Heme Oxygenase-1 Supports Mitochondrial Energy Production and Electron Transport Chain Activity in Cultured Lung Epithelial Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
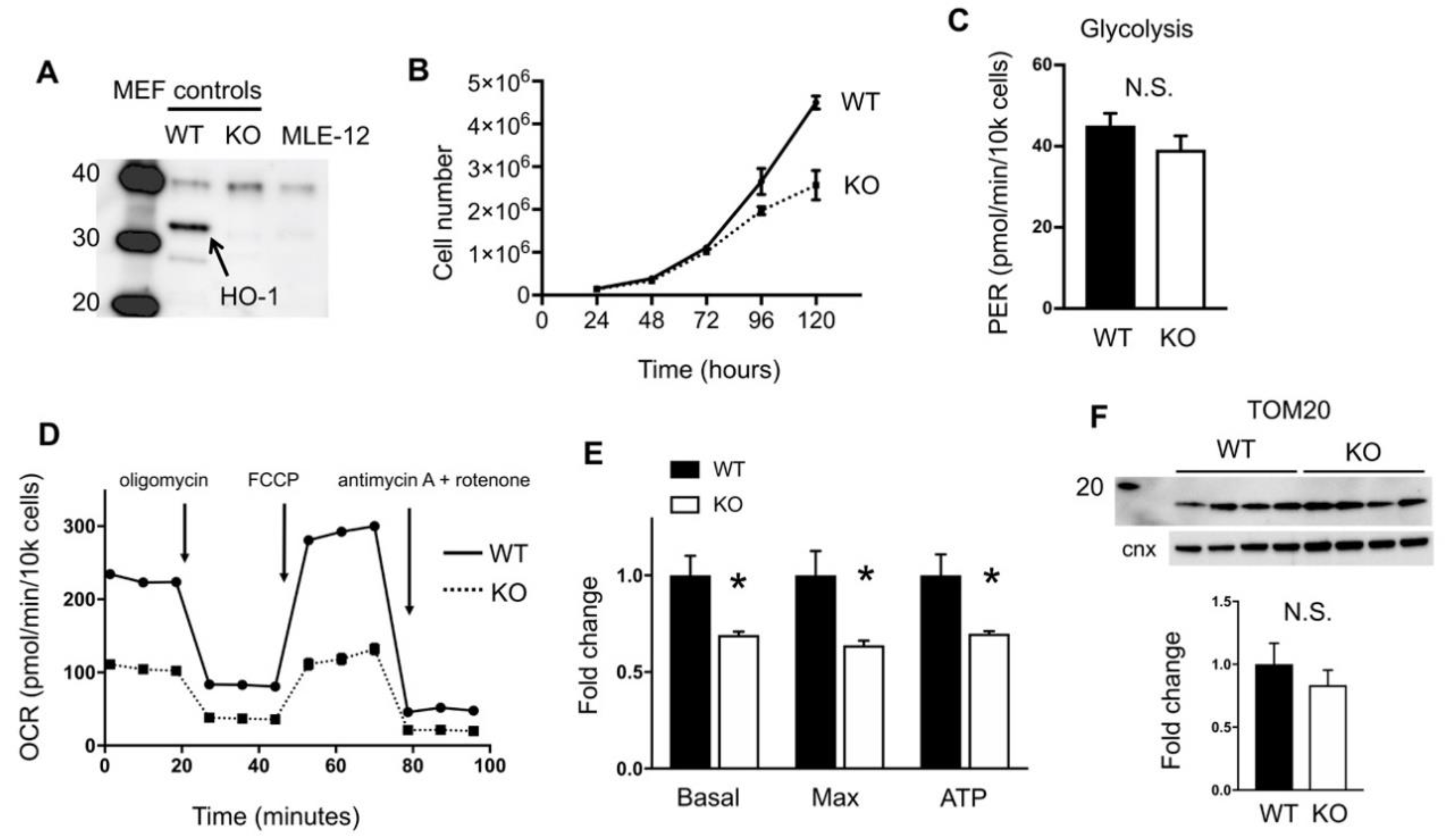
2.1. HO-1 Disruption Slows Growth and Reduces Oxidative Phosphorylation
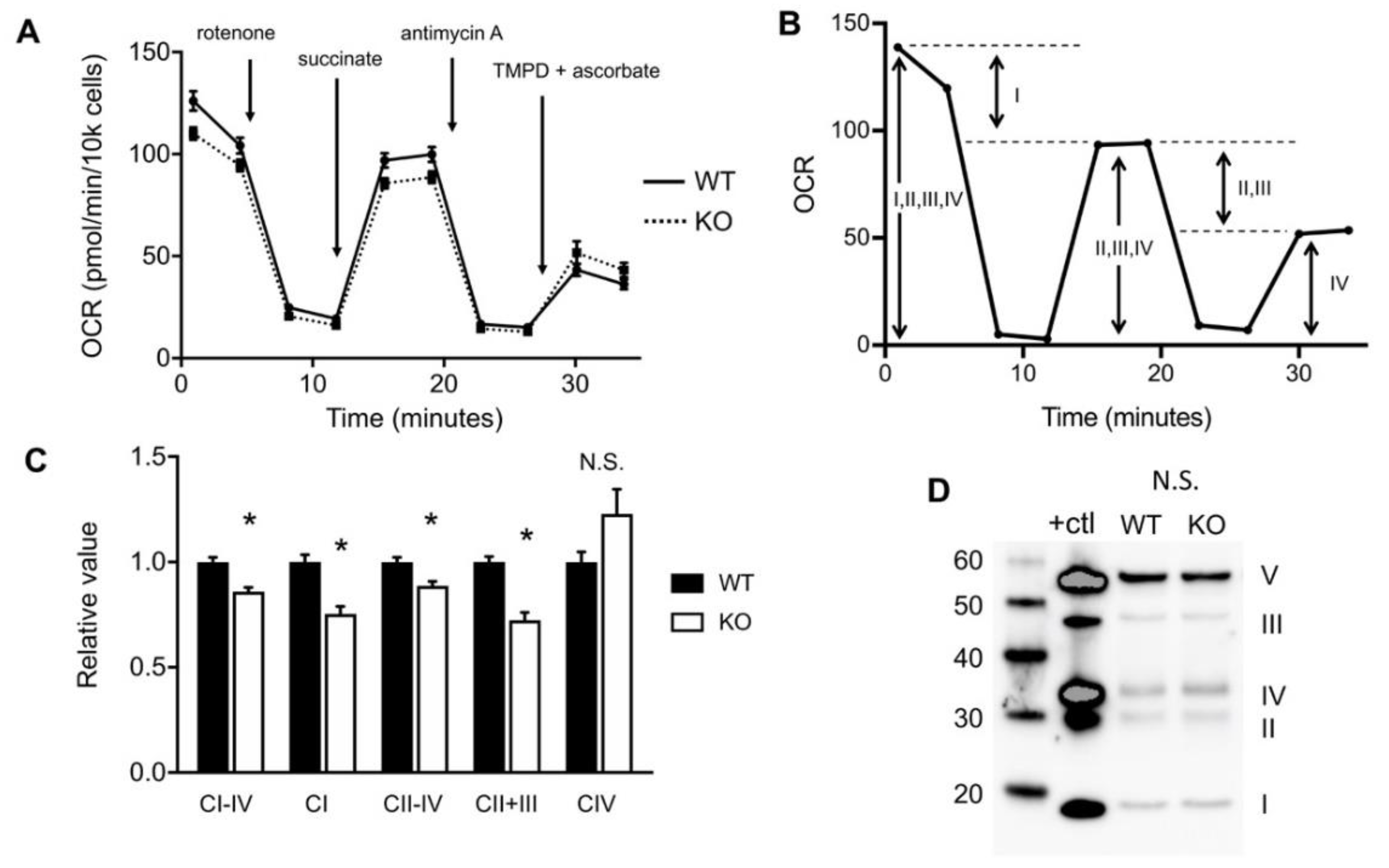
2.2. Loss of HO-1 Restricts Flow of Electrons in the Electron Transport Chain
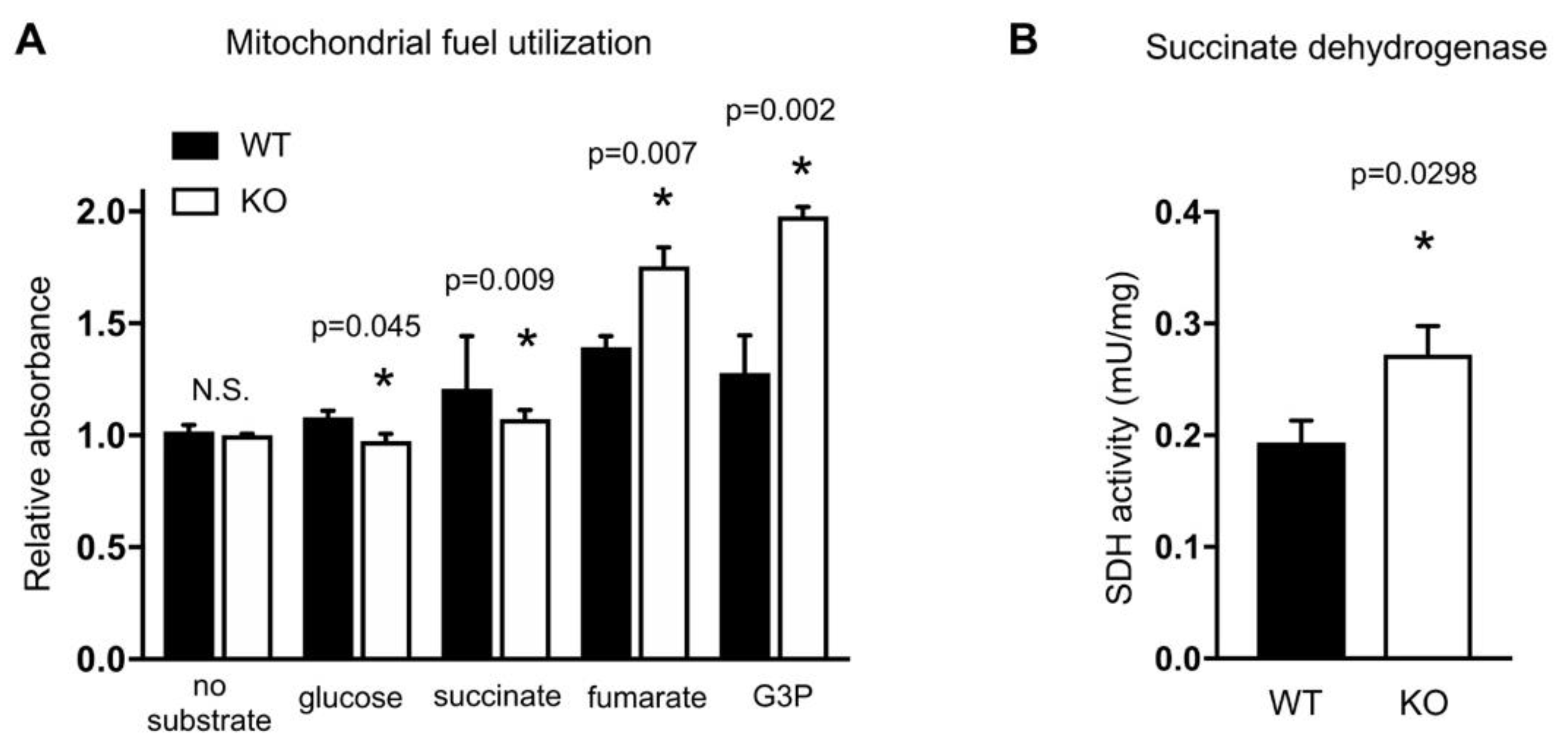
2.3. HO-1 Knockout Cells Have Altered Mitochondrial Fuel Utilization
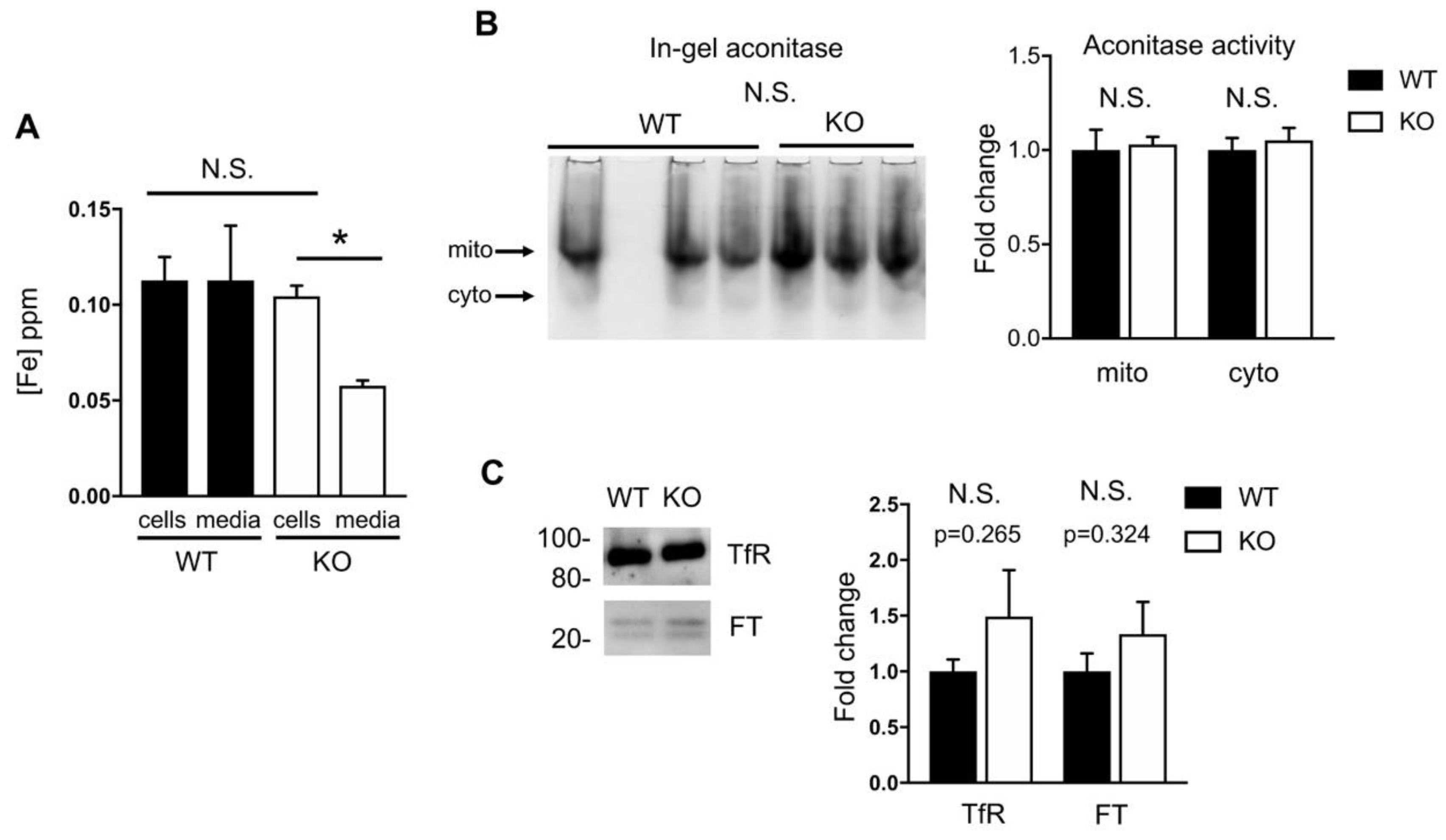
2.4. Iron Homeostasis in HO-1 Knockout MLE-12 Cells is Maintained
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Detection of Proteins
4.3. Mitochondrial Respiration and Glycolysis
4.4. Electron Flow
4.5. Biochemical Assays
4.6. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ETC | Electron transport chain |
| HO-1 | Heme oxygenase 1 |
| OCR | Oxygen consumption rate |
| Oxphos | Oxidative phosphorylation |
| ROS | Reactive oxygen species |
| SDH | Succinate dehydrogenase |
References
- Fraser, S.T.; Midwinter, R.G.; Berger, B.S.; Stocker, R. Heme Oxygenase-1: A Critical Link between Iron Metabolism, Erythropoiesis, and Development. Adv. Hematol. 2011, 2011, 473709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W.; Kim, H.P.; Nakahira, K.; Zuckerbraun, B.S.; Morse, D.; Choi, A.M.K. Protective Functions of Heme Oxygenase-1 and Carbon Monoxide in the Respiratory System. Antioxid. Redox Signal. 2007, 9, 2157–2174. [Google Scholar] [CrossRef]
- Dennery, P.A. Heme Oxygenase in Neonatal Lung Injury and Repair. Antioxid. Redox Signal. 2014, 21, 1881–1892. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Pittalà, V.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Arena, E.; Nabavi, S.M.; Salerno, L. Role of the Nrf2/HO-1 axis in bronchopulmonary dysplasia and hyperoxic lung injuries. Clin. Sci. 2017, 131, 1701–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, B.A.; Crapo, J.D. Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria. J. Biol. Chem. 1981, 256, 10986–10992. [Google Scholar] [PubMed]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [Green Version]
- Sassa, S. Why heme needs to be degraded to iron, biliverdin IXalpha, and carbon monoxide? Antioxid Redox Signal 2004, 6, 819–824. [Google Scholar]
- Abshire, J.R.; Rowlands, C.J.; Ganesan, S.M.; So, P.T.C.; Niles, J.C. Quantification of labile heme in live malaria parasites using a genetically encoded biosensor. Proc. Natl. Acad. Sci. USA 2017, 114, E2068–E2076. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, V.; Wagener, F.; Immenschuh, S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochem. Pharmacol. 2018, 153, 159–167. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Kim, S.F.; Ponka, P. Non-heme induction of heme oxygenase-1 does not alter cellular iron metabolism. J. Biol. Chem. 2007, 282, 10480–10486. [Google Scholar] [CrossRef] [Green Version]
- Hori, R.; Kashiba, M.; Toma, T.; Yachie, A.; Goda, N.; Makino, N.; Soejima, A.; Nagasawa, T.; Nakabayashi, K.; Suematsu, M. Gene Transfection of H25A Mutant Heme Oxygenase-1 Protects Cells against Hydroperoxide-induced Cytotoxicity. J. Biol. Chem. 2002, 277, 10712–10718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suttner, D.M.; Dennery, P.A. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999, 13, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.-H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, A.; Li, M.; Vanella, L.; Kim, N.H.; Rezzani, R.; Rodella, L.F.; Sodhi, K.; Canestraro, M.; Martasek, P.; Peterson, S.J.; et al. Adipocyte heme oxygenase-1 induction attenuates metabolic syndrome in both male and female obese mice. Hypertension 2010, 56, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, A.; Li, M.; Kim, N.H.; Peterson, S.J.; Vanella, L.; Positano, V.; Gastaldelli, A.; Rezzani, R.; Rodella, L.F.; Drummond, G.; et al. Heme oxygenase-1 induction remodels adipose tissue and improves insulin sensitivity in obesity-induced diabetic rats. Hypertension 2009, 53, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Di Noia, M.A.; Van Driesche, S.; Palmieri, F.; Yang, L.-M.; Quan, S.; Goodman, A.I.; Abraham, N. Heme oxygenase-1 enhances renal mitochondrial transport carriers and cytochrome C oxidase activity in experimental diabetes. J. Biol. Chem. 2006, 281, 15687–15693. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, K.T.; Chang, H.-C.; Ardehali, H. Role of heme in cardiovascular physiology and disease. J. Am. Heart Assoc. 2015, 4, e001138. [Google Scholar] [CrossRef] [Green Version]
- Converso, D.P.; Taillé, C.; Carreras, M.C.; Jaitovich, A.; Poderoso, J.J.; Boczkowski, J. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J. 2006, 20, 1236–1238. [Google Scholar] [CrossRef] [Green Version]
- Slebos, D.-J.; Ryter, S.W.; Van Der Toorn, M.; Liu, F.; Guo, F.; Baty, C.J.; Karlsson, J.M.; Watkins, S.C.; Kim, H.P.; Wang, X.; et al. Mitochondrial Localization and Function of Heme Oxygenase-1 in Cigarette Smoke–Induced Cell Death. Am. J. Respir. Cell Mol. Biol. 2007, 36, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Kurumada, T.; Labbe, R.F. Fumarate reductase in the control of heme biosynthesis. Science 1966, 151, 1228–1229. [Google Scholar] [CrossRef]
- Das, K.C. Hyperoxia Decreases Glycolytic Capacity, Glycolytic Reserve and Oxidative Phosphorylation in MLE-12 Cells and Inhibits Complex I and II Function, but Not Complex IV in Isolated Mouse Lung Mitochondria. PLoS ONE 2013, 8, e73358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dylag, A.M.; Haak, J.; Yee, M.; O’Reilly, M.A. Pulmonary mechanics and structural lung development after neonatal hyperoxia in mice. Pediatr. Res. 2020, 87, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear Heme Oxygenase-1 (HO-1) Modulates Subcellular Distribution and Activation of Nrf2, Impacting Metabolic andAnti-oxidant Defenses. J. Boil. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salabei, J.K.; Gibb, A.A.; Hill, B.G. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014, 9, 421–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mráček, T.; Drahota, Z.; Houštěk, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 2013, 1827, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef]
- Podkalicka, P.; Mucha, O.; Kruczek, S.; Biela, A.; Andrysiak, K.; Stepniewski, J.; Mikulski, M.; Gałęzowski, M.; Sitarz, K.; Brzózka, K.; et al. Synthetically Lethal Interactions of Heme Oxygenase-1 and Fumarate Hydratase Genes. Biomolecules 2020, 10, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, C.D.; Jaffrey, S.R.; Sawa, A.; Takahashi, M.; Brady, S.D.; Barrow, R.K.; Tysoe, S.A.; Wolosker, H.; Barañano, D.E.; Doré, S.; et al. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat. Cell Biol. 1999, 1, 152–157. [Google Scholar] [CrossRef]
- Tong, W.-H.; Rouault, T.A. Metabolic regulation of citrate and iron by aconitases: Role of iron-sulfur cluster biogenesis. Biometals 2007, 20, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P.; Beaumont, C.; Richardson, D.R. Function and regulation of transferrin and ferritin. Semin. Hematol. 1998, 35, 35–54. [Google Scholar] [PubMed]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1857, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- May, B.K.; Dogra, S.C.; Sadlon, T.J.; Bhasker, C.R.; Cox, T.C.; Bottomley, S.S. Molecular Regulation of Heme Biosynthesis in Higher Vertebrates. Prog. Nucleic Acid Res. Mol. Biol. 1995, 51, 1–51. [Google Scholar] [PubMed]
- Sassa, S.; Granick, S. Induction of -aminolevulinic acid synthetase in chick embryo liver cells in culture. Proc. Natl. Acad. Sci. USA 1970, 67, 517–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Hayashi, N.; Kikuchi, G. Evidence for the transcriptional inhibition by heme of the synthesis of delta-aminolevulinate synthase in rat liver. Biochem. Biophys. Res. Commun. 1982, 105, 985–990. [Google Scholar] [CrossRef]
- Srivastava, G.; Borthwick, I.A.; Brooker, J.D.; Wallace, J.C.; May, B.K.; Elliott, W.H. Hemin inhibits transfer of pre-delta-aminolevulinate synthase into chick embryo liver mitochondria. Biochem. Biophys. Res. Commun. 1983, 117, 344–349. [Google Scholar] [CrossRef]
- Gutman, M.; Kearney, E.B.; Singer, T.P. Regulation of succinate dehydrogenase activity by reduced coenzymes Q10. Biochemistry 1971, 10, 2726–2733. [Google Scholar]
- Hedblom, A.; Hejazi, S.M.; Canesin, G.; Choudhury, R.; Hanafy, K.A.; Csizmadia, E.; Persson, J.L.; Wegiel, B. Heme detoxification by heme oxygenase-1 reinstates proliferative and immune balances upon genotoxic tissue injury. Cell Death Dis. 2019, 10, 72. [Google Scholar] [CrossRef]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: Effects on macrophage viability and tissue iron distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef] [Green Version]
- Facchinetti, M.M. Heme-Oxygenase-1. Antioxid. Redox Signal. 2020, 32, 1239–1242. [Google Scholar] [CrossRef]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods Enzymol. 2014, 547, 309–354. [Google Scholar] [PubMed]
- Tong, W.-H.; Rouault, T.A. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006, 3, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carr, J.F.; Garcia, D.; Scaffa, A.; Peterson, A.L.; Ghio, A.J.; Dennery, P.A. Heme Oxygenase-1 Supports Mitochondrial Energy Production and Electron Transport Chain Activity in Cultured Lung Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 6941. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186941
Carr JF, Garcia D, Scaffa A, Peterson AL, Ghio AJ, Dennery PA. Heme Oxygenase-1 Supports Mitochondrial Energy Production and Electron Transport Chain Activity in Cultured Lung Epithelial Cells. International Journal of Molecular Sciences. 2020; 21(18):6941. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186941
Chicago/Turabian StyleCarr, Jennifer F., David Garcia, Alejandro Scaffa, Abigail L. Peterson, Andrew J. Ghio, and Phyllis A. Dennery. 2020. "Heme Oxygenase-1 Supports Mitochondrial Energy Production and Electron Transport Chain Activity in Cultured Lung Epithelial Cells" International Journal of Molecular Sciences 21, no. 18: 6941. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186941