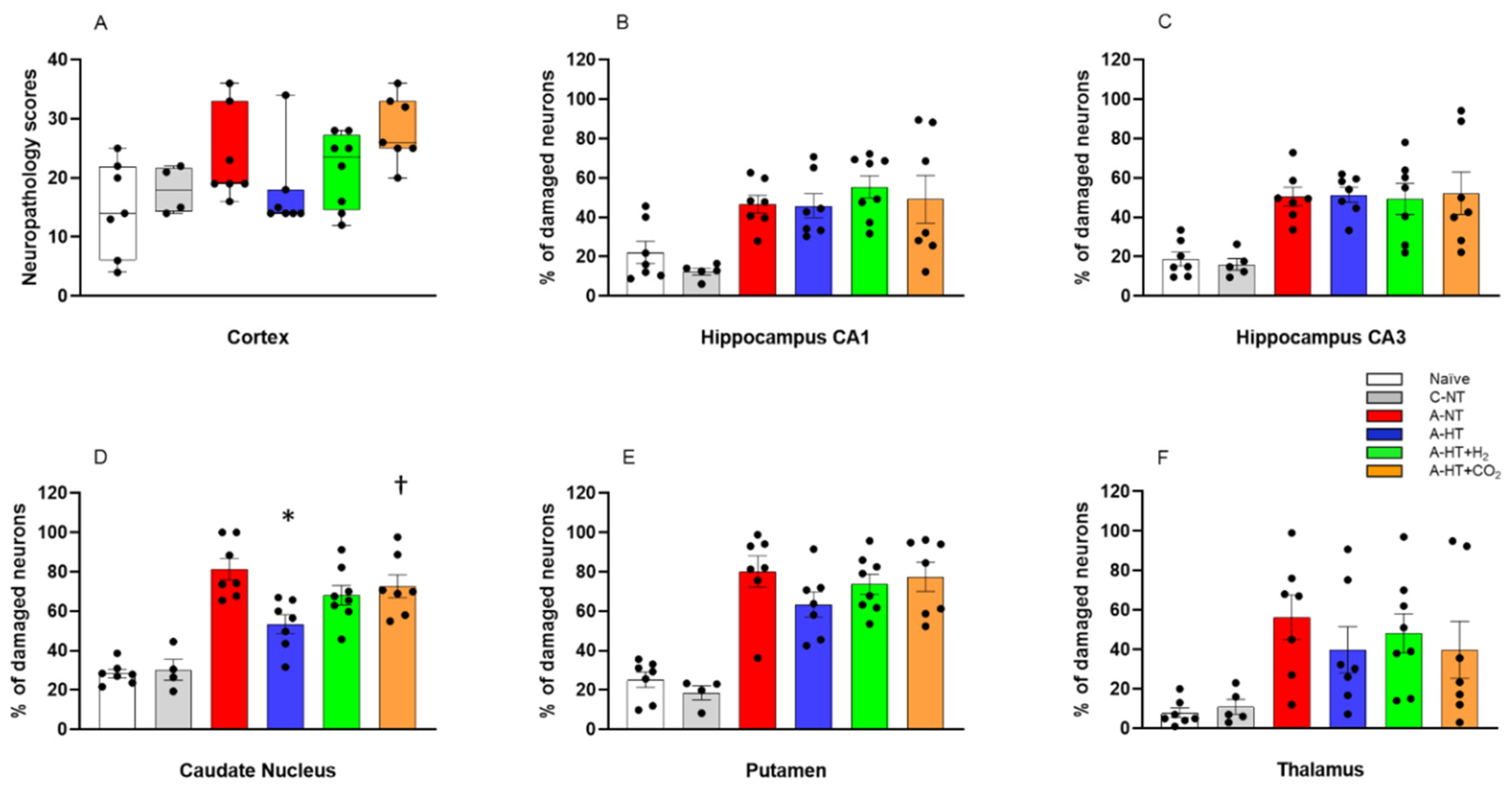
2.3. Neuropathology
2.4. Gene Expression Studies
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Protocol
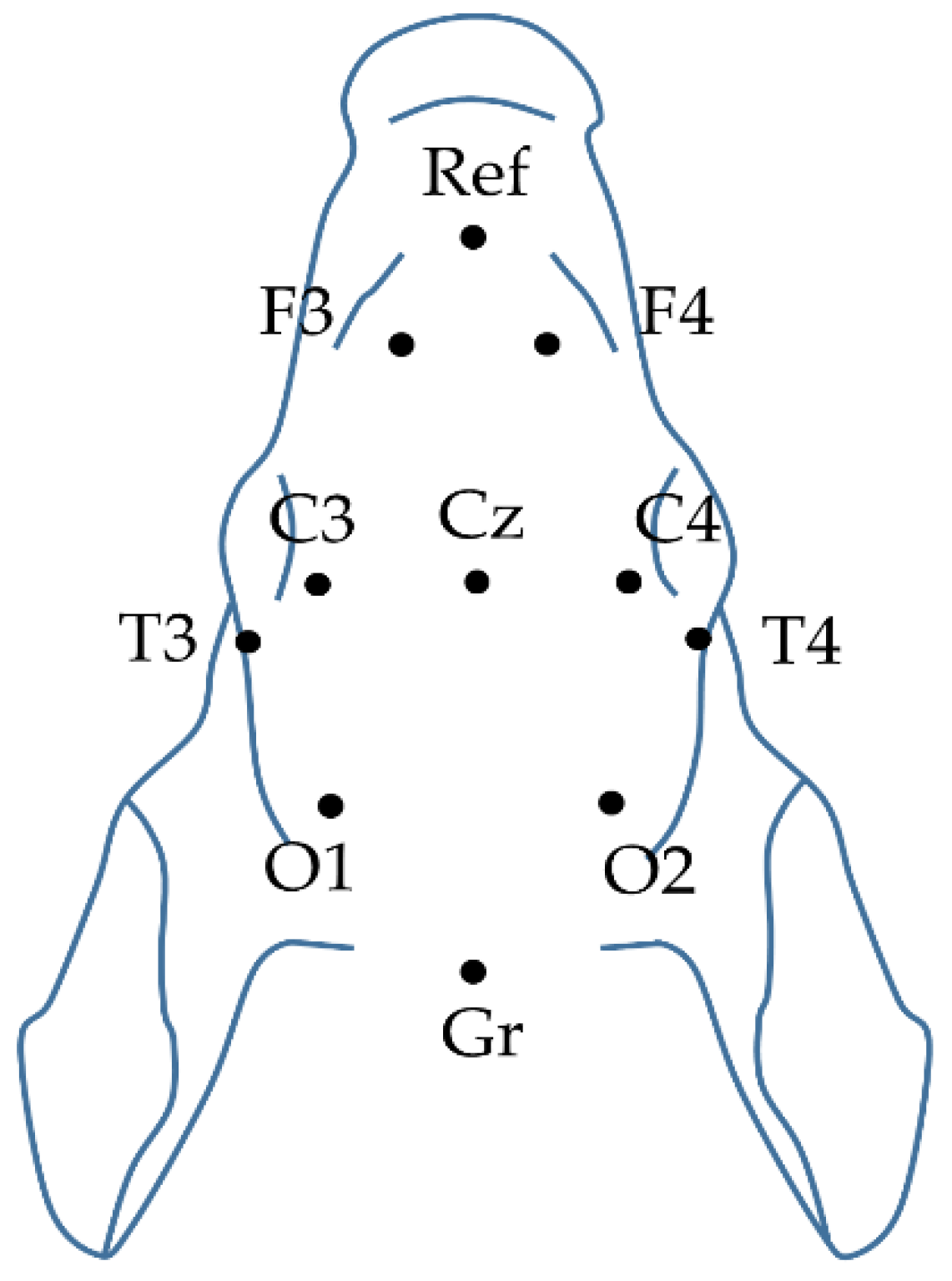
4.2.1. Electroencephalography (EEG)
4.2.2. EEG Spectral Analysis
4.2.3. Cortical H2 Concentration Measurements
4.3. Experimental Groups
4.4. Histology
4.5. Total RNA Extraction and cDNA Synthesis
4.6. Quantitative PCR Analyses
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AIF | apoptosis induction factor |
| Akt | protein kinase B or PKB |
| A-HT | asphyxia-hypothermia |
| A-NT | asphyxia-normothermia |
| BDNF | brain-derived neurotrophic factor |
| C-NT | control-normothermia |
| COX-2 | cyclooxygenase-2 |
| CREB | cAMP response element binding protein |
| Ct | threshold cycles |
| DEPC | diethyl pyrocarbonate |
| DNA | deoxyribonucleic acid |
| ECG | electrocardiogram |
| EEG | electroencephalogram |
| ERK | extracellular signal-regulated kinase |
| FFT | fast Fourier transform |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| HI | hypoxia/ischemia |
| HIE | hypoxic-ischemic encephalopathy |
| HR | heart rate |
| InstSpEnt | instantaneous spectral entropy |
| MABP | mean arterial blood pressure |
| MAPK | mitogen-activated protein kinase |
| mRNA | messenger ribonucleic acid |
| PI-3-K | phosphatidylinositol-3-kinase |
| PPIA | peptidylprolyl isomerase A or cyclophilin A |
| PSD | power spectral density |
| qPCR | quantitative polymerase chain reaction |
| RNA | ribonucleic acid |
| RR | respiration rate |
| SpEnt | spectral entropy |
| TH | therapeutic hypothermia |
| TrkB | tyrosine kinase B |
| VEP | visual evoked potential |
References
- Azzopardi, D.; Brocklehurst, P.; Edwards, D.; Halliday, H.; Levene, M.; Thoresen, M.; Whitelaw, A. The TOBY Study. Whole body hypothermia for the treatment of perinatal asphyxial encephalopathy: A randomised controlled trial. BMC Pediatr. 2008, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassell, K.J.; Ezzati, M.; Alonso-Alconada, D.; Hausenloy, D.J.; Robertson, N.J. New horizons for newborn brain protection: Enhancing endogenous neuroprotection. Arch. Dis. Child Fetal Neonatal Ed. 2015, 100, F541–F552. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Marschitz, M.; Morales, P.; Leyton, L.; Bustamante, D.; Klawitter, V.; Espina-Marchant, P.; Allende, C.; Lisboa, F.; Cunich, G.; Jara-Cavieres, A.; et al. Perinatal asphyxia: Current status and approaches towards neuroprotective strategies, with focus on sentinel proteins. Neurotox. Res. 2011, 19, 603–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, J.A. Intrapartum fetal asphyxia: Definition, diagnosis, and classification. Am. J. Obstet. Gynecol. 1997, 176, 957–959. [Google Scholar] [CrossRef]
- Liu, L.; Oza, S.; Hogan, D.; Perin, J.; Rudan, I.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of child mortality in 2000–2013, with projections to inform post-2015 priorities: An updated systematic analysis. Lancet 2015, 385, 430–440. [Google Scholar] [CrossRef]
- Saugstad, O.D. Reducing Global Neonatal Mortality Is Possible. Neonatology 2011, 99, 250–257. [Google Scholar] [CrossRef]
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 2013, 2013, CD003311. [Google Scholar] [CrossRef]
- Book, S.A.; Bustad, L.K. The fetal and neonatal pig in biomedical research. J. Anim. Sci. 1974, 38, 997–1002. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.S.; Johnson, R.W. The Domestic Piglet: An Important Model for Investigating the Neurodevelopmental Consequences of Early Life Insults. Annu. Rev. Anim. Biosci. 2015, 3, 245–264. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Thoresen, M.; Penrice, J.; Lorek, A.; Cady, E.B.; Wylezinska, M.; Kirkbride, V.; Cooper, C.E.; Brown, G.C.; Edwards, A.D.; Wyatt, J.S.; et al. Mild hypothermia after severe transient hypoxia-ischemia ameliorates delayed cerebral energy failure in the newborn piglet. Pediatr. Res. 1995, 37, 667–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoresen, M.; Tooley, J.; Liu, X.; Jary, S.; Fleming, P.; Luyt, K.; Jain, A.; Cairns, P.; Harding, D.; Sabir, H. Time is brain: Starting therapeutic hypothermia within three hours after birth improves motor outcome in asphyxiated newborns. Neonatology 2013, 104, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, I.; Ishikawa, M.; Takahashi, K.; Watanabe, M.; Nishimaki, K.; Yamagata, K.; Katsura, K.I.; Katayama, Y.; Asoh, S.; Ohta, S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 2007, 13, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Kang, Z.; Liu, W.W.; Luo, X.; Qiang, S.; Zhang, J.H.; Ohta, S.; Sun, X.; Xu, W.; Tao, H.; et al. Hydrogen therapy reduces apoptosis in neonatal hypoxia-ischemia rat model. Neurosci. Lett. 2008, 441, 167–172. [Google Scholar] [CrossRef]
- Domoki, F.; Oláh, O.; Zimmermann, A.; Németh, I.; Tóth-Szűki, V.; Hugyecz, M.; Temesvári, P.; Bari, F. Hydrogen is neuroprotective and preserves cerebrovascular reactivity in asphyxiated newborn pigs. Pediatr. Res. 2010, 68, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Oláh, O.; Tóth-Szűki, V.; Temesvári, P.; Bari, F.; Domoki, F. Delayed neurovascular dysfunction is alleviated by hydrogen in asphyxiated newborn pigs. Neonatology 2013, 104, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Németh, J.; Tóth-Szűki, V.; Varga, V.; Kovács, V.; Remzső, G.; Domoki, F. Molecular hydrogen affords neuroprotection in a translational piglet model of hypoxic-ischemic encephalopathy. J. Physiol. Pharmacol. 2016, 67, 677–689. [Google Scholar]
- Varga, V.; Németh, J.; Oláh, O.; Tóth-Szűki, V.; Kovács, V.; Remzső, G.; Domoki, F. Molecular hydrogen alleviates asphyxia-induced neuronal cyclooxygenase-2 expression in newborn pigs. Acta Pharmacol. Sin. 2018, 39, 1273–1283. [Google Scholar] [CrossRef] [Green Version]
- Htun, Y.; Nakamura, S.; Nakao, Y.; Mitsuie, T.; Nakamura, M.; Yamato, S.; Jinnai, W.; Koyano, K.; Ohta, K.; Morimoto, A.; et al. Hydrogen ventilation combined with mild hypothermia improves short-term neurological outcomes in a 5-day neonatal hypoxia-ischaemia piglet model. Sci. Rep. 2019, 9, 4088. [Google Scholar] [CrossRef] [Green Version]
- Lingappan, K.; Kaiser, J.R.; Srinivasan, C.; Gunn, A.J. Relationship between PCO2 and unfavorable outcome in infants with moderate-to-severe hypoxic ischemic encephalopathy. Pediatr. Res. 2016, 80, 204–208. [Google Scholar] [CrossRef]
- Helmy, M.M.; Ruusuvuori, E.; Watkins, P.V.; Voipio, J.; Kanold, P.O.; Kaila, K. Acid Extrusion via Blood-Brain Barrier Causes Brain Alkalosis and Seizures After Neonatal Asphyxia. Brain 2012, 135, 3311–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pospelov, A.S.; Puskarjov, M.; Kaila, K.; Voipio, J. Endogenous brain-sparing responses in brain pH and PO2 in a rodent model of birth asphyxia. Acta Physiol. 2020, 229, e13467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmigère, F.; Rage, F.; Tapia-Arancibia, L. GABA-glutamate interaction in the control of BDNF expression in hypothalamic neurons. Neurochem. Int. 2003, 42, 353–358. [Google Scholar] [CrossRef]
- Minichiello, L.; Calella, A.M.; Medina, D.L.; Bonhoeffer, T.; Klein, R.; Korte, M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 2002, 36, 121–137. [Google Scholar] [CrossRef] [Green Version]
- Andresen, J.H.; Løberg, E.M.; Wright, M.; Goverud, I.L.; Stray-Pedersen, B.; Saugstad, O.D. Nicotine affects the expression of brain-derived neurotrophic factor mRNA and protein in the hippocampus of hypoxic newborn piglets. J. Perinat. Med. 2009, 37, 553–560. [Google Scholar] [CrossRef]
- D’Cruz, B.J.; Fertig, K.C.; Filiano, A.J.; Hicks, S.D.; DeFranco, D.B.; Callaway, C.W. Hypothermic reperfusion after cardiac arrest augments brain-derived neurotrophic factor activation. J. Cereb. Blood Flow Metab. 2002, 22, 843–851. [Google Scholar] [CrossRef]
- Olson, L.; Faulkner, S.; Lundströmer, K.; Kerenyi, A.; Kelen, D.; Chandrasekaran, M.; Ådén, U.; Olson, L.; Golay, X.; Lagercrantz, H.; et al. Comparison of three hypothermic target temperatures for the treatment of hypoxic ischemia: mRNA level responses of eight genes in the piglet brain. Transl. Stroke Res. 2013, 4, 248–257. [Google Scholar] [CrossRef]
- Randall, G.C. pH values and blood-gas tensions in the normal piglet during the first 48 hours of life. Biol. Neonate 1972, 20, 68–73. [Google Scholar] [CrossRef]
- Iwata, O.; Iwata, S.; Thornton, J.S.; De Vita, E.; Bainbridge, A.; Herbert, L.; Scaravilli, F.; Peebles, D.; Wyatt, J.S.; Cady, E.B.; et al. “Therapeutic time window” duration decreases with increasing severity of cerebral hypoxia-ischaemia under normothermia and delayed hypothermia in newborn piglets. Brain Res. 2007, 1154, 173–180. [Google Scholar] [CrossRef]
- Koehler, R.C.; Yang, Z.J.; Lee, J.K.; Martin, L.J. Perinatal hypoxic-ischemic brain injury in large animal models: Relevance to human neonatal encephalopathy. J. Cereb. Blood Flow Metab. 2018, 38, 2092–2111. [Google Scholar] [CrossRef]
- Davidson, J.O.; Draghi, V.; Whitham, S.; Dhillon, S.K.; Wassink, G.; Bennet, L.; Gunn, A.J. How long is sufficient for optimal neuroprotection with cerebral cooling after ischemia in fetal sheep? J. Cereb. Blood Flow Metab. 2018, 38, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Remzső, G.; Németh, J.; Tóth-Szűki, V.; Varga, V.; Kovács, V.; Domoki, F. NMDA attenuates the neurovascular response to hypercapnia in the neonatal cerebral cortex. Sci. Rep. 2019, 9, 18900. [Google Scholar] [CrossRef] [PubMed]
- Inouye, T.; Shinosaki, K.; Sakamoto, H.; Toi, S.; Ukai, S.; Iyama, A.; Katsuda, Y.; Hirano, M. Quantification of EEG irregularity by use of the entropy of the power spectrum. Electroencephalogr. Clin. Neurophysiol. 1991, 79, 204–210. [Google Scholar] [CrossRef]
- Zhang, A.; Yang, B.; Huang, L. Feature Extraction of EEG Signals Using Power Spectral Entropy. In Proceedings of the 2008 International Conference on BioMedical Engineering and Informatics, Sanya, China, 27–30 May 2008; IEEE: Piscataway, NJ, USA, 2008; Volume 2, pp. 435–439. [Google Scholar]
- Muttitt, S.C.; Taylor, M.J.; Kobayashi, J.S.; MacMillan, L.; Whyte, H.E. Serial visual evoked potentials and outcome in term birth asphyxia. Pediatr. Neurol. 1991, 7, 86–90. [Google Scholar] [CrossRef]
- Feng, J.J.; Wang, W.P.; Guo, S.J.; Liu, Z.W.; Xu, X. Flash visual evoked potentials in preterm infants. Ophthalmology 2013, 120, 489–494. [Google Scholar] [CrossRef]
- Woods, J.R.; Coppes, V.; Brooks, D.E.; Knowles, P.J.; Freeman, M.; Parisi, V.; Omara, P.; Mccarty, G.E. Birth Asphyxia. I. Measurement of Visual Evoked Potential (VEP) in the Healthy Fetus and Newborn Lamb. Pediatr. Res. 1981, 15, 1429–1432. [Google Scholar] [CrossRef] [Green Version]
- Cainelli, E.; Trevisanuto, D.; Cavallin, F.; Manara, R.; Suppiej, A. Evoked potentials predict psychomotor development in neonates with normal MRI after hypothermia for hypoxic-ischemic encephalopathy. Clin. Neurophysiol. 2018, 129, 1300–1306. [Google Scholar] [CrossRef]
- Ichihara, M.; Sobue, S.; Ito, M.; Ito, M.; Hirayama, M.; Ohno, K. Beneficial biological effects and the underlying mechanisms of molecular hydrogen—Comprehensive review of 321 original articles. Med. Gas Res. 2015, 5, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Iketani, M.; Ohsawa, I. Molecular Hydrogen as a Neuroprotective Agent. Curr. Neuropharmacol. 2016, 15, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Tolaymat, Y.; Doré, S.; Griffin, H.W.; Shih, S.; Edwards, M.E.; Weiss, M.D. Inhaled Gases for Neuroprotection of Neonates: A Review. Front. Pediatr. 2020, 7, 558. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, D.; Chen, S. Hydrogen water reduces NSE, IL-6, and TNF-α levels in hypoxic-ischemic encephalopathy. Open Med. 2016, 11, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Kang, Z.; Liu, K.; Liu, W.W.; Li, R.P.; Zhang, J.H.; Luo, X.; Sun, X. Neuroprotective effects of hydrogen saline in neonatal hypoxia-ischemia rat model. Brain Res. 2009, 1256, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Liu, S.; Yuan, L.; Xie, Y.; Li, T.; Wang, L.; Wang, X.; Zhang, T.; Qin, S.; Song, G.; et al. Hydrogen-rich saline mediates neuroprotection through the regulation of endoplasmic reticulum stress and autophagy under hypoxia-ischemia neonatal brain injury in mice. Brain Res. 2016, 1646, 410–417. [Google Scholar] [CrossRef]
- Chu, X.; Cao, L.; Yu, Z.; Xin, D.; Li, T.; Ma, W.; Zhou, X.; Chen, W.; Liu, D.; Wang, Z. Hydrogen-rich saline promotes microglia M2 polarization and complement-mediated synapse loss to restore behavioral deficits following hypoxia-ischemic in neonatal mice via AMPK activation. J. Neuroinflamm. 2019, 16, 104. [Google Scholar] [CrossRef]
- Wu, G.; Chen, Z.; Wang, P.; Zhao, M.; Fujino, M.; Zhang, C.; Zhou, W.; Hirano, S.I.; Li, X.K.; Zhao, L. Hydrogen inhalation protects hypoxic–ischemic brain damage by attenuating inflammation and apoptosis in neonatal rats. Exp. Biol. Med. 2019, 244, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, R.C.; Towfighi, J.; Heitjan, D.F.; Brucklacher, R.M. Carbon Dioxide Protects the Perinatal Brain From Hypoxic-Ischemic Damage: An Experimental Study in the Immature Rat. Pediatrics 1995, 95, 868–874. [Google Scholar]
- Vannucci, R.C.; Towfighi, J.; Brucklacher, R.M.; Vannucci, S.J. Effect of extreme hypercapnia on hypoxic-ischemic brain damage in the immature rat. Pediatr. Res. 2001, 49, 799–803. [Google Scholar] [CrossRef] [Green Version]
- Randall, G.C. The relationship of arterial blood pH and pCO2 to the viability of the newborn piglet. Can. J. Comp. Med. 1971, 35, 141–146. [Google Scholar]
- Randall, G.C.B. Studies on the effect of acute asphyxia on the fetal pig in utero. Biol. Neonate 1979, 36, 63–69. [Google Scholar] [CrossRef]
- Engle, W.D.; Laptook, A.R.; Perlman, J.M. Acute changes in arterial carbon dioxide tension and acid-base status and early neurologic characteristics in term infants following perinatal asphyxia. Resuscitation 1999, 42, 11–17. [Google Scholar] [CrossRef]
- Remzső, G.; Németh, J.; Varga, V.; Kovács, V.; Tóth-Szűki, V.; Kaila, K.; Voipio, J.; Domoki, F. Brain interstitial pH changes in the subacute phase of hypoxic-ischemic encephalopathy in newborn pigs. PLoS ONE 2020, 15, e0233851. [Google Scholar] [CrossRef] [PubMed]
- Pappas, A.; Shankaran, S.; Laptook, A.R.; Langer, J.C.; Bara, R.; Ehrenkranz, R.A.; Goldberg, R.N.; Das, A.; Higgins, R.D.; Tyson, J.E.; et al. Hypocarbia and adverse outcome in neonatal hypoxic-ischemic encephalopathy. J. Pediatr. 2011, 158, 752–758.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E.S.; Cheung, P.-Y.; O’Reilly, M.; LaBossiere, J.; Lee, T.-F.; Cowan, S.; Bigam, D.L.; Schmölzer, G.M. Exhaled CO2 Parameters as a Tool to Assess Ventilation-Perfusion Mismatching during Neonatal Resuscitation in a Swine Model of Neonatal Asphyxia. PLoS ONE 2016, 11, e0146524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szakmar, E.; Kovacs, K.; Meder, U.; Bokodi, G.; Andorka, C.; Lakatos, A.; Szabo, A.J.; Belteki, G.; Szabo, M.; Jermendy, A. Neonatal encephalopathy therapy optimization for better neuroprotection with inhalation of CO2: The HENRIC feasibility and safety trial. Pediatr. Res. 2020, 87, 1025–1032. [Google Scholar] [CrossRef]
- Helmy, M.M.; Tolner, E.A.; Vanhatalo, S.; Voipio, J.; Kaila, K. Brain alkalosis causes birth asphyxia seizures, suggesting therapeutic strategy. Ann. Neurol. 2011, 69, 493–500. [Google Scholar] [CrossRef]
- Tolner, E.A.; Hochman, D.W.; Hassinen, P.; Otáhal, J.; Gaily, E.; Haglund, M.M.; Kubová, H.; Schuchmann, S.; Vanhatalo, S.; Kaila, K. Five percent CO2 is a potent, fast-acting inhalation anticonvulsant. Epilepsia 2011, 52, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Ito, M.; Ohsawa, I. Molecular hydrogen protects against oxidative stress-induced SH-SY5Y neuroblastoma cell death through the process of mitohormesis. PLoS ONE 2017, 12, e0176992. [Google Scholar] [CrossRef] [Green Version]
- Han, B.H.; Holtzman, D.M. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J. Neurosci. 2000, 20, 5775–5781. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Liu, X.; Zhang, N.; Zhu, X.; Liang, H.; Sun, L.; Cheng, Y. Neuroprotective Effects of Brain-Derived Neurotrophic Factor and Noggin-Modified Bone Mesenchymal Stem Cells in Focal Cerebral Ischemia in Rats. J. Stroke Cerebrovasc. Dis. 2016, 25, 410–418. [Google Scholar] [CrossRef]
- Kovács, V.; Tóth-Szűki, V.; Németh, J.; Varga, V.; Remzső, G.; Domoki, F. Active forms of Akt and ERK are dominant in the cerebral cortex of newborn pigs that are unaffected by asphyxia. Life Sci. 2018, 192, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zheng, Z.; Zhu, X.; Shi, Y.; Tian, D.; Zhao, F.; Liu, N.; Hüppi, P.S.; Troy, F.A.; Wang, B. Lactoferrin Promotes Early Neurodevelopment and Cognition in Postnatal Piglets by Upregulating the BDNF Signaling Pathway and Polysialylation. Mol. Neurobiol. 2015, 52, 256–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, P.; Hua, R.; Zhang, Y.; Zhao, H.; Tang, Z.; Mei, X.; Zhang, M.; Cui, J.; Li, C. Hypothermia-induced neuroprotection is associated with reduced mitochondrial membrane permeability in a swine model of cardiac arrest. J. Cereb. Blood Flow Metab. 2013, 33, 928–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Armstrong, J.S.; Lee, J.H.; Bhalala, U.; Kulikowicz, E.; Zhang, H.; Reyes, M.; Moy, N.; Spicer, D.; Zhu, J.; et al. Rewarming from therapeutic hypothermia induces cortical neuron apoptosis in a swine model of neonatal hypoxic-ischemic encephalopathy. J. Cereb. Blood Flow Metab. 2015, 35, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Wassink, G.; Draghi, V.; Dhillon, S.K.; Bennet, L.; Gunn, A.J. Limited benefit of slow rewarming after cerebral hypothermia for global cerebral ischemia in near-term fetal sheep. J. Cereb. Blood Flow Metab. 2019, 39, 2246–2257. [Google Scholar] [CrossRef]
- Tsuchida, T.N.; Wusthoff, C.J.; Shellhaas, R.A.; Abend, N.S.; Hahn, C.D.; Sullivan, J.E.; Nguyen, S.; Weinstein, S.; Scher, M.S.; Riviello, J.J.; et al. American clinical neurophysiology society standardized EEG terminology and categorization for the description of continuous eeg monitoring in neonates: Report of the american clinical neurophysiology society critical care monitoring committee. J. Clin. Neurophysiol. 2013, 30, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Shellhaas, R.A.; Chang, T.; Tsuchida, T.; Scher, M.S.; Riviello, J.J.; Abend, N.S.; Nguyen, S.; Wusthoff, C.J.; Clancy, R.R. The American Clinical Neurophysiology Society’s Guideline on Continuous Electroencephalography Monitoring in Neonates. J. Clin. Neurophysiol. 2011, 28, 611–617. [Google Scholar] [CrossRef] [Green Version]
- American Clinical Neurophysiology Society. Guideline 9B: Guidelines on Visual Evoked Potentials. J. Clin. Neurophysiol. 2006, 23, 138–156. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 623–656. [Google Scholar] [CrossRef]
- Vakkuri, A.; Yli-Hankala, A.; Talja, P.; Mustola, S.; Tolvanen-Laakso, H.; Sampson, T.; Viertiö-Oja, H. Time-frequency balanced spectral entropy as a measure of anesthetic drug effect in central nervous system during sevoflurane, propofol, and thiopental anesthesia. Acta Anaesthesiol. Scand. 2004, 48, 145–153. [Google Scholar] [CrossRef]
- Delorme, A.; Makeig, S. EEGLAB: An open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J. Neurosci. Methods 2004, 134, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Wiesenburg, D.A.; Guinasso, N.L. Equilibrium Solubilities of Methane, Carbon Monoxide, and Hydrogen in Water and Sea Water. J. Chem. Eng. Data 1979, 24, 356–360. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Score | Morphology of Cortical Damage | Ratio of the Most Severe Pattern per Area |
|---|---|---|
| 0 | No damage | |
| 1 | Scattered | <20% |
| 2 | 21–50% | |
| 3 | >50% | |
| 4 | Grouped | <20% |
| 5 | 21–50% | |
| 6 | >50% | |
| 7 | Panlaminar | <20% |
| 8 | 21–50% | |
| 9 | >50% | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, V.; Remzső, G.; Tóth-Szűki, V.; Varga, V.; Németh, J.; Domoki, F. Inhaled H2 or CO2 Do Not Augment the Neuroprotective Effect of Therapeutic Hypothermia in a Severe Neonatal Hypoxic-Ischemic Encephalopathy Piglet Model. Int. J. Mol. Sci. 2020, 21, 6801. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186801
Kovács V, Remzső G, Tóth-Szűki V, Varga V, Németh J, Domoki F. Inhaled H2 or CO2 Do Not Augment the Neuroprotective Effect of Therapeutic Hypothermia in a Severe Neonatal Hypoxic-Ischemic Encephalopathy Piglet Model. International Journal of Molecular Sciences. 2020; 21(18):6801. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186801
Chicago/Turabian StyleKovács, Viktória, Gábor Remzső, Valéria Tóth-Szűki, Viktória Varga, János Németh, and Ferenc Domoki. 2020. "Inhaled H2 or CO2 Do Not Augment the Neuroprotective Effect of Therapeutic Hypothermia in a Severe Neonatal Hypoxic-Ischemic Encephalopathy Piglet Model" International Journal of Molecular Sciences 21, no. 18: 6801. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186801