How Does Replacement of the Axial Histidine Ligand in Cytochrome c Peroxidase by Nδ-Methyl Histidine Affect Its Properties and Functions? A Computational Study


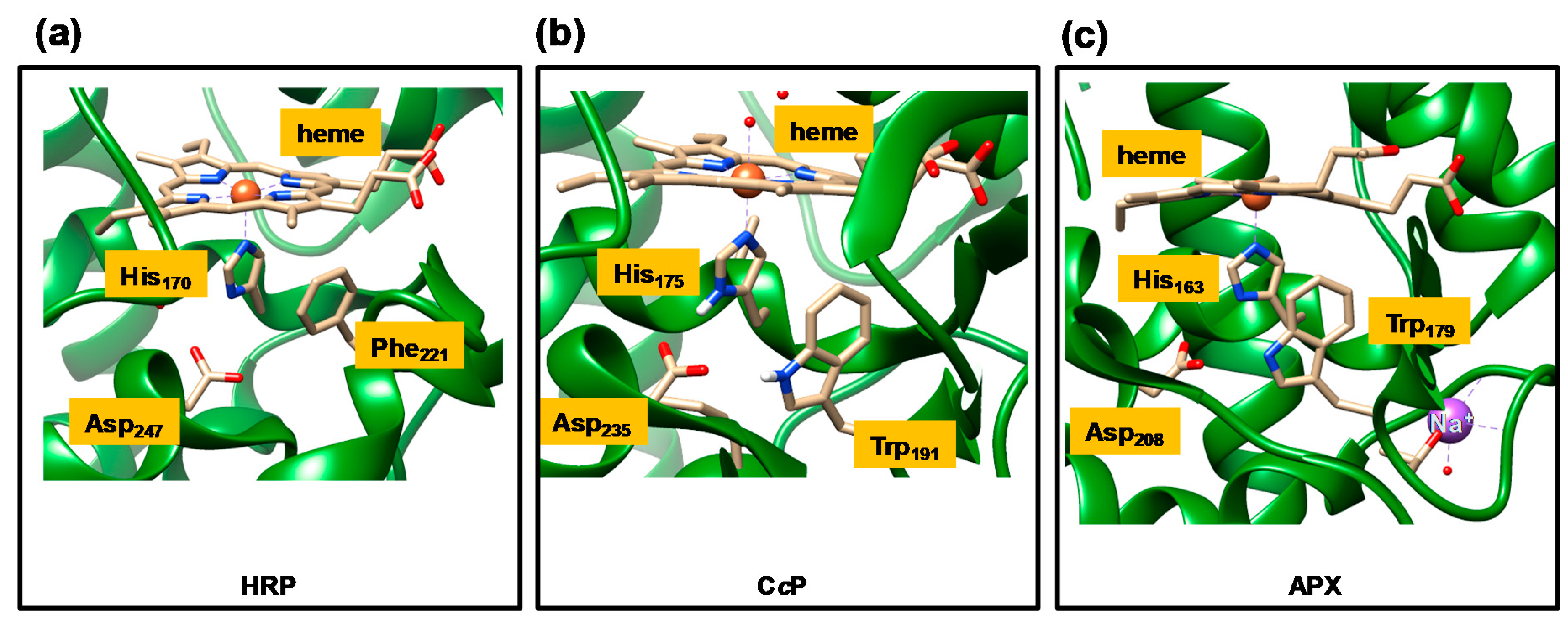{kind=link}
{kind=link}
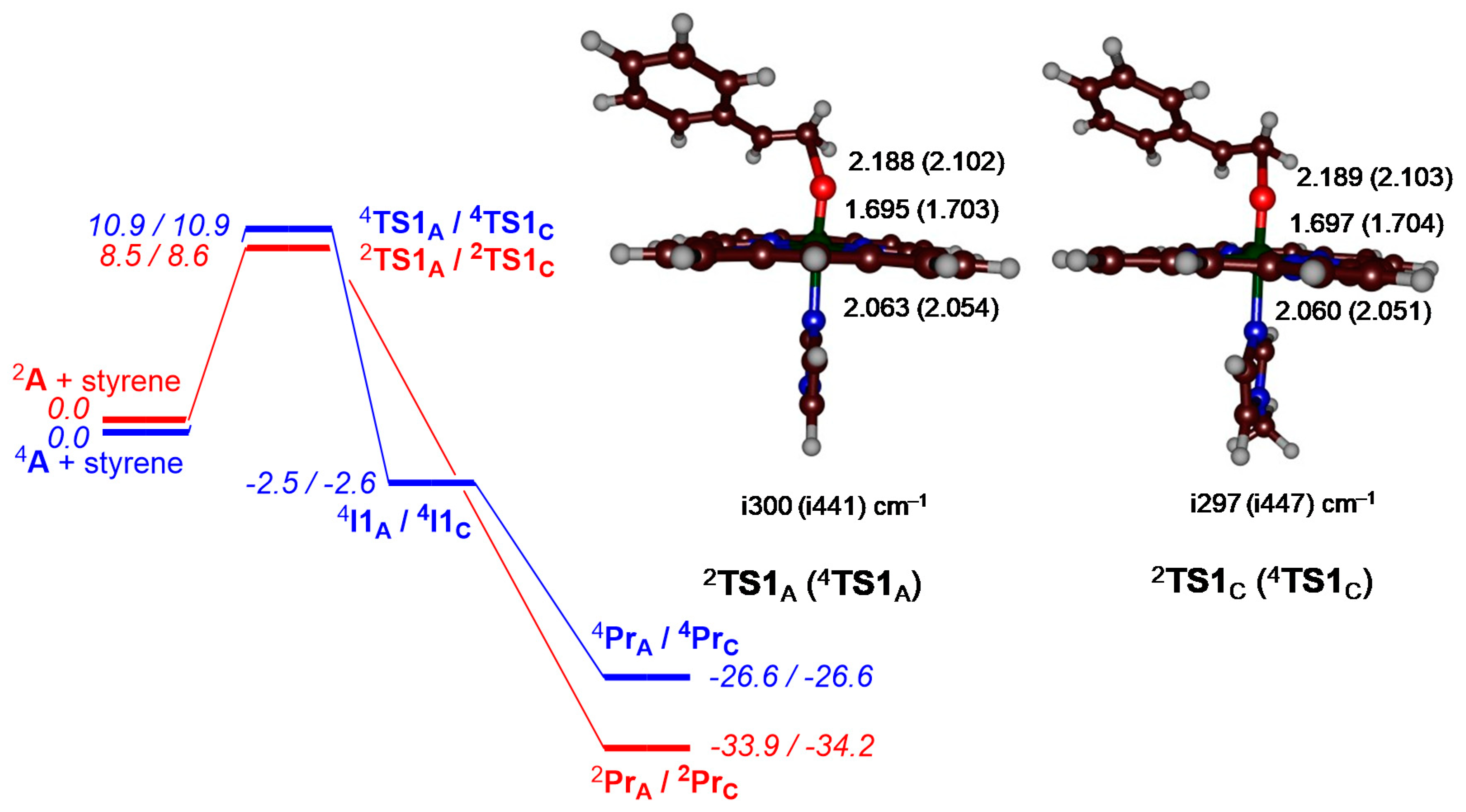{kind=link}
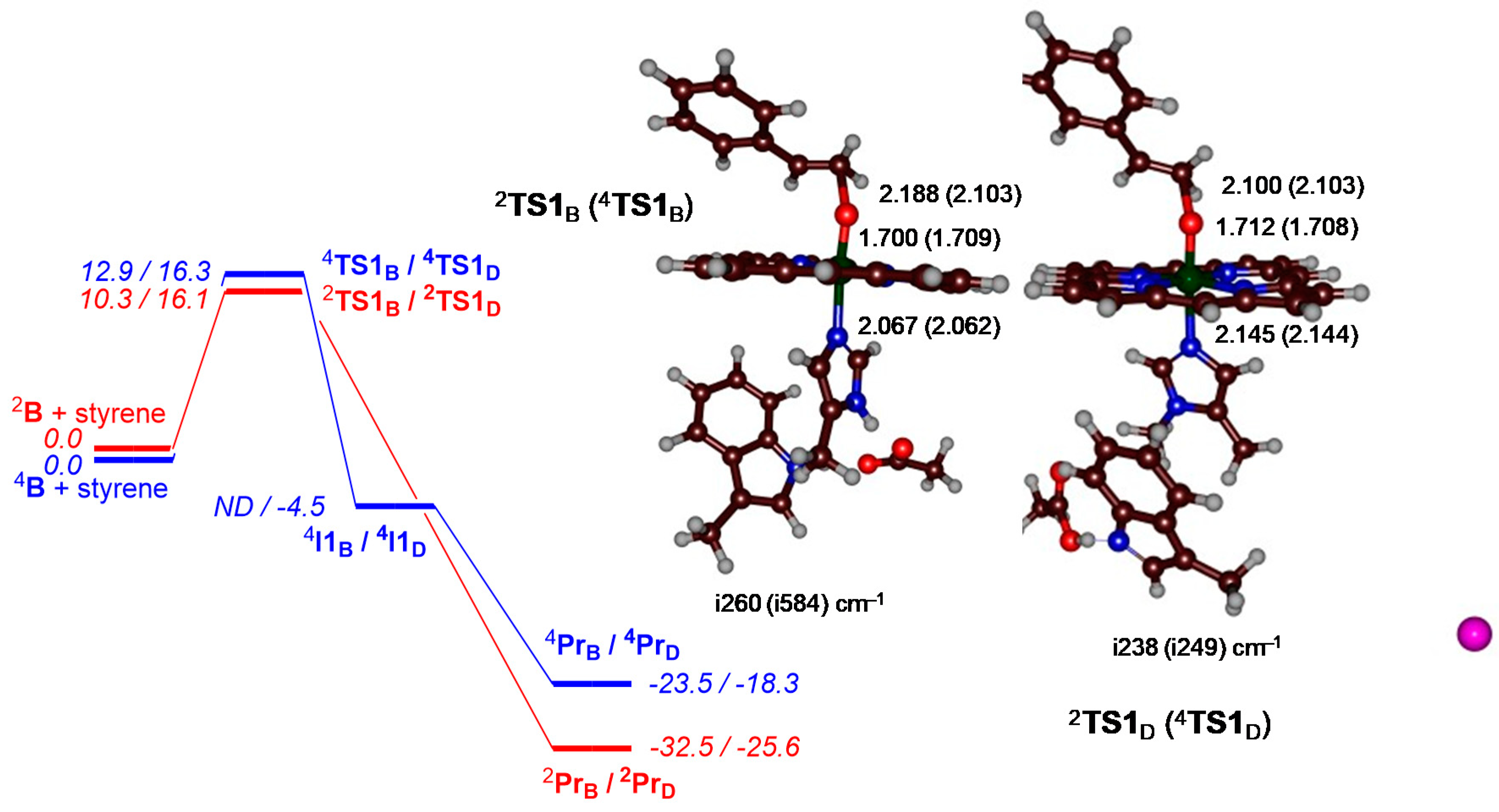{kind=link}
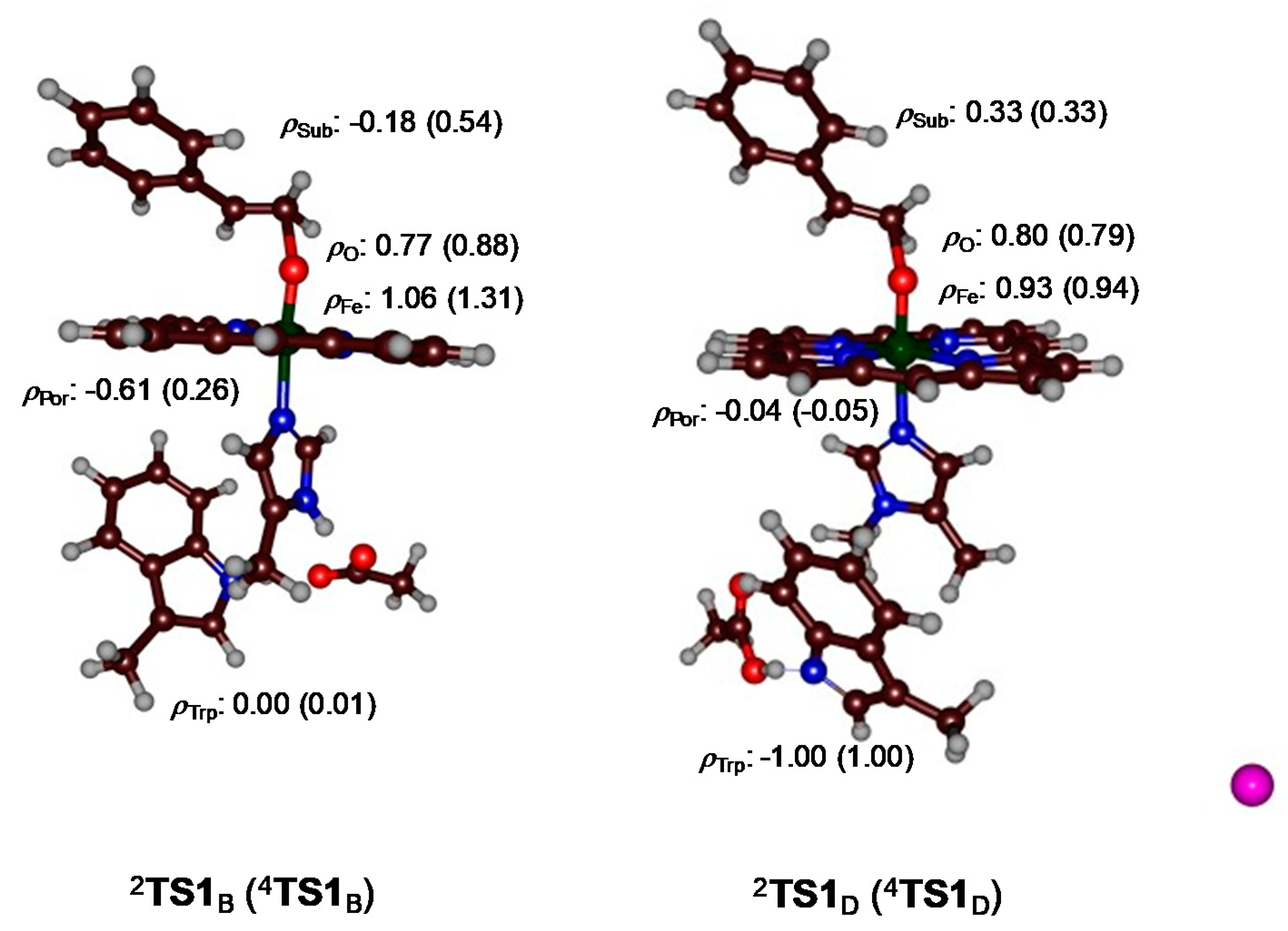{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
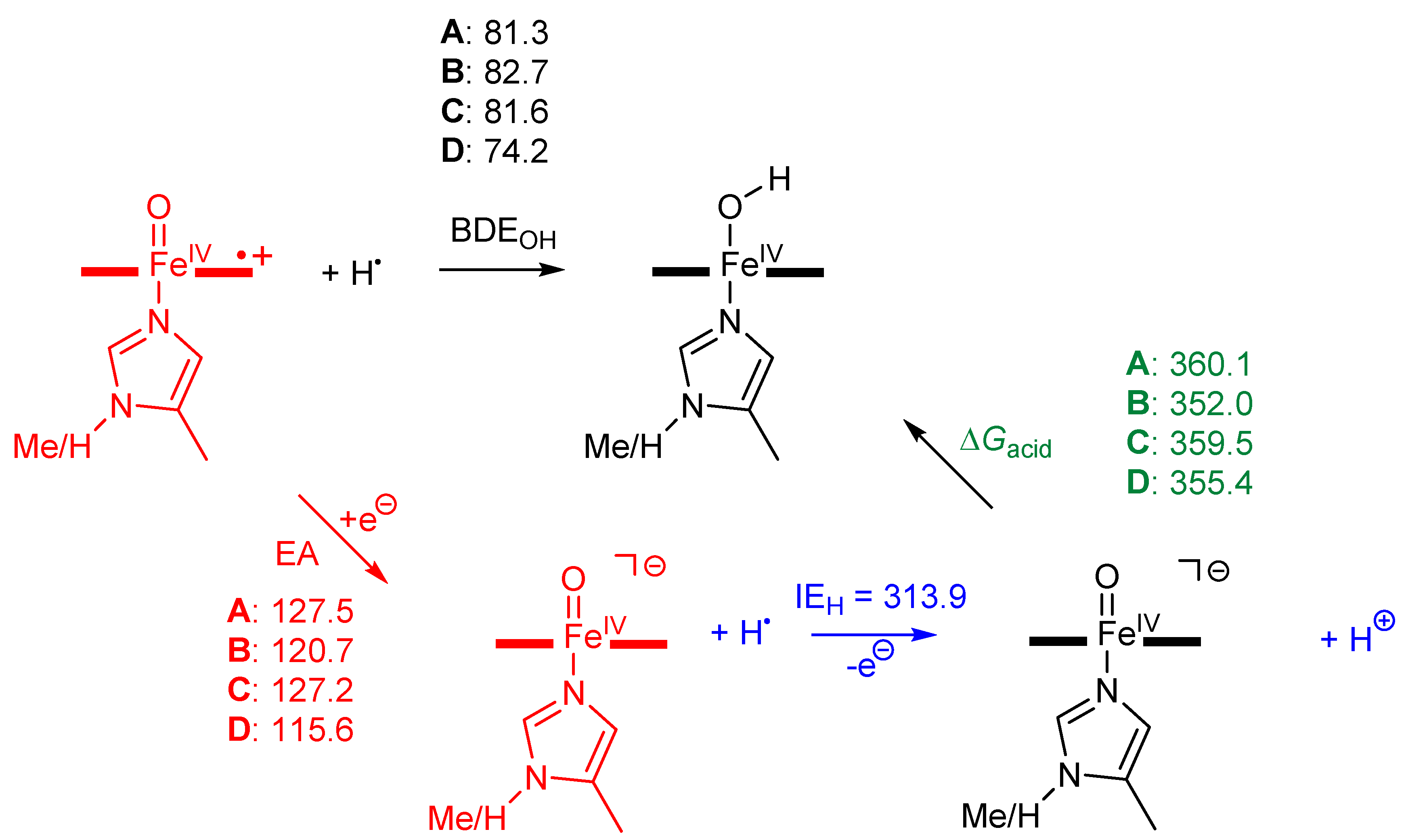
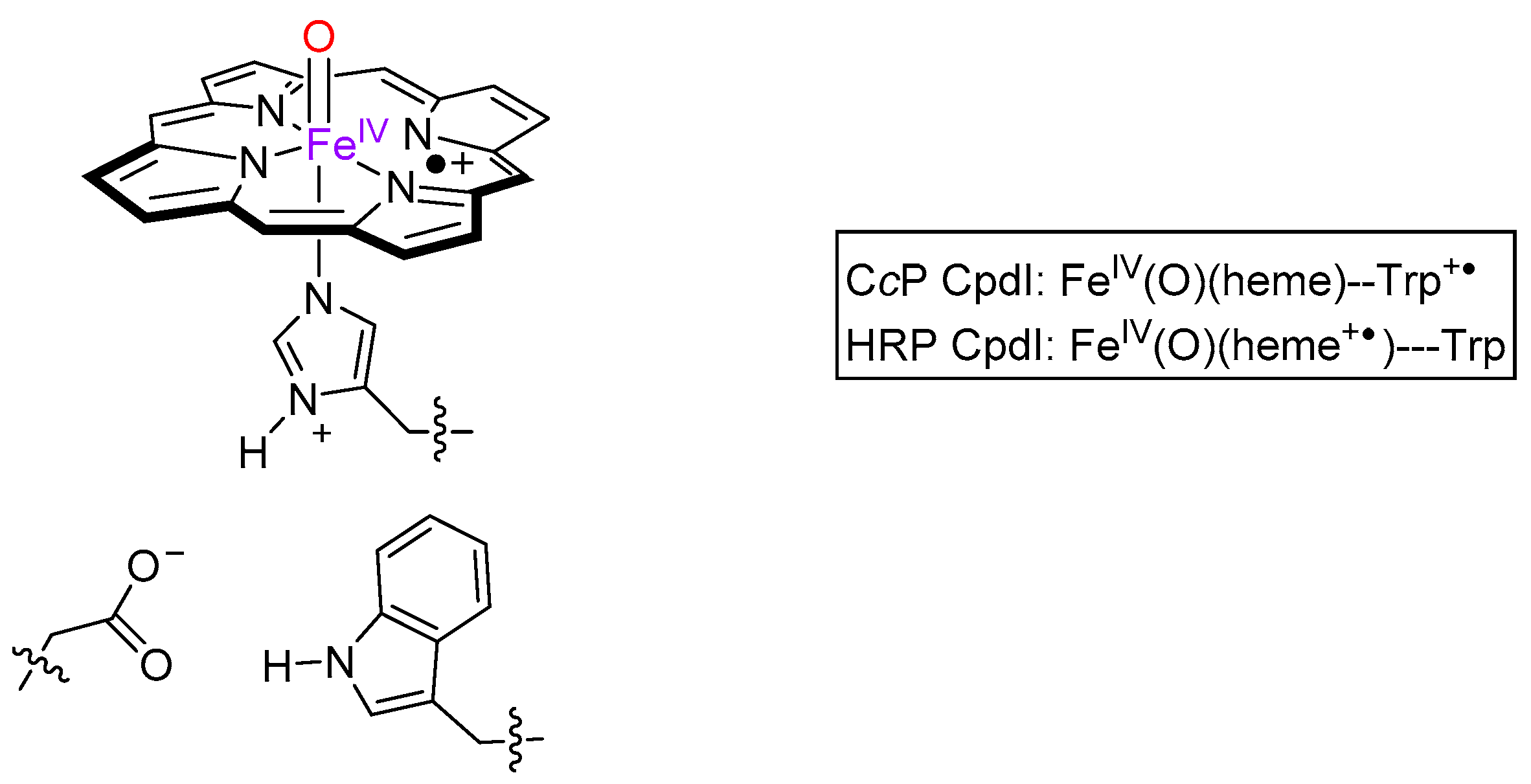
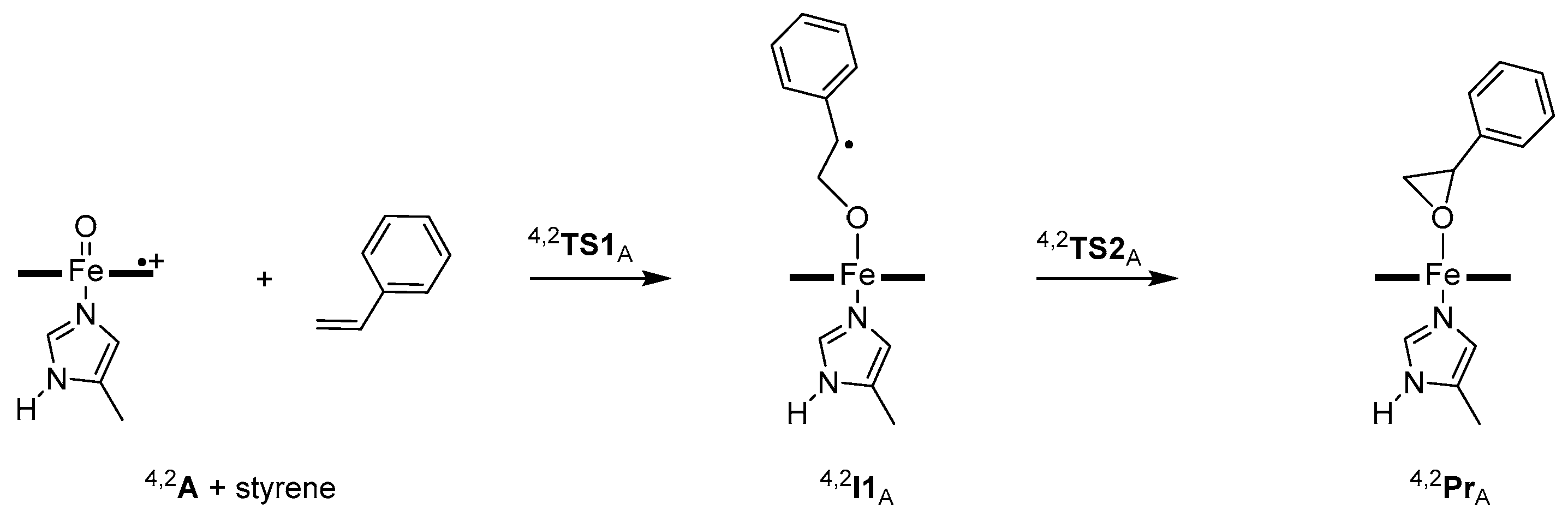
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BDE | Bond dissociation energy |
| CcP | Cytochrome c peroxidase |
| HRP | Horseradish peroxidase |
| ZPE | Zero-point energy |
References
- Kadish, K.M.; Smith, K.M.; Guilard, R. Handbook of Porphyrin Science; World Scientific Publishing Co.: Singapore, 2010. [Google Scholar]
- Dawson, J.H.; Sono, M. Cytochrome P-450 and chloroperoxidase: Thiolate-ligated heme enzymes. Spectroscopic determination of their active site structures and mechanistic implications of thiolate ligation. Chem. Rev. 1987, 87, 1255–1276. [Google Scholar] [CrossRef]
- Smulevich, G.; Feis, A.; Howes, B.D. Fifteen years of Raman spectroscopy of engineered heme containing peroxidases: What have we learned? Acc. Chem. Res. 2005, 38, 433–440. [Google Scholar] [CrossRef]
- Raven, E. Understanding functional diversity and substrate specificity in haem peroxidases: What can we learn from ascorbate peroxidase? Nat. Prod. Rep. 2007, 24, 367–381. [Google Scholar] [CrossRef]
- Zederbauer, M.; Furtmüller, P.G.; Brogioni, S.; Jakopitsch, C.; Smulevich, G.; Obinger, C. Heme to protein linkages in mammalian peroxidases: Impact on spectroscopic, redox and catalytic properties. Nat. Prod. Rep. 2007, 24, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.G.; Cheng, W.-H.; McClung, J.P. Metabolic regulation and function of glutathione peroxidase-1. Ann. Rev. Nutr. 2007, 27, 41–61. [Google Scholar] [CrossRef]
- Khan, A.A.; Rahmani, A.H.; Aldebasi, Y.H.; Aly, S.M. Biochemical and pathological studies on peroxidases—An updated review. Glob. J. Health Sci. 2014, 6, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, G.R.; Pinto, D.C.G.A.; Silva, A.M.S. Horseradish peroxidase (HRP) as a tool in green chemistry. RSC Adv. 2014, 4, 37244–37265. [Google Scholar] [CrossRef]
- Vlasova, I.I. Peroxidase activity of human hemoproteins: Keeping the fire under control. Molecules 2018, 23, 2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, I.; Hanson, L.K.; Walker, F.A.; Fajer, J. Models for compounds I of peroxidases: Axial ligand effects. J. Am. Chem. Soc. 1983, 105, 3296–3300. [Google Scholar] [CrossRef]
- Ikeda-Saito, M.; Kimura, S. Axial ligand coordination in intestinal peroxidase. Arch. Biochem. Biophys. 1990, 283, 351–355. [Google Scholar] [CrossRef]
- Hirst, J.; Wilcox, S.K.; Ai, J.; Moënne-Loccoz, P.; Loehr, T.M.; Goodin, D.B. Replacement of the axial histidine ligand with imidazole in cytochrome c peroxidase. 2. Effects on heme coordination and function. Biochemistry 2001, 40, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.L.; Loew, G.H. Proximal ligand effects on electronic structure and spectra of compound I of peroxidases. J. Porphyr. Phthalocyanines 2001, 5, 334–344. [Google Scholar] [CrossRef]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2887. [Google Scholar] [CrossRef]
- Cai, H.; Guengerich, F.P. Reaction of trichloroethylene and trichloroethylene oxide with cytochrome P450 enzymes: Inactivation and sites of modification. Chem. Res. Toxicol. 2001, 14, 451–458. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2004. [Google Scholar]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef]
- Watanabe, Y.; Nakajima, H.; Ueno, T. Reactivities of oxo and peroxo intermediates studied by hemoprotein mutants. Acc. Chem. Res. 2007, 40, 554–562. [Google Scholar] [CrossRef]
- Munro, A.W.; Girvan, H.M.; McLean, K.J. Variations on a (t)heme—Novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat. Prod. Rep. 2007, 24, 585–609. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [Green Version]
- de Visser, S.P.; Kumar, D. Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature; Royal Society of Chemistry Publishing: Cambridge, UK, 2011; ISBN 978-1-84973-181-2. [Google Scholar]
- Grogan, G. Cytochromes P450: Exploiting diversity and enabling application as biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.H.; Holm, R.H.; Trudell, J.R.; Barth, G.; Linder, R.E.; Bunnenberg, E.; Djerassi, C.; Tang, S.C. Oxidized cytochrome P-450. Magnetic circular dichroism evidence for thiolate ligation in the substrate-bound form. Implications for the catalytic mechanism. J. Am. Chem. Soc. 1976, 98, 3707–3709. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. The role of the proximal ligand in heme enzymes. J. Biol. Inorg. Chem. 1996, 1, 356–359. [Google Scholar] [CrossRef]
- Ogliaro, F.; de Visser, S.P.; Shaik, S. The “push” effect of the thiolate ligand in cytochrome P450: A theoretical gauging. J. Inorg. Biochem. 2002, 91, 554–567. [Google Scholar] [CrossRef]
- Hirst, J.; Wilcox, S.K.; Williams, P.A.; Blankenship, J.; McRee, D.E.; Goodin, D.B. Replacement of the axial histidine ligand with imidazole in cytochrome c peroxidase. 1. Effects on structure. Biochemistry 2001, 40, 1265–1273. [Google Scholar] [CrossRef]
- Bateman, L.; Léger, C.; Goodin, D.B.; Armstrong, F.A. A distal histidine mutant (H52Q) of yeast cytochrome c peroxidase catalyzes the oxidation of H2O2 instead of its reduction. J. Am. Chem. Soc. 2001, 123, 9260–9263. [Google Scholar] [CrossRef]
- Carpena, X.; Vidossich, P.; Schroettner, K.; Calisto, B.M.; Banerjee, S.; Stampler, J.; Soudi, M.; Furtmüller, P.G.; Rovira, C.; Fita, I.; et al. Essential role of proximal histidine-asparagine interaction in mammalian peroxidases. J. Biol. Chem. 2009, 284, 25929–25937. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Sulok, C.D.; Paulat, F.; Lehnert, N.; Twigg, A.I.; Hendrich, M.P.; Schulz, C.E.; Scheidt, W.R. Just a proton: Distinguishing the two electronic states of five-coordinate high-spin iron(II) porphyrinates with imidazole/ate coordination. J. Am. Chem. Soc. 2010, 132, 3737–3750. [Google Scholar] [CrossRef] [Green Version]
- Nastri, F.; Chino, M.; Maglio, O.; Bhagi-Damodaran, A.; Lu, Y.; Lombardi, A. Design and engineering of artificial oxygen-activating metalloenzymes. Chem. Soc. Rev. 2016, 45, 5020–5054. [Google Scholar] [CrossRef]
- Li, G.; Yao, P.; Gong, R.; Li, J.; Liu, P.; Lonsdale, R.; Wu, Q.; Lin, J.; Zhu, D.; Reetz, M.T. Simultaneous engineering of an enzyme’s entrance tunnel and active site: The case of monoamine oxidase MAO-N. Chem. Sci. 2017, 8, 4093–4099. [Google Scholar] [CrossRef] [Green Version]
- Renata, H.; Lewis, R.D.; Sweredoski, M.J.; Moradian, A.; Hess, S.; Wang, Z.J.; Arnold, F.H. Identification of mechanism-based inactivation in P450-catalyzed cyclopropanation facilitates engineering of improved enzymes. J. Am. Chem. Soc. 2016, 138, 12527–12533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavini, P.; Cheong, K.J.; Moitessier, N.; Auclair, K. Active site crowding of cytochrome P450 3A4 as a strategy to alter its selectivity. ChemBioChem 2017, 18, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Pratter, S.M.; Konstantinovics, C.; DiGiuro, C.L.M.; Leitner, E.; Kumar, D.; de Visser, S.P.; Grogan, G.; Straganz, G.D. Inversion of enantio-selectivity of a mononuclear non-heme iron(II)-dependent hydroxylase by tuning the interplay of metal-center geometry and protein structure. Angew. Chem. Int. Ed. 2013, 52, 9677–9681. [Google Scholar] [CrossRef] [PubMed]
- Green, A.P.; Hayashi, T.; Mittle, P.R.E.; Hilvert, D. A chemically programmed proximal ligand enhances the catalytic properties of a heme enzyme. J. Am. Chem. Soc. 2016, 138, 11344–11352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pott, M.; Hayaski, T.; Mori, T.; Mittle, P.R.E.; Green, A.P.; Hilvert, D. A noncanonical proximal heme ligand affords an efficient peroxidase in a globin fold. J. Am. Chem. Soc. 2018, 140, 1535–1543. [Google Scholar] [CrossRef] [Green Version]
- de Visser, S.P. Second-coordination sphere effects on selectivity and specificity of heme and nonheme iron enzymes. Chem. Eur. J. 2020, 26, 5308–5327. [Google Scholar] [CrossRef]
- Berglund, G.I.; Carlsson, G.H.; Smith, A.T.; Szöke, H.; Henriksen, A.; Hajdu, J. The catalytic pathway of horseradish peroxidase at high resolution. Nature 2002, 417, 463–468. [Google Scholar] [CrossRef]
- Goodin, D.B.; McRee, D.E. The Asp-His-iron triad of cytochrome c peroxidase controls the reduction potential electronic structure, and coupling of the tryptophan free radical to the heme. Biochemistry 1993, 32, 3313–3324. [Google Scholar] [CrossRef]
- Sharp, K.H.; Mewies, M.; Moody, P.C.E.; Raven, E.L. Crystal structure of the ascorbate peroxidase–ascorbate complex. Nat. Struct. Biol. 2003, 10, 303–307. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Derat, E.; Shaik, S.; Rovira, C.; Vidossich, P.; Alfonso-Prieto, M. The effect of a water molecule on the mechanism of formation of compound 0 in horseradish peroxidase. J. Am. Chem. Soc. 2007, 129, 6346–6347. [Google Scholar] [CrossRef] [PubMed]
- Vidossich, P.; Alfonso-Prieto, M.; Carpena, X.; Loewen, P.C.; Fita, I.; Rovira, C. Versatility of the electronic structure of compound I in catalase-peroxidases. J. Am. Chem. Soc. 2007, 129, 13436–13446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidossich, P.; Fiorin, G.; Alfonso-Prieto, M.; Derat, E.; Shaik, S.; Rovira, C. On the role of water in peroxidase catalysis: A theoretical investigation of HRP compound I formation. J. Phys. Chem. B 2010, 114, 5161–5169. [Google Scholar] [CrossRef] [PubMed]
- Vidossich, P.; Alfonso-Prieto, M.; Rovira, C. Catalases versus peroxidases: DFT investigation of H2O2 oxidation in models systems and implications for heme protein engineering. J. Inorg. Biochem. 2012, 117, 292–297. [Google Scholar] [CrossRef]
- Sivaraja, M.; Goodin, D.B.; Smith, M.; Hoffman, B.M. Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase compound ES. Science 1989, 245, 738–740. [Google Scholar] [CrossRef]
- Huyett, J.E.; Doan, P.E.; Gurbiel, R.; Houseman, A.L.P.; Sivaraja, M.; Goodin, D.B.; Hoffman, B.M. Compound ES of cytochrome c peroxidase contains a Trp-cation radical: Characterization by continuous wave and pulsed Q-band external nuclear double resonance spectroscopy. J. Am. Chem. Soc. 1995, 117, 9033–9041. [Google Scholar] [CrossRef]
- Colin, J.; Wiseman, B.; Switala, J.; Loewen, P.C.; Ivancich, A. Distinct role of specific tryptophans in facilitating electron transfer or as [Fe(IV)=O Trp•] intermediates in the peroxidase reaction of Bulkholderia pseudomallei catalase-peroxidase: A multifrequency EPR spectroscopy investigation. J. Am. Chem. Soc. 2009, 131, 8557–8563. [Google Scholar] [CrossRef]
- Pipirou, Z.; Guallar, V.; Basran, J.; Metcalfe, C.L.; Murphy, E.J.; Bottrill, A.R.; Mistry, S.C.; Raven, E.L. Peroxide-dependent formation of a covalent link between Trp51 and the heme in cytochrome c peroxidase. Biochemistry 2009, 48, 3593–3599. [Google Scholar] [CrossRef]
- Kathiresan, M.; English, A.M. LC-MS/MS suggests that hole hopping in cytochrome c peroxidase protects its heme from oxidative modification by excess H2O2. Chem. Sci. 2017, 8, 1152–1162. [Google Scholar] [CrossRef] [Green Version]
- de Visser, S.P.; Shaik, S.; Sharma, P.K.; Kumar, D.; Thiel, W. Active species of horseradish peroxidase (HRP) and cytochrome P450: Two electronic chameleons. J. Am. Chem. Soc. 2003, 125, 15779–15788. [Google Scholar] [CrossRef]
- Rydberg, P.; Sigfridsson, E.; Ryde, U. On the role of the axial ligand in heme proteins: A theoretical study. J. Biol. Inorg. Chem. 2004, 9, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Derat, E.; Cohen, S.; Shaik, S.; Altun, A.; Thiel, W. Principal active species of horseradish peroxidase, compound I: A hybrid quantum mechanical/molecular mechanical study. J. Am. Chem. Soc. 2005, 127, 13611–13621. [Google Scholar] [CrossRef]
- Derat, E.; Shaik, S. Two-state reactivity, electromerism, tautomerism, and “surprise” isomers in the formation of compound II of the enzyme horseradish peroxidase from the principal species, compound I. J. Am. Chem. Soc. 2006, 128, 8185–8198. [Google Scholar] [CrossRef]
- Harvey, J.N.; Bathelt, C.M.; Mulholland, A.J. QM/MM modeling of compound I active species in cytochrome P450, cytochrome c peroxidase, and ascorbate peroxidase. J. Comput. Chem. 2006, 27, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Heimdal, J.; Rydberg, P.; Ryde, U. Protonation of the proximal histidine ligand in heme peroxidases. J. Phys. Chem. B 2008, 112, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- Zazza, C.; Sanna, N.; Tatoli, S.; Aschi, M.; Palma, A. Compound I in horseradish peroxidase enzyme: Magnetic state assessment by quadratric configuration interaction calculations. Int. J. Quant. Chem. 2010, 110, 352–357. [Google Scholar] [CrossRef]
- Zazza, C.; Palma, A.; Sanna, N.; Tatoli, S.; Aschi, M. Computational study on compound I redox-active species in horseradish peroxydase enzyme: Conformational fluctuations and solvation effects. J. Phys. Chem. B 2010, 114, 6817–6824. [Google Scholar] [CrossRef]
- Green, M.T. Evidence for sulfur-based radicals in thiolate compound I intermediates. J. Am. Chem. Soc. 1999, 121, 7939–7940. [Google Scholar] [CrossRef]
- Roberts, J.E.; Hoffman, B.M.; Rutter, R.; Hager, L.P. Oxygen-17 ENDOR of horseradish peroxidase compound I. J. Am. Chem. Soc. 1981, 103, 7654–7656. [Google Scholar] [CrossRef]
- Rittle, J.; Green, M.T. Cytochrome P450 Compound I: Capture, characterization, and C-H bond activation kinetics. Science 2010, 330, 933–937. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, Y.; Han, K.; Zhan, C.-G. Fundamental reaction pathways for cytochrome P450-catalyzed 5′-hydroxylation and N-demethylation of nicotine. J. Phys. Chem. B 2010, 114, 9023–9030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirao, H.; Chuanprasit, P.; Cheong, Y.Y.; Wang, X. How is a metabolic iIntermediate formed in the mechanism-based inactivation of cytochrome P450 by using 1,1-dimethylhydrazine: Hydrogen abstraction or nitrogen oxidation? Chem. Eur. J. 2013, 19, 7361–7369. [Google Scholar] [CrossRef] [PubMed]
- Hirao, H.; Cheong, Z.C.; Wang, X. Pivotal role of water in terminating enzymatic function: A density functional theory study of the mechanism-based inactivation of cytochromes P450. J. Phys. Chem. B 2012, 116, 7787–7794. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Li, H. Hydrogen abstraction of camphor catalyzed by cytochrome P450cam: A QM/MM study. J. Phys. Chem. B 2016, 120, 12312–12320. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shi, J.; Liu, Y. Oxidative rearrangement mechanism of pentalenolactone F catalyzed by cytochrome P450 CYP161C2 (PntM). Inorg. Chem. 2018, 57, 8933–8941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y. Mechanical insights into the enzymatic cleavage of double C–C bond in poly(cis-1,4-isoprene) by the latex clearing protein. Inorg. Chem. 2020, 59, 9627–9637. [Google Scholar] [CrossRef]
- Phung, Q.M.; Pierloot, K. Low-lying electromeric states in chloro-ligated iron(IV)-oxo porphyrin as a model for compound I, studied with second-order perturbation theory based on density matrix renormalization group. J. Chem. Theory Comput. 2019, 15, 3033–3043. [Google Scholar] [CrossRef]
- Bhaskar, B.; Bonagura, C.A.; Li, H.; Poulos, T.L. Cation-induced stabilization of the engineered cation-binding loop in cytochrome c peroxidase (CcP). Biochemistry 2002, 41, 2684–2693. [Google Scholar] [CrossRef]
- Wirstam, M.; Blomberg, M.R.A.; Siegbahn, P.E.M. Reaction mechanism of compound I formation in heme peroxidases: A density functional theory study. J. Am. Chem. Soc. 1999, 121, 10178–10185. [Google Scholar] [CrossRef]
- de Visser, S.P. What affects the quartet-doublet energy splitting in peroxidase enzymes? J. Phys. Chem. A 2005, 109, 11050–11057. [Google Scholar] [CrossRef]
- Altarsha, M.; Benighaus, T.; Kumar, D.; Thiel, W. How is the reactivity of cytochrome P450cam affected by Thr252X mutation? A QM/MM study for X = serine, valine, alanine, glycine. J. Am. Chem. Soc. 2009, 131, 4755–4763. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, Y.; Han, K. Recent density functional theory model calculations of drug metabolism by cytochrome P450. Coord. Chem. Rev. 2012, 256, 1137–1150. [Google Scholar] [CrossRef]
- Sainna, M.A.; Kumar, S.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A comprehensive test set of epoxidation rate constants by iron(IV)-oxo porphyrin complexes. Chem. Sci. 2015, 6, 1516–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepp, K.P. Heme isomers substantially affect heme’s electronic structure and function. Phys. Chem. Chem. Phys. 2017, 19, 22355–22362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; de Visser, S.P.; Shaik, S. Multistate reactivity in styrene epoxidation by Compound I of cytochrome P450: Mechanisms of products and side products formation. Chem. Eur. J. 2005, 11, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Karamzadeh, B.; Sastry, G.N.; de Visser, S.P. What factors influence the rate constant of substrate epoxidation by Compound I of cytochrome P450 and analogous iron(IV)-oxo oxidants. J. Am. Chem. Soc. 2010, 132, 7656–7667. [Google Scholar] [CrossRef]
- Kumar, D.; Latifi, R.; Kumar, S.; Rybak-Akimova, E.V.; Sainna, M.A.; de Visser, S.P. Rationalization of the barrier height for para-Z-styrene epoxidation by iron(IV)-oxo porphyrins with variable axial ligands. Inorg. Chem. 2013, 52, 7968–7979. [Google Scholar] [CrossRef]
- Morozov, A.N.; Pardillo, A.D.; Chatfield, D.C. Chloroperoxidase-catalyzed epoxidation of cis-β-methylstyrene: NH–S hydrogen bonds and proximal helix dipole change the catalytic mechanism and significantly lower the reaction barrier. J. Phys. Chem. B 2015, 119, 14350–14363. [Google Scholar] [CrossRef]
- Morozov, A.N.; Chatfield, D.C. How the proximal pocket may influence the enantiospecificities of chloroperoxidase-catalyzed epoxidations of olefins. Int. J. Mol. Sci. 2016, 17, 1297. [Google Scholar] [CrossRef] [Green Version]
- de Visser, S.P.; Ogliaro, F.; Harris, N.; Shaik, S. Multi-state epoxidation of ethene by cytochrome P450: A quantum chemical study. J. Am. Chem. Soc. 2001, 123, 3037–3047. [Google Scholar] [CrossRef]
- de Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. Hydrogen bonding modulates the selectivity of enzymatic oxidation by P450: A chameleon oxidant behavior of Compound I. Angew. Chem. Int. Ed. 2002, 41, 1947–1951. [Google Scholar] [CrossRef]
- de Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. What factors affect the regioselectivity of oxidation by cytochrome P450? A DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 2002, 124, 11809–11826. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; de Visser, S.P.; Sharma, P.K.; Derat, E.; Shaik, S. The intrinsic axial ligand effect on propene oxidation by horseradish peroxidase versus cytochrome P450 enzymes. J. Biol. Inorg. Chem. 2005, 10, 181–189. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P. The axial ligand effect of oxo-iron porphyrin catalysts. How does chloride compare to thiolate? J. Biol. Inorg. Chem. 2006, 11, 168–178. [Google Scholar] [CrossRef]
- de Visser, S.P. Propene activation by the oxo-iron active species of taurine/ketoglutarate dioxygenase (TauD) enzyme. How does the catalysis compare to heme-enzymes? J. Am. Chem. Soc. 2006, 128, 9813–9824. [Google Scholar] [CrossRef]
- de Visser, S.P. What factors influence the ratio of C–H hydroxylation versus C=C epoxidation by a nonheme cytochrome P450 biomimetic? J. Am. Chem. Soc. 2006, 128, 15809–15818. [Google Scholar] [CrossRef]
- Kumar, D.; Tahsini, L.; de Visser, S.P.; Kang, H.Y.; Kim, S.J.; Nam, W. The effect of porphyrin ligands on the regioselective dehydrogenation versus epoxidation of olefins by oxoiron (IV) mimics of cytochrome P450. J. Phys. Chem. A 2009, 113, 11713–11722. [Google Scholar] [CrossRef] [Green Version]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. Compound I reactivity defines alkene oxidation selectivity in cytochrome P450cam. J. Phys. Chem. B 2010, 114, 1156–1162. [Google Scholar] [CrossRef]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 Enzymes: Their structure, reactivity, and selectivity—Modeled by QM/MM calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef]
- de Visser, S.P. Can the replacement of a single atom in the enzyme horseradish peroxidase convert it into a monoxygenase? A density functional study. J. Phys. Chem. B 2006, 110, 20759–20761. [Google Scholar] [CrossRef]
- de Visser, S.P. Preferential hydroxylation over epoxidation catalysis by a horseradish peroxidase mutant: A cytochrome P450 mimic. J. Phys. Chem. B 2007, 111, 12299–12302. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Cohen, S.; de Visser, S.P.; Sharma, P.K.; Kumar, D.; Kozuch, S.; Ogliaro, F.; Danovich, D. The “rebound controversy”: An overview and theoretical modeling of the rebound step in C–H hydroxylation by cytochrome P450. Eur. J. Inorg. Chem. 2004, 2004, 207–226. [Google Scholar] [CrossRef]
- Faponle, A.S.; Quesne, M.G.; de Visser, S.P. Origin of the regioselective fatty acid hydroxylation versus decarboxylation by a cytochrome P450 peroxygenase: What drives the reaction to biofuel production? Chem. Eur. J. 2016, 22, 5478–5483. [Google Scholar] [CrossRef]
- Li, X.-X.; Postils, V.; Sun, W.; Faponle, A.S.; Solà, M.; Wang, Y.; Nam, W.; de Visser, S.P. Reactivity patterns of (protonated) Compound II and Compound I of Cytochrome P450: Which is the better oxidant? Chem. Eur. J. 2017, 23, 6406–6418. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Tan, L.S. Is the bound substrate in nitric oxide synthase protonated or neutral and what is the active oxidant that performs substrate hydroxylation? J. Am. Chem. Soc. 2008, 130, 12961–12974. [Google Scholar] [CrossRef]
- de Visser, S.P. Trends in substrate hydroxylation reactions by heme and nonheme iron(IV)-oxo oxidants give correlations between intrinsic properties of the oxidant with barrier height. J. Am. Chem. Soc. 2010, 132, 1087–1097. [Google Scholar] [CrossRef]
- Colomban, C.; Tobing, A.H.; Mukherjee, G.; Sastri, C.V.; Sorokin, A.B.; de Visser, S.P. Mechanism of oxidative activation of fluorinated aromatic compounds by N-bridged diiron-phthalocyanine. What determines the reactivity? Chem. Eur. J. 2019, 25, 14320–14331. [Google Scholar] [CrossRef]
- Fowler, N.J.; Blanford, C.F.; Warwicker, J.; de Visser, S.P. Prediction of reduction potentials of copper proteins with continuum electrostatics and density functional theory. Chem. Eur. J. 2017, 23, 15436–15445. [Google Scholar] [CrossRef] [Green Version]
- Dixit, V.A.; Warwicker, J.; de Visser, S.P. How do metal ions modulate the rate-determining electron transfer step in Cytochrome P450 reactions? Chem. Eur. J. 2020. [Google Scholar] [CrossRef]
- Bordwell, F.G.; Cheng, J.-P. Substituent effects on the stabilities of phenoxyl radicals and the acidities of phenoxyl radical cations. J. Am. Chem. Soc. 1991, 113, 1736–1743. [Google Scholar] [CrossRef]
- Mayer, J.M. Hydrogen atom abstraction by metal-oxo complexes: Understanding the analogy with organic radical reactions. Acc. Chem. Res. 1998, 31, 441–450. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P. A valence bond modeling of trends in hydrogen abstraction barriers and transition states of hydroxylation reactions catalyzed by cytochrome P450 enzymes. J. Am. Chem. Soc. 2008, 130, 10128–10140. [Google Scholar] [CrossRef] [PubMed]
- Louka, S.; Barry, S.M.; Heyes, D.J.; Mubarak, M.Q.E.; Ali, H.S.; Alkhalaf, L.M.; Munro, A.W.; Scrutton, N.S.; Challis, G.L.; de Visser, S.P. The catalytic mechanism of aromatic nitration by cytochrome P450 TxtE: Involvement of a ferric-peroxynitrite intermediate. J. Am. Chem. Soc. 2020, 142, 15764–15779. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Senthilnathan, D.; Singh, D.; Kumar, D.; Maldivi, P.; Sorokin, A.B.; de Visser, S.P. Origin of the enhanced reactivity of μ-nitrido-bridged diiron(IV)-oxo porphyrinoid complexes over cytochrome P450 Compound I. ACS Catal. 2016, 6, 2230–2243. [Google Scholar] [CrossRef] [Green Version]
- Hunter, E.P.; Lias, S.G. NIST Chemistry Webbook, NIST Standard Reference Database, Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 1998; p. 20899.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–272. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent molecular orbital methods. XII. Further extensions of Gaussian—Type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Kumar, D.; Sastry, G.N.; de Visser, S.P. Effect of the axial ligand on substrate sulfoxidation mediated by iron(IV)-oxo porphyrin cation radical oxidants. Chem. Eur. J. 2011, 17, 6196–6205. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Quesne, M.G.; Neu, H.M.; Cantú Reinhard, F.G.; Goldberg, D.P.; de Visser, S.P. Singlet versus triplet reactivity in an Mn(V)-Oxo species: Testing theoretical predictions against experimental evidence. J. Am. Chem. Soc. 2016, 138, 12375–12386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantú Reinhard, F.G.; Faponle, A.S.; de Visser, S.P. Substrate sulfoxidation by an iron (IV)-oxo complex: Benchmarking computationally calculated barrier heights to experiment. J. Phys. Chem. A 2016, 120, 9805–9814. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Sainna, M.A.; Upadhyay, P.; Balan, G.A.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A systematic account on aromatic hydroxylation by a cytochrome P450 model Compound I: A low-pressure mass spectrometry and computational study. Chem. Eur. J. 2016, 22, 18608–18619. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.W.Z.; Mubarak, M.Q.E.; Green, A.P.; de Visser, S.P. How Does Replacement of the Axial Histidine Ligand in Cytochrome c Peroxidase by Nδ-Methyl Histidine Affect Its Properties and Functions? A Computational Study. Int. J. Mol. Sci. 2020, 21, 7133. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197133
Lee CWZ, Mubarak MQE, Green AP, de Visser SP. How Does Replacement of the Axial Histidine Ligand in Cytochrome c Peroxidase by Nδ-Methyl Histidine Affect Its Properties and Functions? A Computational Study. International Journal of Molecular Sciences. 2020; 21(19):7133. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197133
Chicago/Turabian StyleLee, Calvin W. Z., M. Qadri E. Mubarak, Anthony P. Green, and Sam P. de Visser. 2020. "How Does Replacement of the Axial Histidine Ligand in Cytochrome c Peroxidase by Nδ-Methyl Histidine Affect Its Properties and Functions? A Computational Study" International Journal of Molecular Sciences 21, no. 19: 7133. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197133