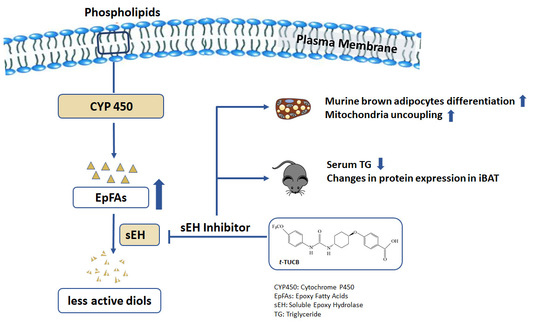
Soluble Epoxide Hydrolase Inhibition by t-TUCB Promotes Brown Adipogenesis and Reduces Serum Triglycerides in Diet-Induced Obesity

 , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
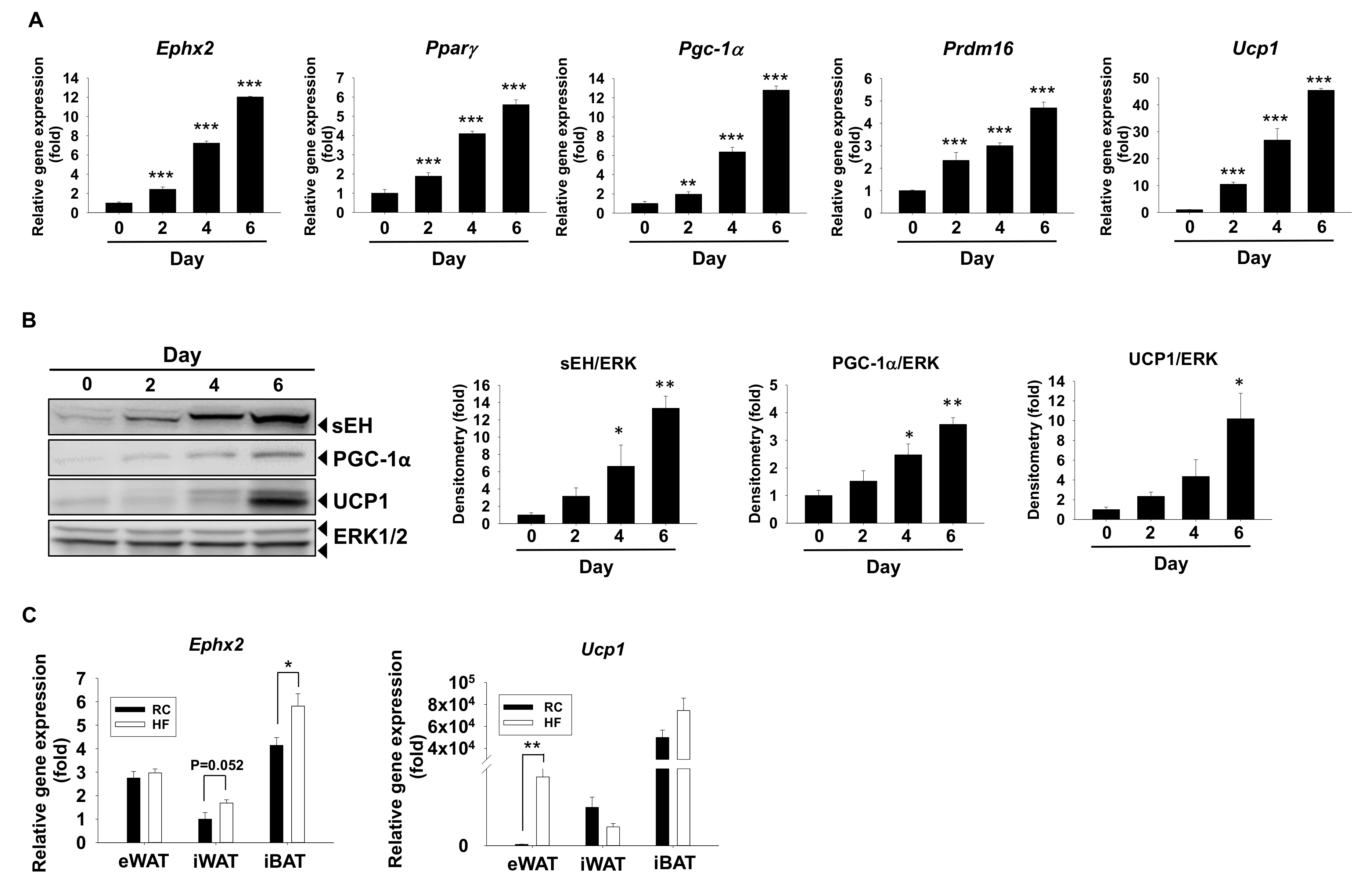
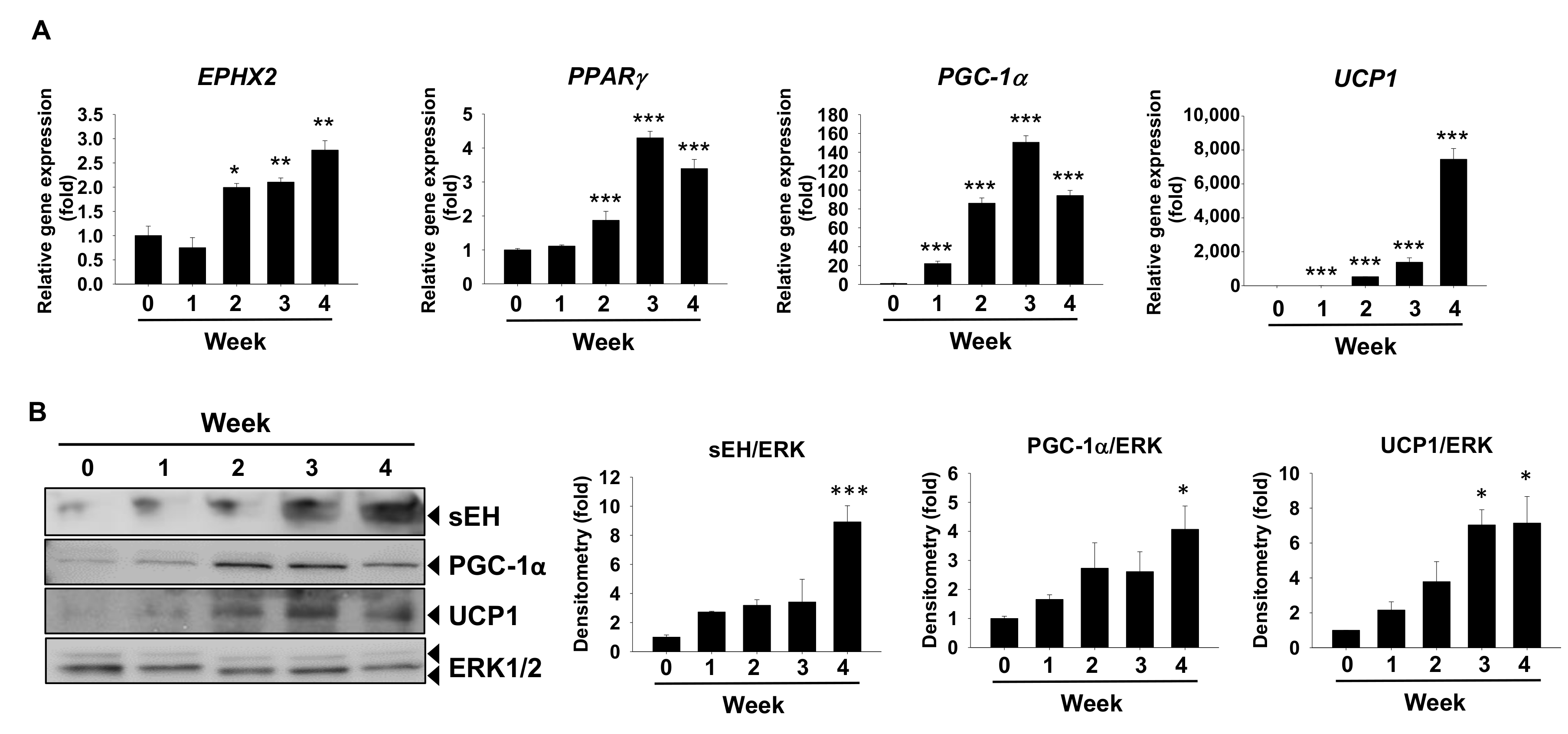
2.1. sEH Expression Is Increased in In Vitro Models of Brown Adipogenesis and the Fat Tissues in Diet-Induced Obesity
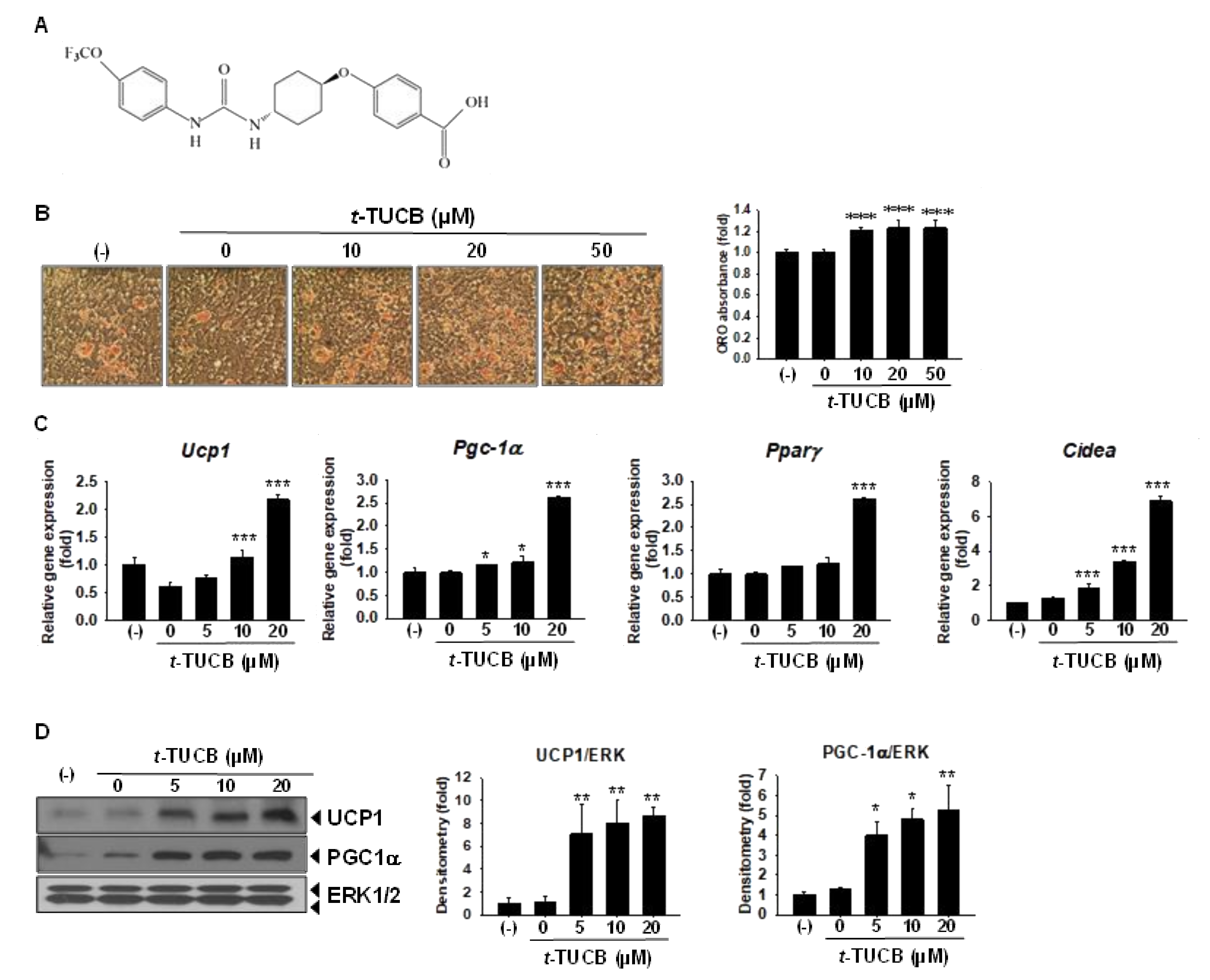
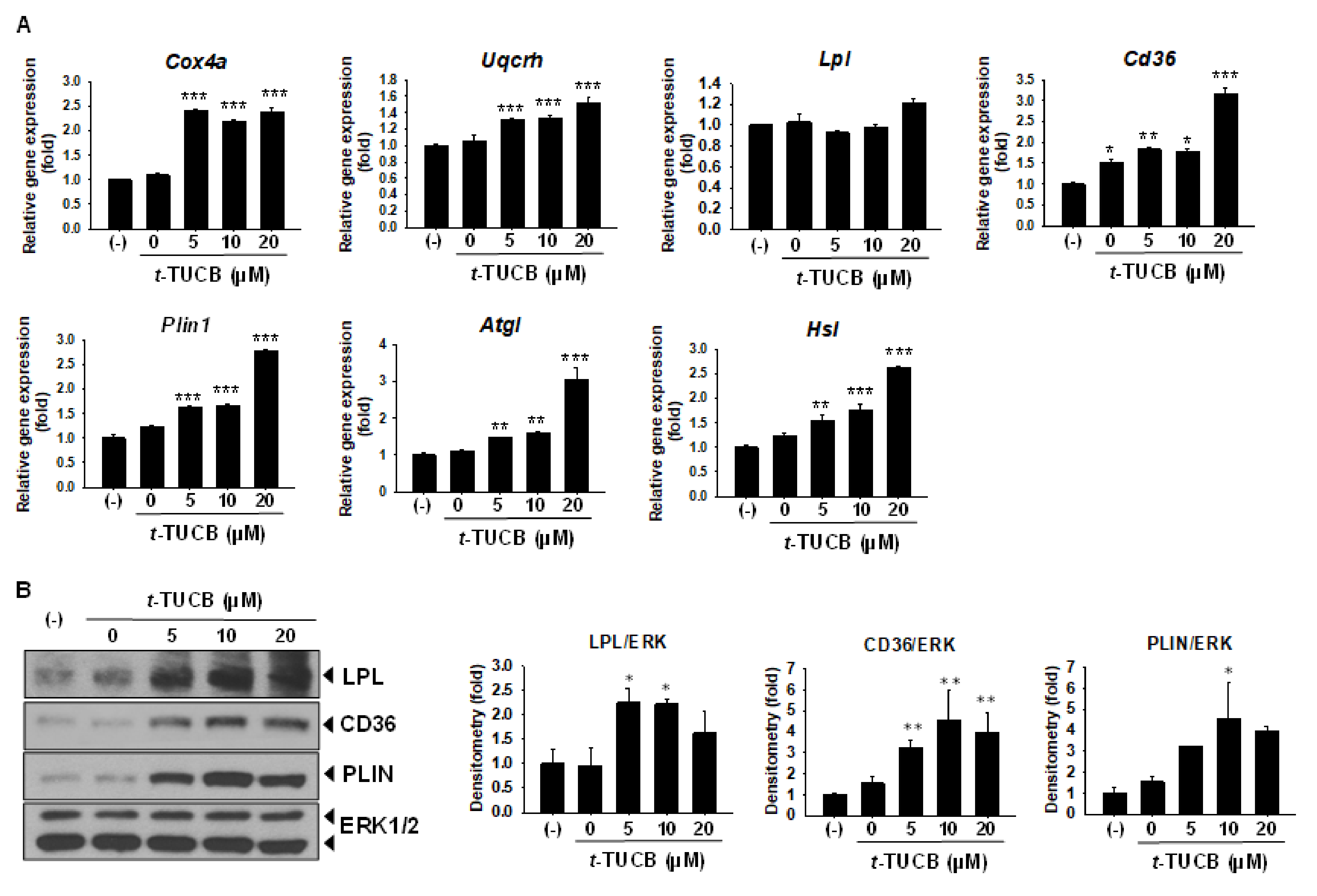
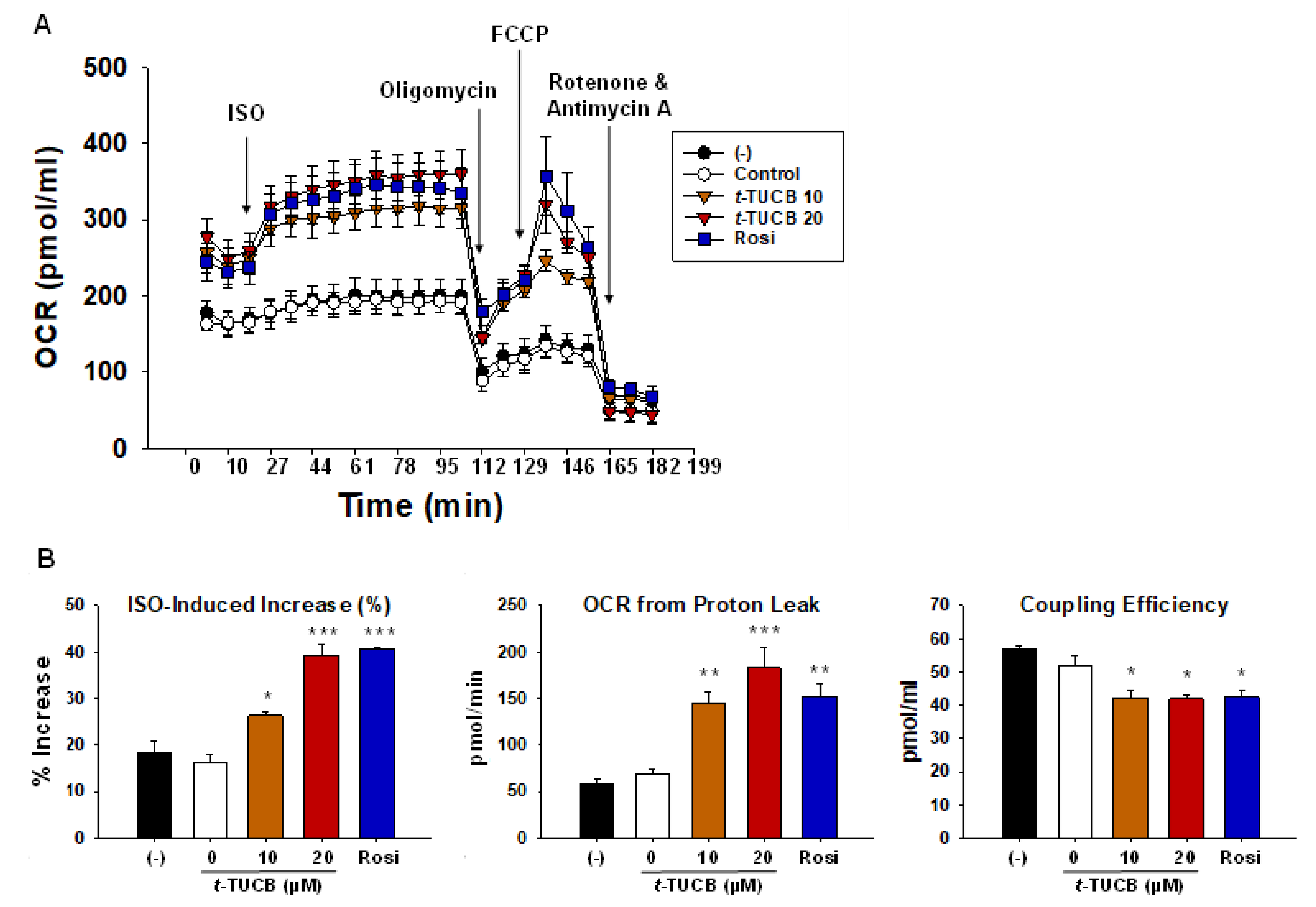
2.2. sEH Inhibition by t-TUCB Promotes Murine Brown Adipogenesis, Upregulates Mitochondrial Respiratory Chain and Lipid Metabolic Genes, and Increases Mitochondrial Uncoupling
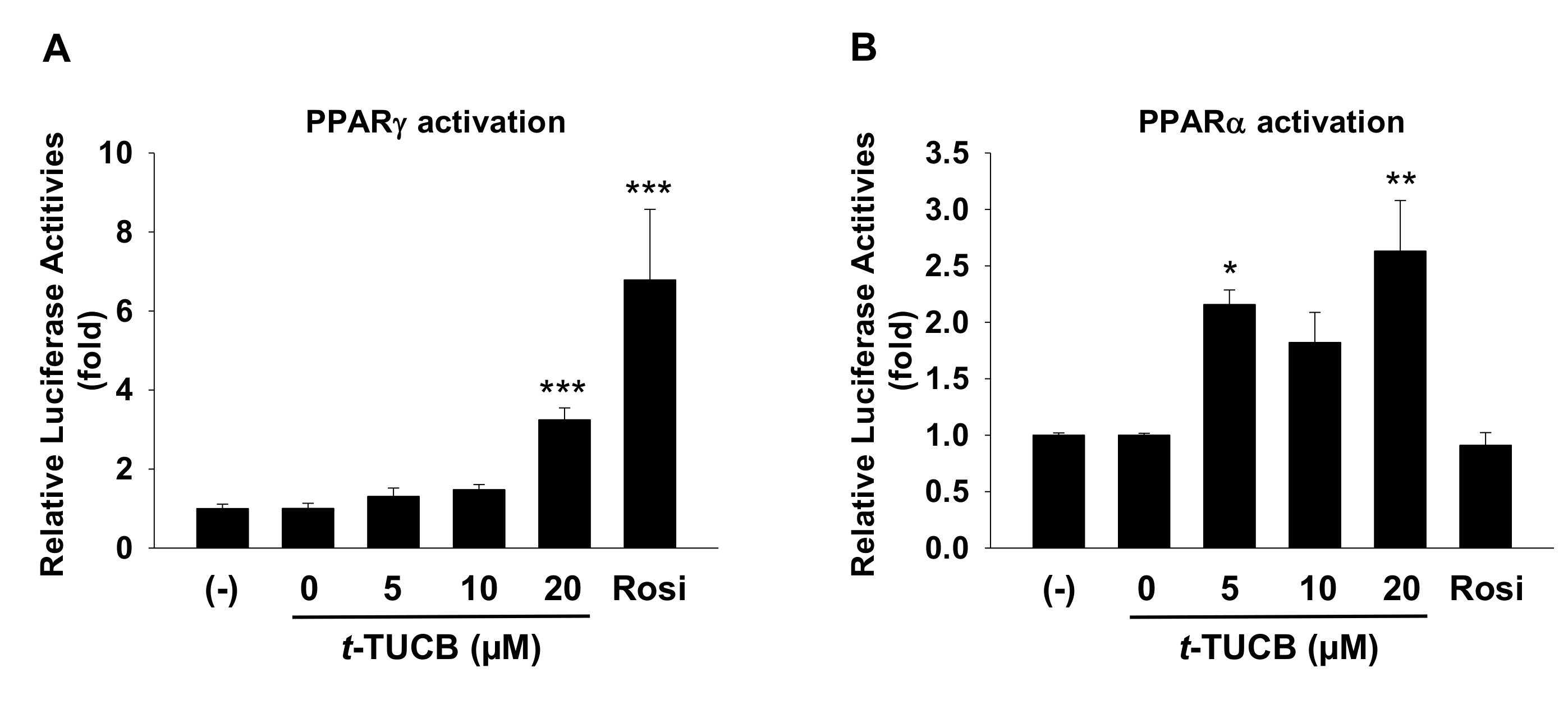
2.3. t-TUCB Activates both PPARγ and PPARα
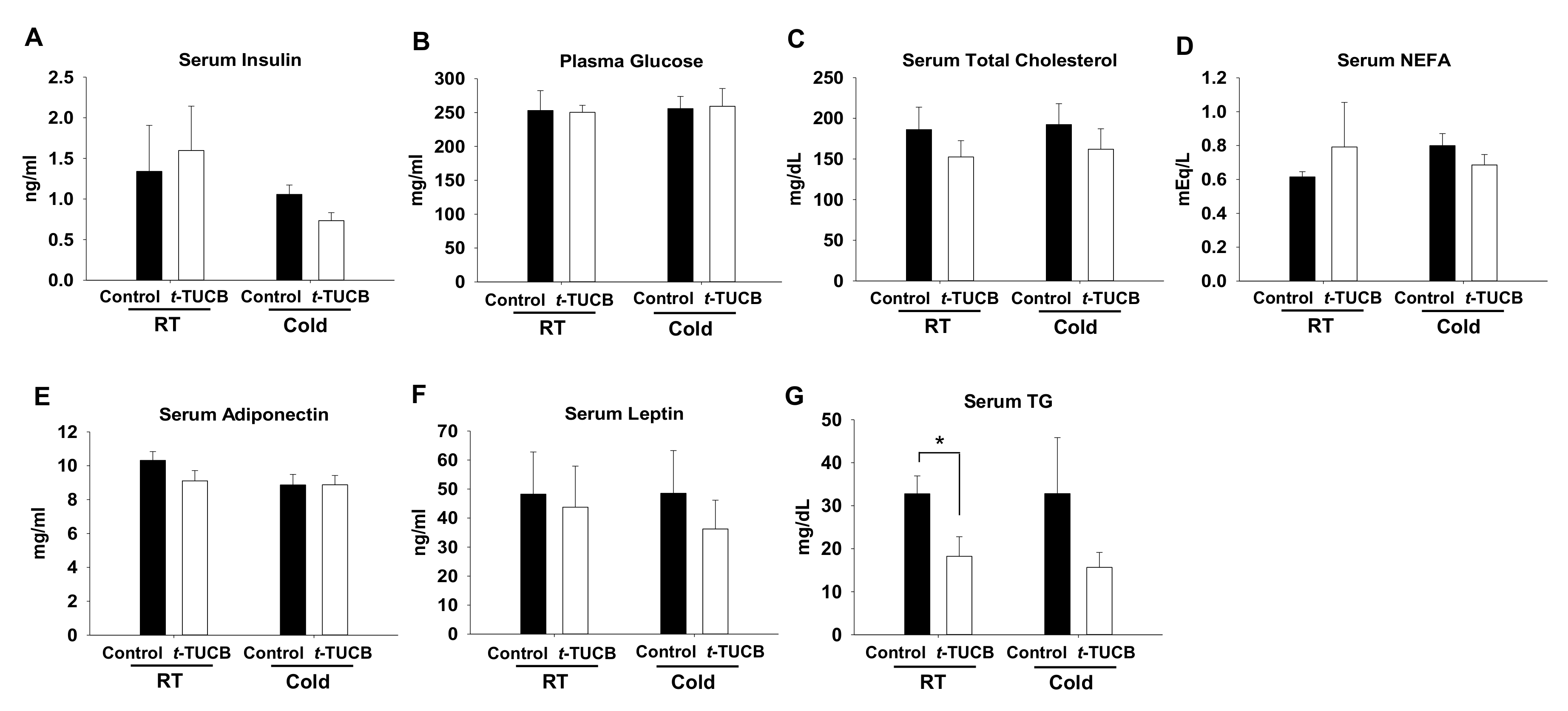
2.4. t-TUCB Delivered via Mini Osmotic Pump Decreases Serum Triglycerides in Diet-Induced Obese Mice
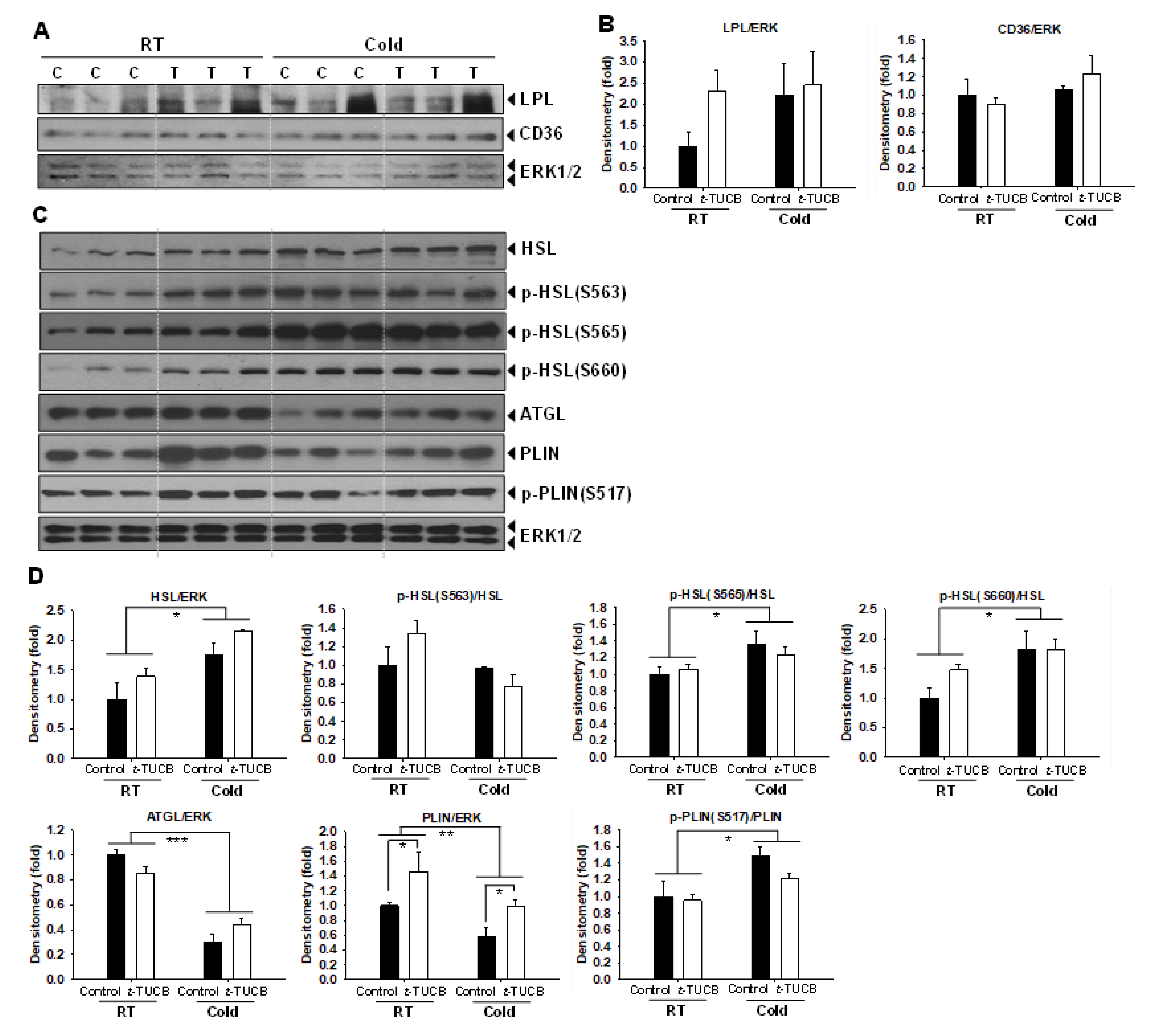
2.5. t-TUCB Delivered via Mini Osmotic Pump Increases Protein Expression of Genes Involved in Lipid Metabolism in the iBAT of Diet-Induced Obese Mice
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatment
4.3. Analysis of Mitochondrial Respiration
4.4. Reporter Gene Assays
4.5. Oxylipin Analysis
4.6. Animal Studies
4.7. Blood Biochemical Analysis
4.8. Western Blot Analysis
4.9. RNA Preparation and Quantitative Real-Time PCR Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| ALA | alpha-linolenic acid |
| ATGL | adipose triglyceride lipase |
| BAT | brown adipose tissue |
| β-gal | beta-galactosidase |
| DHA | docosahexaenoic acid |
| DHET | dihydroxyeicosatrienoic acid |
| DiHDPE | dihydroxydocosapentaenoic acid |
| DiHETE | dihydroxyeicosatetraenoic acid |
| DiHODE | dihydroxyoctadecadienoic acid |
| DiHOME | dihydroxyoctadecenoic acid |
| EET | epoxyeicosatrienoic acid |
| EPA | eicosapentaenoic acid |
| EpDPE | epoxydocosapentaenoic acid |
| EpETE | epoxyeicosatetraenoic acid |
| EpODE | epoxyoctadecadienoic acid |
| EpOME | epoxyoctadecenoic acid |
| ERK | extracellular signal-regulated kinases |
| GTT | glucose tolerance test |
| HSL | hormone sensitive lipase |
| ITT | insulin tolerance test |
| LA | linoleic acid |
| NEFA | non-esterified fatty acids |
| mEH | microsomal epoxide hydrolase |
| OCR | oxygen consumption rate |
| PGC-1α | peroxisome proliferator activator receptor coactivator 1-α |
| PLIN | perilipin |
| PPARα | peroxisome proliferator-activated receptor alpha |
| PPARγ | peroxisome proliferator-activated receptor gamma |
| PRDM16 | PR domain containing 16 |
| Rosi | Rosiglitazone |
| t-TUCB | trans-4-{4-[3-(4-trifluoromethoxy-phenyl)-ureido]-cyclohexyloxy-benzoic acid |
| sEH | soluble epoxide hydrolase |
| UCP1 | uncoupling protein 1 |
| WAT | white adipose tissue |
References
- Cannon, B.; Nedergaard, J. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Cereijo, R.; Giralt, M.; Villarroya, F. Thermogenic brown and beige/brite adipogenesis in humans. Ann. Med. 2014, 47, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, A.; Hoffmann, L.S. Brown, Beige, and White: The New Color Code of Fat and Its Pharmacological Implications. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 207–227. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.-H.; Doria, A.; et al. Identification and Importance of Brown Adipose Tissue in Adult Humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.S.; Taittonen, M.; Laine, J.; Savisto, N.-J.; Enerbäck, S.; et al. Functional Brown Adipose Tissue in Healthy Adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High Incidence of Metabolically Active Brown Adipose Tissue in Healthy Adult Humans: Effects of cold exposure and adiposityeffects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef] [Green Version]
- Yoneshiro, T.; Aita, S.; Matsushita, M.; Kameya, T.; Nakada, K.; Kawai, Y.; Saito, M. Brown Adipose Tissue, Whole-Body Energy Expenditure, and Thermogenesis in Healthy Adult Men. Obesity 2010, 19, 13–16. [Google Scholar] [CrossRef]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.-H.; Khandekar, M.; A Virtanen, K.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Jespersen, N.Z.; Larsen, T.J.; Peijs, L.; Daugaard, S.; Homøe, P.; Loft, A.; De Jong, J.M.A.; Mathur, N.; Cannon, B.; Nedergaard, J.; et al. A Classical Brown Adipose Tissue mRNA Signature Partly Overlaps with Brite in the Supraclavicular Region of Adult Humans. Cell Metab. 2013, 17, 798–805. [Google Scholar] [CrossRef] [Green Version]
- Nedergaard, J.; Cannon, B. How brown is brown fat? It depends where you look. Nat. Med. 2013, 19, 540–541. [Google Scholar] [CrossRef]
- Schulz, T.J.; Huang, P.; Huang, T.L.; Xue, R.; McDougall, L.E.; Townsend, K.L.; Cypess, A.M.; Mishina, Y.; Gussoni, E.; Tseng, Y.-H. Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature 2013, 495, 379–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonet, M.L.; Oliver, P.; Palou, A. Pharmacological and nutritional agents promoting browning of white adipose tissue. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2013, 1831, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.R.; Hammock, B.D. Soluble epoxide hydrolase: Gene structure, expression and deletion. Gene 2013, 526, 61–74. [Google Scholar] [CrossRef] [Green Version]
- De Taeye, B.M.; Morisseau, C.; Coyle, J.; Covington, J.W.; Luria, A.; Yang, J.; Murphy, S.B.; Friedman, D.B.; Hammock, B.B.; Vaughan, D.E. Expression and Regulation of Soluble Epoxide Hydrolase in Adipose Tissue. Obesity 2009, 18, 489–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettaieb, A.; Nagata, N.; AbouBechara, D.; Chahed, S.; Morisseau, C.; Hammock, B.D.; Haj, F.G. Soluble Epoxide Hydrolase Deficiency or Inhibition Attenuates Diet-induced Endoplasmic Reticulum Stress in Liver and Adipose Tissue*. J. Biol. Chem. 2013, 288, 14189–14199. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Dang, H.; Li, D.; Pang, W.; Hammock, B.D.; Zhu, Y. Inhibition of Soluble Epoxide Hydrolase Attenuates High-Fat-Diet–Induced Hepatic Steatosis by Reduced Systemic Inflammatory Status in Mice. PLoS ONE 2012, 7, e39165. [Google Scholar] [CrossRef] [Green Version]
- Roche, C.; Besnier, M.; Cassel, R.; Harouki, N.; Coquerel, D.; Guerrot, D.; Nicol, L.; Loizon, E.; Remy-Jouet, I.; Morisseau, C.; et al. Soluble epoxide hydrolase inhibition improves coronary endothelial function and prevents the development of cardiac alterations in obese insulin-resistant mice. Am. J. Physiol. Circ. Physiol. 2015, 308, H1020–H1029. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-N.; Vincelette, J.; Chen, D.; Gless, R.D.; Anandan, S.-K.; Rubanyi, G.M.; Webb, H.K.; MacIntyre, D.E.; Wang, Y.-X. Inhibition of soluble epoxide hydrolase attenuates endothelial dysfunction in animal models of diabetes, obesity and hypertension. Eur. J. Pharmacol. 2011, 654, 68–74. [Google Scholar] [CrossRef]
- Carmo, J.M.D.; Da Silva, A.A.; Morgan, J.; Wang, Y.-X.; Munusamy, S.; Hall, J.E. Inhibition of soluble epoxide hydrolase reduces food intake and increases metabolic rate in obese mice. Nutr. Metab. Cardiovasc. Dis. 2010, 22, 598–604. [Google Scholar] [CrossRef] [Green Version]
- López-Vicario, C.; Alcaraz-Quiles, J.; García-Alonso, V.; Rius, B.; Hwang, S.H.; Titos, E.; Lopategi, A.; Hammock, B.D.; Arroyo, V.; Claria, J. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: Role for omega-3 epoxides. Proc. Natl. Acad. Sci. USA 2014, 112, 536–541. [Google Scholar] [CrossRef] [Green Version]
- Sanders, W.G.; Morisseau, C.; Hammock, B.D.; Cheung, A.K.; Terry, C.M. Soluble epoxide hydrolase expression in a porcine model of arteriovenous graft stenosis and anti-inflammatory effects of a soluble epoxide hydrolase inhibitor. Am. J. Physiol. Physiol. 2012, 303, C278–C290. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.; Inceoglu, B.; Dong, H.; Yang, J.; Hwang, S.H.; Jones, P.; Morisseau, C.; Hammock, B.D. Comparative efficacy of 3 soluble epoxide hydrolase inhibitors in rat neuropathic and inflammatory pain models. Eur. J. Pharmacol. 2012, 700, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppen, A.; Kalkhoven, E. Brown vs white adipocytes: The PPARγ coregulator story. FEBS Lett. 2010, 584, 3250–3259. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Zhou, A.; Cheung, P.C.K.; Zheng, B.; Zeng, S.; Lin, S. Expression of GPR43 in Brown Adipogenesis Is Enhanced by Rosiglitazone and Controlled by PPARγ/RXR Heterodimerization. PPAR Res. 2018, 2018, 1051074. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 2009, 8, 794–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, Y.; Schmelzer, K.; Lee, T.-S.; Fang, X.; Zhu, Y.; Spector, A.A.; Gill, S.; Morisseau, C.; Hammock, B.D.; et al. The antiinflammatory effect of laminar flow: The role of PPAR, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA 2005, 102, 16747–16752. [Google Scholar] [CrossRef] [Green Version]
- Carobbio, S.; Guénantin, A.-C.; Samuelson, I.; Bahri, M.; Vidal-Puig, A. Brown and beige fat: From molecules to physiology and pathophysiology. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2019, 1864, 37–50. [Google Scholar] [CrossRef]
- Chondronikola, M.; Sidossis, L.S. Brown and beige fat: From molecules to physiology. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2019, 1864, 91–103. [Google Scholar] [CrossRef]
- Kim, D.H.; Vanella, L.; Inoue, K.; Burgess, A.; Gotlinger, K.; Manthati, V.L.; Koduru, S.R.; Zeldin, D.C.; Falck, J.R.; Schwartzman, M.L.; et al. Epoxyeicosatrienoic Acid Agonist Regulates Human Mesenchymal Stem Cell–Derived Adipocytes Through Activation ofHO-1-pAKTSignaling and a Decrease inPPARγ. Stem Cells Dev. 2010, 19, 1863–1873. [Google Scholar] [CrossRef]
- Waldman, M.; Bellner, L.; Vanella, L.; Schragenheim, J.; Sodhi, K.; Singh, S.P.; Lin, D.; Lakhkar, A.D.; Li, J.; Hochhauser, E.; et al. Epoxyeicosatrienoic Acids Regulate Adipocyte Differentiation of Mouse 3T3 Cells, Via PGC-1α Activation, Which Is Required for HO-1 Expression and Increased Mitochondrial Function. Stem Cells Dev. 2016, 25, 1084–1094. [Google Scholar] [CrossRef] [Green Version]
- Bartelt, A.; Bruns, O.T.; Reimer, R.; Hohenberg, H.; Ittrich, H.; Peldschus, K.; Kaul, M.G.; Tromsdorf, U.I.; Weller, H.; Waurisch, C.; et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011, 17, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Lynes, M.D.; O Leiria, L.; Lundh, M.; Bartelt, A.; Shamsi, F.; Huang, T.L.; Takahashi, H.; Hirshman, M.F.; Schlein, C.; Lee, A.; et al. The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nat. Med. 2017, 23, 631–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, S.C.; Christoffolete, M.A.; Ribeiro, M.O.; Miyoshi, H.; Strissel, K.J.; Stancheva, Z.S.; Rogers, N.H.; D’Eon, T.M.; Ii, J.W.P.; Imachi, H.; et al. Perilipin regulates the thermogenic actions of norepinephrine in brown adipose tissue. J. Lipid Res. 2007, 48, 1273–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, T.; Miyoshi, H.; Shimada, K.; Suzuki, A.; Okamatsu-Ogura, Y.; Perfield, J.W.; Kondo, T.; Nagai, S.; Shimizu, C.; Yoshioka, N.; et al. Perilipin Overexpression in White Adipose Tissue Induces a Brown Fat-Like Phenotype. PLoS ONE 2010, 5, e14006. [Google Scholar] [CrossRef]
- Miyoshi, H.; Souza, S.C.; Endo, M.; Sawada, T.; Perfield, J.W.; Shimizu, C.; Stancheva, Z.; Nagai, S.; Strissel, K.J.; Yoshioka, N.; et al. Perilipin overexpression in mice protects against diet-induced obesity. J. Lipid Res. 2009, 51, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Perfield, J.W.; Souza, S.C.; Shen, W.-J.; Zhang, H.; Stancheva, Z.S.; Kraemer, F.B.; Obin, M.S.; Greenberg, A.S. Control of Adipose Triglyceride Lipase Action by Serine 517 of Perilipin A Globally Regulates Protein Kinase A-stimulated Lipolysis in Adipocytes. J. Biol. Chem. 2006, 282, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat Mobilization in Adipose Tissue Is Promoted by Adipose Triglyceride Lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bézaire, V.; Langin, D. Regulation of adipose tissue lipolysis revisited. Proc. Nutr. Soc. 2009, 68, 350–360. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.-J.; Patel, S.; Natu, V.; Kraemer, F.B. Mutational Analysis of Structural Features of Rat Hormone-Sensitive Lipase. Biochemistry 1998, 37, 8973–8979. [Google Scholar] [CrossRef]
- Su, C.-L.; Sztalryd, C.; Holm, C.; Kimmel, A.R.; Londos, C.; Contreras, J.A. Mutational Analysis of the Hormone-sensitive Lipase Translocation Reaction in Adipocytes. J. Biol. Chem. 2003, 278, 43615–43619. [Google Scholar] [CrossRef] [Green Version]
- Daval, M.; Diot-Dupuy, F.; Bazin, R.; Hainault, I.; Viollet, B.; Vaulont, S.; Hajduch, E.; Ferre, P.; Foufelle, F. Anti-lipolytic Action of AMP-activated Protein Kinase in Rodent Adipocytes. J. Biol. Chem. 2005, 280, 25250–25257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Puri, N.; Raffaele, M.; Schragenheim, J.; Singh, S.P.; Bradbury, J.A.; Bellner, L.; Vanella, L.; Zeldin, D.C.; Cao, J.; et al. Ablation of soluble epoxide hydrolase reprogram white fat to beige-like fat through an increase in mitochondrial integrity, HO-1-adiponectin in vitro and in vivo. Prostaglandins Other Lipid Mediat. 2018, 138, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Panigrahy, D.; Mahakian, L.M.; Yang, J.; Liu, J.-Y.; Lee, K.S.S.; Wettersten, H.I.; Ulu, A.; Hu, X.; Tam, S.; et al. Epoxy metabolites of docosahexaenoic acid (DHA) inhibit angiogenesis, tumor growth, and metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 6530–6535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-Y.; Lin, Y.-P.; Qiu, H.; Morisseau, C.; Rose, T.E.; Hwang, S.H.; Chiamvimonvat, N.; Hammock, B.D. Substituted phenyl groups improve the pharmacokinetic profile and anti-inflammatory effect of urea-based soluble epoxide hydrolase inhibitors in murine models. Eur. J. Pharm. Sci. 2013, 48, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Stanford, K.I.; Middelbeek, R.J.; Townsend, K.L.; An, D.; Nygaard, E.B.; Hitchcox, K.M.; Markan, K.; Nakano, K.; Hirshman, M.F.; Tseng, Y.-H.; et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Investig. 2012, 123, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Mandard, S.; Kersten, S. Peroxisome proliferator-activated receptor a target genes. Cell. Mol. Life Sci. 2004, 61, 393–416. [Google Scholar] [CrossRef]
- Rodriguez, M.A.D.L.R.; Kersten, S. Regulation of lipid droplet-associated proteins by peroxisome proliferator-activated receptors. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2017, 1862 (10 Pt B), 1212–1220. [Google Scholar] [CrossRef]
- Xu, D.-Y.; Davis, B.B.; Wang, Z.-H.; Zhao, S.-P.; Wasti, B.; Liu, Z.-L.; Li, N.; Morisseau, C.; Chiamvimonvat, N.; Hammock, B.D. A potent soluble epoxide hydrolase inhibitor, t-AUCB, acts through PPARγ to modulate the function of endothelial progenitor cells from patients with acute myocardial infarction. Int. J. Cardiol. 2013, 167, 1298–1304. [Google Scholar] [CrossRef] [Green Version]
- Morin, C.; Sirois, M.; Echave, V.; Albadine, R.; Rousseau, É. 17,18-Epoxyeicosatetraenoic Acid Targets PPARγ and p38 Mitogen–Activated Protein Kinase to Mediate Its Anti-inflammatory Effects in the Lung: Role of soluble epoxide hydrolase. Am. J. Respir. Cell Mol. Biol. 2010, 43, 564–575. [Google Scholar] [CrossRef]
- Hanif, A.; Edin, M.L.; Zeldin, D.C.; Morisseau, C.; Nayeem, M.A. Effect of Soluble Epoxide Hydrolase on the Modulation of Coronary Reactive Hyperemia: Role of Oxylipins and PPARγ. PLoS ONE 2016, 11, e0162147. [Google Scholar] [CrossRef]
- Pradhan, I.; Ledent, C.; Mustafa, S.J.; Morisseau, C.; Nayeem, M.A. High salt diet modulates vascular response in A2AAR (+/+) and A 2AAR (−/−) mice: Role of sEH, PPARγ, and K ATP channels. Mol. Cell. Biochem. 2015, 404, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, T.E.; Morisseau, C.; Liu, J.-Y.; Inceoglu, B.; Jones, P.D.; Sanborn, J.R.; Hammock, B.D. 1-Aryl-3-(1-acylpiperidin-4-yl)urea Inhibitors of Human and Murine Soluble Epoxide Hydrolase: Structure−Activity Relationships, Pharmacokinetics, and Reduction of Inflammatory Pain. J. Med. Chem. 2010, 53, 7067–7075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, H.-J.; Hwang, S.H.; Morisseau, C.; Yang, J.; Jones, P.D.; Kasagami, T.; Kim, I.-H.; Hammock, B.D. Pharmacokinetic screening of soluble epoxide hydrolase inhibitors in dogs. Eur. J. Pharm. Sci. 2010, 40, 222–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Q.; Ma, M.; Ishima, T.; Morisseau, C.; Yang, J.; Wagner, K.M.; Zhang, J.-C.; Yang, C.; Yao, W.; Dong, C.; et al. Gene deficiency and pharmacological inhibition of soluble epoxide hydrolase confers resilience to repeated social defeat stress. Proc. Natl. Acad. Sci. USA 2016, 113, E1944–E1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettaieb, A.; Koike, S.; Hsu, M.-F.; Ito, Y.; Chahed, S.; Bachaalany, S.; Gruzdev, A.; Calvo-Rubio, M.; Lee, K.S.S.; Inceoglu, B.; et al. Soluble epoxide hydrolase in podocytes is a significant contributor to renal function under hyperglycemia. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861 (11 Pt A), 2758–2765. [Google Scholar] [CrossRef]
- Sura, P.; Sura, R.; EnayetAllah, A.E.; Grant, D.F. Distribution and Expression of Soluble Epoxide Hydrolase in Human Brain. J. Histochem. Cytochem. 2008, 56, 551–559. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Davis, B.B.; Morisseau, C.; Hammock, B.D.; Olson, J.L.; Kroetz, D.L.; Weiss, R.H. Vascular localization of soluble epoxide hydrolase in the human kidney. Am. J. Physiol. Physiol. 2004, 286, F720–F726. [Google Scholar] [CrossRef]
- Klein, J.; Fasshauer, M.; Klein, H.H.; Benito, M.; Kahn, C.R. Novel adipocyte lines from brown fat: A model system for the study of differentiation, energy metabolism, and insulin action. BioEssays 2002, 24, 382–388. [Google Scholar] [CrossRef]
- Shinoda, K.; Luijten, I.H.N.; Hasegawa, Y.; Hong, H.; Sonne, S.B.; Kim, M.; Xue, R.; Chondronikola, M.; Cypess, A.M.; Tseng, Y.-H.; et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat. Med. 2015, 21, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Taxvig, C.; Dreisig, K.; Boberg, J.; Nellemann, C.; Schelde, A.B.; Pedersen, D.; Boergesen, M.; Mandrup, S.; Vinggaard, A.M. Differential effects of environmental chemicals and food contaminants on adipogenesis, biomarker release and PPARγ activation. Mol. Cell. Endocrinol. 2012, 361, 106–115. [Google Scholar] [CrossRef]
- Wang, W.; Yang, J.; Zhang, J.; Wang, Y.; Hwang, S.H.; Qi, W.; Wan, D.; Kim, D.; Sun, J.; Sanidad, K.Z.; et al. Lipidomic profiling reveals soluble epoxide hydrolase as a therapeutic target of obesity-induced colonic inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, 5283–5288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, J.; Ricciardi, C.J.; Esposito, D.; Komarnytsky, S.; Hu, P.; Curry, B.; Brown, P.L.; Gao, Z.; Biggerstaff, J.P.; Chen, J.; et al. Activation of pattern recognition receptors in brown adipocytes induces inflammation and suppresses uncoupling protein 1 expression and mitochondrial respiration. Am. J. Physiol. Physiol. 2014, 306, C918–C930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciardi, C.J.; Bae, J.; Esposito, D.; Komarnytsky, S.; Hu, P.; Chen, J.; Zhao, L. 1,25-Dihydroxyvitamin D3/vitamin D receptor suppresses brown adipocyte differentiation and mitochondrial respiration. Eur. J. Nutr. 2014, 54, 1001–1012. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Overby, H.; Yang, Y.; Xu, X.; Graham, K.; Hildreth, K.; Choi, S.; Wan, D.; Morisseau, C.; Zeldin, D.C.; Hammock, B.D.; et al. Soluble Epoxide Hydrolase Inhibition by t-TUCB Promotes Brown Adipogenesis and Reduces Serum Triglycerides in Diet-Induced Obesity. Int. J. Mol. Sci. 2020, 21, 7039. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197039
Overby H, Yang Y, Xu X, Graham K, Hildreth K, Choi S, Wan D, Morisseau C, Zeldin DC, Hammock BD, et al. Soluble Epoxide Hydrolase Inhibition by t-TUCB Promotes Brown Adipogenesis and Reduces Serum Triglycerides in Diet-Induced Obesity. International Journal of Molecular Sciences. 2020; 21(19):7039. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197039
Chicago/Turabian StyleOverby, Haley, Yang Yang, Xinyun Xu, Katherine Graham, Kelsey Hildreth, Sue Choi, Debin Wan, Christophe Morisseau, Darryl C. Zeldin, Bruce D. Hammock, and et al. 2020. "Soluble Epoxide Hydrolase Inhibition by t-TUCB Promotes Brown Adipogenesis and Reduces Serum Triglycerides in Diet-Induced Obesity" International Journal of Molecular Sciences 21, no. 19: 7039. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197039