The Role of Vesicle Trafficking Defects in the Pathogenesis of Prion and Prion-Like Disorders


1
Calgary Prion Research Unit, Department of Comparative Biology and Experimental Medicine, Faculty of Veterinary Medicine, University of Calgary, Calgary, AB T2N 4Z6, Canada
2
Hotchkiss Brain Institute, University of Calgary, Calgary, AB T2N 4Z6, Canada
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(19), 7016; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197016
Submission received: 20 August 2020
/
Revised: 15 September 2020
/
Accepted: 21 September 2020
/
Published: 23 September 2020
(This article belongs to the Special Issue Prions and Prion Diseases)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Prion diseases are fatal and transmissible neurodegenerative diseases in which the cellular form of the prion protein ‘PrPc’, misfolds into an infectious and aggregation prone isoform termed PrPSc, which is the primary component of prions. Many neurodegenerative diseases, like Alzheimer’s disease, Parkinson’s disease, and polyglutamine diseases, such as Huntington’s disease, are considered prion-like disorders because of the common characteristics in the propagation and spreading of misfolded proteins that they share with the prion diseases. Unlike prion diseases, these are non-infectious outside experimental settings. Many vesicular trafficking impairments, which are observed in prion and prion-like disorders, favor the accumulation of the pathogenic amyloid aggregates. In addition, many of the vesicular trafficking impairments that arise in these diseases, turn out to be further aggravating factors. This review offers an insight into the currently known vesicular trafficking defects in these neurodegenerative diseases and their implications on disease progression. These findings suggest that these impaired trafficking pathways may represent similar therapeutic targets in these classes of neurodegenerative disorders.
1. Prion Diseases
Prion diseases or transmissible spongiform encephalopathies are fatal spongiform neurodegenerative disorders in humans and in other mammals. The classic hallmark of prion diseases is the deposition of amyloid plaques in the brain, which leads to progressive neuronal loss, spongiosis, and astrogliosis [1,2]. They are caused by prions, proteinaceous infectious particles arising upon misfolding of the ubiquitously expressed cellular protein named PrPc into an insoluble and aggregation-prone conformer named PrPSc [3,4]. Prion diseases in humans can have three different etiologies—sporadic, familial or acquired by infection [5,6]. Most cases of human prion diseases are comprised of sporadic Creutzfeldt-Jakob disease (sCJD). Gerstmann Sträussler Scheinker syndrome and fatal familial insomnia (FFI) are inheritable forms of the disease, linked to specific mutations in the PRNP gene encoding the prion protein, e.g., P102L and D178N in combination with methionine at codon 129, respectively. Kuru, identified in some tribes in Papua New Guinea that practiced cannibalism, variant CJD (vCJD), and iatrogenic CJD are forms of the disease acquired by infection. Iatrogenic CJD transmissions have been described upon treatment with cadaveric growth hormone preparations contaminated with prions, as well as by corneal transplants from prion infected individuals [7,8]. Horizontal transmission of vCJD in humans has also been reported through blood transfusions [9]. The major clinical symptoms associated with prion diseases in humans include rapid cognitive decline and ataxia [10].
Historically, the first ever reported prion disease, as early as in the 18th century, is scrapie in sheep and goats [11]. Other animal prion diseases include bovine spongiform encephalopathy (BSE) in cattle and chronic wasting diseases in cervids, transmissible mink encephalopathy, feline spongiform encephalopathy, and the recently described camel prion disease [12,13]. Clinical signs vary depending on the prion strain and the genetic background of the animal but generally involve ataxia, hyperexcitability, and tremors. The zoonotic potential of prion diseases was manifested through the transmission of BSE to humans, giving rise to a novel form of human prion disease called vCJD, in the 1990s, mostly in the UK, claiming more than 200 lives [14,15].
2. Prion Conversion
The characteristic difference between PrPc and PrPSc is that PrPc is predominantly composed of alpha helices, while the infectious isoform PrPSc is composed of β-sheets [16]. The high β-sheet content accounts for the propensity of PrPSc to aggregate and its partial resistance to proteinase K digestion and cellular proteases. It has been proposed that the infectious PrPSc can acquire various conformational arrangements thought to be associated with the prion strain phenomenon [17]. These strains can transmit their particular conformation to the host’s PrPc leading to distinct disease features [18].
The mechanism of PrPc to PrPSc conversion is still ambiguous. Among the proposed models to explain the conversion of PrPc to PrPSc, the most favored model is the nucleated polymerization model [19]. The pathogenic conformer PrPSc forms oligomers, which act as the seed to convert the endogenous PrPc to its infectious conformer which is incorporated into the growing protofibrils and eventually into amyloid-like fibrils. As the fibrils grow, they break into smaller seeds which catalyze additional conversion and elongation of PrPSc fibrils. Hence, fibril fragmentation is the rate limiting step in the pathological process of prion replication [20,21]. Spagnolli et. al. put forward a template-assisted conversion model, where PrPSc exists in a 4-rung β-solenoid structure which acts as the template inducing the self-catalytic conversion of PrPc monomers into PrPSc protofibrils [22]. Many arguments exist in the field favoring the possibility of a cofactor that could aid in prion replication but, so far, no such absolute player has been identified [23,24,25]. The conundrum still remains as the synthesis of infectious prions in the absence of any co-factor has, so far, not been attained experimentally. However, endogenous phospholipids, such as phosphatidylethanolamine, and polyanions, like RNA molecules, have been used for in vitro generation of high titer infectious prions [26,27].
Although clinically and etiologically very diverse, a common characteristic between many neurodegenerative diseases is their causal association with protein misfolding and the aggregation of the respective proteins, such as α-synuclein in Parkinson’s disease (PD), β-amyloid, and tau in Alzheimer’s disease (AD), and huntingtin in Huntington’s diseases (HD), which leads to an impaired protein homeostasis [28]. Unlike these common neurodegenerative disorders, prion diseases occupy a unique position as they can be acquired by natural infection. Despite that the pathological proteins implicated in these diseases are very different, they all exhibit similar molecular mechanisms of pathological conversion, whereby the misfolded form of the protein can act as a seed and catalyze conformational changes of the endogenous protein, fibril elongation and formation of amyloid plaques [29,30,31]. Therefore, these diseases are referred to as prion-like disorders [32]. They also share other commonalities with prion diseases, in particular, cell-to-cell spread of misfolded protein aggregates. This transmission can take place by passive release of the protein aggregates via cell death or by vesicle-mediated exocytosis or exosomes [33,34]. Evidence also shows the existence of direct propagation from cell to cell by tunneling nanotubes [35,36].
3. PrPc Trafficking and Its Role in PrPSc Conversion
The cellular prion protein PrPc is a glycosyl-phosphatidyl-inositol-(GPI) anchored glycoprotein [37]. It is ubiquitously expressed and structurally highly conserved in vertebrates [38]. Endogenous expression of PrPc is highest in neurons, primarily at the synapses, and is more abundant in lymphoid tissue as compared to other tissues. Sub-cellularly, PrPc is predominantly enriched at the plasma membrane in the lipid rafts [39]. It consists of about 250 amino acids depending on the species, with an N-terminal signal peptide that targets it to the lumen of the endoplasmic reticulum (ER) in a co-translational manner. The C-terminal of the protein is modified by the addition of the GPI-anchor, mediated by a C-terminal signal peptide which is cleaved off. The mature form of PrPc that is released from the ER consists of about 209 amino acids, harbors one disulfide bond and is N-glycosylated at two sites. PrPc has a highly conserved hydrophobic domain that connects the flexible unstructured N-terminal domain containing five to six proline and glycine rich copper binding octapeptide repeats with the globular C-terminal domain containing three alpha helices and two short beta strands [40,41]. Like other secretory proteins, PrPc is transported through the Golgi complex, undergoing extensive post translational modifications, such as sialylation, and is anchored to the outer leaflet of the plasma membrane [42,43]. PrPc internalization can occur via clathrin- or caveolae-mediated endocytosis. Clathrin-mediated endocytosis is mediated by non-canonical interaction with the clathrin adaptable proteins, such as LRP1 (low density lipoprotein receptor related protein 1), in which the N-terminal of PrPc is crucial for internalization [44,45,46]. It can then be recycled back to the plasma membrane via recycling endosomes [44]. Caveolae-mediated endocytosis of PrPc has been observed in microglia and neuroblastoma cell lines and is stimulated by copper binding [47]. PrPc ends up in lysosomes, where it is degraded [48].
A seminal study showed that Prnp knockout mice are resistant to scrapie infection, which indicates that the expression of PrPc is the crucial factor necessary for prion propagation [49,50,51]. Furthermore, it is known that the two isoforms directly interact to result in the re-folding of PrPc to PrPSc. Therefore, subcellular trafficking and co-localization of both isoforms in specific subcellular compartments are important to facilitate their physical contact. PrPSc is localized at the plasma membrane and in different intracellular compartments including early endosomes, recycling endosomes, multi vesicular bodies (MVBs)/late endosomes, lysosomes, and perinuclear Golgi regions, coherent with the localization of PrPc [52,53]. Altering the trafficking of PrPc and PrPSc, hence preventing its interactions at the sites of conversions, can be deployed to reduce prion propagation.
Previous studies using mostly persistently prion-infected neuronal cell lines have already indicated that the cell surface localization of PrPc, cholesterol levels, and the endocytic pathway are important for prion conversion (Figure 1). Lowering the temperature to inhibit the endocytosis of PrPc reduces PrPSc levels, manifesting the role of the endocytic pathway in prion conversion [54]. Preventing the lipid raft localization of PrPc by modifying the C-terminal targeting sequence, lowering cellular cholesterol levels, or retaining it in the post ER-Golgi compartment reduces PrPSc propagation, implicating the necessity of the localization of PrPc in the lipid rafts for PrPSc propagation. Cleaving off the surface exposed PrPc with the enzyme phosphatidylinositol-specific phospholipase C (PIPLC) also inhibits prion conversion by interfering with PrPc–PrPSc interactions at the plasma membrane [55,56,57]. Interestingly, transgenic mice expressing a secreted form of PrPc lacking the GPI-anchor accumulate infectious prion plaques in the brain without developing the characteristic clinical scrapie symptoms [58]. Only when higher levels of the GPI-anchorless PrP are expressed, these mice develop prion disease with unique clinical signs [59]. The co-expression of GPI-anchored and GPI-anchorless PrPc accelerates clinical disease, indicating that the GPI-mediated membrane attachment of PrPc may be required for transmitting the toxic signals [58]. PrPc and PrPSc interacting receptors, like glycosaminoglycans (GAGs), laminin receptor precursor (LRP/LR), and LRP1, have a prominent role in the conversion process. It has been demonstrated that cultured cells harboring reduced amounts or modified GAGs, such as heparin sulfate, are resistant to primary prion infection [60,61]. Interfering with lipid raft formation by drugs, such as lovastatin, filipin, and amphotericin B, was capable of reducing PrPSc levels in neuroblastoma cell lines persistently infected with scrapie prion strains, thus emphasizing the role of lipid rafts in PrPSc conversion [47,55,62]. Altering the sub-cellular localization of cholesterol from the plasma membrane to the lysosomes by treating prion-infected neuroblastoma cells with the drug U18666A reduces PrPSc levels. This reduction is restored by rescuing the trafficking of cholesterol to the trans-Golgi network and subsequently to the plasma membrane by over-expression of the Rab9 protein [63].
Several studies modulating endocytic vesicular trafficking demonstrate a reduction in PrPSc levels suggesting multivesicular bodies (MVBs) and endocytic-recycling compartments (ERC) as the intracellular sites of pathogenic prion conversion [53,64,65,66]. Yim et al. [65] show that the knock-down of proteins required for the maturation of MVBs, such as Rab7, and components of the endosomal sorting complexes required for transport (ESCRT) machinery, such as Hrs and Tsg101, resulted in a reduction of PrPSc levels. Knock-down of proteins that are part of the retromer complex, such as vacuolar protein sorting-associated protein 26 (Vps 26), which retrieves cargo away from MVBs resulted in increased PrPSc levels, supporting the conclusion that MVBs could be the site of PrPSc conversion [65]. Manipulating the levels of proteins that are involved in the endocytic recycling also alters PrPSc levels, suggesting that the pathogenic conversion of PrPc can also occur here. Over-expression of dominant negative forms of Rab4 involved in rapid recycling of early endosomes to the plasma membrane does not affect PrPSc, suggesting that this pathway is dispensable for conversion. However, overexpression of a dominant-negative Rab11, a protein critical for slow cargo recycling to the plasma membrane via recycling endosome increases PrPSc levels indicating that the endocytic recycling compartment (ERC) is a site of prion conversion [53,67]. The mode of PrPSc internalization differs depending on the prion strain and the stage of infection [68], raising the question whether all prion strains adopt to the same site for prion conversion. Eventually, the majority of PrPSc accumulates in the lysosomes, where it is partially degraded [53,64].
There is still a knowledge gap about how and what factors could be involved in the cellular conversion of PrPc into the infectious PrPSc isoform. The evidence of the involvement of more than one sub-cellular site in prion conversion as discussed above also questions whether this conversion is a sequential process, where the byproducts of the processing of PrPc/PrPSc at the plasma membrane, MVBs and ERC act hand in hand in the formation of the non-degradable PrPSc that accumulates in the lysosomes. Furthermore, the existence of different strains makes it challenging to define a specific site of conversion, considering the likely existence of strain-specific preferences. In vivo, there is not always a strong correlation between PrPSc deposition and cell death. Follicular dendritic cells in the lymphoid tissues [69] and astrocytes [70] can efficiently amplify PrPSc but does not lead to their cell death. In fact, depleting, specifically, the neuronal prion protein in transgenic mice can prevent PrPSc deposition in the neurons and hinder the clinical manifestations of the disease, such as neurodegeneration, and spongiform changes, despite an effective propagation of PrPSc in other non-neuronal cell lines [71].
Most of the discussed studies were performed in persistently or acutely prion-infected cell lines of neuronal, but also of non-neuronal, origin, which have been established as valuable models to study the cell biology of prion proteins, while, only in a few cases, these findings were confirmed using primary neurons. Limitations of the cell line models of prion propagation include that they do not exhibit neurodegeneration and do not reflect the diversity of neurons as observed in the brain.
Tissue tropism of prion strains is another area that is difficult to study with the existing cell models. This phenomenon is exemplified by the drowsy and the hyper strains of hamster-adapted transmissible mink encephalopathy [72]. The drowsy strain is more restricted to the central nervous system (CNS) and has a lower capability to propagate in the lymphoreticular cells, probably because of the existence of a more efficient clearance mechanism for this strain in the lymphoreticular system [73,74]. However, the hyper strain has a more diverse distribution and can be detected in the CNS, lymphoreticular system, skeletal muscle, nasal secretions, and blood [75,76,77].
Currently, no lymphatic cell-type model exists to study prion replication. Such a model representing another major tissue type propagating prions in an infected host would be desirable to investigate and compare the requirements for and the consequences of prion propagation in different cell types.
4. Consequence of Prion Infection on Vesicular Trafficking
While the above studies demonstrate that the transport and the localization of PrPc and PrPSc to specific sub-cellular compartments are critical for prion propagation, there is evidence that prion infection or the presence of PrPSc aggregates in intracellular vesicles negatively impact and alter endocytic vesicle trafficking.
4.1. Rab GTPases—Coordinators of Vesicular Trafficking
Endocytic vesicular trafficking includes all processes from the internalization of cargo in the form of vesicles from the plasma membrane to the sorting and the transport of these vesicles into different endocytic compartments—to be retrieved by the Golgi complex, or recycled back to the plasma membrane or transported to the lysosomes for degradation. The master regulators in these pathways which help in the trafficking of vesicular cargo to different endocytic compartments and give specific organellar identity to these endocytic compartments are a family of proteins called the Rab GTPases. Rab GTPases, in coordination with their effector proteins and the Sar/Arf family of GTPases, regulate cargo selection and vesicle formation [78].
Rabs are monomeric GTPases and the largest branch of proteins in the Ras superfamily of GTPases. They have a G-domain which can bind guanosine nucleotides and the C-terminal membrane targeting domain is prenylated. Prenylation enables reversible insertion of these proteins into membranes, allowing them to exert their function. Most organelles in the endo-membrane system, like the ER, Golgi, endosomes, and lysosomes, are defined by the presence of characteristic Rab proteins on their surfaces facing the cytosol which facilitate their interaction with other organelle-specific proteins. These include but are not limited to Rab-bound motor proteins and Rab-bound tethering molecules [79,80]. Rab proteins exert their function when activated by binding to GTP, which is mediated by the guanine nucleotide exchange factors (GEFs). The GEF-mediated nucleotide exchange is the primary event that triggers the localization of Rabs to the membranes facilitating interaction with other effector proteins. They are inactive when in a GDP-bound state, and inactivation involves the hydrolysis of GTP through their inherent GTPase activity, which is enhanced by the guanine activating factors (GAP). The GDP-bound Rab proteins are extracted from the membrane and prevented from further activation cycles by their association with GDP Dissociation Inhibitor (GDI) until the GDI displacement factor (GDF) displaces the GDIs and recruits the Rab protein to the membrane for another cycle of activation [81] (Figure 2).
4.2. Rab7
In the endo-lysosomal system, early endosomes are marked by the presence of Rab5 and as they mature to late endosomes and lysosomes, characterized by the increased luminal acidification; there is also an increased recruitment of Rab7 which displaces Rab5 [82]. Rab7 is the main player in the late endosomal maturation and transport along with retromer regulation (Figure 3). The Rab7 effector Rab7 interacting lysosomal protein (RILP) interacts with the HOPS (Homotypic fusion and Protein Sorting) complex to mediate endosome-lysosome fusion. The Rab7-RILP interaction also regulates the assembly and the function of the Vacuolar-ATPase (V-ATPase) for lysosomal acidification. Rab7, by differentially interacting with RILP and FYVE and coiled-coil domain autophagy adaptor-1 (FYCO1), mediates the minus and plus end directed transport of late endosomes, respectively. The retrograde transport of transmembrane cargo to the trans-Golgi network is also regulated by the interaction between Rab7 and sub-units of the retromer complex especially Vps 35 [83].
It is reasonable to connote that the presence of PrPSc aggregates in the endocytic vesicles could disturb the vesicular trafficking pathways in the affected cells. Even though the importance of subcellular trafficking of PrPc and PrPSc in sustaining prion infection has been demonstrated by numerous studies, the impact of prion infection on vesicle trafficking is less clear. Here, we showcase some of the known trafficking impairments identified in prion diseases and draw a contrast on similar trafficking defects known in other prion-like disorders. More importantly, it is a tempting question whether the trafficking defects observed are the cause or the effect of PrPSc propagation and neuronal cell death in these neurodegenerative diseases.
5. Prion Related Vesicular Trafficking Defects
Vesicular trafficking defects associated with pathological changes have been very clearly observed in CJD brains in the histopathological studies—such as enlargement of Rab5 positive early endosomes in regions with mild pathology and enlarged cathepsin D and B immunopositive lysosomes in regions with moderate pathology [84]. Enlarged early endosomes may be due to the accumulation of vesicular cargoes because of impaired exit pathways towards retrograde trafficking and lysosomal degradation. Shim et al. show that the amount of membrane associated Rab7 is significantly reduced in prion infected neuronal cell lines [85]. This reduction in Rab7 could in turn have implications on its physiological functions in the cell where it acts as a key regulator in early-to-late endosomal transition, cargo sorting, lysosomal maturation, neurotrophin transport, lipid metabolism, and autophagy, to name a few of its direct functions [83]. The impaired lysosomal acidification and reduction in the efficiency of lysosomal degradation as reflected by the increased half-life of epidermal growth factor receptor (EGFR) upon prion infection can be linked to the reduced membrane associated Rab7 [85]. These findings further indicate a potential loss of other Rab7 functions, as well in prion-infected neurons (Figure 4).
Another vesicular trafficking pathway affected by prion infection is the retrograde trafficking as observed from the analysis of RNA expression profiles in sporadic CJD brains implicating down regulated gene expression, as well as reduced protein levels of vacuolar protein sorting-associated protein 35 (Vps 35), a subunit of the retromer complex [86]. Prion infection also disturbs post-Golgi trafficking of proteins, like attractin and the α-subunit of insulin receptor (IRα), in 22L and RML/chandler prion infected neuroblastoma cell lines [87]. Reduced levels of functional Rab11 protein were observed in cell lines expressing a mutant prion protein associated with FFI. This was indeed due to the elevated expression levels of Rab GDI, causing an increased fraction of inactive cytosolic Rab11. The enlarged Golgi morphology observed in these mutant prion protein expressing cell lines reflect dysregulated post-Golgi trafficking, a process that is mediated by Rab11 [88].
Studies also show that Rab7 mediated axonal retrograde trafficking of cargo is impaired in motor neurons of prion infected mice. These impairments were observed at the onset of the clinical disease and were not related to neuronal death, allowing the correlation of this phenotype with the clinical signs of the disease, such as hindlimb paralysis and ataxia [89]. Rab5 and Rab7 control the endocytic sorting of neurotrophins, such as brain derived neurotrophic factor (BDNF), along the axonal retrograde pathway [90]. Since prion-infected mice do show a selective impairment in the axonal retrograde vesicle transport, which transport neurotrophins and their receptors, it is highly probable that the retrograde trafficking of BDNF in prion infection is affected and could be a factor leading to neurodegeneration [85,89].
6. Vesicular Trafficking Defects in Prion-Like Disorders
As in the case of prion diseases, one of the earliest characteristics associated with the pre-clinical stages of AD, even before any detectable amount of amyloid deposition is the enlargement of Rab5-positive early endosomes. The pro-protease and mature forms of cathepsin B and D were also detected in these enlarged endosomes, along with cation dependent mannose 6 phosphate receptor (MPR) depicting a failed sorting and trafficking of vesicular cargo along the endo-lysosomal pathway in these AD cases [91,92].
Another striking similarity is the Rab7 loss of function that has been implicated in the pathogenesis of prion diseases. It is interesting to note here that Rab7 recruits the retromer complex through its direct interaction with the N-terminal of Vps 35 [93], and another common trait observed in PD, AD, and sCJD are reduced Vps 35 levels. Mutations in leucine-rich repeat kinase 2 (LRRK2) are associated with late onset PD [94]. Pathogenic LRRK2 reduces Rab7 activity and Vps 35 levels, and consequently delays the transit of cargo from the late endosomes to the lysosomes, resulting in an impaired EGFR degradation [95,96]. Rab7 activation and consequently its membrane association are impaired in autosomal recessive early onset PD linked to a mutation in the parkin gene. This leads to a decreased endosomal tubulation and an impaired retromer pathway due to the lack of recruitment of Vps 35 and sorting nexin 1 (SNX1) in primary dermal fibroblast cells from PD patients carrying a homozygous mutation in the parkin gene [97]. Similarly, Vps 35 protein levels are reduced in post-mortem AD brains [98]. In vitro and in vivo studies revealed that the knock down of Vps 35 exacerbates Aβ formation [99,100]. BDNF is an important neurotrophin that has been extensively studied to play a role in the pathogenesis of AD [101]. It has also been shown that the mutant huntingtin alters the retrograde transport of BDNF-tropomysoin related kinase B (TrkB) complexes in the striatal dendrites which subsequently cause neurodegeneration [102,103,104].
Altogether, these studies highlight the role of a dysregulated vesicular trafficking in the pathogenesis of prion and prion like disorders. In general, the endocytic pathway is affected, resulting in an impeded transport of the vesicular cargo to the lysosomes for degradation and to the retromer pathway, where the cargo is retrieved from early endosomes for subsequent transport to the Golgi complex or to the plasma membrane. Among the vesicular cargo are factors, such as BDNF, that are critical for neuronal function, suggesting similarities in the hypothesized mechanisms of neurodegeneration. Hence, such similarities offer the opportunities for exploring common potential therapeutic targets to develop medications for the treatment of these neurodegenerative disorders. A limitation of the above discussed studies is that most of them have been carried out in cell lines or utilized brains from terminal disease stages. Therefore, these results do not account for a potential progressive pattern of a dysfunction in the vesicular trafficking machinery, which appeals for a time-course study to correlate the onset of vesicular trafficking defects with neurodegeneration. An advantage of the cell culture models is that they allow to explore the direct consequences of protein aggregation on the physiology of neurons which is critical to define strategies to counteract such impairments.
7. Therapeutic Strategies in Prion and Prion-Like Disorders
The pathogenicity and the infectivity of prion diseases are associated with the accumulation of PrPSc aggregates. One of the most promising strategies adopted are the methods that can prevent the interaction between the endogenous protein and its misfolded isoform. These include the down-regulation, re-routing, or intracellular retention of the cellular PrPc itself via RNA interference or peptide aptamers [105,106,107]. Another approach is re-routing the PrPc from the potential sites of conversion by using drugs, such as suramin, which retain them in the Golgi and activate post-ER quality control mechanisms [56]. Sulfated glycans, such as pentosan polysulfate (PPS) and dextran sulfate, can prevent the formation of PrPSc in cell culture models [108] and scrapie-infected mice [109]. On contrary, in cell-free conversion models, sulfated glycans, such as PPS, can stimulate the formation of PrPSc [110]. These conflicting results can be explained by the different effects PPS has on the PrPc on the cell surface, as compared to the PrPc in in vitro conversion. Among the effects that PPS has in cultured cells is that it enhances the endocytosis of PrPc, thereby reducing the amount of the surface localized PrPc prone to conversion [111]. It also competitively inhibits the binding of PrPc with other GAGs or cofactors that can aid in the pathogenic conversion [108]. However, in in vitro conversion assays, sulfated glycans appear to act as cofactors, favoring conversion by inducing a conformational change in PrPc upon direct interaction [108]. Since lipid rafts are the main sites of PrPc localization, using cholesterol lowering drugs, such as lovastatin [55] and squalestatin [112], or cholesterol binding drugs, like filipin [47] and amphotericin B [62], can abrogate proper raft formation and hence reduce PrPSc formation.
Understanding the cellular biology of prion infection is crucial for devising therapeutic strategies for prion diseases. It has been demonstrated that prion propagation can only be sustained when the rate of PrPSc degradation does not exceed the rate of new conversion [113]. Autophagy is a major clearance mechanism of aggregated proteins in AD, PD, HD, and prion diseases [114], suggesting that stimulation of this pathway results in an enhanced degradation of the aggregated proteins. Treatment with resveratrol has shown to attenuate the aggregation of Aβ, α-synuclein, and mutant huntingtin in the animal models of AD, PD, and HD, respectively [115,116,117]. In prion diseases, a significant reduction in PrPSc aggregates has been attained by mammalian Target of rapamycin (mTOR) dependent or independent induction of autophagy by using small molecules, like imatinib mesylate [118,119], lithium, rapamycin, trehalose, and sirtuin [120,121,122]. Stimulating autophagy not only enhances PrPSc degradation but also prevents the exosomal release of PrPSc and hence the lateral transmission of prions to the neighboring cells [123].
An impairment seen in both prion and prion-like disorders is the trafficking defects in response to protein aggregation which possibly contribute to neurodegeneration. The retromer subunit Vps 35 is one of the most critical proteins involved in the retrograde trafficking. Vps 35 mutations or deficiencies have been implicated in several neurodegenerative diseases, such as AD, PD, and prion diseases. In mouse models of PD, an increased expression of Vps 35 reduces α-synuclein accumulation and also provide neuroprotection [124]. Pharmacological chaperones that stabilize the retromer complex and increase its half-life have been deployed as a therapeutic strategy to limit the pathogenic processing of amyloid precursor protein (APP) by directing it away from the endosomes in cultured hippocampal neurons [125]. Deficient levels of Vps 35 are also observed in prion diseases [86]. Reduced Vps 35 levels might serve as an impediment in the cell to degrade the PrPSc due to the vital role it plays in the lysosomal degradation pathway, as it is involved in the transport of lysosomal hydrolases, such as sortilin and cation independent 6 mannose phosphate receptor (CI-MPR) [126,127,128]. Vps 35 plays a major role in the trafficking of WASP and Scar homologue (WASH )complex and ATG9a protein, which has a role in the degradation pathways, such as macroautophagy [129]. It also regulates the chaperone mediated autophagy by retrieving the LAMP2A receptor to the lysosomes [130]. Hence, this calls for an active research in this unexplored area of the therapeutic potential of Vps 35 in prion diseases.
Ineffective lysosomal degradation is another important yet underexplored area regarding therapeutic solutions for these neurodegenerative diseases. Attenuated lysosomal degradation can be attributed to various factors, such as lysosomal rupture, due to the accumulation of α-synuclein aggregates in PD [131] or due to the incomplete maturation of the lysosomes [132]. Lysosomal maturation is associated with a decreasing pH gradient from early endosomes to lysosomes and also by their positioning in the neurons where they are more localized to the soma as compared to the distal parts of the axons [133]. Decreased lysosomal acidification related to Rab7 dysfunction has been reported in prion infection [85] and in early onset AD [134,135]. Abnormal transport and positioning of lysosomes have been implicated as one of the histopathological changes in AD and HD which might be a result of inefficient lysosomal maturation [91,136]. Since loss of LRRK2 or Rab7 also reduce Vps 35 levels in PD [96], it is reasonable to hypothesize that the loss of Rab7 could act as the upstream factor for the reduced levels of Vps 35 observed in AD and prion diseases. Along this line, it might be interesting to explore Rab7-based therapy as a potential treatment of these neurodegenerative diseases to combat impaired vesicular trafficking and lysosomal acidification. It has already been shown that cholesterol transport inhibiting amphiphiles, such as U18666A, reduce aberrant APP processing by retaining the localization of presinilins (PS) in Rab7-positive late endosomes. This shows that altering the sub-cellular cholesterol levels or Rab7 based therapeutic approaches to increase the transport of PS to Rab7-positive late endosomes can be adopted to reduce pathogenic Aβ processing [137]. Rab7 and Rab9 based approaches have already been adapted to correct lipid trafficking defects in Niemann-Pick type C (NP-C) lipid storage disease in fibroblast cell lines. By the over-expression of these proteins, it was possible to restore the normal trafficking of accumulated sphingolipids from the lysosomes to the Golgi [138]. Charcot-Marie Tooth 2B is an autosomal dominant neuropathy due to the partial loss of function of the Rab7 protein and stimulating enhanced Rab7 function may have utility for its treatment [139]. Rab7 based gene therapy approaches or small molecular interventions that can restore the normal physiological functions of the protein can be explored in the future as a cure to these above discussed diseases, including prion diseases [140].
8. Concluding Remarks
A very interesting point to ponder is whether the trafficking defects are the cause or an effect in these above discussed neurodegenerative diseases. In many spontaneous cases of prion diseases or AD, it appears that the amyloidogenic aggregates cause defects in trafficking without any obvious links to any known genetic mutations. On the other hand, many cases exist with a genetic link to the diseases, such as the mutation associated with LRRK2 in the late onset of PD; hence, it is a very pellucid fact that this defective mutation could affect vesicle trafficking and in fact trigger the onset of the disease, which subsequently results in the deposition of α-synuclein aggregates.
Regardless, manipulating, altering, or restoring the defective trafficking pathways in these neurodegenerative diseases is an arena that needs more active research, which could potentially open new doors to therapeutics and to understand the causal links of neurodegeneration associated with the vesicular trafficking defects.
Author Contributions
Conceptualization, P.C. and S.G.; writing—original draft preparation, P.C.; writing—review and editing, S.G.; visualization, P.C.; supervision, S.G.; project administration, S.G.; funding acquisition, P.C. and S.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Canada Research Chairs, Alberta Prion Research Institute: 201600035 to S.G. and University of Calgary Eyes High Graduate Student Fellowship to P.C.
Acknowledgments
We are grateful to Sheng Chun (Chris) Chang for proof-reading the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef]
- Cohen, F.E.; Pan, K.M.; Huang, Z.; Baldwin, M.; Fletterick, R.J.; Prusiner, S.B. Structural clues to prion replication. Science 1994, 264, 530–531. [Google Scholar] [CrossRef]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. Natural and experimental prion diseases of humans and animals. Curr. Opin. Neurobiol. 1992, 2, 638–647. [Google Scholar] [CrossRef]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtner, M.; Gelpi, E.; Kiechl, S.; Knoflach, M.; Zangerl, A.; Gotwald, T.; Willeit, J.; Maier, H.; Ströbel, T.; Unterberger, U. Iatrogenic Creutzfeldt–Jakob disease 22 years after human growth hormone therapy: Clinical and radiological features. J. Neurol. Neurosurg. Psychiatry 2008, 79, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Duffy, P.; Wolf, J.; Collins, G.; DeVoe, A.G.; Streeten, B.; Cowen, D. Possible Person-to-Person Transmission of Creutzfeldt-Jakob disease. N. Engl. J. Med. 1974, 290, 692–693. [Google Scholar]
- Peden, A.H.; Head, M.W.; Diane, L.R.; Jeanne, E.B.; James, W.I. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 2004, 364, 527–529. [Google Scholar] [CrossRef]
- Collinge, J. Prion diseases of humans and animals: Their causes and molecular basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.; Bradley, R. 1755 and all that: A historical primer of transmissible spongiform encephalopathy. BMJ 1998, 317, 1688–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigurdson, C.J.; Miller, M.W. Other animal prion diseases. Br. Med. Bull. 2003, 66, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Babelhadj, B.; Bari, M.A.D.; Pirisinu, L.; Chiappini, B.; Gaouar, S.B.S.; Riccardi, G.; Marco, S.; Agrimi, U.; Nonno, R.; Vacari, G. Prion Disease in Dromedary Camels, Algeria. Emerg. Infect. Dis. 2018, 24, 1029. [Google Scholar] [CrossRef] [Green Version]
- Will, R.G.; Ironside, J.W.; Zeidler, M.; Estibeiro, K.; Cousens, S.N.; Estibeiro, K.; Alperovitch, A.; Pocchiarri, M.; Hofman, A.; Smith, P.G. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996, 347, 921–925. [Google Scholar] [CrossRef]
- Lasmézas, C.I.; Deslys, J.-P.; Demaimay, R.; Adjou, K.T.; Lamoury, F.; Dormont, D.; Robain, O.; Ironside, J.; Hauw, J.J. BSE transmission to macaques. Nature 1996, 381, 743–744. [Google Scholar] [CrossRef]
- Vázquez-Fernández, E.; Vos, M.R.; Afanasyev, P.; Cebey, L.; Sevillano, A.M.; Vidal, E.; Rossa, I.; Renault, L.; Ramos, A.; Peters, P.J.; et al. The Structural Architecture of an Infectious Mammalian Prion Using Electron Cryomicroscopy. PLoS Pathogens. 2016, 12, e1005835. [Google Scholar] [CrossRef]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the Conformation of the Pathologic Isoform of the Prion Protein Enciphering and Propagating Prion Diversity. Science 1996, 274, 2079–2082. [Google Scholar] [CrossRef] [Green Version]
- Morales, R.; Abid, K.; Soto, C. The prion strain phenomenon: Molecular basis and unprecedented features. Biochim. Biophys. Acta 2007, 1772, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Lansbury, P.T.; Caughey, B. The chemistry of scrapie infection: Implications of the ‘ice 9′ metaphor. Chem. Biol. 1995, 2, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Caughey, B.W.; Dong, A.; Bhat, K.S.; Ernst, D.; Hayes, S.F.; Caughey, W.S. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 1991, 30, 7672–7680. [Google Scholar] [CrossRef]
- Stöhr, J.; Weinmann, N.; Wille, H.; Kaimann, T.; Nagel-Steger, L.; Birkmann, E.; Panza, G.; Prusiner, S.B.; Eigen, M.; Riesner, E.; et al. Mechanisms of prion protein assembly into amyloid. Proc. Natl. Acad. Sci. USA 2008, 105, 2409–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spagnolli, G.; Rigoli, M.; Orioli, S.; Sevillano, A.M.; Faccioli, P.; Wille, H.; Biasini, E.; Requena, J.R. Full atomistic model of prion structure and conversion. PLoS Pathog. 2019, 15, e1007864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abid, K.; Morales, R.; Soto, C. Cellular Factors Implicated in Prion Replication. FEBS Lett. 2010, 584, 2409–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.B.; Wang, D.W.; Wang, F.; Noble, G.P.; Ma, J.; Woods, V.L.; Li, S.; Supattapone, S. Cofactor Molecules Induce Structural Transformation During Infectious Prion Formation. Structure 2013, 21, 2061–2068. [Google Scholar] [CrossRef] [Green Version]
- Deleault, N.R.; Walsh, D.J.; Piro, J.R.; Wang, F.; Wang, X.; Ma, J.; Rees, J.R.; Supattapone, S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. USA 2012, 109, E1938–E1946. [Google Scholar] [CrossRef] [Green Version]
- Burke, C.M.; Walsh, D.J.; Steele, A.D.; Agrimi, U.; Bari, M.A.D.; Watts, J.C.; Supattapone, S. Full restoration of specific infectivity and strain properties from pure mammalian prion protein. PLoS Pathog. 2019, 15, e1007662. [Google Scholar] [CrossRef]
- Deleault, N.R.; Piro, J.R.; Walsh, D.J.; Wang, F.; Ma, J.; Geoghegan, J.C.; Supattapone, S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. USA 2012, 109, 8546–8551. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Matsuda, N. Proteostasis and neurodegeneration: The roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body–Like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the Inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [Green Version]
- Ren, P.-H.; Lauckner, J.E.; Kachirskaia, I.; Heuser, J.E.; Melki, R.; Kopito, R.R. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 2009, 11, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-J.; Patel, S.; Lee, S.-J. Intravesicular Localization and Exocytosis of α-Synuclein and its Aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.T.; Chenouard, N.; Chaumont, F.; Martino, A.; Enninga, J.; et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Victoria, G.S.; Zurzolo, C. The spread of prion-like proteins by lysosomes and tunneling nanotubes: Implications for neurodegenerative diseases. J. Cell Biol. 2017, 216, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Wopfner, F.; Weidenhöfer, G.; Schneider, R.; von Brunn, A.; Gilch, S.; Schwarz, T.F.; Werner, T.; Schätzl, H.M. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J. Mol. Biol. 1999, 289, 1163–1178. [Google Scholar] [CrossRef]
- Moya, K.L.; Salès, N.; Hässig, R.; Créminon, C.; Grassi, J.; Di Giamberardino, L. Immunolocalization of the cellular prion protein in normal brain. Microsc. Res. Tech. 2000, 50, 58–65. [Google Scholar] [CrossRef]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef]
- Riek, R.; Hornemann, S.; Wider, G.; Billeter, M.; Glockshuber, R.; Wüthrich, K. NMR structure of the mouse prion protein domain PrP(121–231). Nature 1996, 382, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Taraboulos, A.; Raeber, A.J.; Borchelt, D.R.; Serban, D.; Prusiner, S.B. Synthesis and trafficking of prion proteins in cultured cells. Mol. Biol. Cell 1992, 3, 851–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, N.; Baldwin, M.A.; Hecker, R.; Pan, K.M.; Burlingame, A.L.; Prusiner, S.B. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry 1992, 31, 5043–5053. [Google Scholar] [CrossRef] [PubMed]
- Sunyach, C.; Jen, A.; Deng, J.; Fitzgerald, K.T.; Frobert, Y.; Grassi, J.; McCaffrey, M.W.; Morris, R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003, 22, 3591–3601. [Google Scholar] [CrossRef] [PubMed]
- Shyng, S.L.; Heuser, J.E.; Harris, D.A. A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. J. Cell Biol. 1994, 125, 1239–1250. [Google Scholar] [CrossRef]
- Taylor, D.R.; Hooper, N.M. The low-density lipoprotein receptor-related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem. J. 2007, 402, 17–23. [Google Scholar] [CrossRef]
- Marella, M.; Lehmann, S.; Grassi, J.; Chabry, J. Filipin Prevents Pathological Prion Protein Accumulation by Reducing Endocytosis and Inducing Cellular PrP Release. J. Biol. Chem. 2002, 277, 25457–25464. [Google Scholar] [CrossRef] [Green Version]
- Peters, P.J.; Mironov, A.; Peretz, D.; van Donselaar, E.; Leclerc, E.; Erpel, S.; DeArmond, S.J.; Burton, D.R.; Williamson, R.A.; Vey, M.; et al. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J. Cell Biol. 2003, 162, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Büeler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.-A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Sailer, A.; Büeler, H.; Fischer, M.; Aguzzi, A.; Weissmann, C. No propagation of prions in mice devoid of PrP. Cell 1994, 77, 967–968. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Groth, D.; Serban, A.; Koehler, R.; Foster, D.; Torchia, M.; Burton, D.; Yang, S.L.; DeArmond, S.J. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc. Natl. Acad. Sci. USA 1993, 90, 10608–10612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinley, M.P.; Taraboulos, A.; Kenaga, L.; Serban, D.; Stieber, A.; DeArmond, S.J.; Prusiner, S.B.; Gonatas, N. Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab. Investig. 1991, 65, 622–630. [Google Scholar] [PubMed]
- Marijanovic, Z.; Caputo, A.; Campana, V.; Zurzolo, C. Identification of an Intracellular Site of Prion Conversion. PLoS Pathog. 2009, 5, e1000426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchelt, D.R.; Taraboulos, A.; Prusiner, S.B. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 1992, 267, 16188–16199. [Google Scholar] [PubMed]
- Taraboulos, A.; Scott, M.; Semenov, A.; Avrahami, D.; Laszlo, L.; Prusiner, S.B. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 1995, 129, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilch, S.; Winklhofer, K.F.; Groschup, M.H.; Nunziante, M.; Lucassen, R.; Spielhaupter, C.; Muranyi, W.; Riesner, D.; Tatzelt, J.; Schätzl, H.M. Intracellular re-routing of prion protein prevents propagation of PrPSc and delays onset of prion disease. EMBO J. 2001, 20, 3957–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enari, M.; Flechsig, E.; Weissmann, C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl. Acad. Sci. USA 2001, 98, 9295–9299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesebro, B.; Trifilo, M.; Race, R.; Meade-White, K.; Teng, C.; LaCasse, R.; Raymond, L.; Favara, C.; Baron, G.; Priola, S.; et al. Anchorless Prion Protein Results in Infectious Amyloid Disease Without Clinical Scrapie. Science 2005, 308, 1435–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesebro, B.; Race, B.; Meade-White, K.; LaCasse, R.; Race, R.; Klingeborn, M.; Striebel, J.; Dorward, D.; McGovern, G.; Jeffrey, M. Fatal Transmissible Amyloid Encephalopathy: A New Type of Prion Disease Associated with Lack of Prion Protein Membrane Anchoring. PLoS Pathog. 2010, 6, e1000800. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.; Graßmann, A.; Bester, R.; Hossinger, A.; Möhl, C.; Paulsen, L.; Groschup, M.H.; Schätzl, H.M.; Vorberg, I. Modulation of Glycosaminoglycans Affects PrP Sc Metabolism but Does Not Block PrP Sc Uptake. J. Virol. 2015, 89, 9853–9864. [Google Scholar] [CrossRef] [Green Version]
- Horonchik, L.; Tzaban, S.; Ben-Zaken, O.; Yedidia, Y.; Rouvinski, A.; Papy-Garcia, D.; Barritault, D.; Vlodavsky, I.; Taraboulos, A. Heparan Sulfate is a Cellular Receptor for Purified Infectious Prions. J. Biol. Chem. 2005, 280, 17062–17067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangé, A.; Nishida, N.; Milhavet, O.; McMahon, H.E.M.; Casanova, D.; Lehmann, S. Amphotericin B Inhibits the Generation of the Scrapie Isoform of the Prion Protein in Infected Cultures. J. Virol. 2000, 74, 3135–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilch, S.; Bach, C.; Lutzny, G.; Vorberg, I.; Schätzl, H.M. Inhibition of cholesterol recycling impairs cellular PrPSc propagation. Cell. Mol. Life Sci. 2009, 66, 3979–3991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goold, R.; McKinnon, C.; Rabbanian, S.; Collinge, J.; Schiavo, G.; Tabrizi, S.J. Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J. Cell Sci. 2013, 126, 3552–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, Y.-I.; Park, B.-C.; Yadavalli, R.; Zhao, X.; Eisenberg, E.; Greene, L.E. The multivesicular body is the major internal site of prion conversion. J. Cell Sci. 2015, 128, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, T.; Suzuki, A.; Shimizu, T.; Watarai, M.; Hasebe, R.; Horiuchi, M. Characterization of intracellular localization of PrPSc in prion-infected cells using a mAb that recognizes the region consisting of aa 119–127 of mouse PrP. J. Gen. Virol. 2012, 93, 668–680. [Google Scholar] [CrossRef]
- Béranger, F.; Mangé, A.; Goud, B.; Lehmann, S. Stimulation of PrPC Retrograde Transport toward the Endoplasmic Reticulum Increases Accumulation of PrPSc in Prion-infected Cells. J. Biol. Chem. 2002, 277, 38972–38977. [Google Scholar] [CrossRef] [Green Version]
- Fehlinger, A.; Wolf, H.; Hossinger, A.; Duernberger, Y.; Pleschka, C.; Riemschoss, K.; Liu, S.; Bester, R.; Paulsen, L.; Priola, S.A.; et al. Prion strains depend on different endocytic routes for productive infection. Sci. Rep. 2017, 7, 6923. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.L.; Stewart, K.; Ritchie, D.L.; Mabbott, N.A.; Williams, A.; Fraser, H.; Morrison, W.I.; Bruce, M.E. Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat. Med. 1999, 5, 1308–1312. [Google Scholar] [CrossRef]
- Raeber, A.J.; Race, R.E.; Brandner, S.; Priola, S.A.; Sailer, A.; Bessen, R.A.; Mucke, L.; Manson, J.; Aguzzi, A.; Oldstone, M.B.; et al. Astrocyte-specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO J. 1997, 16, 6057–6065. [Google Scholar] [CrossRef] [Green Version]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klöhn, P.-C.; Brandner, S.; Collinge, J. Depleting Neuronal PrP in Prion Infection Prevents Disease and Reverses Spongiosis. Science 2003, 302, 871–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessen, R.A.; Marsh, R.F. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J. Gen. Virol. 1992, 73, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Bessen, R.A.; Martinka, S.; Kelly, J.; Gonzalez, D. Role of the Lymphoreticular System in Prion Neuroinvasion from the Oral and Nasal Mucosa. J. Virol. 2009, 83, 6435–6445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shikiya, R.A.; Langenfeld, K.A.; Eckland, T.E.; Trinh, J.; Holec, S.A.M.; Mathiason, C.K.; Kincaid, A.E.; Bartz, J.C. PrPSc formation and clearance as determinants of prion tropism. PLoS Pathog. 2017, 13, e1006298. [Google Scholar] [CrossRef] [PubMed]
- Bessen, R.A.; Shearin, H.; Martinka, S.; Boharski, R.; Lowe, D.; Wilham, J.M.; Caughey, B.; Wiley, J.A. Prion Shedding from Olfactory Neurons into Nasal Secretions. PLoS Pathog. 2010, 6, e1000837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy, E.R.; Bartz, J.C.; Kincaid, A.E.; Bessen, R.A. Prion Infection of Skeletal Muscle Cells and Papillae in the Tongue. J. Virol. 2004, 78, 6792–6798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elder, A.M.; Henderson, D.M.; Nalls, A.V.; Hoover, E.A.; Kincaid, A.E.; Bartz, J.C.; Mathiason, C.K. Immediate and Ongoing Detection of Prions in the Blood of Hamsters and Deer following Oral, Nasal, or Blood Inoculations. J. Virol. 2015, 89, 7421–7424. [Google Scholar] [CrossRef] [Green Version]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in Membrane Traffic and Cell Physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.R. Rab GTPases: Specifying and deciphering organelle identity and function. Trends Cell Biol. 2001, 11, 487–491. [Google Scholar] [CrossRef]
- Zhen, Y.; Stenmark, H. Cellular functions of Rab GTPases at a glance. J. Cell Sci. 2015, 128, 3171–3176. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.P.; Goody, R.S. Molecular control of Rab activity by GEFs, GAPs and GDI. Small GTPases 2018, 9, 5–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poteryaev, D.; Datta, S.; Ackema, K.; Zerial, M.; Spang, A. Identification of the switch in early-to-late endosome transition. Cell 2010, 141, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, F.; Bucci, C. Multiple Roles of the Small GTPase Rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Kovács, G.G.; Gelpi, E.; Ströbel, T.; Ricken, G.; Nyengaard, J.R.; Bernheimer, H.; Budka, H. Involvement of the Endosomal-Lysosomal System Correlates with Regional Pathology in Creutzfeldt-Jakob Disease. J. Neuropathol. Exp. Neurol. 2007, 66, 628–636. [Google Scholar] [CrossRef] [Green Version]
- Shim, S.Y.; Karri, S.; Law, S.; Schatzl, H.M.; Gilch, S. Prion infection impairs lysosomal degradation capacity by interfering with rab7 membrane attachment in neuronal cells. Sci. Rep. 2016, 6, 21658. [Google Scholar] [CrossRef] [Green Version]
- Bartoletti-Stella, A.; Corrado, P.; Mometto, N.; Baiardi, S.; Durrenberger, P.F.; Arzberger, T.; Reynolds, R.; Kretzschmar, H.; Capellari, S.; Parchi, P. Analysis of RNA Expression Profiles Identifies Dysregulated Vesicle Trafficking Pathways in Creutzfeldt-Jakob Disease. Mol. Neurobiol. 2019, 56, 5009–5024. [Google Scholar] [CrossRef]
- Uchiyama, K.; Muramatsu, N.; Yano, M.; Usui, T.; Miyata, H.; Sakaguchi, S. Prions disturb post-Golgi trafficking of membrane proteins. Nat. Commun. 2013, 4, 1846. [Google Scholar] [CrossRef] [Green Version]
- Massignan, T.; Biasini, E.; Lauranzano, E.; Veglianese, P.; Pignataro, M.; Fioriti, L.; Harris, D.A.; Salmona, M.; Chiesa, R.; Bonetto, V. Mutant Prion Protein Expression Is Associated with an Alteration of the Rab GDP Dissociation Inhibitor α (GDI)/Rab11 Pathway. Mol. Cell. Proteom. 2010, 9, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Ermolayev, V.; Cathomen, T.; Merk, J.; Friedrich, M.; Härtig, W.; Harms, G.S.; Klein, M.A.; Flechsig, E. Impaired Axonal Transport in Motor Neurons Correlates with Clinical Prion Disease. PLoS Pathog. 2009, 5, e1000558. [Google Scholar] [CrossRef]
- Deinhardt, K.; Salinas, S.; Verastegui, C.; Watson, R.; Worth, D.; Hanrahan, S.; Bucci, C.; Schiavo, G. Rab5 and Rab7 Control Endocytic Sorting along the Axonal Retrograde Transport Pathway. Neuron 2006, 52, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic Pathway Abnormalities Precede Amyloid β Deposition in Sporadic Alzheimer’s Disease and Down Syndrome. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Barnett, J.L.; Pieroni, C.; Nixon, R.A. Increased Neuronal Endocytosis and Protease Delivery to Early Endosomes in Sporadic Alzheimer’s Disease: Neuropathologic Evidence for a Mechanism of Increased β-Amyloidogenesis. J. Neurosci. 1997, 17, 6142–6151. [Google Scholar] [CrossRef] [PubMed]
- Priya, A.; Kalaidzidis, I.V.; Kalaidzidis, Y.; Lambright, D.; Datta, S. Molecular Insights into Rab7-Mediated Endosomal Recruitment of Core Retromer: Deciphering the Role of Vps26 and Vps35. Traffic 2015, 16, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Nichols, W.C.; Pankratz, N.; Hernandez, D.; Paisán-Ruíz, C.; Jain, S.; Halter, C.A.; Michaels, V.E.; Reed, T.; Rudolph, A.; Shults, A.W.; et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 2005, 365, 410–412. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Rivero-Ríos, P.; Fdez, E.; Blanca Ramírez, M.; Ferrer, I.; Aiastui, A.; Munain, A.L.; Hilfiker, S. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum. Mol. Genet. 2014, 23, 6779–6796. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; McCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 2013, 77, 425–439. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Trajkovic, K.; Tsunemi, T.; Krainc, D. Parkin Modulates Endosomal Organization and Function of the Endo-Lysosomal Pathway. J. Neurosci. 2016, 36, 2425–2437. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, A.; Flores, I.; Zhang, H.; Yu, R.; Staniszewski, A.; Planel, E.; Herman, M.; Ho, L.; Kreber, R.; Honig, L.S.; et al. Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Aβ accumulation. Proc. Natl. Acad. Sci. USA 2008, 105, 7327–7332. [Google Scholar] [CrossRef] [Green Version]
- Small, S.A.; Kent, K.; Pierce, A.; Leung, C.; Kang, M.S.; Okada, H.; Honig, L.; Vonsattel, J.P.; Kim, T.W. Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann. Neurol. 2005, 58, 909–919. [Google Scholar] [CrossRef]
- Wen, L.; Tang, F.-L.; Hong, Y.; Luo, S.-W.; Wang, C.-L.; He, W.; Shen, C.; Jung, J.U.; Xiong, F.; Lee, D.; et al. VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J. Cell Biol. 2011, 195, 765–779. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H. Brain-derived neurotrophic factor exerts neuroprotective actions against amyloid β-induced apoptosis in neuroblastoma cells. Exp. Ther. Med. 2014, 8, 1891–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L.F. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caviston, J.P.; Zajac, A.L.; Tokito, M.; Holzbaur, E.L.F. Huntingtin coordinates the dynein-mediated dynamic positioning of endosomes and lysosomes. Mol. Biol. Cell 2011, 22, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant Huntingtin Alters Retrograde Transport of TrkB Receptors in Striatal Dendrites. J. Neurosci. 2013, 33, 6298–6309. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Relano-Gines, A.; Resina, S.; Brillaud, E.; Casanova, D.; Vincent, C.; Hamela, C.; Poupeau, S.; Laffont, M.; Gabelle, A.; et al. Systemic Delivery of siRNA Down Regulates Brain Prion Protein and Ameliorates Neuropathology in Prion Disorder. PLoS ONE 2014, 9, e88797. [Google Scholar] [CrossRef]
- Gilch, S.; Kehler, C.; Schatzl, H.M. Peptide aptamers expressed in the secretory pathway interfere with cellular PrPSc formation. J. Mol. Biol. 2007, 371, 362–373. [Google Scholar] [CrossRef]
- Kong, Q. RNAi: A novel strategy for the treatment of prion diseases. J. Clin. Investig. 2006, 116, 3101–3103. [Google Scholar] [CrossRef] [Green Version]
- Caughey, B.; Raymond, G.J. Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol. 1993, 67, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Beringue, V.; Adjou, K.T.; Lamoury, F.; Maignien, T.; Deslys, J.-P.; Race, R.; Dormont, D. Opposite Effects of Dextran Sulfate 500, the Polyene Antibiotic MS-8209, and Congo Red on Accumulation of the Protease-Resistant Isoform of PrP in the Spleens of Mice Inoculated Intraperitoneally with the Scrapie Agent. J. Virol. 2000, 74, 5432–5440. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.; Xiong, L.-W.; Horiuchi, M.; Raymond, L.; Wehrly, K.; Chesebro, B.; Caughey, B. Sulfated glycans and elevated temperature stimulate PrPSc-dependent cell-free formation of protease-resistant prion protein. EMBO J. 2001, 20, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Shyng, S.-L.; Lehmann, S.; Moulder, K.L.; Harris, D.A. Sulfated Glycans Stimulate Endocytosis of the Cellular Isoform of the Prion Protein, PrPC, in Cultured Cells. J. Biol. Chem. 1995, 270, 30221–30229. [Google Scholar] [PubMed] [Green Version]
- Bate, C.; Salmona, M.; Diomede, L.; Williams, A. Squalestatin Cures Prion-infected Neurons and Protects Against Prion Neurotoxicity. J. Biol. Chem. 2004, 279, 14983–14990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaemmaghami, S.; Phuan, P.-W.; Perkins, B.; Ullman, J.; May, B.C.H.; Cohen, F.E.; Prusiner, S.B. Cell division modulates prion accumulation in cultured cells. Proc. Natl. Acad. Sci. USA 2007, 104, 17971–17976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; Zhao, D.; Khan, S.H.; Yang, L. Role of autophagy in prion protein-induced neurodegenerative diseases. Acta Biochim. Biophys. Sin. 2013, 45, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated Protein Kinase Signaling Activation by Resveratrol Modulates Amyloid-β Peptide Metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-Activated AMPK/SIRT1/Autophagy in Cellular Models of Parkinson’s Disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Ertmer, A.; Gilch, S.; Yun, S.-W.; Flechsig, E.; Klebl, B.; Stein-Gerlach, M.; Klein, M.A.; Schätzl, H.M. The Tyrosine Kinase Inhibitor STI571 Induces Cellular Clearance of PrPSc in Prion-infected Cells. J. Biol. Chem. 2004, 279, 41918–41927. [Google Scholar] [CrossRef] [Green Version]
- Valent, P. Imatinib-resistant chronic myeloid leukemia (CML): Current concepts on pathogenesis and new emerging pharmacologic approaches. Biologics 2007, 1, 433–448. [Google Scholar]
- Heiseke, A.; Aguib, Y.; Riemer, C.; Baier, M.; Schätzl, H.M. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J. Neurochem. 2009, 109, 25–34. [Google Scholar] [CrossRef]
- Aguib, Y.; Heiseke, A.; Gilch, S.; Riemer, C.; Baier, M.; Schätzl, H.M.; Ertmer, A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009, 5, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.-K.; Moon, M.-H.; Bae, B.-C.; Lee, Y.-J.; Seol, J.-W.; Kang, H.-S.; Kim, J.S.; Kang, S.J.; Park, S.Y. Autophagy induced by resveratrol prevents human prion protein-mediated neurotoxicity. Neurosci. Res. 2012, 73, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Abdulrahman, B.A.; Abdelaziz, D.H.; Schatzl, H.M. Autophagy regulates exosomal release of prions in neuronal cells. J. Biol. Chem. 2018, 293, 8956–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhungel, N.; Eleuteri, S.; Li, L.; Kramer, N.J.; Chartron, J.W.; Spencer, B.; Kosberg, K.; Fields, J.A.; Stafa, K.; Adame, A.; et al. Parkinson’s Disease Genes VPS35 and EIF4G1 Interact Genetically and Converge on α-Synuclein. Neuron 2015, 85, 76–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecozzi, V.J.; Berman, D.E.; Simoes, S.; Vetanovetz, C.; Awal, M.R.; Patel, V.M.; Schneider, R.T.; Petsko, G.A.; Ringe, D.; Small, S.A. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014, 10, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Follett, J.; Norwood, S.J.; Hamilton, N.A.; Mohan, M.; Kovtun, O.; Tay, S.; Zhe, Y.; Wood, S.A.; Mellick, G.D.; Silburn, P.A.; et al. The Vps35 D620N Mutation Linked to Parkinson’s Disease Disrupts the Cargo Sorting Function of Retromer. Traffic 2014, 15, 230–244. [Google Scholar] [CrossRef]
- Arighi, C.N.; Hartnell, L.M.; Aguilar, R.C.; Haft, C.R.; Bonifacino, J.S. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 2004, 165, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Eleuteri, S.; Albanese, A. VPS35-Based Approach: A Potential Innovative Treatment in Parkinson’s Disease. Front. Neurol. 2019, 10, 1272. [Google Scholar] [CrossRef]
- Zavodszky, E.; Seaman, M.N.J.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef]
- Tang, F.-L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.-M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.C. VPS35 in Dopamine Neurons is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy that is Critical for -Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef]
- Freeman, D.; Cedillos, R.; Choyke, S.; Lukic, Z.; McGuire, K.; Marvin, S.; Burrage, A.M.; Sudholt, S.; Rana, A.; O’Connor, C.; et al. Alpha-Synuclein Induces Lysosomal Rupture and Cathepsin Dependent Reactive Oxygen Species Following Endocytosis. PLoS ONE 2013, 8, e62143. [Google Scholar] [CrossRef] [PubMed]
- Malik, B.R.; Maddison, D.C.; Smith, G.A.; Peters, O.M. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain 2019, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Overly, C.C.; Hollenbeck, P.J. Dynamic Organization of Endocytic Pathways in Axons of Cultured Sympathetic Neurons. J. Neurosci. 1996, 16, 6056–6064. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Peterhoff, C.M.; Schmidt, S.D.; Terio, N.B.; Duff, K.; Beard, M.; Mathews, P.M.; Nixon, R.A. Presenilin Mutations in Familial Alzheimer Disease and Transgenic Mouse Models Accelerate Neuronal Lysosomal Pathology. J. Neuropathol. Exp. Neurol. 2004, 63, 821–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.Y.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca2+ Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [Green Version]
- Erie, C.; Sacino, M.; Houle, L.; Lu, M.L.; Wei, J. Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington’s disease. Eur. J. Neurosci. 2015, 42, 1941–1951. [Google Scholar] [CrossRef] [Green Version]
- Runz, H.; Rietdorf, J.; Tomic, I.; de Bernard, M.; Beyreuther, K.; Pepperkok, R.; Hartmann, T. Inhibition of Intracellular Cholesterol Transport Alters Presenilin Localization and Amyloid Precursor Protein Processing in Neuronal Cells. J. Neurosci. 2002, 22, 1679–1689. [Google Scholar] [CrossRef]
- Choudhury, A.; Dominguez, M.; Puri, V.; Sharma, D.K.; Narita, K.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Investig. 2002, 109, 1541–1550. [Google Scholar] [CrossRef]
- Cherry, S.; Jin, E.J.; Özel, M.N.; Lu, Z.; Agi, E.; Wang, D.; Jung, W.H.; Epstein, D.; Meinertzhagen, I.A. Charcot-Marie-Tooth 2B mutations in rab7 cause dosage-dependent neurodegeneration due to partial loss of function. eLife 2013, 2, e01064. [Google Scholar] [CrossRef]
- Stein, M.-P.; Dong, J.; Wandinger-Ness, A. Rab proteins and endocytic trafficking: Potential targets for therapeutic intervention. Adv. Drug Deliv. Rev. 2003, 55, 1421–1437. [Google Scholar] [CrossRef]
Figure 1.
Trafficking of a cellular form of the prion protein (PrPc) and an infectious and aggregation prone isoform (PrPSc) and the potential inhibitors of the pathogenic conversion at the different stages of trafficking. (1) Cellular PrPc is synthesized in the endoplamic reticulum (ER), transported to the Golgi complex, and then is finally localized at the plasma membrane in the lipid rafts. (2) Preventing the surface localization or the accessibility of PrPc at the plasma membrane (PM) is one strategy adopted to reduce PrPSc levels. This can be done by treating cells with suramin, which retains the PrPc in the Golgi or by abrogating the proper lipid raft formation by treating with amphotericin B, filipin, or lovastatin, which prevent the localization of PrPc in the lipid rafts and subsequently prevent PrPSc formation. (3). Phosphoinositide phospholipase C (PIPLC)-mediated cleavage of PrPc or the removal of glycosaminoglycans (GAGs) by lyases, reduce PrPSc levels. (4). Preventing the endocytosis of PrPc/PrPSc by lowering the temperature and (5) preventing the maturation of the multi-vesicular bodies can also reduce PrPSc propagation. (6) In contrast, inhibiting the retrograde trafficking to Golgi increases PrPSc levels. (7) Eventually, PrPSc accumulates in lysosomes where it is partially degraded.
Figure 1.
Trafficking of a cellular form of the prion protein (PrPc) and an infectious and aggregation prone isoform (PrPSc) and the potential inhibitors of the pathogenic conversion at the different stages of trafficking. (1) Cellular PrPc is synthesized in the endoplamic reticulum (ER), transported to the Golgi complex, and then is finally localized at the plasma membrane in the lipid rafts. (2) Preventing the surface localization or the accessibility of PrPc at the plasma membrane (PM) is one strategy adopted to reduce PrPSc levels. This can be done by treating cells with suramin, which retains the PrPc in the Golgi or by abrogating the proper lipid raft formation by treating with amphotericin B, filipin, or lovastatin, which prevent the localization of PrPc in the lipid rafts and subsequently prevent PrPSc formation. (3). Phosphoinositide phospholipase C (PIPLC)-mediated cleavage of PrPc or the removal of glycosaminoglycans (GAGs) by lyases, reduce PrPSc levels. (4). Preventing the endocytosis of PrPc/PrPSc by lowering the temperature and (5) preventing the maturation of the multi-vesicular bodies can also reduce PrPSc propagation. (6) In contrast, inhibiting the retrograde trafficking to Golgi increases PrPSc levels. (7) Eventually, PrPSc accumulates in lysosomes where it is partially degraded.

Figure 2.
Rab activation cycle. GDP/GTP exchange factors (GEFs) mediate Rab activation by replacing the GDP with GTP. The GTP bound Rabs can interact with their effector proteins and mediate vesicular trafficking. Rabs are in an inactive state when bound to GDP, and this involves the hydrolysis of GTP by GTPase activating protein (GAP). The GDP bound Rab7 is prevented from further activation cycle by their association with GDP dissociation inhibitor (GDI). GDI displacement factor (GDF) displaces GDI and makes it available for another cycle of activation.
Figure 2.
Rab activation cycle. GDP/GTP exchange factors (GEFs) mediate Rab activation by replacing the GDP with GTP. The GTP bound Rabs can interact with their effector proteins and mediate vesicular trafficking. Rabs are in an inactive state when bound to GDP, and this involves the hydrolysis of GTP by GTPase activating protein (GAP). The GDP bound Rab7 is prevented from further activation cycle by their association with GDP dissociation inhibitor (GDI). GDI displacement factor (GDF) displaces GDI and makes it available for another cycle of activation.

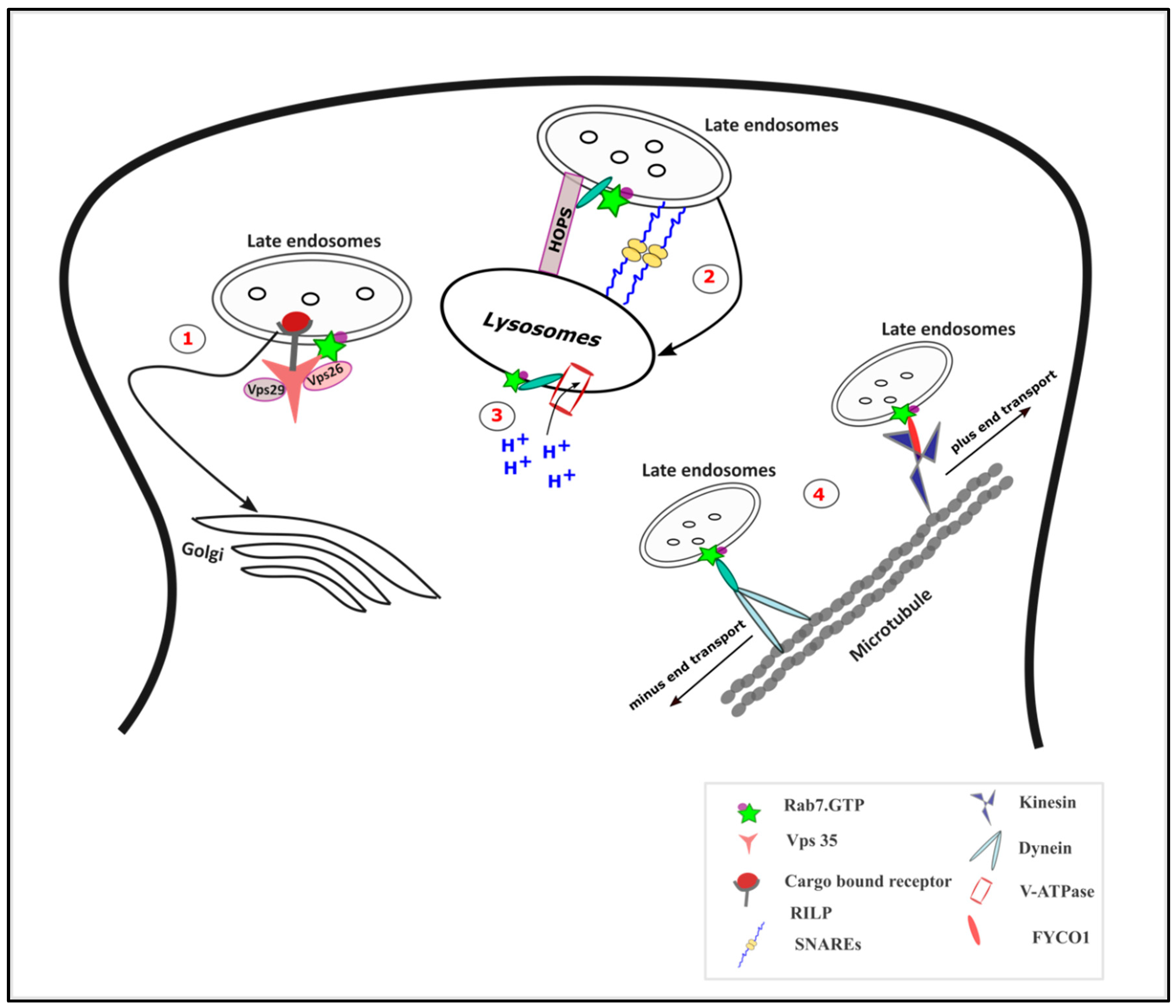
Figure 3.
Functions of Rab7 in vesicular trafficking: (1) The retrograde transport of cargo to the Golgi complex is mediated by Rab7 via its interaction with the subunits of retromer complex especially vacuolar protein sorting-associated protein 35 (Vps 35). (2) Rab7-interacting lysosomal protein (RILP) interaction with the Homotypic fusion and Protein Sorting (HOPS) complex mediates the endosome-lysosome fusion along with the SNAREs. (3) The Rab7-RILP complex regulates the function of Vacuolar-ATPase (V-ATPase) and hence in lysosomal acidification. (4) Rab7, by interacting with RILP, mediates the minus end transport of the late endosomes, while its interaction with FYVE and coiled-coil domain autophagy adaptor-1 (FYCO1) mediates the plus end directed transport.
Figure 3.
Functions of Rab7 in vesicular trafficking: (1) The retrograde transport of cargo to the Golgi complex is mediated by Rab7 via its interaction with the subunits of retromer complex especially vacuolar protein sorting-associated protein 35 (Vps 35). (2) Rab7-interacting lysosomal protein (RILP) interaction with the Homotypic fusion and Protein Sorting (HOPS) complex mediates the endosome-lysosome fusion along with the SNAREs. (3) The Rab7-RILP complex regulates the function of Vacuolar-ATPase (V-ATPase) and hence in lysosomal acidification. (4) Rab7, by interacting with RILP, mediates the minus end transport of the late endosomes, while its interaction with FYVE and coiled-coil domain autophagy adaptor-1 (FYCO1) mediates the plus end directed transport.

Figure 4.
Potential Rab7 mediated impairments upon prion infection. Active levels of Rab7 are reduced upon prion infection which implicates an impaired function of Rab7 in the endocytic vesicular trafficking. The following functions of Rab7 in trafficking could be impaired in prion infection: (1) LDL trafficking and consequently cholesterol metabolism; (2) the interaction of Rab7 with RILP1, resulting in decreased lysosomal maturation; (3) Rab7-RILP interaction with oxysterol-binding protein-related protein 1L ORP1L, resulting in inefficient cholesterol egress from the lysosomes; and (4) Rab7 interaction with Vps 35 resulting in defective retrograde trafficking.
Figure 4.
Potential Rab7 mediated impairments upon prion infection. Active levels of Rab7 are reduced upon prion infection which implicates an impaired function of Rab7 in the endocytic vesicular trafficking. The following functions of Rab7 in trafficking could be impaired in prion infection: (1) LDL trafficking and consequently cholesterol metabolism; (2) the interaction of Rab7 with RILP1, resulting in decreased lysosomal maturation; (3) Rab7-RILP interaction with oxysterol-binding protein-related protein 1L ORP1L, resulting in inefficient cholesterol egress from the lysosomes; and (4) Rab7 interaction with Vps 35 resulting in defective retrograde trafficking.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cherry, P.; Gilch, S. The Role of Vesicle Trafficking Defects in the Pathogenesis of Prion and Prion-Like Disorders. Int. J. Mol. Sci. 2020, 21, 7016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197016
AMA Style
Cherry P, Gilch S. The Role of Vesicle Trafficking Defects in the Pathogenesis of Prion and Prion-Like Disorders. International Journal of Molecular Sciences. 2020; 21(19):7016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197016
Chicago/Turabian StyleCherry, Pearl, and Sabine Gilch. 2020. "The Role of Vesicle Trafficking Defects in the Pathogenesis of Prion and Prion-Like Disorders" International Journal of Molecular Sciences 21, no. 19: 7016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197016
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.