Multi-Compartment, Early Disruption of cGMP and cAMP Signalling in Cardiac Myocytes from the mdx Model of Duchenne Muscular Dystrophy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. cGMP Signalling in the Cytosol
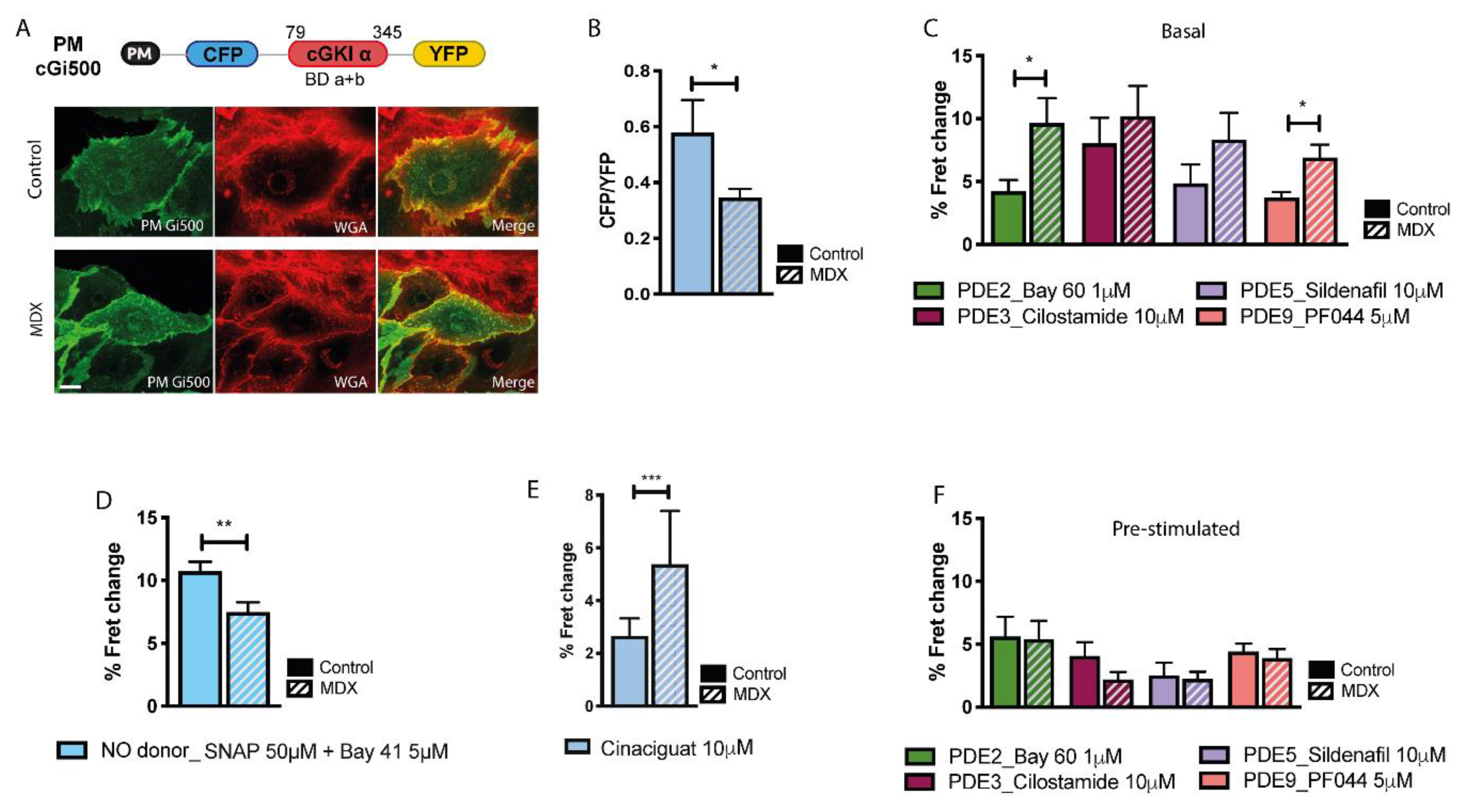
2.2. cGMP Signalling at the Plasmalemma
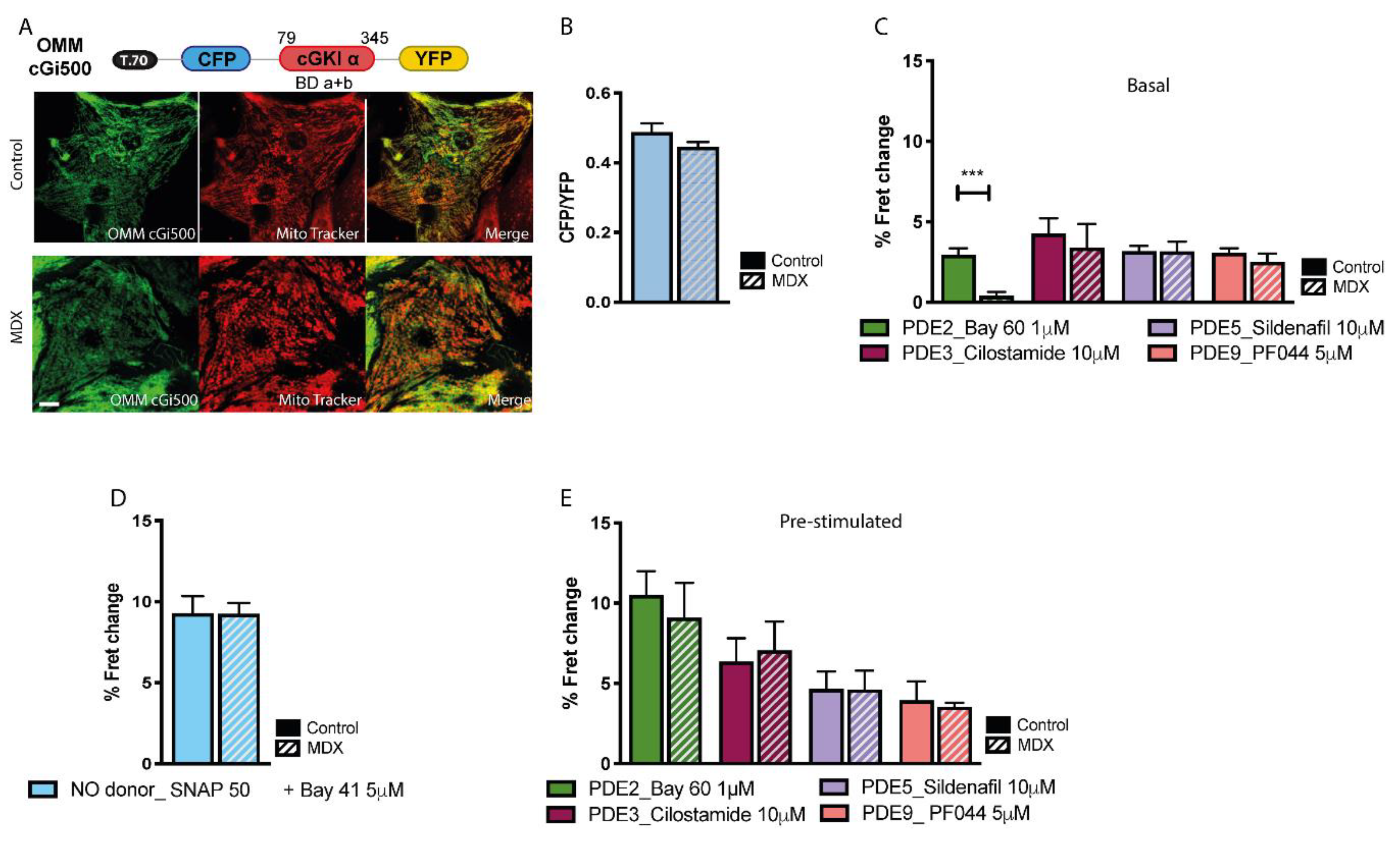
2.3. cGMP Signalling at the OMM
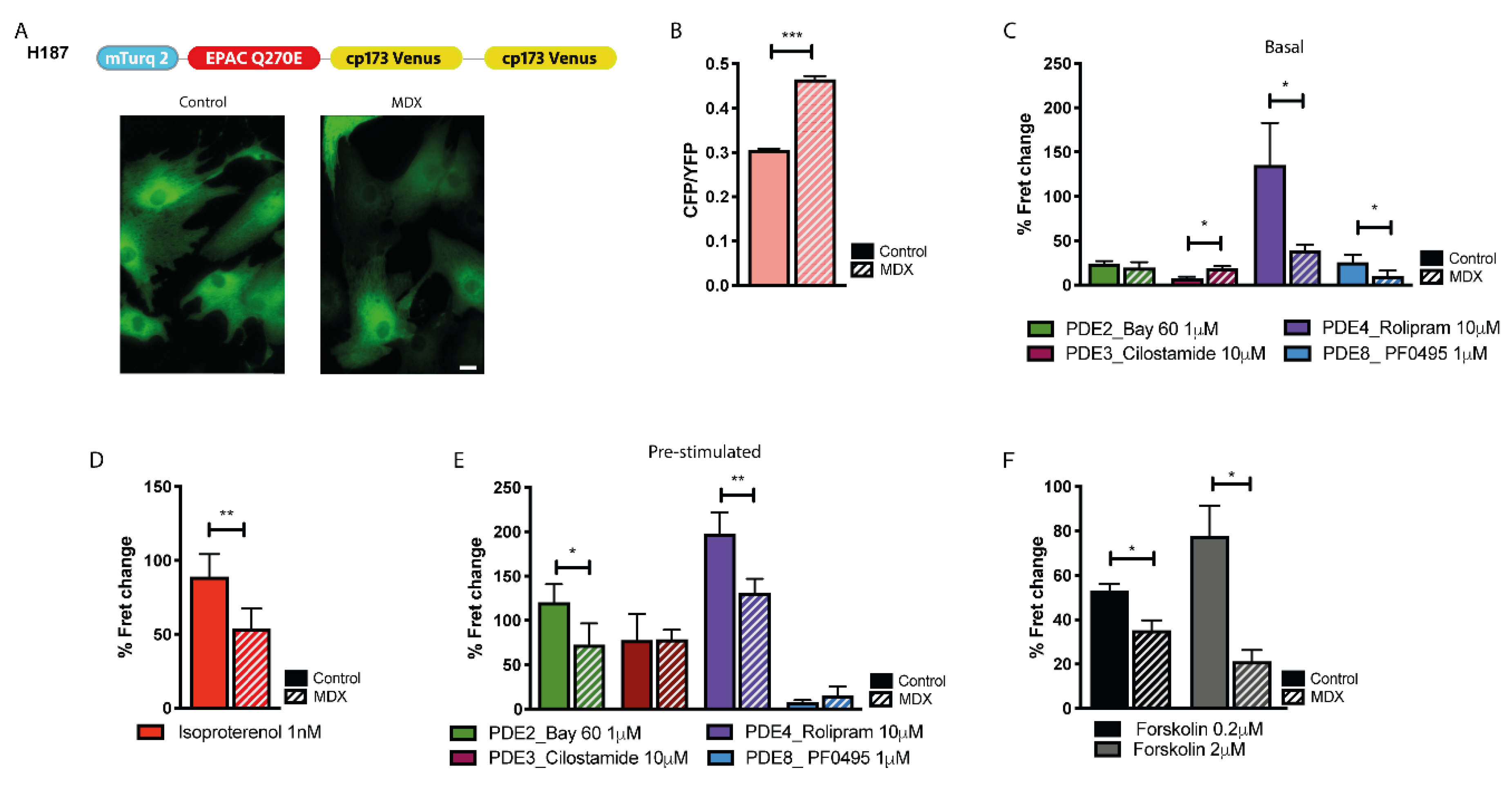
2.4. cAMP Signalling in the Bulk Cytosol
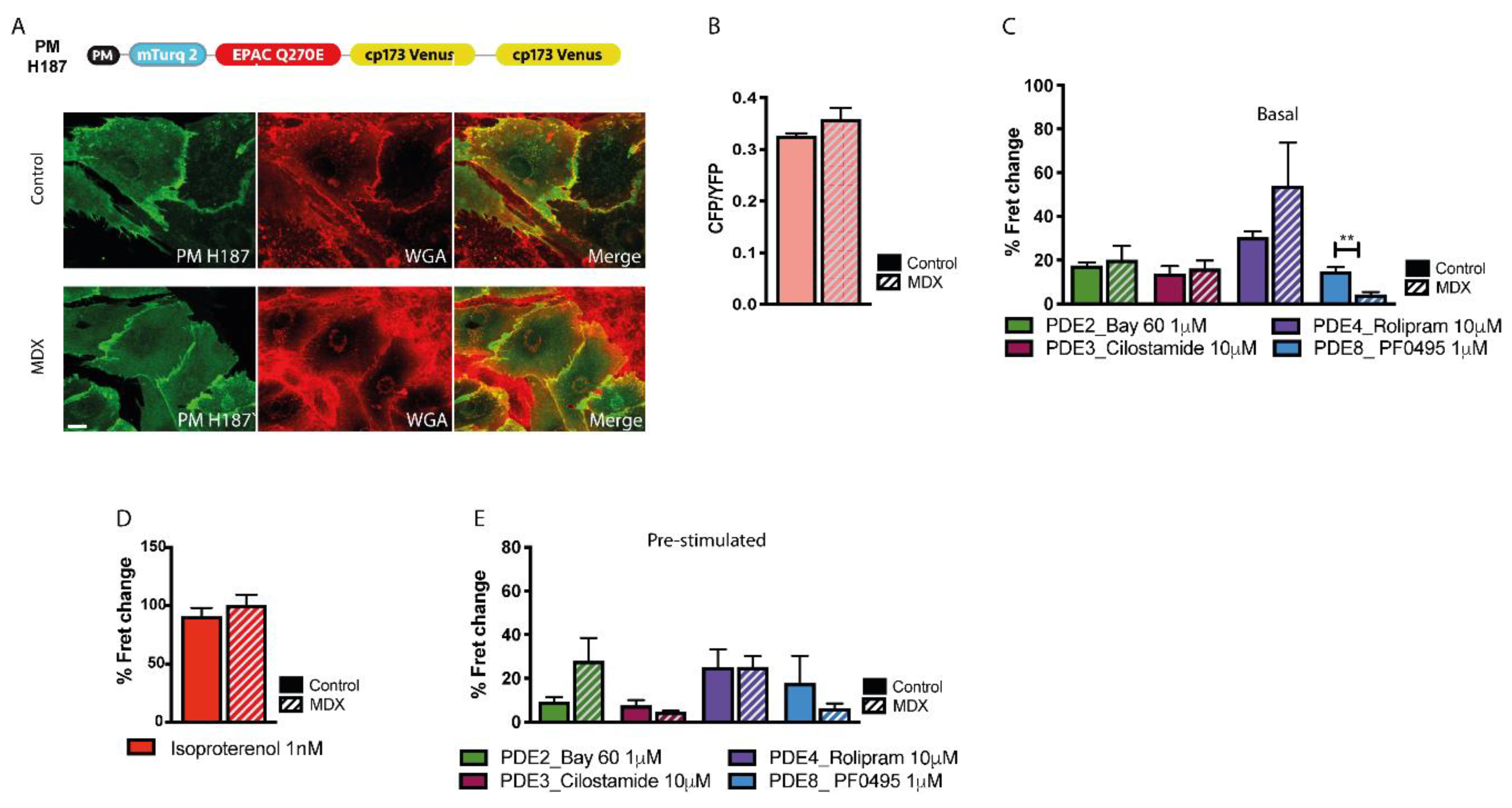
2.5. cAMP Signalling at the Plasmalemma
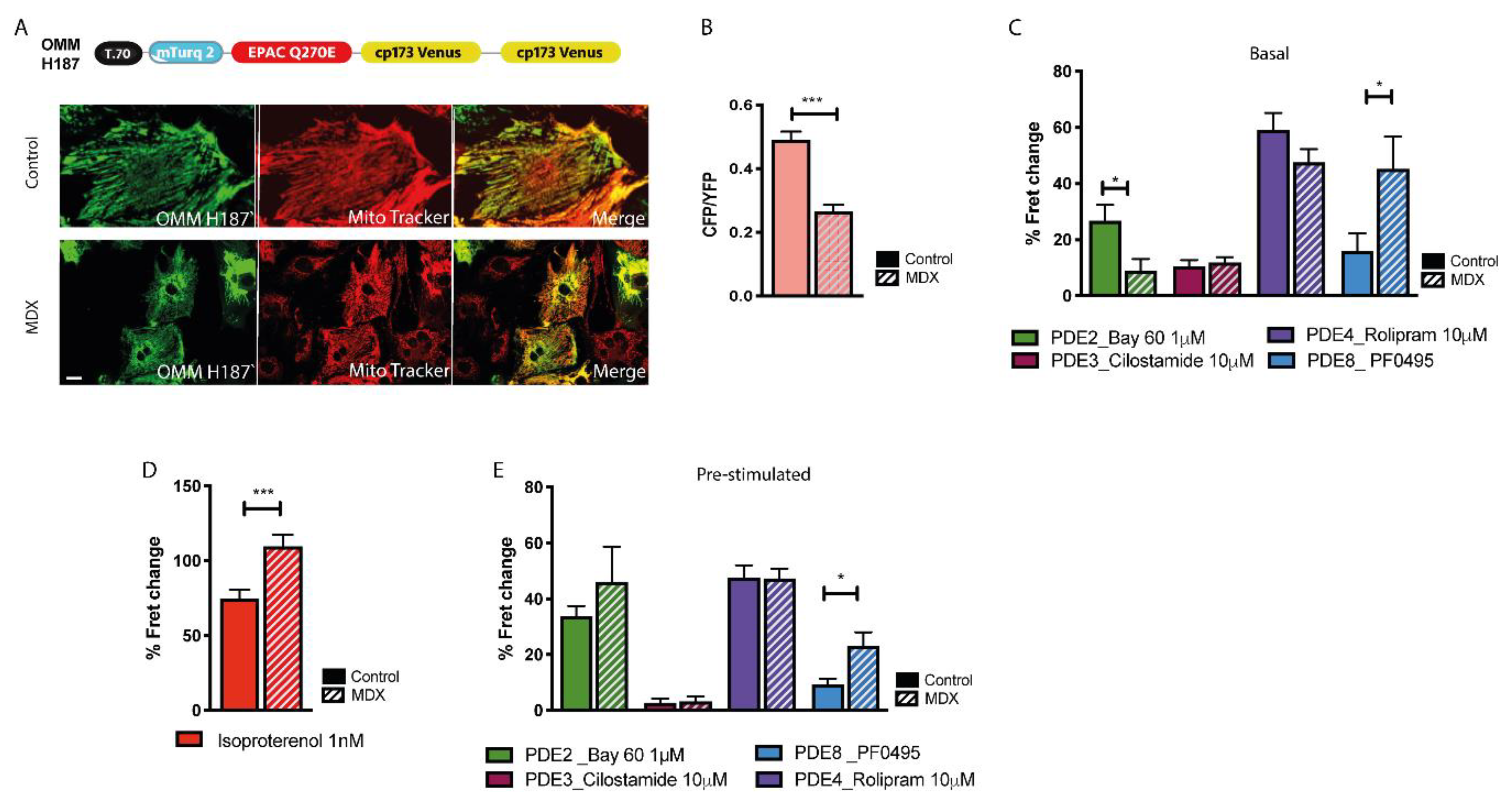
2.6. cAMP Signalling at the OMM
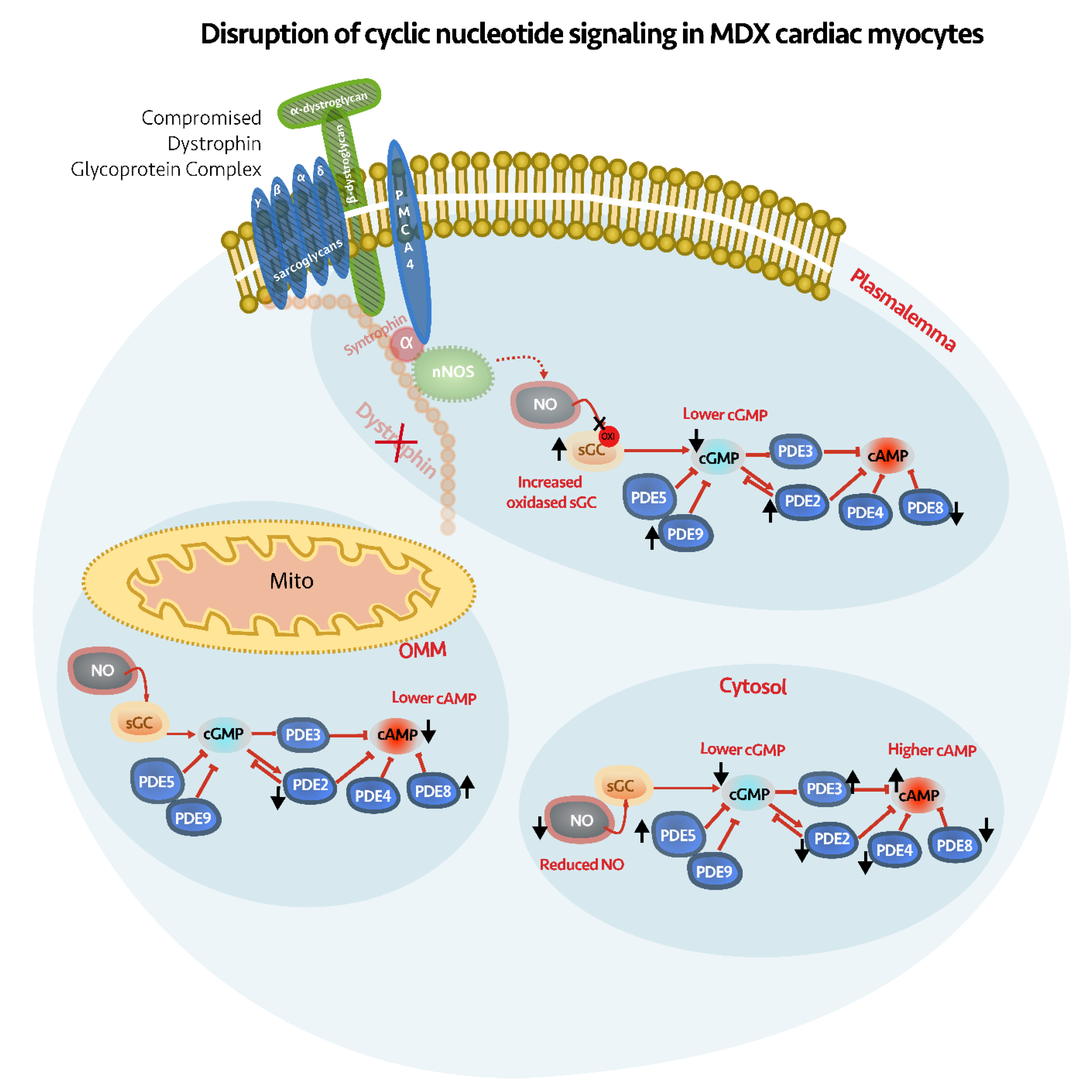
3. Discussion
4. Materials and Methods
4.1. Generation of Targeted FRET-Based Sensors
4.2. Determination of FRET Sensors Dynamic Range
4.3. Neonatal Mouse Ventricular Cardiomyocyte Isolation
4.4. Viral Infection
4.5. FRET Imaging
4.6. Confocal Imaging
4.7. qPCR
4.8. Western Blotting
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Müller, C.R.; Lindlöf, M.; Kaariainen, H. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar] [PubMed]
- Meyers, T.A.; Townsend, D. Cardiac pathophysiology and the future of cardiac therapies in Duchenne muscular dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef] [Green Version]
- Gieron-Korthals, M.; Fernandez, R. New Developments in Diagnosis, Treatment, and Management of Duchenne Muscular Dystrophy. Adv. Pediatr. 2020, 67, 183–196. [Google Scholar] [CrossRef]
- Houang, E.M.; Sham, Y.Y.; Bates, F.S.; Metzger, J.M. Muscle membrane integrity in Duchenne muscular dystrophy: Recent advances in copolymer-based muscle membrane stabilizers. Skelet. Muscle 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, D.G.; Gervasio, O.L.; Yeung, E.W.; Whitehead, N.P. Calcium and the damage pathways in muscular dystrophy. Can. J. Physiol. Pharm. 2010, 88, 83–91. [Google Scholar] [CrossRef]
- Van Westering, T.L.E.; Betts, C.A.; Wood, M.J.A. Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy. Molecules 2015, 20, 8823–8855. [Google Scholar] [CrossRef]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne muscular dystrophy: Insights from cellular studies. J. Mol. Cell. Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Kokkonen, K.; Kass, D.A. Nanodomain regulation of cardiac cyclic nucleotide signaling by phosphodiesterases. Annu. Rev. Pharm. Toxicol. 2017, 57, 455–479. [Google Scholar] [CrossRef]
- Chang, W.-J.; Iannaccone, S.T.; Lau, K.S.; Masters, B.S.; McCabe, T.J.; McMillan, K.; Padre, R.C.; Spencer, M.J.; Tidball, J.G.; Stull, J.T. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. USA 1996, 93, 9142–9147. [Google Scholar] [CrossRef] [Green Version]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Thomas, G.D.; Yue, Y.; Yang, H.T.; Li, D.; Long, C.; Judge, L.; Bostick, B.; Chamberlain, J.S.; Terjung, R.L. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Investig. 2009, 119, 624–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Zhao, J.; Yue, Y.; Wasala, N.B.; Duan, D. Partial restoration of cardiac function with ΔPDZ nNOS in aged mdx model of Duchenne cardiomyopathy. Hum. Mol. Genet. 2014, 23, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- McDonough, P.M.; Glembotski, C.C. Induction of atrial natriuretic factor and myosin light chain-2 gene expression in cultured ventricular myocytes by electrical stimulation of contraction. J. Biol. Chem. 1992, 267, 11665–11668. [Google Scholar]
- Mawatari, S.; Schonberg, M.; Olarte, M. Biochemical abnormalities of erythrocyte membranes in Duchenne dystrophy: Adenosine triphosphatase and adenyl cyclase. Arch. Neurol. 1976, 33, 489–493. [Google Scholar] [CrossRef]
- Cerri, C.G.; Willner, J.H.; Miranda, A.F. Adenylate cyclase in Duchenne fibroblasts. J. Neurol. Sci. 1982, 53, 181–185. [Google Scholar] [CrossRef]
- Canal, N.; Frattola, L.; Smirne, S. The metabolism of cyclic-3′-5′-adenosine monophosphate (cAMP) in diseased muscle. J. Neurol. 1975, 208, 259–265. [Google Scholar] [CrossRef]
- Bers, D.M.; Xiang, Y.K.; Zaccolo, M. Whole-cell cAMP and PKA activity are epiphenomena, nanodomain signaling matters. Physiology 2019, 34, 240–249. [Google Scholar] [CrossRef]
- Shaulsky, G.; Fuller, D.; Loomis, W.F. A cAMP-phosphodiesterase controls PKA-dependent differentiation. Development 1998, 125, 691–699. [Google Scholar]
- Zaccolo, M. Spatial control of cAMP signalling in health and disease. Curr. Opin. Pharm. 2011, 11, 649–655. [Google Scholar] [CrossRef] [Green Version]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuckey, D.J.; Carr, C.A.; Camelliti, P.; Tyler, D.J.; Davies, K.E.; Clarke, K. In vivo MRI characterization of progressive cardiac dysfunction in the mdx mouse model of muscular dystrophy. PLoS ONE 2012, 7, e28569. [Google Scholar] [CrossRef] [PubMed]
- Russwurm, M.; Mullershausen, F.; Friebe, A.; Jäger, R.; Russwurm, C.; Koesling, D. Design of fluorescence resonance energy transfer (FRET)-based cGMP indicators: A systematic approach. Biochem. J. 2007, 407, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandeput, F.; Krall, J.; Ockaili, R.; Salloum, F.N.; Florio, V.; Corbin, J.D.; Francis, S.H.; Kukreja, R.C.; Movsesian, M.A. cGMP-hydrolytic activity and its inhibition by sildenafil in normal and failing human and mouse myocardium. J. Pharmacol. Exp. Ther. 2009, 330, 884–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, T.J.; Mumby, M.C.; Beavo, J.A. Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J. Biol. Chem. 1982, 257, 1973–1979. [Google Scholar] [PubMed]
- Tsai, E.J.; Liu, Y.; Koitabashi, N.; Bedja, D.; Danner, T.; Jasmin, J.-F.; Lisanti, M.P.; Friebe, A.; Takimoto, E.; Kass, D.A. Pressure-overload–induced subcellular Relocalization/oxidation of soluble guanylyl cyclase in the heart modulates enzyme stimulation. Circ. Res. 2012, 110, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Resh, M.D. Fatty acylation of proteins: New insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta (Bba)-Mol. Cell Res. 1999, 1451, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Surmeli, N.B.; Marletta, M.A. Insight into the rescue of oxidized soluble guanylate cyclase by the activator cinaciguat. Chembiochem 2012, 13, 977–981. [Google Scholar] [CrossRef] [Green Version]
- Lefkimmiatis, K.; Leronni, D.; Hofer, A.M. The inner and outer compartments of mitochondria are sites of distinct cAMP/PKA signaling dynamics. J. Cell Biol. 2013, 202, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Klarenbeek, J.; Goedhart, J.; van Batenburg, A.; Groenewald, D.; Jalink, K. Fourth-generation epac-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: Characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS ONE 2015, 10, e0122513. [Google Scholar] [CrossRef] [Green Version]
- Susheela, A.K.; Kaul, R.D.; Sachdeva, K.; Singh, N. Adenyl cyclase activity in Duchenne dystrophic muscle. J. Neurol. Sci. 1975, 24, 361–363. [Google Scholar] [CrossRef]
- Burdyga, A.; Surdo, N.C.; Monterisi, S.; Di Benedetto, G.; Grisan, F.; Penna, E.; Pellegrini, L.; Zaccolo, M.; Bortolozzi, M.; Swietach, P. Phosphatases control PKA-dependent functional microdomains at the outer mitochondrial membrane. Proc. Natl. Acad. Sci. USA 2018, 115, E6497–E6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surdo, N.C.; Berrera, M.; Koschinski, A.; Brescia, M.; Machado, M.R.; Carr, C.; Wright, P.; Gorelik, J.; Morotti, S.; Grandi, E. FRET biosensor uncovers cAMP nano-domains at β-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dombernowsky, N.W.; Ölmestig, J.N.E.; Witting, N.; Kruuse, C. Role of neuronal nitric oxide synthase (nNOS) in Duchenne and Becker muscular dystrophies–Still a possible treatment modality? Neuromuscul. Disord. 2018, 28, 914–926. [Google Scholar]
- Bloom, T.J. Cyclic nucleotide phosphodiesterase isozymes expressed in mouse skeletal muscle. Can. J. Physiol. Pharmacol. 2002, 80, 1132–1135. [Google Scholar] [PubMed]
- Adamo, C.M.; Dai, D.-F.; Percival, J.M.; Minami, E.; Willis, M.S.; Patrucco, E.; Froehner, S.C.; Beavo, J.A. Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 19079–19083. [Google Scholar]
- Barst, R.J.; Ivy, D.D.; Gaitan, G.; Szatmari, A.; Rudzinski, A.; Garcia, A.E.; Sastry, B.K.S.; Pulido, T.; Layton, G.R.; Serdarevic-Pehar, M. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension. Circulation 2012, 125, 324–334. [Google Scholar]
- Leung, D.G.; Herzka, D.A.; Thompson, W.R.; He, B.; Bibat, G.; Tennekoon, G.; Russell, S.D.; Schuleri, K.H.; Lardo, A.C.; Kass, D.A. Sildenafil does not improve cardiomyopathy in D uchenne/B ecker muscular dystrophy. Ann. Neurol. 2014, 76, 541–549. [Google Scholar]
- Tetsi, L.; Charles, A.-L.; Georg, I.; Goupilleau, F.; Lejay, A.; Talha, S.; Maumy-Bertrand, M.; Lugnier, C.; Geny, B. Effect of the Phosphodiesterase 5 Inhibitor Sildenafil on Ischemia-Reperfusion-Induced Muscle Mitochondrial Dysfunction and Oxidative Stress. Antioxidants 2019, 8, 93. [Google Scholar]
- Uaesoontrachoon, K.; Quinn, J.L.; Tatem, K.S.; Van Der Meulen, J.H.; Yu, Q.; Phadke, A.; Miller, B.K.; Gordish-Dressman, H.; Ongini, E.; Miglietta, D. Long-term treatment with naproxcinod significantly improves skeletal and cardiac disease phenotype in the mdx mouse model of dystrophy. Hum. Mol. Genet. 2014, 23, 3239–3249. [Google Scholar] [CrossRef] [Green Version]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S. Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Ascah, A.; Khairallah, M.; Daussin, F.; Bourcier-Lucas, C.; Godin, R.; Allen, B.G.; Petrof, B.J.; Des Rosiers, C.; Burelle, Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H144–H153. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Xi, L.; Kukreja, R.C. Phosphodiesterase-5 Inhib. Sildenafil Preconditions Adult Card. Myocytes Against Necrosis Apoptosis. Essent. Role Nitric Oxide Signal. J. Biol. Chem. 2005, 280, 12944–12955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlhaas, M.; Nickel, A.G.; Bergem, S.; Casadei, B.; Laufs, U.; Maack, C. Endogenous nitric oxide formation in cardiac myocytes does not control respiration during β-adrenergic stimulation. J. Physiol. 2017, 595, 3781–3798. [Google Scholar] [CrossRef] [PubMed]
- Percival, J.M.; Siegel, M.P.; Knowels, G.; Marcinek, D.J. Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Genet. 2013, 22, 153–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, S.; Zhang, X.; Li, J.; Ai, X.; Zhang, L.; Yu, D.; Ge, S.; Peng, Y.; Chen, X. Blunted cardiac beta-adrenergic response as an early indication of cardiac dysfunction in Duchenne muscular dystrophy. Cardiovasc. Res. 2014, 103, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Monterisi, S.; Favia, M.; Guerra, L.; Cardone, R.A.; Marzulli, D.; Reshkin, S.J.; Casavola, V.; Zaccolo, M. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J. Cell Sci. 2012, 125, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Straub, V.; Bittner, R.E.; Léger, J.J.; Voit, T. Direct visualization of the dystrophin network on skeletal muscle fiber membrane. J. Cell Biol. 1992, 119, 1183–1191. [Google Scholar] [CrossRef]
- Porter, G.A.; Dmytrenko, G.M.; Winkelmann, J.C.; Bloch, R.J. Dystrophin colocalizes with beta-spectrin in distinct subsarcolemmal domains in mammalian skeletal muscle. J. Cell Biol. 1992, 117, 997–1005. [Google Scholar] [CrossRef]
- Fisher, D.A.; Smith, J.F.; Pillar, J.S.; Denis, S.H.S.; Cheng, J.B. Isolation and characterization of PDE8A, a novel human cAMP-specific phosphodiesterase. Biochem. Biophys. Res. Commun. 1998, 246, 570–577. [Google Scholar] [CrossRef]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [Green Version]
- Diviani, D.; Osman, H.; Delaunay, M.; Kaiser, S. The role of A-kinase anchoring proteins in cardiac oxidative stress. Biochem. Soc. Trans. 2019, 47, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J. Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef] [Green Version]
- Terrin, A.; Di Benedetto, G.; Pertegato, V.; Cheung, Y.-F.; Baillie, G.; Lynch, M.J.; Elvassore, N.; Prinz, A.; Herberg, F.W.; Houslay, M.D. PGE1 stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: Role of compartmentalized phosphodiesterases. J. Cell Biol. 2006, 175, 441–451. [Google Scholar] [CrossRef]
- Koschinski, A.; Zaccolo, M. A novel approach combining real-time imaging and the patch-clamp technique to calibrate FRET-based reporters for cAMP in their cellular microenvironment. In cAMP Signaling; Springer, Humana Press: New York, NY, USA, 2015; pp. 25–40. [Google Scholar]
- Yang, Y.; Guo, T.; Oda, T.; Chakraborty, A.; Chen, L.; Uchinoumi, H.; Knowlton, A.A.; Fruen, B.R.; Cornea, R.L.; Meissner, G. Cardiac myocyte Z-line calmodulin is mainly RyR2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ. Res. 2014, 114, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Abràmoff, M.D.; Magalhães, P.J.; Ram, S.J. Image processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brescia, M.; Chao, Y.-C.; Koschinski, A.; Tomek, J.; Zaccolo, M. Multi-Compartment, Early Disruption of cGMP and cAMP Signalling in Cardiac Myocytes from the mdx Model of Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 7056. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197056
Brescia M, Chao Y-C, Koschinski A, Tomek J, Zaccolo M. Multi-Compartment, Early Disruption of cGMP and cAMP Signalling in Cardiac Myocytes from the mdx Model of Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2020; 21(19):7056. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197056
Chicago/Turabian StyleBrescia, Marcella, Ying-Chi Chao, Andreas Koschinski, Jakub Tomek, and Manuela Zaccolo. 2020. "Multi-Compartment, Early Disruption of cGMP and cAMP Signalling in Cardiac Myocytes from the mdx Model of Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 21, no. 19: 7056. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197056