Gold Nanoparticle Formation via X-ray Radiolysis Investigated with Time-Resolved X-ray Liquidography

 , , , , , , and
, , , , , , and
Abstract
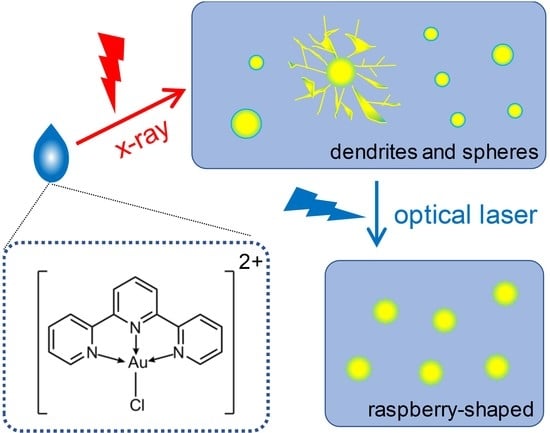
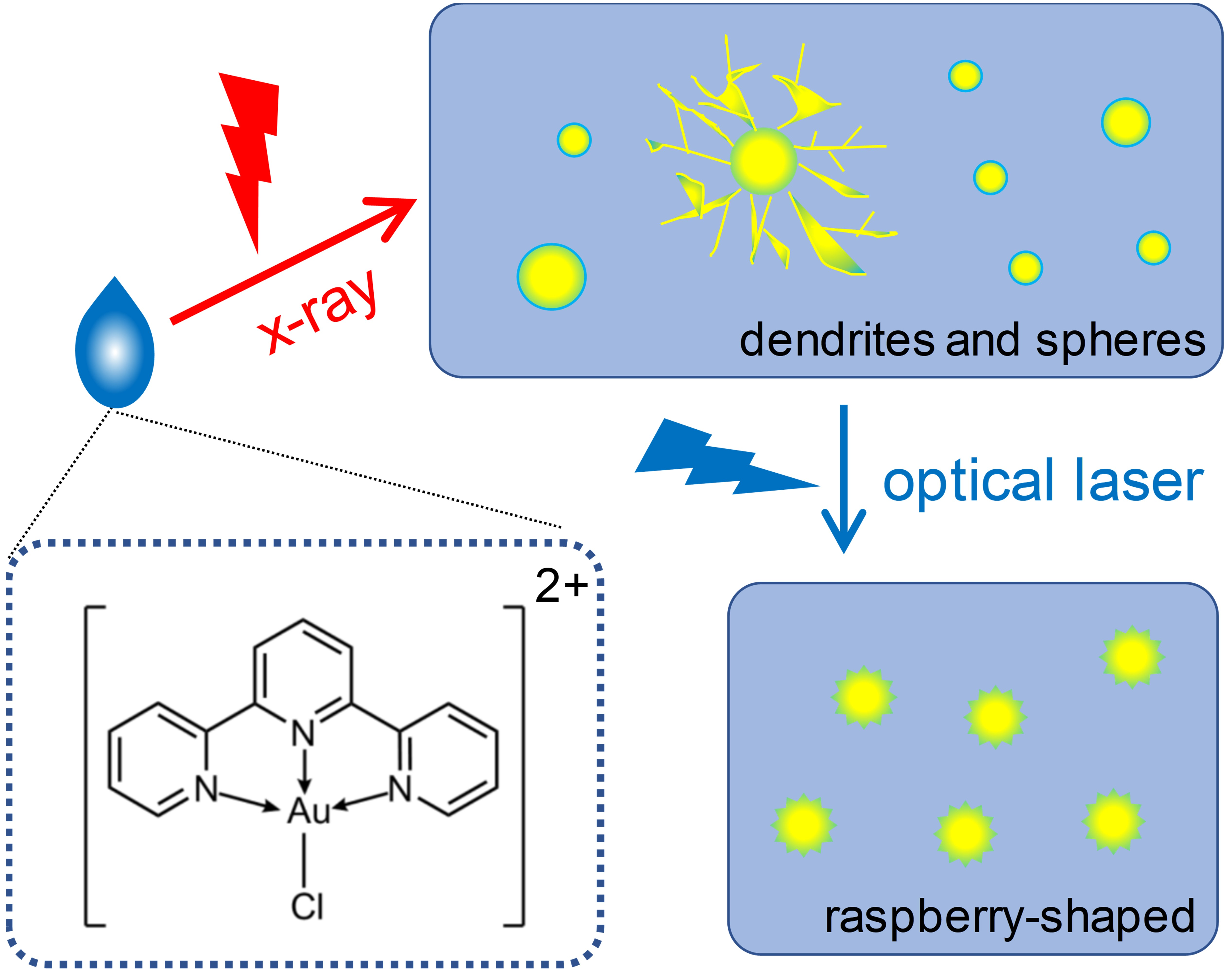
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
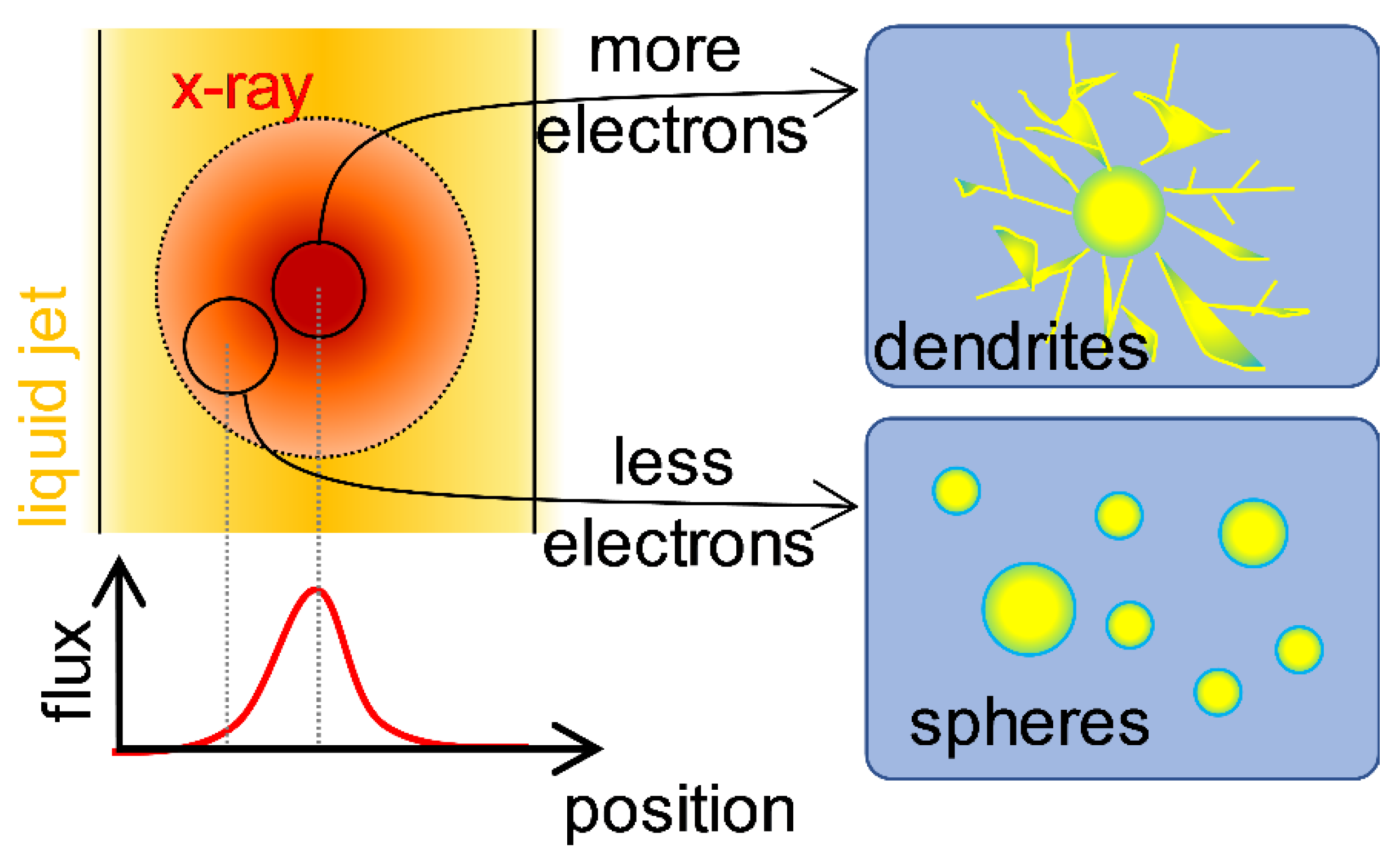
2.1. Formation of AuNPs Induced by X-ray Radiolysis
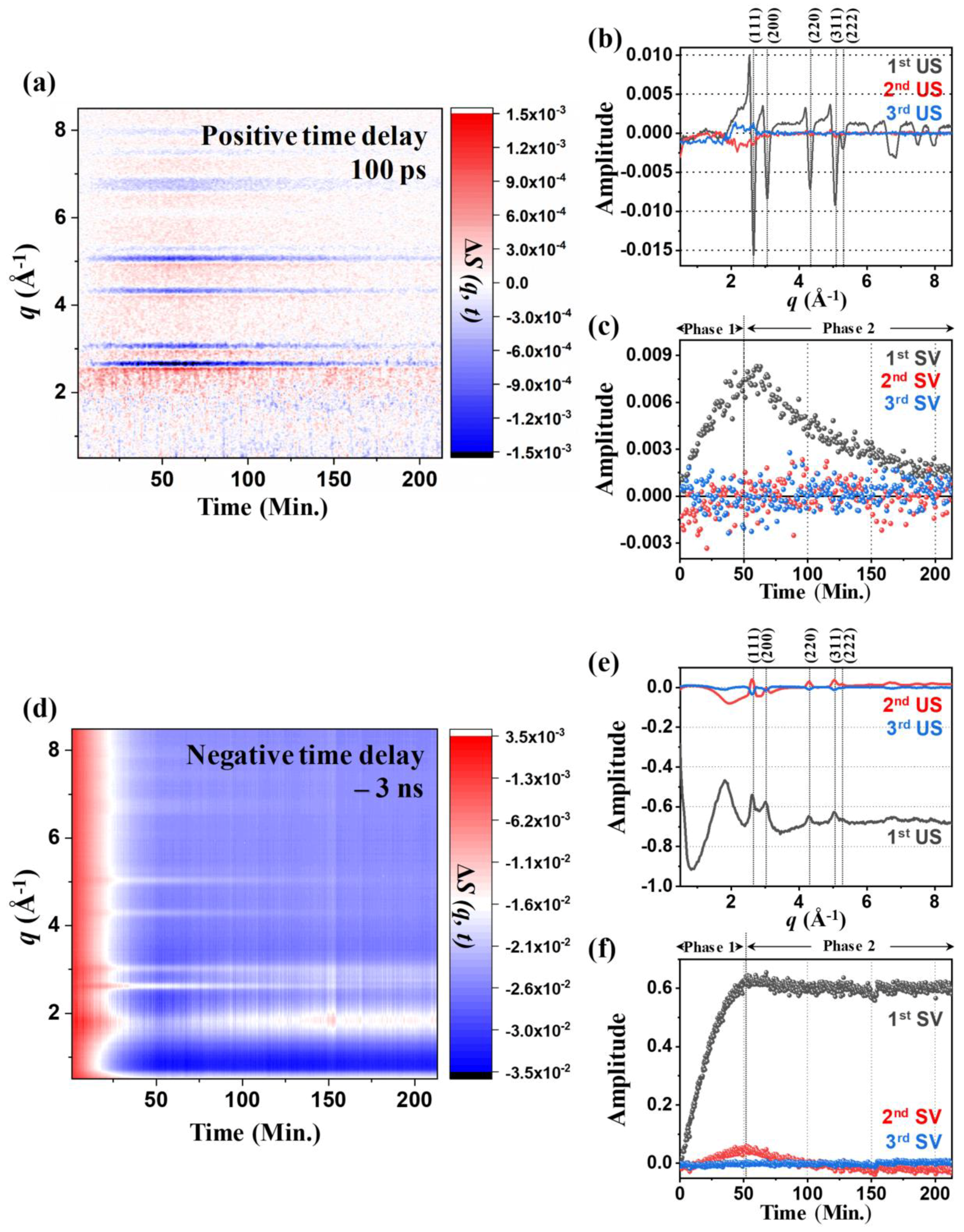
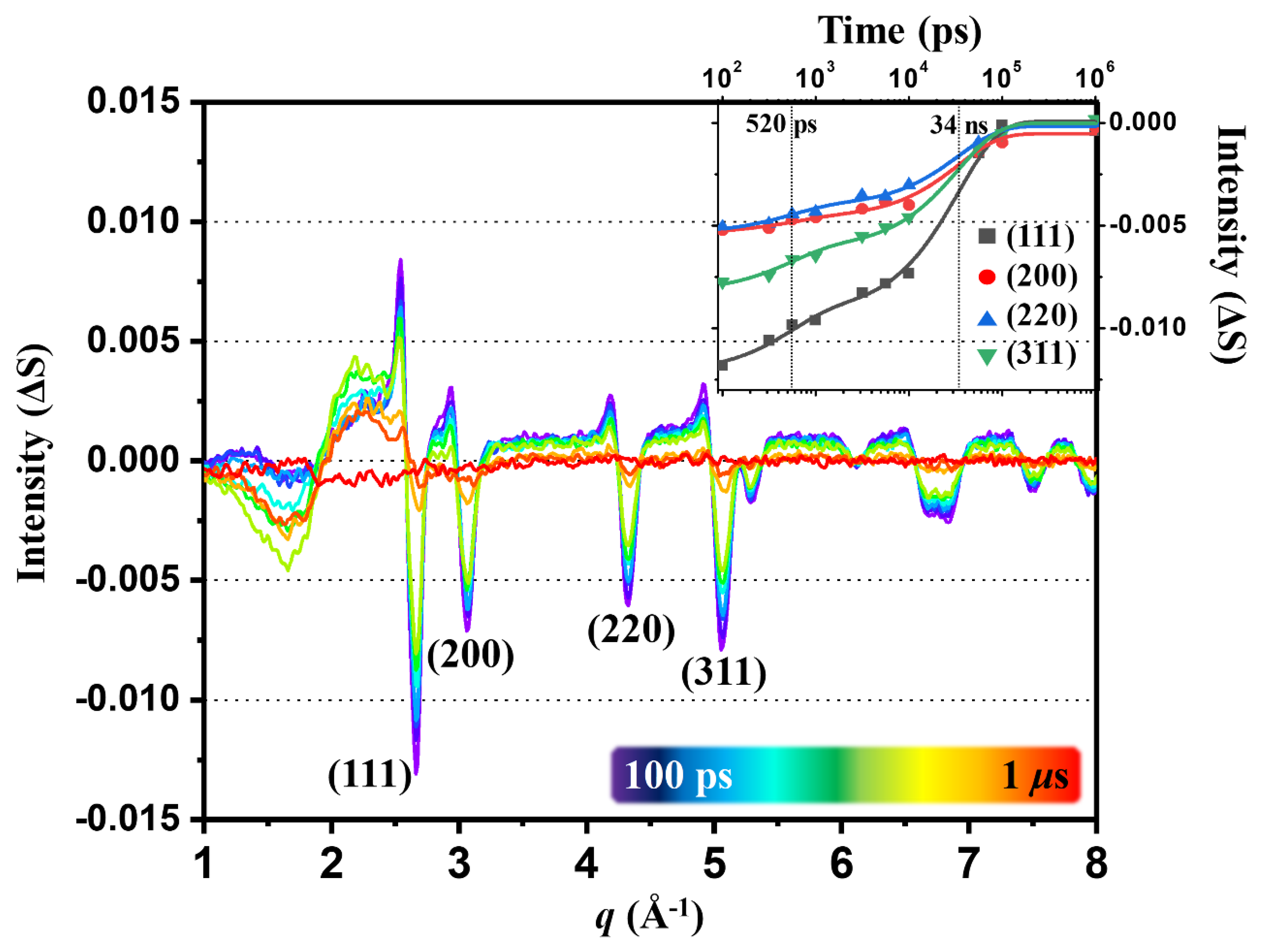
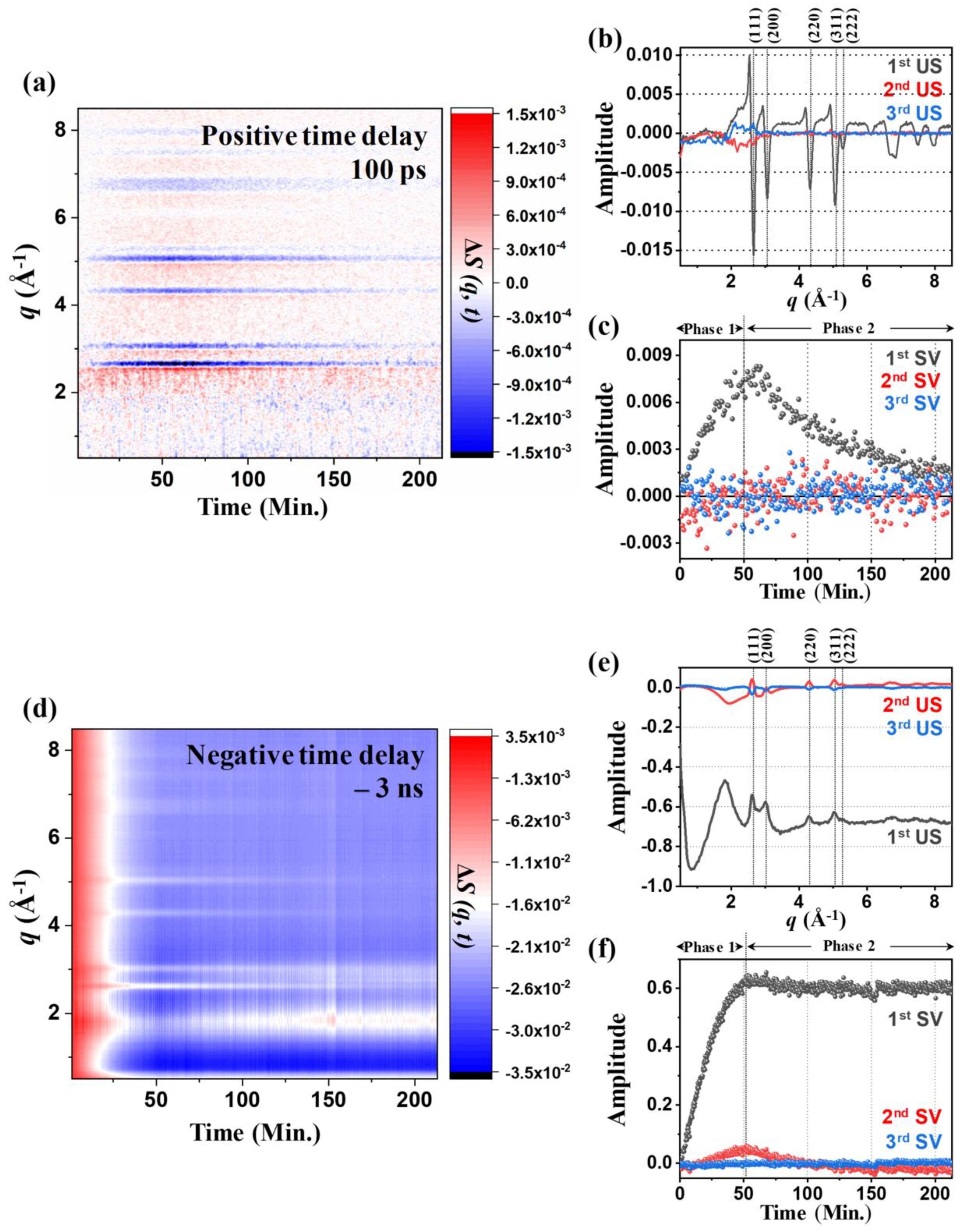
2.2. Kinetics of the 100 PS TRXL Data
2.3. Kinetics of Static Difference X-ray Scattering (SDXS) Data and Comparison with the Kinetics of TRXL Data
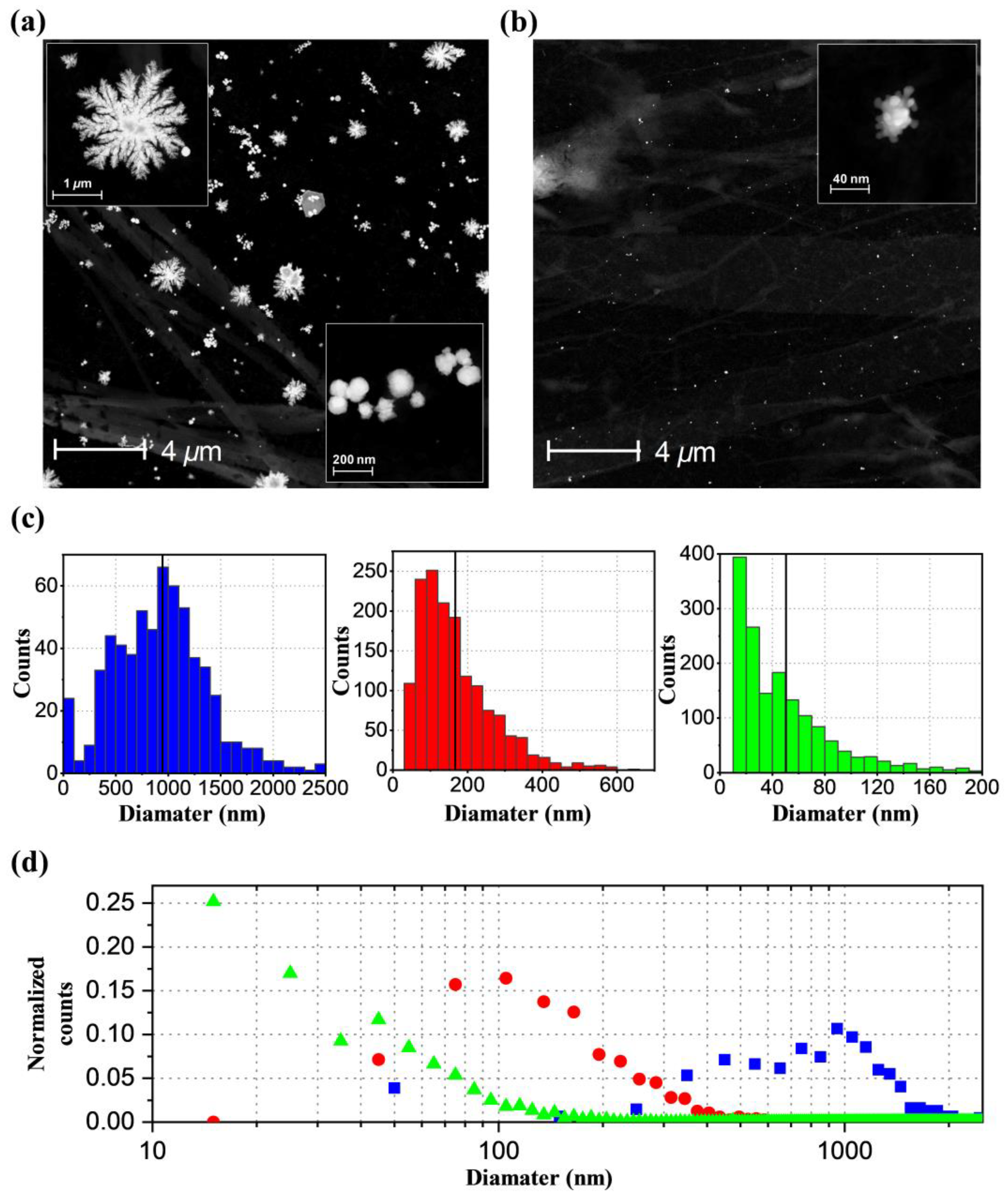
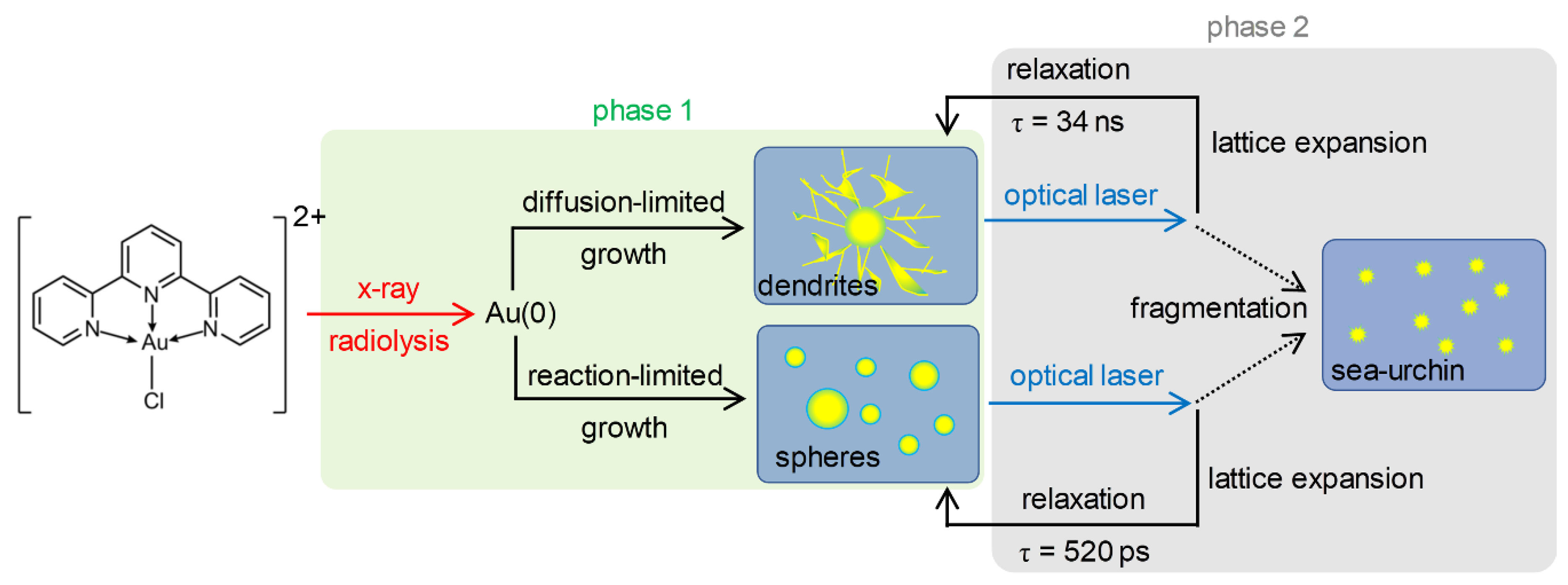
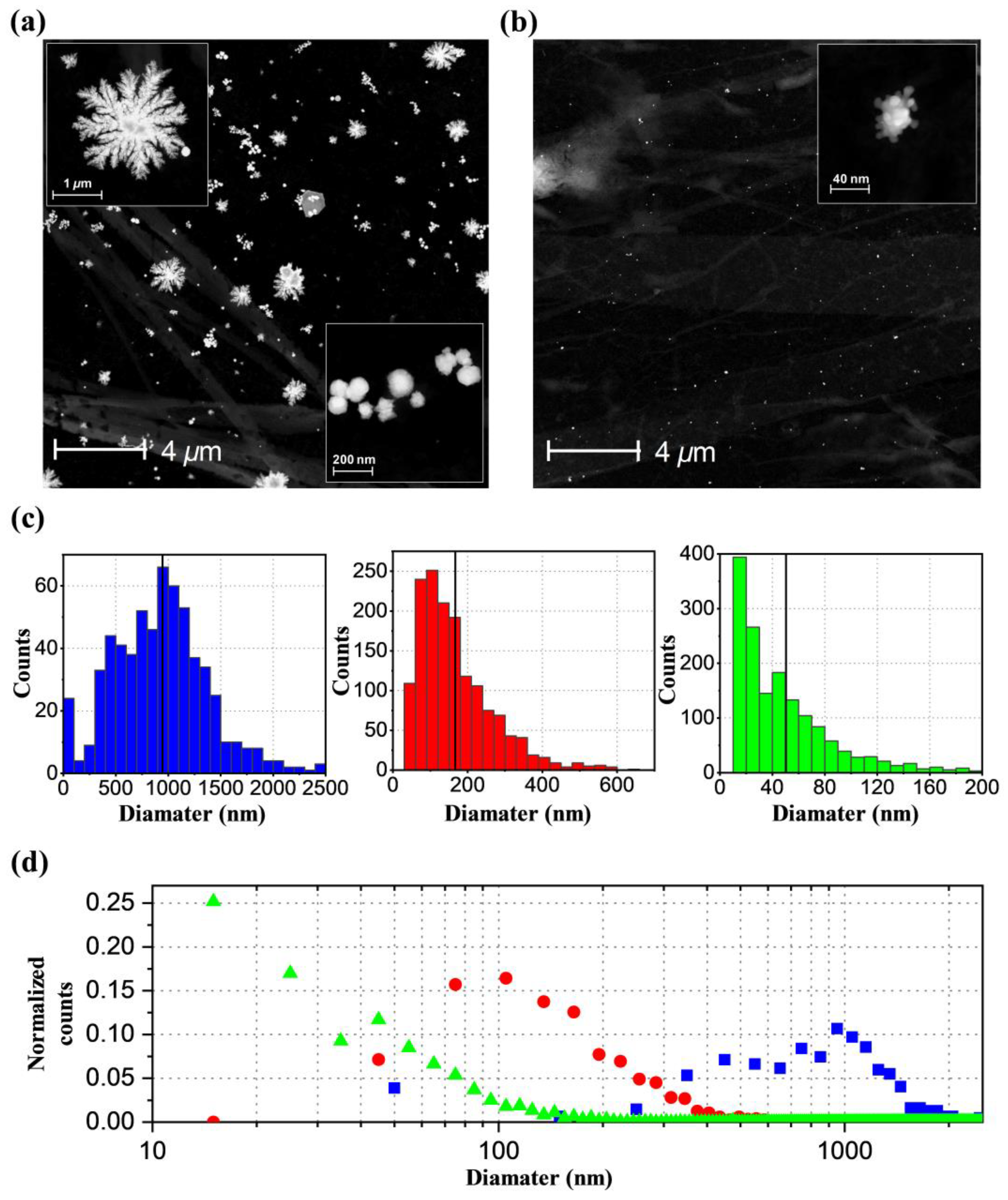
2.4. TEM Images and Formation Mechanism of AuNPs
2.5. Lattice Dynamics and Ablation of AuNPs
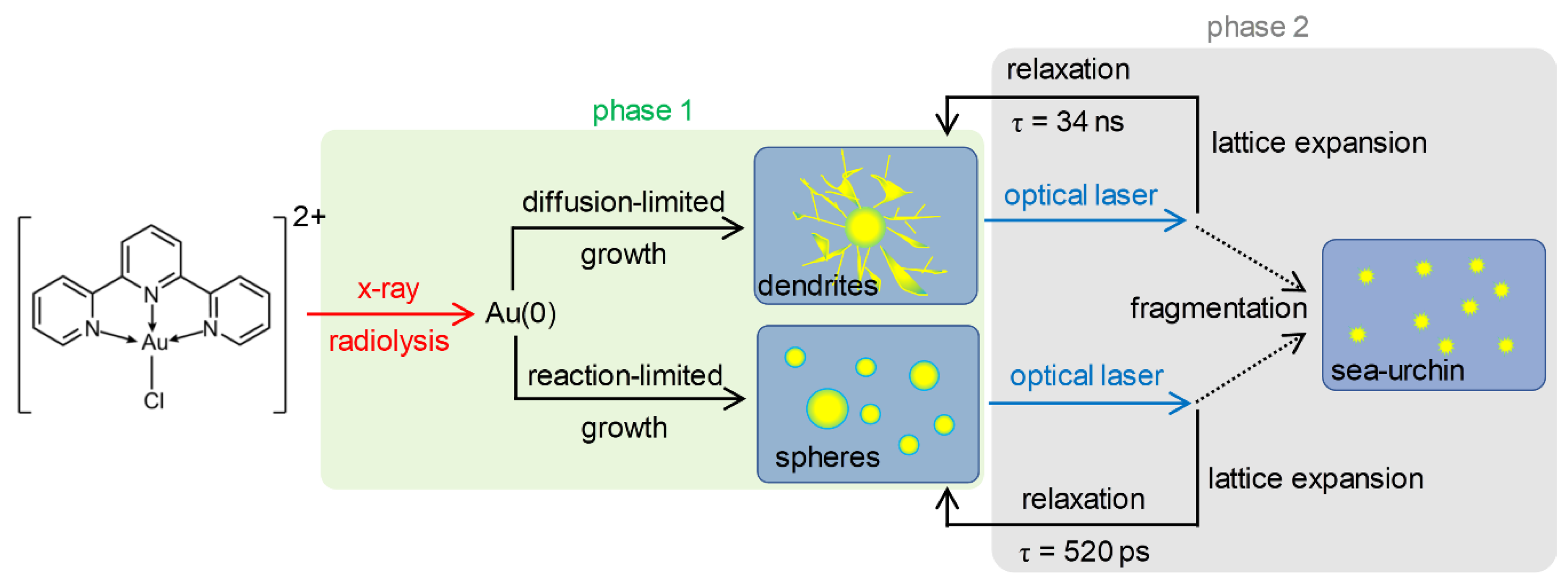
2.6. Overall Picture
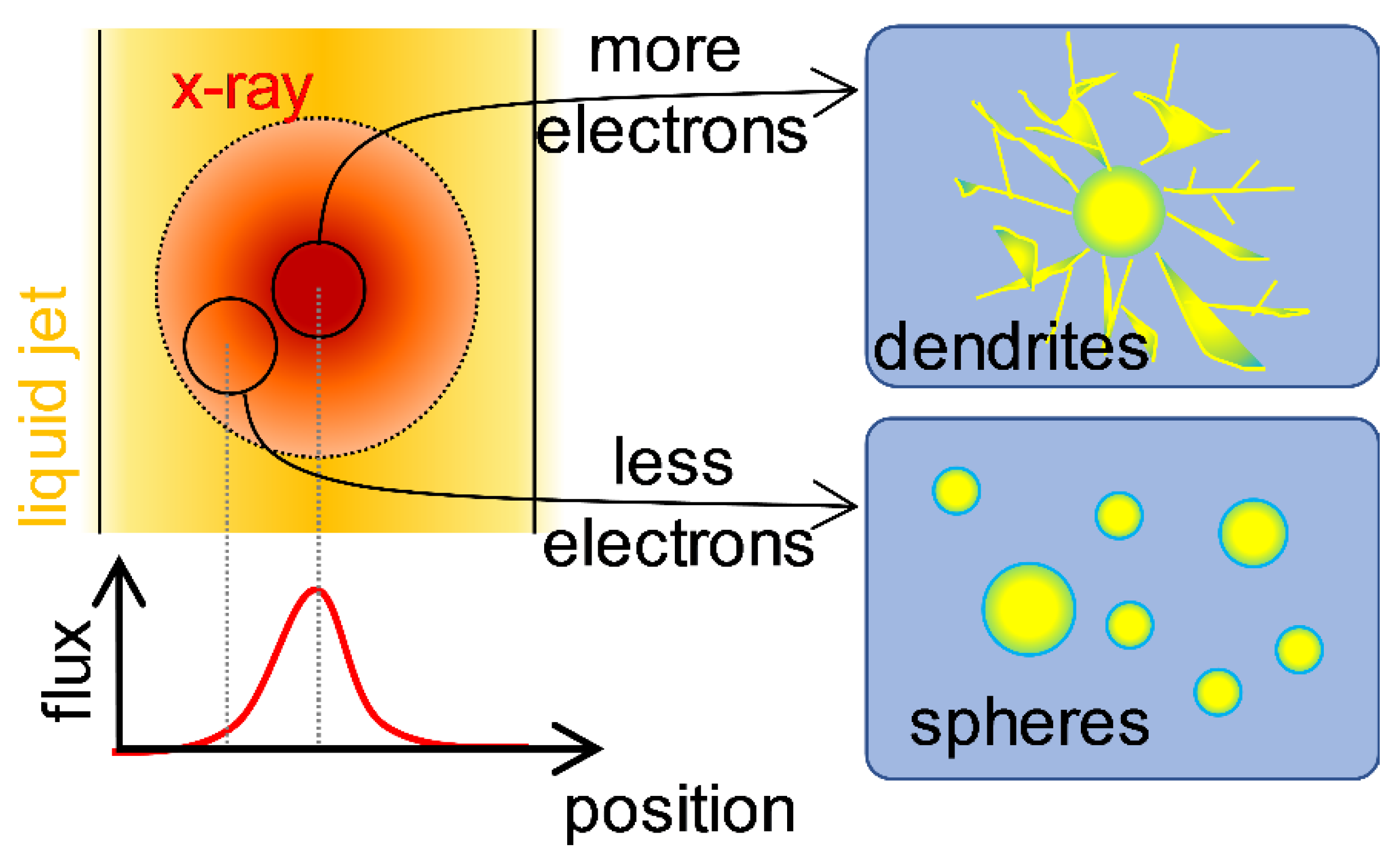
2.7. Determining Factor for Nanoparticle Formation by X-ray Radiolysis
3. Materials and Methods
3.1. Preparation of [Au(tpy)Cl]Cl2
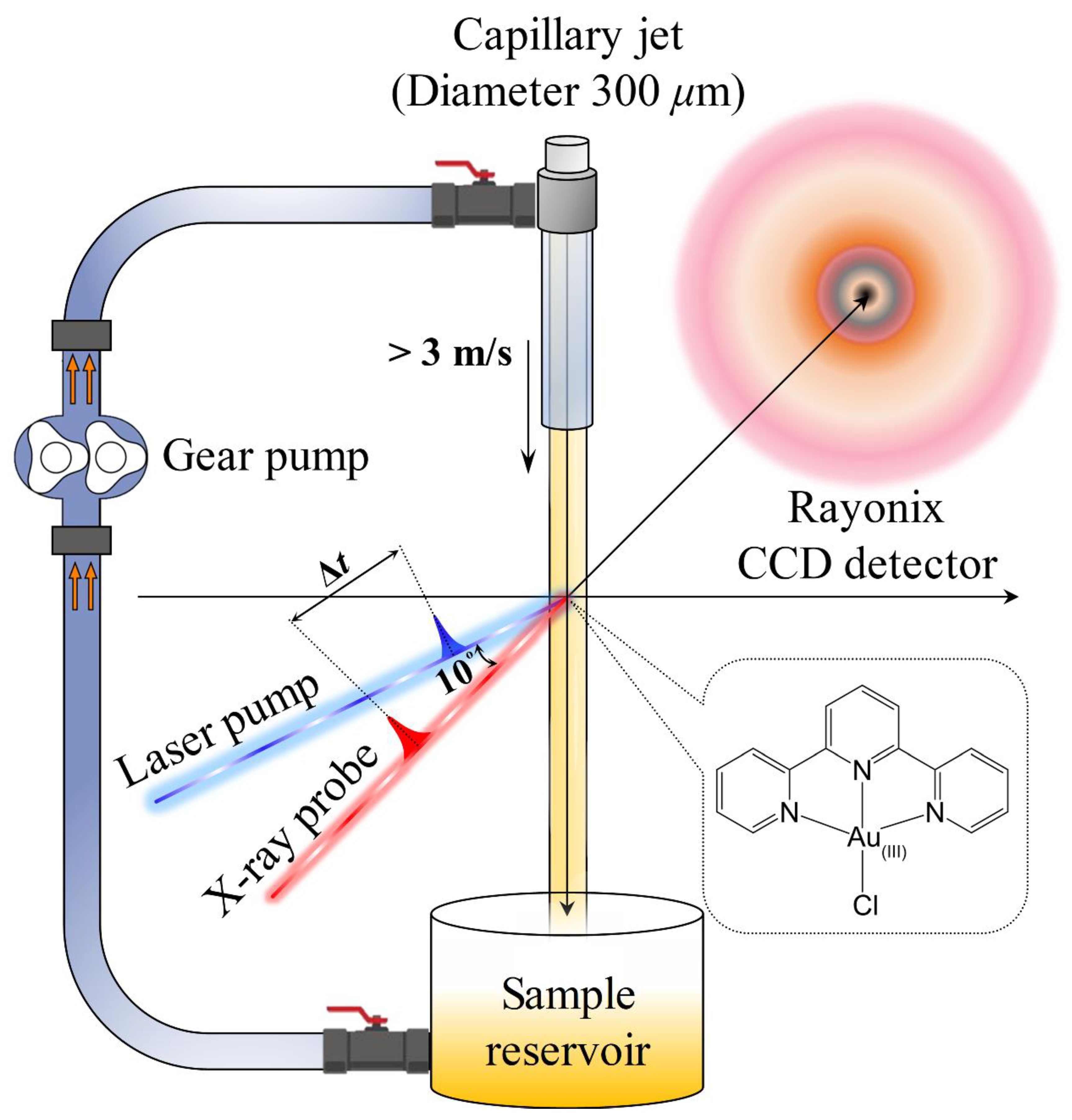
3.2. Time-Resolved X-ray Solution Scattering
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AuNPs | Gold nanoparticles |
| [Au(tpy)Cl]2+ | Chloro(2,2′,2″-terpyridine)gold(III) ion |
| tpy | 2,2′:6′,2″-Terpyridine |
| TRXL | Time-resolved X-ray liquidography |
| XFEL | X-ray free-electron laser |
| TEM | Transmission electron microscopy |
| PXRD | Powder X-ray diffraction |
| RP | Reduction potential |
| HP | Hydration potential |
| SVD | Singular value decomposition |
| rSV | Right singular vector |
| lSV | Left singular vector |
| fcc | Face-centered cubic |
| SDXS | Static difference X-ray scattering |
| E° | Standard reduction potentials |
| DFT | Density functional theory |
| ESRF | European Synchrotron Radiation Facility |
References
- Kim, J.G.; Kim, T.W.; Kim, J.; Ihee, H. Protein Structural Dynamics Revealed by Time-Resolved X-ray Solution Scattering. Acc. Chem. Res. 2015, 48, 2200–2208. [Google Scholar] [CrossRef]
- Biasin, E.; van Driel, T.B.; Kjær, K.S.; Dohn, A.O.; Christensen, M.; Harlang, T.; Chabera, P.; Liu, Y.; Uhlig, J.; Pápai, M.; et al. Femtosecond X-ray Scattering Study of Ultrafast Photoinduced Structural Dynamics in Solvated [Co(terpy)2]2+. Phys. Rev. Lett. 2016, 117, 013002. [Google Scholar] [CrossRef] [Green Version]
- Kjær, K.S.; Driel, T.B.V.; Harlang, T.C.B.; Kunnus, K.; Biasin, E.; Ledbetter, K.; Hartsock, R.W.; Reinhard, M.E.; Koroidov, S.; Li, L.; et al. Finding intersections between electronic excited state potential energy surfaces with simultaneous ultrafast X-ray scattering and spectroscopy. Chem. Sci. 2019, 10, 5749–5760. [Google Scholar] [CrossRef] [Green Version]
- Haldrup, K.; Levi, G.; Biasin, E.; Vester, P.; Laursen, M.G.; Beyer, F.; Kjær, K.S.; Brandt van Driel, T.; Harlang, T.; Dohn, A.O.; et al. Ultrafast X-ray Scattering Measurements of Coherent Structural Dynamics on the Ground-State Potential Energy Surface of a Diplatinum Molecule. Phys. Rev. Lett. 2019, 122, 063001. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Wulff, M.; Bratos, S.; Petersen, J.; Guerin, L.; Leicknam, J.C.; Carnmarata, M.; Kong, Q.; Kim, J.; Moller, K.B.; et al. Filming the Birth of Molecules and Accompanying Solvent Rearrangement. J. Am. Chem. Soc. 2013, 135, 3255–3261. [Google Scholar] [CrossRef] [PubMed]
- Arnlund, D.; Johansson, L.C.; Wickstrand, C.; Barty, A.; Williams, G.J.; Malmerberg, E.; Davidsson, J.; Milathianaki, D.; DePonte, D.P.; Shoeman, R.L.; et al. Visualizing a protein quake with time-resolved X-ray scattering at a free-electron laser. Nat. Methods 2014, 11, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, J.G.; Nozawa, S.; Sato, T.; Oang, K.Y.; Kim, T.; Ki, H.; Jo, J.; Park, S.; Song, C.; et al. Direct observation of bond formation in solution with femtosecond X-ray scattering. Nature 2015, 518, 385–389. [Google Scholar] [CrossRef]
- Ki, H.; Oang, K.Y.; Kim, J.; Ihee, H. Ultrafast X-ray Crystallography and Liquidography. Annu. Rev. Phys. Chem. 2017, 68, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Nozawa, S.; Kim, H.; Choi, E.H.; Sato, T.; Kim, T.W.; Kim, K.H.; Ki, H.; Kim, J.; Choi, M.; et al. Mapping the Emergence of Molecular Vibrations Mediating Bond Formation. Nature 2020, 582, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Lee, S.J.; Jo, J.; Kim, J.G.; Ki, H.; Kim, C.W.; Cho, K.H.; Choi, J.; Lee, J.H.; Wulff, M.; et al. Protein Folding From Heterogeneous Unfolded State Revealed by Time-Resolved X-ray Solution Scattering. Proc. Natl. Acad. Sci. USA 2020, 117, 14996. [Google Scholar] [CrossRef]
- Kim, K.H.; Oang, K.Y.; Kim, J.; Lee, J.H.; Kim, Y.; Ihee, H. Direct observation of myoglobin structural dynamics from 100 picoseconds to 1 microsecond with picosecond X-ray solution scattering. Chem. Commun. 2011, 47, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Levantino, M.; Schiro, G.; Lemke, H.T.; Cottone, G.; Glownia, J.M.; Zhu, D.L.; Chollet, M.; Ihee, H.; Cupane, A.; Cammarata, M. Ultrafast myoglobin structural dynamics observed with an X-ray free-electron laser. Nat. Commun. 2015, 6, 6772. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Malmerberg, E.; Westenhoff, S.; Katona, G.; Cammarata, M.; Wohri, A.B.; Johansson, L.C.; Ewald, F.; Eklund, M.; Wulff, M.; et al. Structural dynamics of light-driven proton pumps. Structure 2009, 17, 1265–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.S.; Dashdorj, N.; Schotte, F.; Graber, T.; Henning, R.; Anfinrud, P. Protein structural dynamics in solution unveiled via 100-ps time-resolved x-ray scattering. Proc. Natl. Acad. Sci. USA 2010, 107, 7281–7286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, P.L.; Lovett, J.E.; Carl, P.J.; Cammarata, M.; Lee, J.H.; Jung, Y.O.; Ihee, H.; Timmel, C.R.; van Thor, J.J. The short-lived signaling state of the photoactive yellow protein photoreceptor revealed by combined structural probes. J. Am. Chem. Soc. 2011, 133, 9395–9404. [Google Scholar] [CrossRef] [Green Version]
- Ihee, H. Visualizing solution-phase reaction dynamics with time-resolved X-ray liquidography. Acc. Chem. Res. 2009, 42, 356–366. [Google Scholar] [CrossRef]
- Ihee, H.; Wulff, M.; Kim, J.; Adachi, S. Ultrafast X-ray scattering: Structural dynamics from diatomic to protein molecules. Int. Rev. Phys. Chem. 2010, 29, 453–520. [Google Scholar] [CrossRef]
- Kim, T.K.; Lee, J.H.; Wulff, M.; Kong, Q.Y.; Ihee, H. Spatiotemporal kinetics in solution studied by time-resolved X-ray liquidography (solution scattering). ChemPhysChem 2009, 10, 1958–1980. [Google Scholar] [CrossRef]
- Eustis, S.; El-Sayed, M.A. Why Gold Nanoparticles Are More Precious Than Pretty Gold: Noble Metal Surface Plasmon Resonance and Its Enhancement of the Radiative and Nonradiative Properties of Nanocrystals of Different Shapes. Chem. Soc. Rev. 2006, 35, 209–217. [Google Scholar] [CrossRef]
- Nehl, C.L.; Hafner, J.H. Shape-Dependent Plasmon Resonances of Gold Nanoparticles. J. Mater. Chem. 2008, 18, 2415–2419. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Liao, J.; Shao, X.; Li, Q.; Lin, Y. The Effect of Shape on Cellular Uptake of Gold Nanoparticles in the Forms of Stars, Rods, and Triangles. Sci. Rep. 2017, 7, 3827. [Google Scholar] [CrossRef]
- Dong, Y.C.; Hajfathalian, M.; Maidment, P.S.N.; Hsu, J.C.; Naha, P.C.; Si-Mohamed, S.; Breuilly, M.; Kim, J.; Chhour, P.; Douek, P.; et al. Effect of Gold Nanoparticle Size on Their Properties as Contrast Agents for Computed Tomography. Sci. Rep. 2019, 9, 14912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Heo, J.; Kim, M.; Lee, Y.W.; Han, S.W. Size-Controlled Synthesis of Monodisperse Gold Nanooctahedrons and Their Surface-Enhanced Raman Scattering Properties. Chem. Phys. Lett. 2009, 468, 245–248. [Google Scholar] [CrossRef]
- Kim, F.; Connor, S.; Song, H.; Kuykendall, T.; Yang, P. Platonic Gold Nanocrystals. Angew. Chem. Int. Ed. 2004, 43, 3673–3677. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.; Yoo, C.I.; Chung, I.S.; Park, S.M.; Ryu, S.; Song, H. Shape Adjustment Between Multiply Twinned and Single-Crystalline Polyhedral Gold Nanocrystals: Decahedra, Icosahedra, and Truncated Tetrahedra. J. Phys. Chem. C 2008, 112, 2469–2475. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; An, K.; Hwang, Y.; Park, J.-G.; Noh, H.-J.; Kim, J.-Y.; Park, J.-H.; Hwang, N.-M.; Hyeon, T. Ultra-Large-Scale Syntheses of Monodisperse Nanocrystals. Nat. Mater. 2004, 3, 891–895. [Google Scholar] [CrossRef]
- Goulet, P.J.G.; Lennox, R.B. New Insights into Brust−Schiffrin Metal Nanoparticle Synthesis. J. Am. Chem. Soc. 2010, 132, 9582–9584. [Google Scholar] [CrossRef]
- Kimling, J.; Maier, M.; Okenve, B.; Kotaidis, V.; Ballot, H.; Plech, A. Turkevich Method for Gold Nanoparticle Synthesis Revisited. J. Phys. Chem. B 2006, 110, 15700–15707. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Wang, C.-H.; Hwu, Y.-K.; Je, J.-H. Synchrotron X-ray Synthesis of Colloidal Gold Particles for Drug Delivery. Mater. Chem. Phys. 2006, 100, 72–76. [Google Scholar] [CrossRef]
- Gachard, E.; Remita, H.; Khatouri, J.; Keita, B.; Nadjo, L.; Belloni, J. Radiation-Induced and Chemical Formation of Gold Clusters. New J. Chem. 1998, 22, 1257–1265. [Google Scholar] [CrossRef]
- Henglein, A.; Meisel, D. Radiolytic Control of the Size of Colloidal Gold Nanoparticles. Langmuir 1998, 14, 7392–7396. [Google Scholar] [CrossRef]
- Ershov, B.G.; Gordeev, A.V. A Model for Radiolysis of Water and Aqueous Solutions of H2, H2O2 and O2. Radiat. Phys. Chem. 2008, 77, 928–935. [Google Scholar] [CrossRef]
- Abedini, A.; Bakar, A.A.A.; Larki, F.; Menon, P.S.; Islam, M.S.; Shaari, S. Recent Advances in Shape-Controlled Synthesis of Noble Metal Nanoparticles by Radiolysis Route. Nanoscale Res. Lett. 2016, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, A.; Okada, I.; Fukuoka, T.; Sakurai, I.; Utsumi, Y. Synthesis of Metallic Nanoparticles Through X-ray Radiolysis Using Synchrotron Radiation. Jpn. J. Appl. Phys. 2016, 55, 055502. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Matsumoto, T.; Okada, I.; Sakurai, I.; Utsumi, Y. Surface-Enhanced Raman Scattering Active Gold Nanostructure Fabricated by Photochemical Reaction of Synchrotron Radiation. Mater. Chem. Phys. 2015, 160, 205–211. [Google Scholar] [CrossRef]
- Plech, A.; Kotaidis, V.; Lorenc, M.; Wulff, M. Thermal Dynamics in Laser Excited Metal Nanoparticles. Chem. Phys. Lett. 2005, 401, 565–569. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Keller, D.; Rossell, M.D.; Erni, R. Formation of Au Nanoparticles in Liquid Cell Transmission Electron Microscopy: From a Systematic Study to Engineered Nanostructures. Chem. Mater. 2017, 29, 10518–10525. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, A.; Nagata, K.; Taguchi, N.; Iwase, A.; Hori, F. Synthesis of Au Nanorods by Using Gamma-Ray Irradiation. Jpn. J. Appl. Phys. 2012, 51, 11PH01. [Google Scholar] [CrossRef]
- Abedini, A.; Susthitha Menon, P.; Daud, A.R.; Hamid, M.A.A.; Shaari, S. Radiolytic Formation of Highly Luminescent Triangular Ag Nanocolloids. J. Radioanal. Nucl. Chem. 2016, 307, 985–991. [Google Scholar] [CrossRef]
- Plech, A.; Kotaidis, V.; Siems, A.; Sztucki, M. Kinetics of the X-ray Induced Gold Nanoparticle Synthesis. Phys. Chem. Chem. Phys. 2008, 10, 3888–3894. [Google Scholar] [CrossRef]
- Meader, V.K.; John, M.G.; Rodrigues, C.J.; Tibbetts, K.M. Roles of Free Electrons and H2O2 in the Optical Breakdown-Induced Photochemical Reduction of Aqueous [AuCl4]−. J. Phys. Chem. A 2017, 121, 6742–6754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore Tibbetts, K.; Tangeysh, B.; Odhner, J.H.; Levis, R.J. Elucidating Strong Field Photochemical Reduction Mechanisms of Aqueous [AuCl4]−: Kinetics of Multiphoton Photolysis and Radical-Mediated Reduction. J. Phys. Chem. A 2016, 120, 3562–3569. [Google Scholar] [CrossRef] [PubMed]
- Odhner, J.H.; Moore Tibbetts, K.; Tangeysh, B.; Wayland, B.B.; Levis, R.J. Mechanism of Improved Au Nanoparticle Size Distributions Using Simultaneous Spatial and Temporal Focusing for Femtosecond Laser Irradiation of Aqueous KAuCl4. J. Phys. Chem. C 2014, 118, 23986–23995. [Google Scholar] [CrossRef]
- Tsuji, T.; Yahata, T.; Yasutomo, M.; Igawa, K.; Tsuji, M.; Ishikawa, Y.; Koshizaki, N. Preparation and Investigation of the Formation Mechanism of Submicron-Sized Spherical Particles of Gold Using Laser Ablation and Laser Irradiation in Liquids. Phys. Chem. Chem. Phys. 2013, 15, 3099–3107. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimkutty, S.; Wagener, P.; Menzel, A.; Plech, A.; Barcikowski, S. Nanoparticle Formation in a Cavitation Bubble After Pulsed Laser Ablation in Liquid Studied with High Time Resolution Small Angle X-ray Scattering. Appl. Phys. Lett. 2012, 101, 103104. [Google Scholar] [CrossRef] [Green Version]
- Marin, M.L.; McGilvray, K.L.; Scaiano, J.C. Photochemical Strategies for the Synthesis of Gold Nanoparticles From Au(III) and Au(I) Using Photoinduced Free Radical Generation. J. Am. Chem. Soc. 2008, 130, 16572–16584. [Google Scholar] [CrossRef]
- Plech, A.; Kotaidis, V.; Grésillon, S.; Dahmen, C.; Von Plessen, G. Laser-Induced Heating and Melting of Gold Nanoparticles Studied by Time-Resolved X-ray Scattering. Phys. Rev. B 2004, 70, 195423. [Google Scholar] [CrossRef]
- Hu, M.; Hartland, G.V. Heat Dissipation for Au Particles in Aqueous Solution: Relaxation Time versus Size. J. Phys. Chem. B 2002, 106, 7029–7033. [Google Scholar] [CrossRef]
- Plech, A.; Kotaidis, V.; Lorenc, M.; Boneberg, J. Femtosecond Laser Near-Field Ablation from Gold Nanoparticles. Nat. Phys. 2006, 2, 44–47. [Google Scholar] [CrossRef]
- Werner, D.; Furube, A.; Okamoto, T.; Hashimoto, S. Femtosecond Laser-Induced Size Reduction of Aqueous Gold Nanoparticles: In Situ and Pump−Probe Spectroscopy Investigations Revealing Coulomb Explosion. J. Phys. Chem. C 2011, 115, 8503–8512. [Google Scholar] [CrossRef]
- Ichiyanagi, K.; Sekiguchi, H.; Nozawa, S.; Sato, T.; Adachi, S.-I.; Sasaki, Y.C. Laser-Induced Picosecond Lattice Oscillations in Submicron Gold Crystals. Phys. Rev. B 2011, 84, 024110. [Google Scholar] [CrossRef]
- Besner, S.; Kabashin, A.V.; Meunier, M. Fragmentation of Colloidal Nanoparticles by Femtosecond Laser-Induced Supercontinuum Generation. Appl. Phys. Lett. 2006, 89, 233122. [Google Scholar] [CrossRef]
- Kumar, A.; Walker, J.A.; Bartels, D.M.; Sevilla, M.D. A Simple Ab Initio Model for the Hydrated Electron That Matches Experiment. J. Phys. Chem. A 2015, 119, 9148–9159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollis, L.S.; Lippard, S.J. Aqueous Chemistry of (2,2′,2″-Terpyridine)Gold(III). Preparation and Structures of Chloro(2,2′,2″-Terpyridine)Gold Dichloride Trihydrate ([Au(Terpy)Cl]Cl2·3H2O) and the Mixed Valence Gold(I)-Gold(III) Salt Bis[Chloro(2,2′,2″-Terpyridine)Gold] Tris(Dichloroaurate) Tetrachloroaurate ([Au(Terpy)Cl]2[AuCl2]3[AuCl4]). J. Am. Chem. Soc. 1983, 105, 4293–4299. [Google Scholar]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ki, H.; Park, S.; Eom, S.; Gu, J.; Kim, S.; Kim, C.; Ahn, C.W.; Choi, M.; Ahn, S.; Ahn, D.-S.; et al. Gold Nanoparticle Formation via X-ray Radiolysis Investigated with Time-Resolved X-ray Liquidography. Int. J. Mol. Sci. 2020, 21, 7125. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197125
Ki H, Park S, Eom S, Gu J, Kim S, Kim C, Ahn CW, Choi M, Ahn S, Ahn D-S, et al. Gold Nanoparticle Formation via X-ray Radiolysis Investigated with Time-Resolved X-ray Liquidography. International Journal of Molecular Sciences. 2020; 21(19):7125. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197125
Chicago/Turabian StyleKi, Hosung, Sungjun Park, Seunghwan Eom, Jain Gu, Siin Kim, Changwon Kim, Chi Woo Ahn, Minseo Choi, Sena Ahn, Doo-Sik Ahn, and et al. 2020. "Gold Nanoparticle Formation via X-ray Radiolysis Investigated with Time-Resolved X-ray Liquidography" International Journal of Molecular Sciences 21, no. 19: 7125. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197125