Microtubule Dysfunction: A Common Feature of Neurodegenerative Diseases


Unit of Neuromuscular and Neurodegenerative Disorders, Genetics and Rare Diseases Research Division, Bambino Gesù Children’s Hospital, IRCCS, 00146 Rome, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(19), 7354; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197354
Submission received: 8 September 2020
/
Revised: 24 September 2020
/
Accepted: 1 October 2020
/
Published: 5 October 2020
(This article belongs to the Section Molecular Neurobiology)
Abstract
:Neurons are particularly susceptible to microtubule (MT) defects and deregulation of the MT cytoskeleton is considered to be a common insult during the pathogenesis of neurodegenerative disorders. Evidence that dysfunctions in the MT system have a direct role in neurodegeneration comes from findings that several forms of neurodegenerative diseases are associated with changes in genes encoding tubulins, the structural units of MTs, MT-associated proteins (MAPs), or additional factors such as MT modifying enzymes which modulating tubulin post-translational modifications (PTMs) regulate MT functions and dynamics. Efforts to use MT-targeting therapeutic agents for the treatment of neurodegenerative diseases are underway. Many of these agents have provided several benefits when tested on both in vitro and in vivo neurodegenerative model systems. Currently, the most frequently addressed therapeutic interventions include drugs that modulate MT stability or that target tubulin PTMs, such as tubulin acetylation. The purpose of this review is to provide an update on the relevance of MT dysfunctions to the process of neurodegeneration and briefly discuss advances in the use of MT-targeting drugs for the treatment of neurodegenerative disorders.
1. Introduction
Microtubules (MTs) are structurally and functionally important components of the cell cytoskeleton, providing most of the spatial organization in the cell, and regulating different cellular functions such as intracellular trafficking, cell division, and organelle positioning.
They are characterized by a great chemical and functional heterogeneity, and are thus subjected to highly dynamic regulatory mechanisms that modulate MT composition, tubulin chemical modification, and MT association with other proteins. The result of these regulations is the generation of MT arrays with different morphology and degrees of stability that are able to adapt to specific functions in accordance with the cell context.
This modulation is particularly important in neurons, whose structures and functions strongly depend on the correct functioning of MTs. It is therefore not surprising that MT dysfunctions can be the cause or can contribute to the onset of neurodegenerative processes. Specific mutations in tubulin genes, dysfunctions of MT-associated proteins (MAPs), and alterations in the levels of tubulin PTMs are indeed associated with different forms of neurodegenerative diseases. This discovery has recently allowed the development of compounds aimed to counteract neurodegeneration by restoring the correct functioning of MTs. Although, recently, MT-oriented approaches currently represent one of the most promising strategies in the field of neurodegenerative diseases as several works have demonstrated that these compounds are effectively able to prevent or partially rescue MT dysfunctions in different animal and cellular models and that some of them have been translated into clinical trials. These results lead us to believe that in the near future, more preclinical approaches will be translated to patients, improving survival and their quality of life.
2. Microtubule Structural and Functional Complexity
MTs are composed of heterodimers of α–β-tubulin that align head to tail to form linear protofilaments which associate laterally to form a hollow, polar cylinder [1]. The directional alignment of polarized α–β heterodimers into MTs make the structures intrinsically polarized because of the presence two distinct ends: a minus end that exhibits an α-tubulin subunit and the opposite plus end that exposes a β-tubulin subunit (Figure 1).
In a solution of purified tubulin, MT elongation can occur to both ends [2]. In cells, MT growth is significantly more rapid at the plus end and occurs through the addition of α–β tubulin heterodimers in which each subunit of β-tubulin binds a GTP molecule. After heterodimer addition, the GTP is hydrolyzed to GDP and prolonged exposure of a terminal GDP at the α–β heterodimer of the plus end leads to MT depolymerization [3].
Within the cell, MTs do not reach a steady state length but undergo continual assembly and disassembly; the co-existence of growing and shrinking MTs in the same conditions is known as “dynamic instability”. Conversion from growth to shrinkage is defined “catastrophe”, whereas the switch from shrinkage to growth is called “rescue”. This “non equilibrium” in MT population is critical for several cell functions, such as cell division or during or the spatial arrangements of the cell [4].
MTs also undergo “treadmilling”, a dynamic process in which the plus end of the filament grows in length while the other one shrinks due to the removal of tubulin molecules bound to GTP from the minus end that travel to the plus end of the same MT [5].
In most cell types, MTs are nucleated at the centrosomal region in the MT-organizing center (MTOC), where a previously formed γ-tubulin ring complex provides a base for the start of filament extension [6]. The γ-tubulin ring complex has been proposed to form a ring-like template that binds the α-tubulin subunit exposed at the MT minus end. Thus, MTs grow with a defined polarity, with their minus ends anchored on the ring to γ-tubulin subunits and the plus end of the MT extending into the cytoplasm [7].
MTs constitute a heterogeneous and dynamic filament network with great chemical and functional complexity.
This complexity is produced by three regulatory mechanisms: (i) differential incorporation of alternative isotypes of α and β tubulin into MTs; (ii) PTMs of these isotypes; (iii) MT interaction with MAPs. These mechanisms do not act separately but can influence each other. Each of these will be briefly discussed below and in the specific context of neuronal cells.
In many organisms, both α and β tubulins are encoded by multiple genes at different loci [8,9]. Most of them, such TUBB2A, TUBB2B, and TUBA1A, TUBA1B, TUBA1C, are clustered in the genome and are presumably the products of genomic duplications [10]. In vitro experiments have shown that tubulin isotypes have different effects on MT dynamics [11,12,13,14], suggesting that they can differentially regulate the MT network in vivo. It was assumed that tubulin isotypes can assemble into discrete MT species that carry out unique functions [15]. This concept, known as the “multi tubulin hypothesis”, has long been quite controversial. It was demonstrated that all tubulin isotypes freely co-polymerize into heterogeneous MTs and that different α- and β-tubulin isotypes in fungi were functionally interchangeable [16,17]. However, several findings have shown that highly specialized MTs, such as ciliary axonemes [18,19], neuronal MTs [20,21], and MTs of the marginal band of platelets [22,23] depend on specific tubulin isotypes. Axonemal MTs of Drosophila olfactory neurons are highly enriched in β1- and β4-tubulins [24]. β6-Tubulin, instead, is essential for the formation of the platelet marginal band and the loss of this isoform results in thrombocytopenia [23]. Studies in Drosophila melanogaster have shown that specific tubulin isotypes are not functionally equivalent and possess distinctive properties depending on cell type and specific stages of development [25,26]. Hoyle and colleagues have demonstrated that in Drosophila male testes, the function of β2-tubulin is not rescued by the expression of β3-tubulin, and that the latter acts in a dominant-negative manner when co-expressed at levels exceeding 20% of the total tubulin. This finding indicates that while β2-tubulin regulates generic functions in Drosophila male testes, β3-tubulin has been evolved to ensure different MT functions in fly [24]. Similarly, in mouse tubb3−/− neurons the replacement of β3-tubulin by other β-tubulin isoforms cannot overcome its requirement in peripheral nerve regeneration, suggesting that the functions of the different β-tubulins are not interchangeable in neurons [27].
Tubulin isotypes also seem to directly determine MT structure by defining its protofilament numbers. In vitro experiments have shown that dimers of α1B-tubulin and β3-tubulin form typical MTs composed by 13 protofilaments, while heterodimers of α1β-tubulin and β2B-tubulin preferentially assemble into MTs composed by 14 protofilaments [28]. Moreover, in touch receptor neurons of Caenorhabditis elegans, MEC-12 α-tubulin and MEC-7 β-tubulin assemble to form 15 protofilaments [29,30,31]. Interestingly, MEC-12 α-tubulin is the only tubulin subjected to acetylation in C. elegans. This modification seems to be crucial for the assembly of these characteristic 15-protofilament MTs and the absence of αTA1, the enzyme that catalyzes MEC-12 α-tubulin acetylation, causes the formation of MTs with a lower protofilament number in C. elegans sensory neurons [32].
This finding introduces another important regulatory mechanism of MTs, which is that of tubulin PTMs. Tubulin isotypes are indeed further diversified through a large number of chemically induced PTMs. Some of these, such acetylation, are ubiquitous protein modifications, while others, such polyglutamylation, even appear to be unique to tubulins [33]. Almost all PTMs are reversible modifications and with the exception of acetylation, that occurs on the luminal surface of MTs, they take all place on the carboxy-terminal domain of tubulins [34]. Different PTMs occur concurrently on MTs; they can be scattered or concentrated in distinct domains, and depending on their localization and combination, create different structural patterns that regulate specific MT functions.
Most tubulin PTMs occur on axonemes, the MT-based core structures of eukaryotic cilia and flagella, and alterations of their levels have been found to impair the proper maintenance and functioning of these structures [35,36,37,38].
Analogous to the model of the “histone code” on chromatin, it was proposed that PTMs on the MT surface “write” a biochemical “tubulin code” that is “read” by specific proteins [39].
The specialized functions of MT are also determined by a large number of protein partners collectively known as MAPs. MAPs were first discovered as proteins that bind and stabilize MTs. The huge number of MAPs identified in the last years has indeed revealed a more complex picture of their functional properties. They constitute a class of heterogeneous regulators which perform the most disparate functions within the cells, such as the modulation of MT stability and dynamics, the directional transport of cargo, or the regulation of MT nucleation.
Thus, each of these three regulatory mechanisms adds a level of complexity to MT properties and, together with the others, establishes and regulates the specialized behavior of MTs. The result of these regulations is the creation of spatial and temporal patterns of MT network within the cell. This modulation is particularly important in neurons: the precise organization of MTs, including their composition, modification, spacing, and interaction, is essential for the correct morphology and function of these cells.
3. Microtubule Organization in Neurons
Neurons are highly compartmentalized and polarized cells whose structure and functions rely on MTs. Neuronal MT arrays are very heterogeneous and consist of single MT with different lengths and stability [40,41,42,43]. Long MTs have a dual function: they act as structural backbone that opposes to axon and dendrite retraction and as tracks for the transport of membrane-bound vesicles and organelles [44]. Indeed, short MTs are highly dynamic elements that move along axons, where they act as MT nucleation sites and sources for the generation of free α- and β-tubulin subunits that are used for the elongation of growing MTs [45,46].
Differently from non-neuronal cells that present uniformly oriented MTs anchored at the MTOC, neuronal MTs are discontinuous along axonal and dendritic processes. In young neurons, MTs are nucleated at the centrosome as short polymers that are rapidly released and transported into neurites [47]. The release of MTs from the MTOC is critical for the axonal growth and inhibition of katanin, the enzyme that severs MTs from the centrosome, suppresses axonal elongation [48].
From the early stage of maturation of developing neurons, centrosomes lose their function as MT-organizing centers and MTs are nucleated through non-centrosomal mechanisms, such as at the Golgi outposts [49,50] or from the lateral surface of pre-existing MTs [51,52].
The organization of neuronal MTs is tightly regulated in axons and dendrites. In axons, MTs have uniform polarity orientation, with the plus ends are directed away from the cell body [53], while in proximal dendrites, MTs are organized in antiparallel bundles oriented with their plus ends pointing away or toward the soma [54] (Figure 1). This orientation is, in part, regulated by kinesin and dynein molecular motors that transport nucleated MTs in the different neuronal compartments sliding along existing MTs [55,56,57]. Dynein and kinesin depletion disrupts the axonal bidirectional transport of plus end and minus end out MTs and the dendritic transport of minus end out MTs, respectively, resulting in impaired MT orientation in these compartments [55,57,58,59,60]. Preservation of the distinct patterns of MT polarity in axons and dendrites ensures the maintenance of the morphological and structural differences between these compartments.
For example, dendrites are restricted to small regions around the cell body while axons can extend to great distances. This happens because vesicular organelles that expand the cell membrane have a unidirectional vector to the tip of the growing axon via their plus end distal MTs [47]. MT orientation in axon and dendrites also affect their organelle compartmentalization. Dendrites contain cellular organelles, such as endoplasmic reticulum, polyribosome, and Golgi outposts that are rare in axons [61]. This occurs because these organelles are transported by minus end directed motor proteins that move from the soma to the minus end out MTs, which are exclusively present in dendrites [62].
At the end of the axon, MT arrays extend deep into the growth cone when bundles splay and adopt characteristic distributions (splayed, captured, looped, and bundled) [63,64] (Figure 1). The major function of MTs into growth cone is that to drive its directed movement; MT distribution on one rather than another side into the growth cone decides where it will turn [63].
4. Neuronal Microtubules: Regulative Mechanisms
4.1. Incorporation of Specific Tubulin Isotypes
Neuronal MTs rely on specific tubulin isotypes. Except for some of them that have been intensely characterized during mouse brain development, mainly through experiments of RNA in situ hybridization and most recently through the generation of transgenic mouse models, the spatial and temporal expression patterns of neuronal tubulin isotypes are still not completely understood, principally due to the difficulties in tracking protein expression due to their high homology.
Among the different tubulin isotypes expressed in the brain, β3-tubulin (encoded by TUBB3 human gene and tubb3 mouse gene) is considered a specific marker for neuronal MTs and has been largely characterized [65,66]. Its expression is primarily limited to post-mitotic neurons in development and adulthood [65] and is greatest during the periods of axon guidance and maturation [67]. During the embryonic stages, it is earlier expressed in both peripheral and central nervous system (at E9.0–E9.5) and later in the forming neocortex and retina (at E10.5–E12.5). Indeed, during the postnatal period, β3-tubulin is expressed in neurons of the layer II–VI and in the inner granular cell layer and Purkinje cells of the cerebellum. β3-tubulin expression is also maintained in the postnatal period in regions of the central nervous system where adult neurogenesis occurs, such as cells of the olfactory bulb and dentate gyrus [66].
Purified MTs assembled from β3-tubulin are less resistant to depolymerization and spend less time in paused states than those composed of β2- and β4-tubulin or a mixture of all three isotypes [68]. These findings suggest that neuronal MTs are more dynamic, a concept that was initially hypothesized after the observation that brain MT depleted of β3-tubulin showed an increased assembly speed in vitro [69]. According to these in vitro findings, the lack of β3-tubulin in tubb3 knockout mouse model decreases the dynamics of MTs in growth cones and reduces the neurite outgrowth rate in vivo [27]. Depending on the causative mutation, TUBB3 mutations are associated with a broad range of neurological conditions, including malformations of cortical development (i.e., polymicrogyria, gyral disorganization, basal ganglia anomalies, abnormal corpus callosum, hypoplastic brainstem, dysplastic cerebellar vermis), a congenital cranial dysinnervation disorder known as congenital fibrosis of the extraocular muscles type 3 (CFEOM3), and axonal sensorimotor neuropathy [70,71,72,73,74].
Other tubulin isotypes, such as α-tubulin1A (encoded by human TUBA1A and mouse tuba1a), α8-tubulin (encoded by human TUBA8 and mouse tuba8 ), β-tubulin2B (encoded by human TUBB2B and mouse tubb2b), β-tubulin2A (encoded by human TUBB2A and mouse tubb2a), the class I β-tubulin (encoded by TUBB human gene and tubb5 mouse gene), α-tubulin4A (encoded by human TUBA4A and mouse tuba4a), and β-tubulin4B (encoded by human TUBA4B and mouse tuba4b) are reported to be expressed in brain.
The neurological disorders caused by mutations in genes encoding these tubulins as well as in TUBB3 are known as “tubulinopathies” [75,76,77]. The disease spectra associated with these different tubulin isotypes are overlapping but distinct, reflecting functional specification of the tubulin multigene family.
Among the β-tubulin isotypes, class I β-tubulin is the highest expressed in developmental mouse brain [78]. In situ hybridization and immunocytochemical experiments, conducted respectively on the brain tissue of wild type and tubb5 transgenic mice, have revealed predominant expression of this tubulin in neuronal progenitors and post-mitotic neurons during fetal cortical development [78]. Perturbation of tubb5 impairs differentiation of neuronal cortex neurons and the maturation of dendritic spines in mouse [79]. Patients with TUBB mutations exhibit neurological features including microcephaly, polymicrogyria, and dysmorphic basal ganglia which can occur concurrently to the “Michelin tire baby syndrome”, characterized by congenital symmetrical circumferential skin creases and facial abnormalities [78,80,81,82,83].
Similarly to class I β-tubulin, β-tubulin 2B is highly expressed in progenitors and post-mitotic neurons during mouse cortical development [84]. Its expression decreases in postnatal cortex and persists only in a population of post-mitotic neurons in 8-week-old retina [84]. Mutations in TUBB2B principally cause polymicrogyria, pachygyria, microcephaly, and axon guidance defects [85,86,87,88,89].
Much less is known about the spatial and temporal expression of β-tubulin2A. This tubulin isotype represents ~30% of all β-tubulins in human brain [90]. Using real-time PCR experiments, Breuss and colleagues have demonstrated lower expression of tubb2a during brain development, which reached its highest level at E14.5, E16.5, and around the time of birth [78]. This finding suggests that this tubulin carries out relatively minor functions during brain development, in accordance with the mild brain phenotypes observed in patients harboring TUBB2A mutations [91,92,93] and the recent finding that the loss of tubb2a leads to relatively mild cortical malformations in mice [10]. Accordingly, we have recently described a novel TUBB2A mutation associated with a phenotype presenting peripheral sensory motor polyneuropathy without cortical malformation [94].
It is captivating to hypothesize that expression of TUBB2A in brain may be maintained, if not even higher in the postnatal period, as reported for TUBA4A, which is associated with a later age disease phenotype without presenting brain malformations.
TUBA4A brain expression levels have been reported to dramatically increase (>50-fold) with age. Further, RNA-Seq experiments have shown that TUBA4A is the highest β-tubulin expressed in human motor cortex after birth, reaching its maximum expression between 4 months and 1 year and at 8, 19, and 23 years after the birth [90]. This expression pattern may explain why, differently from congenital tubulin mutations, mutations in TUBA4A cause amyotrophic lateral sclerosis (ALS), a late-onset disease characterized by adult-onset upper and lower motor neuron degeneration [95].
Much less is known about TUBB4A expression data. In human brain, Hersheson and colleagues [96] have found that this gene has the highest expression in the cerebellum, followed by putamen and white matter. Furthermore, similar to TUBA4A, expression of TUBB4A increases over time in human motor cortex, reaching its maximum expression at 11 and between 19 and 30 years [95]. Interestingly, mutations in this gene are responsible for the torsion dystonia type 4 (DYT4) [96,97], an adult-onset generalized dystonia (also known as “whispering dystonia”). Mutations in TUBB4A are also associated with hypomyelination and atrophy of the basal ganglia and cerebellum (H-ABC), a rare hypomyelinating disease of infancy and childhood [98,99,100], and with an allelic clinical variant of congenital or childhood-onset hereditary spastic paraplegia [101,102,103].
By contrast, TUBA1A is the most prevalent α-tubulin gene expressed in neurons [75,104,105]. Expression studies on its mRNA have shown that that this isotype is the most prevalent α-tubulin expressed in embryonic nervous system, representing more than 95% of total α-tubulin mRNA [106,107], consistent with its pathogenic role in migration disorders such as pachygyria, lissencephaly, and polymicrogyria [104,108,109,110,111,112,113,114]. The loss of Tuba1a in mouse is perinatal lethal and leads to significant forebrain dysmorphology [10].
Lastly, TUBA8 represents an atypical tubulin isotype. It is the most divergent α-tubulin isotype, showing a protein sequence very similar to that of mouse Tuba8 and higher than the other human α-tubulins [115]. It is expressed during brain development, displaying an expression pattern similar to that of tubb2b in the cortical plate and subplate at E15.5–E16.5, and in the postnatal period where it is highly expressed in the cortical layers II–III and V and in the subplate. Its high expression in post-migratory neurons suggests a role for tuba8 in local adjustments of cell position after arrival in the cortical plate [116]. Differently from all tubulin mutations that present an autosomal dominant inheritance, TUBA8 mutations have an autosomal recessive inheritance and result in polymicrogyria and optic nerve hypoplasia [116].
In conclusion, the complex expression patterns of each of these genes reveals a specific function of tubulin isotypes, in particular, neuronal cell types at specific stages of brain development and during the postnatal period; understanding the exact role of each tubulin isotype in brain still requires further efforts and may be able to explain the variability of neurological phenotypes associated with this class of genes.
4.2. Tubulin PTMs
More than that of any other cell type, neuronal MTs are enriched in a variety of PTMs. PTMs differ between neuronal compartments and vary during neuronal differentiation, leading to the widely accepted hypothesis that specific patterns of PTMs are accumulated at a specific stage of neuron maturation and locally control MT functions.
We will focalize on four PTMs, which are acetylation, tyrosination, detyrosination, and polyglutamylation of tubulin, which have received great attention in recent years, particularly in the context of neuronal cells.
4.2.1. Acetylation
With the exception of TUBA8, all α-tubulin isotypes undergo acetylation on lysine 40 (K40), a residue located in the lumen of MT. This is a reversible PTM catalyzed by the tubulin acetyltransferase αTAT1 [117], and is removed by the histone deacetylase 6 (HDAC6) and Sirtuin-2 (SIRT2) [118,119].
αTAT1 preferentially catalyzes the acetylation of MT in its polymerized state; this finding has opened the question of how this enzyme reaches the K40 inside the lumen of the MT. Currently, the most reliable models are that αTAT1 enters inside the MT lumen via irregularities in the MT structure [120,121] or via the opened MT ends [122]. The switching of protofilament numbers and the loss of tubulin dimers from MTs support the first model; different studies have, indeed, proposed that αTAT1 may use these transient defects to locally enter inside MTs and catalyze the acetylation of K40 [121]. The second and most accepted model is instead supported by recent studies which have shown that αTAT1 binds with high affinity to the MT opening ends [122]. Moreover, MT opening ends directly contact different cellular structures enriched in αTAT1, such as focal adhesions or clathrin-coated pits, facilitating entry of the enzyme [123].
Tubulin acetylation has been proposed to stabilize long-lived MTs, preventing their breakage induced by mechanical stress [124,125]. MTs of mechanosensory neurons, which experience repeated mechanical stresses, are typically highly acetylated [124]. Elimination of MT acetylation by either mutating K40 or deleting the gene encoding αTAT1 impairs touch sensitivity [124]. High levels of acetylated α-tubulin have also been observed in cells derived from Wallerian degeneration slow mice, which are resistant to paclitaxel-mediated axonal degeneration [126,127].
In vitro experiments have shown that acetylation of tubulin enhances MT flexibility and confers resilience against deformations induced by repeated mechanical stresses, possibly by weakening inter-protofilament interactions and making MTs more pliable during bending [125]. Thus, in contrast to other PTMs that, as we will see, act through the recruitment of MAPs, intraluminal acetylation directly regulates MT structure [125].
In neurons, acetylated MTs are not uniformly distributed. In mature neurons, acetylated MTs are present in lower amounts in the cell body and growth cone and are enriched at the proximal site of the axon, controlling axon branching [128]. Acetylated MTs are also observed in the distal axon and presynaptic regions, consistent with the increase in stable MTs at stable neuronal connections [129] (Figure 2). Instead, in young neurons, acetylated MTs localize to the proximal site of the neurite and in the cell body, connecting neurite outgrowth, while they are absent in the growth cone and in lower amounts in minor neurites [129] (Figure 2). It has likewise been shown that acetylation of MTs increases the recruitment of molecular motors [130,131]. In neuronal cells treated with HDAC inhibitors that increase MT acetylation, the kinesin-1-mediated transport into neurite tips is improved [130,132]. Biochemical experiments have shown that kinesin-1 selectively binds acetylated MTs; however, considering the lumen localization of K40, how this PTM influences kinesin binding is still unclear. It has been recently proposed that acetylated MTs, being enriched in high curve regions, can expose a greater number of available kinesin-binding sites and enhance kinesin run lengths [133].
4.2.2. Detyrosination and Tyrosination
The detyrosination–tyrosination cycle of α-tubulin is the best characterized tubulin modification. These modifications occur in α-tubulin isotypes and consist of the addition and removal of a C-terminal tyrosine residue. With the exception of TUBA4A and TUBA8, that present a C-terminal phenylalanine, all α-tubulin isotypes expose a C-terminal tyrosine residue, which can be removed by detyrosination and re-added by tyrosination [134]. Thus, detyrosination is the initial step that triggers the detyrosination–tyrosination cycle. Tyrosination is catalyzed by the tubulin tyrosine ligase (TTL), while the carboxypeptidase that catalyzes detyrosination has not yet been discovered [33]. Detyrosinated tubulin can be further converted to Δ2-tubulin through deglutamylation of the penultimate glutamic acid residue. Removal of the penultimate glutamic acid residue is irreversible, resulting in the exclusion of Δ2-tubulin from the reversible cycle of detyrosination–tyrosination [135]. Multiple enzymes are capable of removing the penultimate glutamate residue to create Δ2-tubulin, including all six cytoplasmic carboxypeptidases (CCP1–6) [136,137,138,139].
In cultured cells, tyrosinated α-tubulin is observed in labile, newly formed MTs whereas detyrosinated tubulin is enriched in stable MT with long turnover [140,141]. In cells with very long-living MTs, detyrosinated tubulin is finally converted into Δ2-tubulin.
Tyrosinated MTs are primarily observed in developing neurons, where they are located in the distal region of axons, contiguous with the growth cone and in the cell body. Detyrosinated MTs are instead distributed in the growing axon and minor neurites of young neurons. Moreover, levels of detyrosinated MTs are high in the axon shaft and in differentiated neurites of mature neurons, while Δ2-tubulin is restricted to very stable MTs and seems to occur during the final stages of neuronal differentiation [129,141,142] (Figure 2).
4.2.3. Polyglutamylation
Polyglutamylation consists of the addition of one or more glutamate residues to the carboxy-terminal tail of both α- and β-tubulin [143,144,145,146].
The addition of strings of glutamines on tubulins consist of two steps: the initiation step, during which the first residue of glutamate is added on the carboxy-terminal domain of tubulin, and the elongation step that leads to the formation of a polyglutamyl side chain [144].
This PTM is catalyzed by nine members of the TTLL family (TTL1, -2, -4, -5, -6, -7, -9, -11, and -13), each of which exhibits a preference for the tubulin isoform (α vs β) and for the initiation or elongation steps. TTLL7 preferentially polyglutamylates β-tubulin isoforms while TTLL5 and -6 prefer α-tubulin as a substrate [145]. Moreover, TTLL1, -4, -5, and -7 catalyze the addition of the first residue of glutamate, while glutamate chain elongation is mediated by TTLL6, -9, -11, and -13 [145].
Polyglutamylation is a reversible PTM, and the removal of glutamate residues is catalyzed by CCP enzymes 1, 4, 5, and 6 [137,138].
Although the mechanistic role of polyglutamylation in neurons is still unclear, different studies have reported that polyglutamylation can affect MAPs interactions. The clearest examples come from MAP1, MAP2, tau, and the severing enzymes spastin and katanin [147,148,149,150,151]. Affinity of MAP1B, MAP2, and tau increase progressively for tubulins bearing 1–3 glutamyl units while decreasing for tubulin having longer chains [149,150]. Spastin and katanin are instead preferentially activated by long glutamate side chains [147,148,151]. In addition, this PTM is spatially and temporally regulated in neurons. Glutamylation of α-tubulin is abundant in young neurons whereas glutamylation of β-tubulin increases during postnatal development [152]. Moreover, polyglutamylated α-tubulin is enriched within axons while polyglutamylated β-tubulin accumulates in MAP2-enriched subcellular compartments, where it is required for the growth of MAP2-positive neurites [153] (Figure 2).
4.3. Interaction with MAPs
Both the function and organization of MTs, especially neuronal MTs, depends on different MAPs.
Based on their mode of action, MAPs can be roughly divided into five groups. The first group contains the motile MAPs, which use the energy of ATP hydrolysis to walk along MTs. They display unidirectional movement along MTs and include kinesins that move toward the plus end, and are responsible for the anterograde transport, and dyneins that move toward the minus end of MTs and are responsible for the retrograde transport [154]. The second group is that of the MT end-tracking proteins (TIPs) which are distinguished by their specific accumulation at the plus ends (+TIPs) or minus ends of MTs (−TIPs). +TIPs typically target growing but not shrinking MT plus ends where they act mainly as MT-stabilizing factors or link MT ends to various cellular structures, such as the cell cortex or kinetochore [155]. Indeed, −TIPs include protein factors that recognize MT minus ends and regulate MT nucleation, stability or interactions with cellular partners [156].
The third group includes MT-severing enzymes, such as katanin and spastin, that generate internal breaks into MTs [151], while the fourth group comprise MT nucleators, such as the γ-tubulin ring complex which directly modulates MT numbers and nucleation [157] and MT-anchoring proteins, such as the centrosomal proteins ninein and Kif3a that regulate MT aster anchoring [158,159].
Lastly, the fifth group includes the structural MAPs that bind the MT lattice and stabilize or crosslink MTs [160]. These MAPs are highly expressed in the brain and show distinct distribution and expression patterns [161]. Performing such disparate functions, MAPs constitute one of the most complex class of regulators and compensation mechanisms exist among them, as demonstrated by the viability and the absence of a strong phenotype in MAP knockout mice [162,163,164,165,166,167].
The structural MAPs are probably the most intensely studied in the context of neurodegeneration. They are classified in two categories, the type I (MAP1a/1b family) and the type II (MAP2/tau family), with MAP2 and tau the major MAPs expressed in the central nervous system [168].
Both MAP2 and tau exist as alternative spliced isoforms sharing a conserved carboxy-terminal domain containing MT-binding repeats which are involved in the binding of MTs, and an amino-terminal projection domain of varying size [169]. They are natively unfolded molecules which adopt specific conformations upon binding to their targets [170]. MAP2 family is composed of two high-molecular-weight proteins and two low-molecular-weight proteins which are developmentally regulated in brain [169,171,172,173,174,175,176].
Tau has six spliced isoforms [170,171] that differ for the presence of three MT-binding repeats or four MT-binding repeats. In addition, these isoforms are developmentally regulated in brain [177,178,179,180].
Both tau and MAP2 bind longitudinally along the outer ridges of MT protofilaments and exhibit MT-stabilizing activities [181].
Tau is mainly found in axon of neurons where it promotes MT assembly and bundle formation [168,182,183,184]. In vitro experiments have demonstrated that tau promotes tubulin assembly and ring formation [185]. It was also found to increase the time that MTs spent in the pause state, stabilizing MTs in the steady state of assembly [186]. Panda and colleagues showed that a low ratio of tau to tubulin can decrease the treadmilling rate of MTs by 70% in vitro [187]. This is considered one of the principal mechanisms through which tau stabilizes axonal MTs, which are particularly susceptible to treadmilling since their minus ends are not anchored to the centrosome.
Moreover, it was demonstrated that by binding to the interface between the tubulin dimers, tau controls the spacing between MTs in the bundle via its projection domain [188,189]. Upon binding, tau also induces a local distortion in the protofilament, changing the spontaneous curvature of MTs and shifting the distribution of protofilament number [190]. This phenomenon suggests that tau can also act as allosteric regulator, influencing the binding of additional MAPs on the outer surface of MTs [190].
On the contrary, MAP2 localizes to dendrites of mature neurons [191] where MT stabilization and growth are required. MAP2 is distributed at the crossbridge between dendritic MTs or between dendritic MTs and other cytoskeletal components [192,193]. Because of the association of MAP2 with MTs, they become stabilized and their stiffness is increased, thus giving rise to longer polymers [194,195,196,197]. MAP2 depletion inhibits neurite formation and outgrowth of cerebellar macroneurons and carcinoma cell lines [198] and disrupts MT spacing and dendritic arbor complexity in the mouse brain [199,200].
Both MAP2 and tau contain a great number of serines and threonines, and their biological activities are regulated by the degree of their phosphorylation at these sites. Low levels of phosphorylation do not interfere with their binding to MTs while high levels of phosphorylation decrease their association. As we will discuss in the next section, this phenomenon has important pathological implications in the case of tau: hyperphosphorylation of tau make this protein more susceptible to aggregating into insoluble inclusions that destabilize MTs and make neurons more vulnerable and prone to degenerating.
5. Microtubule Dysfunction and Neurodegeneration
The impairment of MT functions is considered the central physiopathological mechanism of several neurodegenerative diseases. In this section, we will briefly describe how abnormalities in MT regulative pathways impair MT properties and functions, leading to neurodegeneration.
5.1. Tubulin Dysfunction: Primary Tubulinopathies
As previously reported, mutations in tubulin genes are associated with a wide spectrum of neurological disorders collectively known as “tubulinopathies” and mostly characterized by cortical malformations. Despite tubulinopathies primarily causing neurodevelopmental diseases, in the last ten years, several studies have shown that specific tubulin isotypes or even mutations in specific residues of tubulin isotypes can also cause neurodegenerative profiles. How tubulin mutations impair tubulin functions and neuron activities, causing neurodegeneration or neurodevelopmental defects, is still not clear and certainly represents an intriguing area of investigation. To date, four tubulin isotypes, TUBB3, TUBB2A, TUBA4A, and TUBB4A, have been associated with neurodegenerative phenotypes [71,72,94,95,96,97,98,101,102,103,201,202] (Table 1).
TUBB3 was the first tubulin isoform to be associated with a neurodegenerative phenotype characterized by adult-onset axonal sensory motor polyneuropathy [71,72].
To date three neurodegeneration-causing mutations (D417N, D417H, and E410K) have been identified in TUBB3; all disrupt the MT interaction with kinesin motor proteins [71,203], inducing the accumulation of kinesins in axon tips and perturbing the axonal transport of synaptic vesicles and mitochondria in neurons [71,101]. Both D417 and E410 are highly conserved residues located in the H12 helix of β3-tubulin, and the substitutions affecting these amino acid positions are predicted to change the net negative charge of this domain, which is critical for the binding of kinesin motor proteins [71,101].
Interestingly, the D417N substitution has also recently been identified in TUBB2A in a patient affected by progressive spastic ataxia and sensory motor axonal neuropathy [94].
Consistent with such a critical role of D417 residue in kinesin binding and axonal transport, co-immunoprecipitation experiments have shown that the mutant TUBB2A was unable to bind KIF1A, a neuron-specific kinesin required for transport of synaptic vesicle precursors. Moreover, in vitro experiments have demonstrated that D417N substitution in TUBB2A causes cell cycle defects, impairing MT dynamics in the mitotic spindle [94]. Similar defects have previously been described in several neurodegenerative disorders [204,205,206,207,208] and were proposed to generate defective aneuploid neurons that are more prone to degeneration [209,210].
Moreover, in 2014, a large-scale analysis of exome-wide variant burdens involving 363 cases of familial amyotrophic lateral sclerosis (FALS) identified six pathogenic variants in TUBA4A (G43V, R215C, R320C, R320H, A383T, and W407X) [95]. All mutations disrupt MT dynamics and stability through a dominant-negative mechanism. Furthermore, the R320C/H, A383T, and W407 mutations in TUBA4A were also reported to impair α–β heterodimer formation [95]. Interestingly, the W407X mutation, that removes the last 41 amino acids of the C-terminal domain of TUBA4A, completely abolishes the incorporation of tubulin into MTs. Mutant TUBA4A harboring the W407X substitution shows aggregation propensities and, analogous to other ALS-associated mutant proteins, forms small cytoplasmatic inclusions. The biological relevance of these aggregates is not yet clear; it was proposed that these inclusions may act in trapping MAPs into aggregates or overloading the ubiquitin proteasome system [95].
Instead, depending on their position, mutations in TUBB4A may cause three neurodegenerative disorders: (i) hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC), a rare hypomyelinating disease of infancy and childhood [98,99,100,201,211,212,213,214,215,216,217,218] (ii) hereditary spastic paraplegia [96,97,98]; (iii) DYT4 with whispering dysphonia and normal MRI [96,97,98].
To date, 23 H-ABC-linked TUBB4A mutations have been described. In patients harboring these mutations, a lack of age-appropriate myelin formation is accompanied by progressive neuronal atrophy of specific brain regions, namely the basal ganglia and cerebellum. The most common H-ABC-related mutation is the D249N substitution [98,201,202]. D249 is located within the T7 loop and is predicted to form a salt bridge with the R2 located in the amino-terminal domain of TUBB4A. This interaction is critical for the positioning of the T7-loop and allows it to contact the GTP of α-tubulin in the α–β heterodimer [201].
According to the neuropathological phenotype characterized by the loss of putaminal neurons and oligodendrocytes [98,99,201,211,212], D249N mutation has been reported to affect both neuronal and oligodendroglial precursor cells in vitro and in a H-ABC mouse model [219,220].
Moreover, Vulinovic and colleagues have demonstrated that D249 substitution caused greater incorporation of the mutant TUBB4A into MTs and reduced the MT affinity for the kinesin KIF5 [221]. Accordingly, engineered D249N iPSCs present impaired mitochondrial axonal transport, characterized by reduced mitochondrial movement and increased mitochondrial speed, with the latter probably the consequence of a compensatory mechanism [221].
The H190Y and D353V mutations in TUBB4A have instead been described in patients affected by congenital-onset spastic paraplegia, which can occur concurrently with mild hypomyelination [101,102,103]. Functional data associated with these two mutations are still not reported.
Lastly, the heterozygous mutations p.R2G and p.A271T in TUBB4A have been associated with DYT4 [96,97].
p.R2G affects a highly conserved residue in the amino-terminal β-tubulin tetrapeptide Met–Arg–Glu–Ile (MREI), an autoregulatory domain that modulates the stability of polysome-bound tubulin messenger RNAs [222]. Yen and colleagues have been previously demonstrated that p.R2G substitution abrogates the MREI autoregulatory ability to destabilize β-tubulin mRNA, affecting the balance of tubulin subunits and interfering with MT proper assembly [222].
Differently from the previous study of Yen et al., Lohmann and colleagues detected a consistent decrease in mutant TUBB4A transcript levels of different cell types obtained from a patient harboring the p.R2G mutation [97]. Further studies will be necessary to explain this discrepancy and describe the mechanisms through which p.R2G mutation affects MT biochemical and functional properties. Recently, both p.R2G and p.A271T substitutions were reported to diminish TUBB4A ability to interact with α-tubulin, altering the MT network and inhibiting the neuronal growth process [223].
Overall, these findings highlight the specialized behavior of tubulin isotypes: depending on the affected isoforms and their location, tubulin mutations impair different MT functions, affecting specific cell types.
5.2. Tubulin PTM Imbalance
Several findings have shown that the disruption or dysregulation of PTMs are implicated in neurodegeneration processes [224,225,226,227,228,229,230,231,232].
Imbalance of α-tubulin acetylation and concomitant axonal transport defects have been found to be linked to different neurodegenerative diseases [224,225,226,227,228].
In the brains of patients affected by Alzheimer’s disease (AD), the levels of acetylated α-tubulin were found to be reduced in neurons containing neurofibrillary tangles [228]. Moreover, the levels of the deacetylating enzyme HDAC6 have been reported to be significantly increased in relevant degenerating brain regions of AD patients and AD animal models [229,230,231].
The loss of HADC6 in transgenic APP/PS1 mice, a model of AD, has been reported to restore the level of α-tubulin acetylation and ameliorate AD cognitive pathology [230].
Similarly, null mutants of HDAC6 rescue tau-mediated MT defects in both muscle and neurons of a drosophila model of tauopathies in which human tau is overexpressed in muscle cells [232].
Reduced levels of tubulin acetylation and defective axonal transports have also been described in motor neurons of patients affected by Charcot–Marie–Tooth disease (CMT) [227], a hereditary motor and sensory peripheral neuropathy. Accordingly, decreased levels of α-tubulin acetylation as well as axonal transport defects were also reported in peripheral neurons of mutant HSPB1-transgenic mice, a murine model of CMT2F [224].
Interestingly, in these cases, HDAC6 inhibition has also been reported to reverse mitochondrial axonal movement defects of CMT motor neurons [227] and to improve the phenotype of mutant SOD1 mice [225].
Reduced levels of acetylated tubulin have also been observed in the brains of patients affected by Huntington disease (HD), and the pharmacological treatment of mouse striatal cells derived from transgenic HD mice with compounds that increment tubulin acetylation stimulates MT-dependent transport of BDNF and prevents the alterations observed in HD mutant cells [131].
Imbalance of acetylated tubulin together with tyrosinated tubulin has also been observed in brain tissues of Parkin knockout mice which exhibit motor dysfunction and dopaminergic neuronal loss [233]. Dopaminergic neurons of young age brain tissues show an early accumulation of tyrosinated tubulin and decreased levels of acetylated tubulin, while at the later ages, the situation is overturned [234].
Tubulin glutamylation has been recently found to be involved in neurodegeneration as well.
The excessive polyglutamylation of tubulin has been linked to the rapid degeneration of Purkinje cells in pcd mice which present an inactivating mutation in the deglutamylating enzyme CCP1 [235] and in Ccp1flox/flox L7-cre mice in which the CCP1 gene is specifically deleted in Purkinje cells [236].
Both mice displayed massive cerebellar atrophy, abnormally accumulate glutamylated tubulin in degenerating neurons, and display aberrant mitochondria axonal transport [237].
In Ccp1−/− neuron, hyperglutamylation of tubulin was reported to impair motility of several cargo items such as mitochondria, lysosomes, Lamp1-positive endosomes, and BDNF vesicles [238].
Importantly, Magiera and colleagues have demonstrated that hyperglutamylation of tubulin, rather than MT-severing enzymes that specifically target polyglutamylated MTs, is responsible for neurodegeneration since the concomitant deletion of the main brain glutamylase enzyme TTLL1, but not of the severing enzyme spastin, is able to prevent neuronal cell death [236].
In 2018, 6 loss-of-function mutations in human CCP1 were described for 13 patients affected by infantile-onset progressive neurodegeneration mainly involving the cerebellum, spinal motor neurons, and peripheral nerves. The disease-associated mutation lead to the absence of functional CCP1, determining the accumulation of polyglutamylated tubulin and the loss of Δ2-tubulin [239].
Interestingly, CCP1-deficient mice recapitulate several key features of the human disease, including abnormal tubulin polyglutamylation, cerebellar atrophy, and the involvement of spinal motor neurons and peripheral nerves [239].
Recently Vu and colleagues have reported high levels of polyglutamylated and Δ2 tubulin in a kainate-induced epileptic seizure mouse model and the slow-developing AD brain of patients [240].
5.3. MAP Dysfunction
Several findings have widely demonstrated that dysfunctions in MT-interacting proteins trigger neurodegeneration via defective regulation of MTs [241,242,243].
AD is probably the best example of neurodegenerative disease stemming from MAP dysfunction.
It is a member of a family of neurodegenerative disorders called tauopathies [244]; they are diseases of the nervous system in which tau becomes abnormally phosphorylated and accumulates in insoluble inclusions within brain neurons and often glia [245,246]. These tau accumulations are referred to as neurofibrillary tangles when found in the neuronal soma, and neuropil threads when found in dendritic processes [247,248]. Unlike classic tauopathies, AD is also characterized by the extracellular accumulation of β-amyloid (αβ) as senile plaques in specific brain regions [249]. Several lines of evidence indicate that αβ accumulation initiates tau phosphorylation [250,251,252,253,254]. Indeed, the exposure of primary neurons to oligomers of Aβ cause mislocalization of Tau in dendrites and the MT breakdown in dendrites invaded by Tau [254].
It has been demonstrated that Tau hyperphosphorylation promotes tau sequestration into neuronal inclusion and impairs MT stability [255,256].
Hyperphosphorylated tau isolated from the brain homogenate of AD patients is not able to promote MT assembly in vitro and its MT assembly activity is recovered only upon tau dephosphorylation with alkaline phosphatase treatment [257]. Moreover, hyperphosphorylated tau displays prion-like activity, sequestrating normal phosphorylated tau and other MAPs, such as MAP1 and MAP2, and destroying pre-assembled MTs in vitro [257,258].
To date, more than 40 phosphorylation sites have been identified in tau protein isolated from AD brain [259] and, depending on their position, have been reported to have a different impact on the pathogenic role of tau. In vitro kinetic studies have demonstrated that Ser199/Ser202/Thr205, Thr212, Thr231/Ser235, Ser262/Ser356, and Ser422 are the critical phosphorylation sites that convert tau to an inhibitory molecule that sequesters normal MAPs from MTs. Phosphorylation sites also seem to differently reduce tau affinity for binding MTs: phosphorylation of tau at Ser262, Thr231, and Ser235, for example, inhibits its MT affinity binding by ~35%, ~25%, and ~10%, respectively [259].
Transgenic mouse models of AD display fewer MTs [257,258,259,260,261], increased MT hyperdynamicity, axonal dystrophy, and reduced fast axonal transport [262,263]. The ectopic expression of human tau in Drosophila melanogaster results in decreased MT density, increased MT fragments, and increased amounts of satellite boutons at neuromuscular junctions [222]. Similarly, there is also evidence of MT deficits in the AD brain [228,256,264]. Ultra-morphometric analysis conducted on brain biopsies from AD patients documented a reduced MT density in pyramidal neurons [264,265]. Moreover, Zhang and colleagues [265] documented a decrease in the total level of α-tubulin and of α-tubulin-positive axonal processes in the neurons of AD patients.
In addition to AD, MT dysfunction has been considered an important contributor also to the pathogenesis of Parkinson’s disease (PD), a neurodegenerative condition characterized by the loss of dopaminergic neurons from substantia nigra and the presence in these cells of intracellular inclusions of α-synuclein (α-syn) [266].
Mutations in genes encoding the MT-interacting proteins Parkin, leucine-rich repeat kinase 2 (LRRK2), and α-syn indeed cause autosomal forms of parkinsonism [267].
By directly or indirectly binding tubulin, these proteins have been reported to modulate MT stability and function [268,269,270,271,272].
Mutations in PRKN gene, encoding the Parkin protein, are the most common cause of autosomal recessive juvenile parkinsonism and a major contributor to familial and sporadic early-onset PD [273,274]. Parkin is an E3 ubiquitin ligase that catalyzes the covalent attachment of ubiquitin to specific substrates, targeting them for proteasomal degradation [275]. This protein co-purifies with tubulin and was found in highly purified tubulin preparations [270]. Ren and colleagues have been reported that Parkin bound to α–β tubulin heterodimers with high affinity and that this interaction enhanced the ubiquitination and degradation of tubulins. In the same study, the authors demonstrated that Parkin colocalized with MTs in HEK293T cells and that the point mutations K161N, T240R, and C431F, associated with PD, abolished tubulin ubiquitination [268]. In 2005, Yang and colleagues reported that Parkin stabilized MT by binding three independent MT-binding domains (linker, RING1, and RING2) and that the overexpression of these domain as well as wild-type Parkin significantly attenuated colchicine-induced MT depolymerization in COS-7 cells [270]. Moreover, the authors demonstrated that the MT-binding activity of Parkin and its E3 ligase activity are independent since PD-linked mutations K161N, T240R, and C431F were able to impair its E3 ligase activity but not MT binding and stabilization [270]. Cartelli and colleagues have demonstrated that primary fibroblasts obtained from PD-affected patients exhibited reduced MT mass and higher MT destabilization and that overexpression of wild type Parkin restored the control phenotype [276]. Accordingly, experiments performed in both murine and human midbrain dopaminergic neurons showed that mutations or exon deletion in PRKN caused MT destabilization, abolishing the stabilizing effect of Parkin against MT-destabilizing toxins [277,278].
LRRK2 is another example of a microtubule-interacting protein associated with familial PD [279].
LRKK2 is a large multidomain protein and has been found associated with MTs [269].
LRRK2 was reported to co-immunoprecipitate with β-tubulin from both wild-type mouse brain and LRRK2-overexpressing HEK293T cells [280]. Moreover GFP-tagged LRRK2 was found to co-localize with β-tubulin in HEK-293 cells and the endogenous LRKK2 was found to colocalize with tubulin in primary hippocampal neurons [226,281].
Law and colleagues demonstrated that LRRK2 directly binds the carboxy-terminal domain of TUBB, TUBB4, and TUBB6 and that the binding specificity is determined by lysine 362 and alanine 364 of these β-tubulin isotypes. Interestingly, this interaction takes place at the luminal MT surface close to the K40 acetylation site of α-tubulin, a residue exposed in dynamic MT populations which present a more open and flexible conformation [269]. Consistent with this finding, endogenous LRRK2 displayed preferential localization to dynamic MTs within the growth cone [269]. Accordingly, mouse embryonic fibroblasts derived from LRRK2 knockout mice displayed increased MT acetylation, indicating that LRRK2 may interfere with tubulin acetylation [269].
LRRK2 was further reported to phosphorylate β-tubulin purified from bovine brain [280]. In vitro co-incubation of bovine brain tubulins with LRRK2 increased MT formation and stability in the presence of MAPs. Consistent with these data, Gillardon and colleagues detected higher levels of soluble tubulin in brain lysates from LRRK2 knockout mice and shorter neurites in cell cultures of LRRK2-deficient neurons [280]. However, MacLeod and colleagues demonstrated that LRRK2 depletion in cortical neurons increased neurite length after 2 weeks [282]. Hence, further experiments will be necessary to explain the nature of LRRK2 and tubulin interaction and clarify this discrepancy.
LRRK2 was recently proposed also to control tau release from MT by mediating its phosphorylation [283,284]. Indeed, in a PD mouse model carrying the P301L mutation in LRRK2, the expression of mutant LRRK2 significantly increased hyperphosphorylated tau deposition [284].
By contrast, missense mutation and duplication and triplication of the SNCA gene encoding α-Syn underlies autosomal forms of parkinsonism [285].
α-Syn is a small, soluble unfolded protein that localizes predominantly to presynaptic terminals of neurons [286]. Although little is known about the mechanisms through which α-Syn acts in PD, different studies have recently proposed that this protein may act as a functional MAP by directly binding MTs [272,287] and regulating their stability. However, the effect of this interaction has been quite controversial, since different studies have reported that α-Syn, promotes MT stabilization, while others suggested the opposite effect [287,288].
Recently, Cartelli and colleagues have demonstrated that α-Syn interacts with pre-formed MTs in co-sedimentation experiments, co-purifies with brain tubulin and forms a specific complex with the tubulin α2β2 tetramer [271]. In the same study, the authors demonstrated that upon interaction with the tubulin α2β2 tetramer, α-Syn acquires a helical structure and becomes able to govern multiple steps of MT assembly and dynamics, such as nucleation, growth rate, and catastrophe frequencies, in a purified system as well as in a neuronal cell model [271], and that the PD-linked point mutations p.A30P, A53T, and E46K corrupt these functions and lead to tubulin aggregation [271].
Moreover, very recently, Cartelli and Cappeletti demonstrated that α-Syn is also able to regulate the partitioning between tubulin dimers and MTs in differentiated PC12 cells [289].
6. Microtubule-Targeting Agents
The awareness that MT dysfunctions are associated with several neurodegenerative disorders has promoted, in the last 20 years, numerous efforts to search for therapeutic compounds to correct MT defects related to neurodegeneration.
Currently, two main lines of intervention exist: the first is centered on drugs that promote MT stability and assembly by directly binding tubulin, while the second relies on drugs that target tubulin enzymes which impact on MT functional properties by modifying tubulin PTMs. In this section, we will briefly discuss the most relevant therapeutic compounds developed for each of the two lines of intervention (Table 2).
6.1. Microtubule Stabilizing Compounds
The concept of utilizing MT-stabilizing drugs for the treatment of neurodegenerative diseases was tested for the first time in a tau transgenic mouse model of tauopathies in which the mouse prion protein (PrP) drives the overexpression of the shortest human brain tau isoform, T44, in the neurons of the central nervous system (PrP T44 mouse). At 3 to 12 months old, Prp T44 mice exhibit tau pathology, including accumulation of tau inclusions and insoluble hyperphosphorylated tau, reduced MT numbers, and impaired fast axonal transport in the spinal motor neurons that project outside the blood–brain barrier [290].
Zhang and colleagues [290] addressed the therapeutic potential of the MT-stabilizer paclitaxel.
Paclitaxel is a member of the taxane family, and initially emerged as an anticancer drug [305].
It promotes MT assembly in vitro [306] by binding within the lumen of the MT at a site in the β-tubulin subunit which is commonly referred to as the taxane-binding site [307,308]. Paclitaxel binding prevents compaction at the tubulin dimer interface and results in more stable lateral interactions, reducing the stochastic switching of MTs between growth and shrinkage [309].
Intravenous administration of paclitaxel in PrP T44 mice restored axonal transport in spinal axons and reduced the motor phenotype [290]. Moreover, beneficial effects of paclitaxel have been mechanistically linked to the stabilization of MTs because its administration increased the levels of stable detyrosinated tubulin as well as the numbers of MTs in the ventral root axons of transgenic mice [290].
Despite these beneficial effects, paclitaxel turns out to be an unsuitable therapeutic candidate for diseases affecting the brain because of its limited brain bioavailability. Paclitaxel does not cross the blood–brain barrier and its effectiveness in improving the phenotype of PrP T44 mouse depends on its uptake at neuromuscular junctions and subsequent retrograde transport to the spinal cord [290].
More recent studies have focused on the therapeutic effect of the MT-stabilizer epithilone D (Epo D). Epo D is a taxol-derived small molecule which prevents MT disassembly by interacting with β-tubulin at the taxane-binding site. This compound is probably the most promising MT stabilizer due to its excellent blood–brain barrier penetration and prolonged brain retention [310]. Indeed, it is selectively accumulated in the central nervous system, reducing its unwanted effects in the peripheral nervous system or elsewhere in the body [310].
The treatment of different lines of tau mouse models with Epo D has improved a number of central nervous system outcomes, increasing MT density, axonal integrity, and neuronal survival [263,291,310]. Moreover, in both young and old PS19 tauopathy models in which tau pathology is developing or well established, Epo D reversed behavioral and cognitive deficits, cleared tau pathology, and curbed neuron loss [291]. Epo D has also recently undergone evaluation in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced mouse model of PD [292]. MPTP-treated mice showed an impairment of axonal transport in dopaminergic axons and changes in tubulin PTMs that were normalized following the treatment with Epo D. Moreover, repeated daily administrations of Epo D attenuated the loss of nigral dopaminergic neurons induced by MPTP [292]. Epo D has also been reported to restore the levels of acetylated α-tubulin and rescue the MT-based intracellular transport of peroxisomes in stem cells derived from patients affected by hereditary spastic paraplegia [293]. Fan and colleagues have demonstrated that very low doses of Epo D restored peroxisome speeds by increasing the number of stable MTs, and ameliorated patient cell sensitivity to hydrogen peroxide [293].
In 2012, Epo D has progressed to phase 1b clinical testing, in short 2–3 months studies in AD and/or tauopathy patients (NCT 01492374). Unfortunately, no results have been published, and drug development efforts for AD have halted. However, a new trial was since started, and the results are expected shortly (NCT 01966666).
Additional brain-penetrant MT-stabilizing products have been tested in recent years.
Triazolopyrimidines (TPDs) are members of the vinca alkaloids which interact with MTs at a site that is distinct from the taxane-binding site, and generally targeted by agents such as vinblastine. The TPD binding of MTs stabilizes the longitudinal contacts between tubulin subunits and prevents MT disassembly [311]. In 2018, Zhang and colleagues tested CNDR-51657, a prototype of TPD, on a PS19 transgenic mouse model of tauopathies which expresses the P301S mutant form of human tau and shows the progressive accumulation of neurofibrillary tangles with age [294]. This study has demonstrated that low doses of CNDR-51657 significantly improved MT density in hippocampal neurons and reduced axonal dystrophy with a resulting reduction in tau pathology. Importantly, no adverse effects were observed in compound-treated mice, including no change in white blood cell counts [294].
Recently, Fanara and colleagues [293] have reported that the MT-modulating agent noscapine attenuates the deficits in axonal transport and improves MT stability in a mutant SOD1 transgenic mouse model of ALS which manifests MT hyperdynamicity in axons from the spinal cord, sciatic nerve, and cortex [295]. The noscapine mechanism of action is still not completely clear; it has been reported to specifically bind to tubulin and modulate MT dynamics by reducing the growing and shortening rates and increasing the percentage of time that MTs spend in the attenuated state [295].
However, among non-taxane MT-stabilizing agents, davunetide (also known as NAP or CP201) has reached the most advanced clinical development status. It is an 8 aa peptide neuropeptide (NAPVSIPQ) derived from the activity-dependent neuroprotective protein [312]. Although still elusive, the mechanism of action of NAP is believed to involve MTs. Affinity chromatography coupled with mass spectrometry identified tubulin as the major NAP-binding proteins in neurons and glial cells. Moreover, NAP decorates MTs in cultured cells and has been reported to enhance in vitro MT assembly in the presence of nocodazole [313]. However, Yenjerla and colleagues demonstrated that increased concentrations of NAP failed to induce tubulin polymerization in a purified system [314]. NAP was first tested on ApoE knockout mice, a mouse model for AD, and was reported to improve its short-term memory [296]. Recent studies have demonstrated that this compound enhances axonal transport and improves cognitive performance in both mouse models of AD and ALS disease [297,315]. Interestingly, NAP is brain penetrant when administered either by intranasal delivery (AL-108) [316,317], intravenously (AL-208) [318], or intraperitoneally [319].
NAP represents a potential therapeutic target for tauopathies; Magen and colleagues have reported that NAP blocks phosphorylation of tau on Ser262, thus increasing MT affinity for tau [317]. It is therefore likely that reduction in hyperphosphorylated tau by NAP is central for its mechanism [320,321]. NAP is in a phase II trial (NCT 01056965) to assess its effects on mild cognitive impairment in AD. Earlier analysis showed that NAP was well tolerated by AD patients with no obvious side effects, and there was some improvement in individual memory tasks but not for composite memory score [298,299].
6.2. Tubulin PTM-Targeting Compounds
The second line of intervention points to restore specific tubulin PTMs by modulating the activity of enzymes that directly regulate their level. At present, the major approach points to increase the levels of tubulin acetylation, which have been found to be reduced in several neurodegenerative diseases, through the inhibition of HDAC6 and Sirt2, the enzymes responsible for the deacetylation of α-tubulin.
The use of compounds that inhibit the enzyme HDAC6 represent, at the moment, a particularly promising therapeutic strategy. A few HDAC6 inhibitors are available. These compounds have been tested in several cellular and animal models of neurodegenerative diseases exerting neuroprotection. However, it is important to specify that almost all these classes of compounds, in addition to inhibiting HDAC6, also exert their actions on other HDACs. Moreover, the same HDAC6, besides the action to deacetylate α-tubulin, participates in other cellular processes, such as the transport and autophagic clearance of misfolded proteins [322]. Thus, the effect of these inhibitors is not always specific for tubulin acetylation and may involve additional processes.
In this section, we will focus our attention only on the effects that these compounds exert on tubulin acetylation.
The first HDAC6 inhibitor to be investigated was the natural compound trichostatin (TSA) [323,324]. It is a non selective HDAC inhibitor that acts by binding to the zinc ion at the HDAC active site of most zinc-dependent HDACs, including HDAC6 [325]. Its in vitro supplementation has been reported to lead to the hyperacetylation of tubulin in mammalian cells [324]. The pharmacological effects of TSA have been well established in several neurodegenerative diseases of the central nervous system. TSA was tested in an LRRK2 Drosophila model of PD. Systematic administration of TSA fully restored fly locomotor behavior and increased tubulin acetylation, improving axonal transport both in vitro and in vivo [223]. The same study showed that in vitro TSA treatments of rat cortical neurons expressing mutant LRKK2 increase tubulin acetylation and restore axonal transport [226].
Moreover, Hanson and colleagues have recently demonstrated that TSA prevents the decrease in tubulin acetylation precluding axon fragmentation and axonal loss in mouse cortical neurons exposed to kainic acid, an excitotoxic agent commonly used to induce epilepsy in rodents [300].
Together with tubastatin A (TubA), a selective inhibitor of HDAC6, TSA was reported to correct mitochondria axonal transport defects caused by HSPB1 mutations and rescue the CMT phenotype of symptomatic mutant HSPB1 mice [218].
TubA is a synthetic compound, first tested in primary cortical neuron cultures where it was found to induce elevated levels of acetylated α-tubulin, but not histone, consistent with its HDAC6 selectivity [326]. In a transgenic mouse model of tau deposition, TubA administration was reported to increase α-tubulin acetylation in the brain and revert behavioral defects such as memory impairment and hyperactivity [301]. Further, TSA was reported to increase vesicular transport of brain-derived neurotrophic factor by increasing the acetylation of α-tubulin in striatal precursor cells of a HD model [131].
AGK2, by contrast, is a selective inhibitor of the deacetylase Sirt2. In vitro experiments have shown that AGK2 increases the acetylation levels of tubulin heterodimers purified from bovine brain. Moreover, AGK2 treatment results in a dose-dependent increase in acetylation of soluble tubulin monomers and polymerized microtubules of HeLa cells without affecting their viability [302]. Moreover, in this study, the authors demonstrated that AGK2 prevents dopaminergic cell death both in vitro and in a Drosophila model of PD [302].
AK7, an additional inhibitor of SIRT2, has also been reported to increase acetylation of α-tubulin [303]. AK7 treatment exerts neuroprotective effect in models of PD and prevents in vivo dopamine depletion and dopaminergic neuron loss in MPTP-treated mice [304]. Moreover, its administration improved motor function, extended survival, and reduced brain atrophy in two genetic mouse models of HD [304].
7. Conclusions
Within the cell, MTs are among the most complex structures both in terms of chemical and functional properties. This complexity is generated through the combinatorial action of regulative mechanisms which determinate the temporal and tissue expression of different tubulin isotypes, their chemical modification as well as their interaction with proteins and cellular effectors.
Alterations of these regulative mechanisms impair MT functions. It has been widely demonstrated that MT dysfunction can contribute to, or be the cause of neurodegenerative processes.
This observation has received great attention in the design of novel therapeutic approaches aimed to counteract neurodegeneration by rescuing MT alterations. Compounds that modulate MT stability or restore the levels of tubulin acetylation have provided several benefits in cellular and animal models of different neurodegenerative diseases and some of them have been translated into clinical trials. These results have aroused great enthusiasm, encouraging research in this field. However, a better understanding of MT dysfunctions is necessary, such as through the analysis of the dynamic state of MTs in different neurodegenerative diseases. Indeed, although MT-stabilizing compounds can restore MT stability, excessively high doses of these drugs can disrupt MT integrity, promoting its over-stabilization. Instead, regarding the use of inhibitors for tubulin deacetylases, these drugs have multiple targets, and there is the need to develop compounds with greater selectivity.
Overall, despite these limits, MT-focused therapy actually represents an innovative and promising approach for the treatment of neurodegenerative diseases and may help to prevent or delay neurodegeneration in patients, improving their quality of life.
Author Contributions
Conceptualization: A.S. Writing—Original Draft. Preparation: A.S.: Writing—Review and Editing: All the authors. Supervision: A.S. All authors have read and agreed to the published version of the manuscript.
Funding
Ricerca 5 per mille Ospedale Pediatrico Bambino Gesù, IRCCS.
Acknowledgments
Authors of this publication are members of the European Reference Network for Rare Neurological Diseases—Project ID No 739510.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| MT | microtubule |
| MAPs | MT-associated proteins |
| PTMs | post-translational modifications |
| MTOC | MT-organizing center |
| CFMO3 | congenital fibrosis of the extraocular muscles type 3 |
| DYT4 | torsion dystonia type4 |
| ALS | amyotrophic lateral sclerosis |
| K40 | lysine 40 |
| HDAC6 | histone deacetylase 6 |
| TTL | tubulin tyrosine ligase |
| CCP | cytoplasmic carboxypeptidases |
| TIP | end-tracking protein |
| FALS | familial amyotrophic lateral sclerosis |
| H-ABC | atrophy of the basal ganglia and cerebellum |
| MREI | tetrapeptide Met–Arg–Glu–Ile |
| AD | Alzheimer’s disease |
| CMT PD | Charcot–Marie–Tooth disease Parkinson’s disease |
| αβ α-syn LRRK2 PrP | β-amyloid α-synuclein leucine-rich repeat kinase 2 mouse prion protein |
| Epo D | epithilone D |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| TPD | triazolopyrimidines |
| NAP | davunetide |
| TSA | trichostatin |
| TubA | tubastatin A |
References
- Horio, T.; Murata, T. The role of dynamic instability in microtubule organization. Front. Plant Sci. 2014, 5, 511. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Akhmanova, A. Microtubule tip-interacting proteins: A view from both ends. Curr. Opin. Cell Biol. 2011, 23, 94–101. [Google Scholar] [CrossRef]
- Piedra, F.A.; Kim, T.; Garza, E.S.; Geyer, E.A.; Burns, A.; Ye, X.; Rice, L.M. GDP-to-GTP exchange on the microtubule end can contribute to the frequency of catastrophe. Mol. Biol. Cell 2016, 27, 3515–3525. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, M.W.; Mitchison, T. Microtubule dynamics. Nature 1986, 324, 621. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.A.; O’Brien, E.T.; Pryer, N.K.; Soboeiro, M.F.; Voter, W.A.; Erickson, H.P.; Salmon, E.D. Dynamic instability of individual microtubules analyzed by video light microscopy: Rate constants and transition frequencies. J. Cell Biol. 1988, 107, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Van Haren, J.; Wittmann, T. Microtubule plus end dynamics—Do we know how microtubules grow? Cells boost microtubule growth by promoting distinct structural transitions at growing microtubule ends. Bioessays 2019, 41, e1800194. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, H.; Luo, Y.; Zhou, S.; An, L.; Wang, C.; Jin, Q.; Zhou, M.; Xu, J.R. Molecular evolution and functional divergence of tubulin superfamily in the fungal tree of life. Sci. Rep. 2014, 4, 6746. [Google Scholar] [CrossRef] [Green Version]
- Ludueña, R.F. A hypothesis on the origin and evolution of tubulin. Int. Rev. Cell Mol. Biol. 2013, 302, 41–185. [Google Scholar]
- Bittermann, E.; Abdelhamed, Z.; Liegel, R.P.; Menke, C.; Timms, A.; Beier, D.R.; Stottmann, R.W. Differential requirements of tubulin genes in mammalian forebrain development. PLoS Genet. 2019, 15, e1008243. [Google Scholar] [CrossRef] [Green Version]
- Ludueńa, R.F.; Roach, M.C.; Prasad, V.; Banerjee, M.; Koiso, Y.; Li, Y.; Iwasaki, S. Interaction of ustiloxin A with bovine brain tubulin. Biochem. Pharmacol. 1994, 47, 1593–1599. [Google Scholar] [CrossRef]
- Derry, W.B.; Wilson, L.; Khan, I.A.; Luduena, R.F.; Jordan, M.A. Taxol differentially modulates the dynamics of microtubules assembled from unfractionated and purified beta-tubulin isotypes. Biochemistry 1997, 36, 3554–3562. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.J.; Gupta, M.L.; Suprenant, K.A.; Himes, R.H. The two alpha-tubulin isotypes in budding yeast have opposing effects on microtubule dynamics in vitro. EMBO Rep. 2003, 4, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vemu, A.; Atherton, J.; Spector, J.O.; Moores, C.A.; Roll-Mecak, A. Tubulin isoform composition tunes microtubule dynamics. Mol. Biol. Cell 2017, 28, 3564–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulton, C.; Simpson, P. Selective synthesis and utilization of flagellar tubulin. The multi-tubulin hypothesis. In Cell Motility; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1976; pp. 987–1005. [Google Scholar]
- Schatz, P.J.; Solomon, F. Genetically essential and nonessential alpha-tubulin genes specify functionally interchangeable proteins. Mol. Cell Biol. 1986, 6, 3722–3733. [Google Scholar] [CrossRef] [Green Version]
- Lopata, M.A.; Cleveland, D.W. In vivo microtubules are copolymers of available beta-tubulin isotypes: Localization of each of six vertebrate beta-tubulin isotypes using polyclonal antibodies elicited by synthetic peptide antigens. J. Cell Biol. 1987, 105, 1707–1720. [Google Scholar] [CrossRef]
- Renthal, R.; Schneider, B.G.; Miller, M.M.; Ludueña, R.F. Beta IV is the major beta-tubulin isotype in bovine cilia. Cell Motil. Cytoskelet. 1993, 25, 19–29. [Google Scholar] [CrossRef]
- Silva, M.; Morsci, N.; Nguyen, K.C.Q.; Rizvi, A.; Rongo, C.; Hall, D.H.; Barr, M.M. Cell-specific α-tubulin isotype regulates ciliary microtubule ultrastructure, intraflagellar transport, and extracellular vesicle biology. Curr. Biol. 2017, 27, 968–980. [Google Scholar] [CrossRef] [Green Version]
- Denoulet, P.; Eddé, B.; Gros, F. Differential expression of several neurospecific beta-tubulin mRNAs in the mouse brain during development. Gene 1986, 50, 289–297. [Google Scholar] [CrossRef]
- Joshi, H.C.; Cleveland, D.W. Differential utilization of beta-tubulin isotypes in differentiating neurites. J. Cell Biol. 1989, 109, 663–673. [Google Scholar] [CrossRef]
- Wang, D.; Villasante, A.; Lewis, S.A.; Cowan, N.J. The mammalian beta-tubulin repertoire: Hematopoietic expression of a novel, heterologous beta-tubulin isotype. J. Cell Biol. 1986, 103, 1903–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwer, H.D.; Lecine, P.; Tiwari, S.; Italiano, J.E., Jr.; Hartwig, J.H.; Shivdasani, R.A. A lineage-restricted and divergent beta-tubulin isoform is essential for the biogenesis, structure and function of blood platelets. Curr. Biol. 2001, 11, 579–586. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.G.; Turner, F.R.; Hutchens, J.A.; Raff, E.C. Axoneme-specific beta-tubulin specialization: A conserved C-terminal motif specifies the central pair. Curr. Biol. 2001, 11, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Hoyle, H.D.; Raff, E.C. Two drosophila beta tubulin isoforms are not functionally equivalent. J. Cell Biol. 1990, 111, 1009–1026. [Google Scholar] [CrossRef]
- Matthews, K.A.; Rees, D.; Kaufman, T.C. A functionally specialized alpha-tubulin is required for oocyte meiosis and cleavage mitoses in Drosophila. Development 1993, 117, 977–991. [Google Scholar]
- Latremoliere, A.; Cheng, L.; DeLisle, M.; Wu, C.; Chew, S.; Hutchinson, E.B.; Sheridan, A.; Alexandre, C.; Latremoliere, F.; Sheu, S.H.; et al. Neuronal-specific TUBB3 is not required for normal neuronal function but is essential for timely axon regeneration. Cell Rep. 2018, 24, 1865–1879.e9. [Google Scholar] [CrossRef] [Green Version]
- Ti, S.C.; Alushin, G.M.; Kapoor, T.M. Human β-tubulin isotypes can regulate microtubule protofilament number and stability. Dev. Cell 2018, 47, 175–190.e5. [Google Scholar] [CrossRef] [Green Version]
- Chalfie, M.; Thomson, J.N. Organization of neuronal microtubules in the nematode Caenorhabditis elegans. J. Cell Biol. 1979, 82, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Chalfie, M.; Au, M. Genetic control of differentiation of the Caenorhabditis elegans touch receptor neurons. Science 1989, 243, 1027–1033. [Google Scholar] [CrossRef]
- Savage, C.; Hamelin, M.; Culotti, J.G.; Coulson, A.; Albertson, D.G.; Chalfie, M. mec-7 is a beta-tubulin gene required for the production of 15-protofilament microtubules in Caenorhabditis elegans. Genes Dev. 1989, 3, 870–881. [Google Scholar] [CrossRef] [Green Version]
- Topalidou, I.; Keller, C.; Kalebic, N.; Nguyen, K.C.; Somhegyi, H.; Politi, K.A.; Heppenstall, P.; Hall, D.H.; Chalfie, M. Genetically separable functions of the MEC-17 tubulin acetyltransferase affect microtubule organization. Curr. Biol. 2012, 22, 1057–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magiera, M.M.; Janke, C. Post-translational modifications of tubulin. Curr. Biol. 2014, 24, R351–R354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirajuddin, M.; Rice, L.M.; Vale, R.D. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat. Cell Biol. 2014, 16, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch Grau, M.; Gonzalez Curto, G.; Rocha, C.; Magiera, M.M.; Marques Sousa, P.; Giordano, T.; Spassky, N.; Janke, C. Tubulin glycylases and glutamylases have distinct functions in stabilization and motility of ependymal cilia. J. Cell Biol. 2013, 202, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangs, F.K.; Schrode, N.; Hadjantonakis, A.K.; Anderson, K.V. Lineage specificity of primary cilia in the mouse embryo. Nat. Cell Biol. 2015, 17, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Ran, J.; Yang, Y.; Li, D.; Liu, M.; Zhou, J. Deacetylation of α-tubulin and cortactin is required for HDAC6 to trigger ciliary disassembly. Sci. Rep. 2015, 5, 12917. [Google Scholar] [CrossRef]
- Konno, A.; Ikegami, K.; Konishi, Y.; Yang, H.J.; Abe, M.; Yamazaki, M.; Sakimura, K.; Yao, I.; Shiba, K.; Inaba, K.; et al. Ttll9-/-mice sperm flagella show shortening of doublet 7, reduction of doublet 5 polyglutamylation and a stall in beating. J. Cell Sci. 2016, 129, 2757–2766. [Google Scholar] [CrossRef] [Green Version]
- Gadadhar, S.; Bodakuntla, S.; Natarajan, K.; Janke, C. The tubulin code at a glance. J. Cell Sci. 2017, 130, 1347–1353. [Google Scholar] [CrossRef] [Green Version]
- Kapitein, L.C.; Hoogenraad, C.C. Building the Neuronal Microtubule Cytoskeleton. Neuron 2015, 87, 492–506. [Google Scholar] [CrossRef] [Green Version]
- Conde, C.; Caceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Tsukita, S.; Usukura, J.; Tsukita, S.; Ishikawa, H. The cytoskeleton in myelinated axons: A freeze-etch replica study. Neuroscience 1982, 7, 2135–2147. [Google Scholar] [CrossRef]
- Yu, W.; Baas, P.W. Changes in microtubule number and length during axon differentiation. J. Neurosci. 1994, 14, 2818–2829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, I.; Voelzmann, A.; Liew, Y.T.; Costa-Gomes, B.; Prokop, A. The model of local axon homeostasis—Explaining the role and regulation of microtubule bundles in axon maintenance and pathology. Neural Dev. 2019, 14, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, E.W.; Baas, P.W. Microtubules in neurons as information carriers. J. Neurochem. 2014, 129, 235–239. [Google Scholar] [CrossRef] [Green Version]
- Baas, P.W.; Rao, A.N.; Matamoros, A.J.; Leo, L. Stability properties of neuronal microtubules. Cytoskeleton (Hoboken) 2016, 73, 442–460. [Google Scholar] [CrossRef] [Green Version]
- Baas, P.W.; Lin, S. Hooks and comets: The story of microtubule polarity orientation in the neuron. Dev. Neurobiol. 2011, 71, 403–418. [Google Scholar] [CrossRef] [Green Version]
- Karabay, A.; Yu, W.; Solowska, J.M.; Baird, D.H.; Baas, P.W. Axonal growth is sensitive to the levels of katanin, a protein that severs microtubules. J. Neurosci. 2004, 24, 5778–5788. [Google Scholar] [CrossRef]
- Muroyama, A.; Lechler, T. Microtubule organization, dynamics and functions in differentiated cells. Development 2017, 144, 3012–3021. [Google Scholar] [CrossRef] [Green Version]
- Ori-McKenney, K.M.; Jan, L.Y. Golgi outposts shape dendrite morphology by functioning as sites of acentrosomal microtubule nucleation in neurons. Neuron 2012, 76, 921–930. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Huertas, C.; Freixo, F.; Viais, R.; Lacasa, C.; Soriano, E.; Lüders, J. Non-centrosomal nucleation mediated by augmin organizes microtubules in post-mitotic neurons and controls axonal microtubule polarity. Nat. Commun. 2016, 7, 12187. [Google Scholar] [CrossRef]
- Cunha-Ferreira, I.; Chazeau, A.; Buijs, R.R.; Stucchi, R.; Will, L.; Pan, X.; Adolfs, Y.; van der Meer, C.; Wolthuis, J.C.; Kahn, O.I.; et al. The HAUS complex is a key regulator of non-centrosomal microtubule organization during neuronal development. Cell Rep. 2018, 24, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.C.; Roegiers, F.; Rolls, M.M. Microtubules have opposite orientation in axons and dendrites of Drosophila neurons. Mol. Biol. Cell 2008, 19, 4122–4129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, K.W.; Schätzle, P.; Tortosa, E.; Pagès, S.; Holtmaat, A.; Kapitein, L.C.; Hoogenraad, C.C. Dendrites in vitro and in vivo contain microtubules of opposite polarity and axon formation correlates with uniform plus-end-out microtubule orientation. J. Neurosci. 2016, 36, 1071–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Castillo, U.; Winding, M.; Lu, W.; Gelfand, V.I. Interplay between kinesin-1 and cortical dynein during axonal outgrowth and microtubule organization in Drosophila neurons. eLife 2015, 4, e10140. [Google Scholar] [CrossRef]
- Craig, E.M.; Yeung, H.T.; Rao, A.N.; Baas, P.W. Polarity sorting of axonal microtubules: A computational study. Mol. Biol. Cell 2017, 28, 3271–3285. [Google Scholar] [CrossRef]
- Rao, A.N.; Patil, A.; Black, M.M.; Craig, E.M.; Myers, K.A.; Yeung, H.T.; Baas, P.W. Cytoplasmic dynein transports axonal microtubules in a polarity-sorting manner. Cell Rep. 2017, 19, 2210–2219. [Google Scholar] [CrossRef] [Green Version]
- Sharp, D.J.; Kuriyama, R.; Essner, R.; Baas, P.W. Expression of a minus-end-directed motor protein induces Sf9 cells to form axon-like processes with uniform microtubule polarity orientation. J. Cell Sci. 1997, 110, 2373–2380. [Google Scholar]
- Yu, W.; Sharp, D.J.; Kuriyama, R.; Mallik, P.; Baas, P.W. Inhibition of a mitotic motor compromises the formation of dendrite-like processes from neuroblastoma cells. J. Cell Biol. 1997, 136, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Cook, C.; Sauter, C.; Kuriyama, R.; Kaplan, P.L.; Baas, P.W. Depletion of a microtubule-associated motor protein induces the loss of dendritic identity. J. Neurosci. 2000, 20, 5782–5791. [Google Scholar] [CrossRef] [Green Version]
- Gurel, P.S.; Hatch, A.L.; Higgs, H.N. Connecting the cytoskeleton to the endoplasmic reticulum and Golgi. Curr. Biol. 2014, 24, R660–R672. [Google Scholar] [CrossRef] [Green Version]
- Black, M.M.; Baas, P.W. The basis of polarity in neurons. Trends Neurosci. 1989, 12, 211–214. [Google Scholar] [CrossRef]
- Tanaka, E.M.; Kirschner, M.W. Microtubule behavior in the growth cones of living neurons during axon elongation. J. Cell Biol. 1991, 115, 345–363. [Google Scholar] [CrossRef] [PubMed]
- Geraldo, S.; Gordon-Weeks, P.R. Cytoskeletal dynamics in growth-cone steering. J. Cell Sci. 2009, 122, 3595–3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsetos, C.D.; Legido, A.; Perentes, E.; Mörk, S.J. Class III beta-tubulin isotype: A key cytoskeletal protein at the crossroads of developmental neurobiology and tumor neuropathology. J. Child Neurol. 2004, 19, 531. [Google Scholar]
- Liu, L.; Geisert, E.E.; Frankfurter, A.; Spano, A.J.; Jiang, C.X.; Yue, J.; Dragatsis, I.; Goldowitz, D. A transgenic mouse class-III beta tubulin reporter using yellow fluorescent protein. Genesis 2007, 45, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.Q.; Oblinger, M.M. Differential regulation of beta III and other tubulin genes during peripheral and central neuron development. J. Cell Sci. 1992, 103, 643–651. [Google Scholar]
- Panda, D.; Miller, H.P.; Banerjee, A.; Ludueña, R.F.; Wilson, L. Microtubule dynamics in vitro are regulated by the tubulin isotype composition. Proc. Natl. Acad. Sci. USA 1994, 91, 11358–11362. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Roach, M.C.; Trcka, P.; Ludueña, R.F. Increased microtubule assembly in bovine brain tubulin lacking the type III isotype of beta-tubulin. J. Biol. Chem. 1990, 265, 1794–1799. [Google Scholar]
- Poirier, K.; Saillour, Y.; Bahi-Buisson, N.; Jaglin, X.H.; Fallet-Bianco, C.; Nabbout, R.; Castelnau-Ptakhine, L.; Roubertie, A.; Attie-Bitach, T.; Desguerre, L.; et al. Mutations in the neuronal ß-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum. Mol. Genet. 2010, 19, 4462–4473. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Baris, H.N.; Wu, C.; Rudolph, G.; Van Maldergem, L.; He, W.; Chan, W.M.; Andrews, C.; Demer, J.L.; Robertson, R.L.; et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 2010, 140, 74–87. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.B.; Lee, J.H.; Park, H.J.; Choi, Y.R.; Hyun, Y.S.; Park, J.H.; Koo, H.; Chung, K.W.; Choi, B.O. A family with axonal sensorimotor polyneuropathy with TUBB3 mutation. Mol. Med. Rep. 2015, 11, 2729–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumura, S.; Kato, M.A.; Kawamura, K.; Tsuzuki, A.; Tsutsumi, H. mutation in the tubulin-encoding TUBB3 gene causes complex cortical malformations and unilateral hypohidrosis. Child Neurol. Open. 2016, 3, 2329048X16665758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitman, M.C.; Andrews, C.; Chan, W.M.; Tischfield, M.A.; Stasheff, S.F.; Brancati, F.; Ortiz-Gonzalez, X.; Nuovo, S.; Garaci, F.; MacKinnon, S.E.; et al. Two unique TUBB3 mutations cause both CFEOM3 and malformations of cortical development. Am. J. Med. Genet. A 2016, 170, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallet-Bianco, C.; Laquerrière, A.; Poirier, K.; Razavi, F.; Guimiot, F.; Dias, P.; Loeuillet, L.; Lascelles, K.; Beldjord, C.; Carion, N.; et al. Mutations in tubulin genes are frequent causes of various foetal malformations of cortical development including microlissencephaly. Acta Neuropathol. Commun. 2014, 25, 2–69. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.S. Tubulinopathies and their brain malformation syndromes: Every TUB on its own bottom. Epilepsy Curr. 2015, 15, 65–67. [Google Scholar] [CrossRef]
- Romaniello, R.; Arrigoni, F.; Fry, A.E.; Bassi, M.T.; Rees, M.I.; Borgatti, R.; Pilz, D.T.; Cushion, T.D. Tubulin genes and malformations of cortical development. Eur. J. Med. Genet. 2018, 61, 744–754. [Google Scholar] [CrossRef]
- Breuss, M.; Heng, J.I.; Poirier, K.; Tian, G.; Jaglin, X.H.; Qu, Z.; Braun, A.; Gstrein, T.; Ngo, L.; Haas, M.; et al. Mutations in the β-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2012, 2, 1554–1562. [Google Scholar] [CrossRef] [Green Version]
- Ngo, L.; Haas, M.; Qu, Z.; Li, S.S.; Zenker, J.; Teng, K.S.; Gunnersen, J.M.; Breuss, M.; Habgood, M.; Keays, D.A.; et al. TUBB5 and its disease-associated mutations influence the terminal differentiation and dendritic spine densities of cerebral cortical neurons. Hum. Mol. Genet. 2014, 23, 5147–5158. [Google Scholar] [CrossRef] [Green Version]
- Isrie, M.; Breuss, M.; Tian, G.; Hansen, A.H.; Cristofoli, F.; Morandell, J.; Kupchinsky, Z.A.; Sifrim, A.; Rodriguez-Rodriguez, C.M.; Dapena, E.P. Mutations in either TUBB or MAPRE2 cause circumferential skin creases Kunze type. Am. J. Hum. Genet. 2015, 97, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Dentici, M.L.; Terracciano, A.; Bellacchio, E.; Capolino, R.; Novelli, A.; Digilio, M.C.; Dallapiccola, B. Intrafamiliar clinical variability of circumferential skin creases Kunze type caused by a novel heterozygous mutation of N-terminal TUBB gene. Clin. Genet. 2018, 93, 1223–1228. [Google Scholar] [CrossRef]
- Madrigal, I.; Rabionet, R.; Alvarez-Mora, M.I.; Sanchez, A.; Rodríguez-Revenga, L.; Estivill, X.; Mila, M. Spectrum of clinical heterogeneity of β-tubulin TUBB5 gene mutations. Gene 2019, 695, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Sferra, A.; Petrini, S.; Bellacchio, E.; Nicita, F.; Scibelli, F.; Dentici, M.L.; Alfieri, P.; Cestra, G.; Bertini, E.S.; Zanni, G. TUBB variants underlying different phenotypes result in altered vesicle trafficking and microtubule dynamics. Int. J. Mol. Sci. 2020, 21, 1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuss, M.; Morandell, J.; Nimpf, S.; Gstrein, T.; Lauwers, M.; Hochstoeger, T.; Braun, A.; Chan, K.; Sánchez Guajardo, E.R.; Zhang, L.; et al. The expression of Tubb2b undergoes a developmental transition in murine cortical neurons. J. Comp. Neurol. 2015, 523, 2161–2186. [Google Scholar] [CrossRef] [PubMed]
- Jaglin, X.H.; Poirier, K.; Saillour, Y.; Buhler, E.; Tian, G.; Bahi-Buisson, N.; Fallet-Bianco, C.; Phan-Dinh-Tuy, F.; Kong, X.P.; Bomont, P.; et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat. Genet. 2009, 41, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Cederquist, G.Y.; Luchniak, A.; Tischfield, M.A.; Peeva, M.; Song, Y.; Menezes, M.P.; Chan, W.M.; Andrews, C.; Chew, S.; Jamieson, R.V.; et al. An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation. Hum. Mol. Genet. 2012, 21, 5484–5499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrini, R.; Mei, D.; Cordelli, D.M.; Pucatti, D.; Franzoni, E.; Parrini, E. Symmetric polymicrogyria and pachygyria associated with TUBB2B gene mutations. Eur. J. Hum. Genet. 2012, 20, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, R.; Tonelli, A.; Arrigoni, F.; Baschirotto, C.; Triulzi, F.; Bresolin, N.; Bassi, M.T.; Borgatti, R. A novel mutation in the β-tubulin gene TUBB2B associated with complex malformation of cortical development and deficits in axonal guidance. Dev. Med. Child Neurol. 2012, 54, 765–769. [Google Scholar] [CrossRef]
- Wang, H.; Li, S.; Li, S.; Jiang, N.; Guo, J.; Zhang, W.; Zhong, M.; Xie, J. De novo mutated TUBB2B associated pachygyria diagnosed by medical exome sequencing and long-range PCR. Fetal Pediatr. Pathol. 2019, 38, 63–71. [Google Scholar] [CrossRef]
- Leandro-García, L.J.; Leskelä, S.; Landa, I.; Montero-Conde, C.; López-Jiménez, E.; Letón, R.; Cascón, A.; Robledo, M.; Rodríguez-Antona, C. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton (Hoboken) 2010, 67, 214–223. [Google Scholar] [CrossRef]
- Cushion, T.D.; Paciorkowski, A.R.; Pilz, D.T.; Mullins, J.G.; Seltzer, L.E.; Marion, R.W.; Tuttle, E.; Ghoneim, D.; Christian, S.L.; Chung, S.K.; et al. De novo mutations in the beta-tubulin gene TUBB2A cause simplified gyral patterning and infantile-onset epilepsy. Am. J. Hum. Genet. 2014, 94, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Ejaz, R.; Lionel, A.C.; Blaser, S.; Walker, S.; Scherer, S.W.; Babul-Hirji, R.; Marshall, C.R.; Stavropoulos, D.J.; Chitayat, D. De novo pathogenic variant in TUBB2A presenting with arthrogryposis multiplex congenita, brain abnormalities, and severe developmental delay. Am. J. Med. Genet. A 2017, 173, 2725–2730. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Li, J.; Wu, Y.; Jiang, Y. De novo mutations of TUBB2A cause infantile-onset epilepsy and developmental delay. J. Hum. Genet. 2020, 65, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Sferra, A.; Fattori, F.; Rizza, T.; Flex, E.; Bellacchio, E.; Bruselles, A.; Petrini, S.; Cecchetti, S.; Teson, M.; Restaldi, F.; et al. Defective kinesin binding of TUBB2A causes progressive spastic ataxia syndrome resembling sacsinopathy. Hum. Mol. Genet. 2018, 27, 1892–1904. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Ticozzi, N.; Fallini, C.; Gkazi, A.S.; Topp, S.; Kenna, K.P.; Scotter, E.L.; Kost, J.; Keagle, P.; Miller, J.W.; et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 2014, 84, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hersheson, J.; Mencacci, N.E.; Davis, M.; MacDonald, N.; Trabzuni, D.; Ryten, M.; Pittman, A.; Paudel, R.; Kara, E.; Fawcett, K.; et al. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann. Neurol. 2013, 73, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, K.; Wilcox, R.A.; Winkler, S.; Ramirez, A.; Rakovic, A.; Park, J.S.; Arns, B.; Lohnau, T.; Groen, J.; Kasten, M.; et al. Whispering dysphonia (DYT4 dystonia) is caused by a mutation in the TUBB4 gene. Ann. Neurol. 2013, 73, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.M.; Polder, E.; Vanderver, A.; Naidu, S.; Schiffmann, R.; Fisher, K.; Raguž, A.B.; Blumkin, L.; van Berkel, C.G.; H-ABC Research Group; et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: Further delineation of the phenotype and genotype-phenotype correlation. Brain 2014, 137, 1921–1930. [Google Scholar] [CrossRef]
- Pizzino, A.; Pierson, T.M.; Guo, Y.; Helman, G.; Fortini, S.; Guerrero, K.; Saitta, S.; Murphy, J.L.; Padiath, Q.; Xie, Y. TUBB4A de novo mutations cause isolated hypomyelination. Neurology 2014, 83, 898–902. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Ondo, Y.; Shimojima, K.; Osaka, H.; Yamamoto, T. A novel TUBB4A mutation G96R identified in a patient with hypomyelinating leukodystrophy onset beyond adolescence. Hum. Genome Var. 2017, 4, 17035. [Google Scholar] [CrossRef] [Green Version]
- Kancheva, D.; Chamova, T.; Guergueltcheva, V.; Mitev, V.; Azmanov, D.N.; Kalaydjieva, L.; Tournev, I.; Jordanova, A. Mosaic dominant TUBB4A mutation in an inbred family with complicated hereditary spastic paraplegia. Mov. Disord. 2015, 30, 854–858. [Google Scholar] [CrossRef]
- Nicita, F.; Bertini, E.; Travaglini, L.; Armando, M.; Aiello, C. Congenital-onset spastic paraplegia in a patient with TUBB4A mutation and mild hypomyelination. J. Neurol. Sci. 2016, 368, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Sagnelli, A.; Magri, S.; Farina, L.; Chiapparini, L.; Marotta, G.; Tonduti, D.; Consonni, M.; Scigliuolo, G.M.; Benti, R.; Pareyson, D. Early-onset progressive spastic paraplegia caused by a novel TUBB4A mutation: Brain MRI and FDG-PET findings. J. Neurol. 2016, 263, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Poirier, K.; Fourniol, F.; Saillour, Y.; Valence, S.; Lebrun, N.; Hully, M.; Bianco, C.F.; Boddaert, N.; Elie, C.; et al. The wide spectrum of tubulinopathies: What are the key features for the diagnosis? Brain 2014, 137, 1676–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiken, J.; Buscaglia, G.; Bates, E.A.; Moore, J.K. The α-Tubulin gene TUBA1A in brain development: A key ingredient in the neuronal isotype blend. J. Dev. Biol. 2017, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Lewis, S.A.; Lee, M.G.; Cowan, N.J. Five mouse tubulin isotypes and their regulated expression during development. J. Cell Biol. 1985, 101, 852–861. [Google Scholar] [CrossRef] [Green Version]
- Miller, F.D.; Naus, C.C.; Durand, M.; Bloom, F.E.; Milner, R.J. Isotypes of alpha-tubulin are differentially regulated during neuronal maturation. J. Cell Biol. 1987, 105, 3065–3073. [Google Scholar] [CrossRef]
- Keays, D.A.; Tian, G.; Poirier, K.; Huang, G.J.; Siebold, C.; Cleak, J.; Oliver, P.L.; Fray, M.; Harvey, R.J.; Molnár, Z. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 2007, 128, 45–57. [Google Scholar] [CrossRef]
- Poirier, K.; Keays, D.A.; Francis, F.; Saillour, Y.; Bahi, N.; Manouvrier, S.; Fallet-Bianco, C.; Pasquier, L.; Toutain, A.; Tuy, F.P.; et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum. Mutat. 2007, 28, 1055–1064. [Google Scholar] [CrossRef]
- Jansen, A.C.; Oostra, A.; Desprechins, B.; De Vlaeminck, Y.; Verhelst, H.; Régal, L.; Verloo, P.; Bockaert, N.; Keymolen, K.; Seneca, S. TUBA1A mutations: From isolated lissencephaly to familial polymicrogyria. Neurology 2011, 76, 988–992. [Google Scholar] [CrossRef]
- Zanni, G.; Colafati, G.S.; Barresi, S.; Randisi, F.; Talamanca, L.F.; Genovese, E.; Bellacchio, E.; Bartuli, A.; Bernardi, B.; Bertini, E. Description of a novel TUBA1A mutation in Arg-390 associated with asymmetrical polymicrogyria and mid-hindbrain dysgenesis. Eur. J. Paediatr. Neurol. 2013, 17, 361–365. [Google Scholar] [CrossRef]
- Poirier, K.; Saillour, Y.; Fourniol, F.; Francis, F.; Souville, I.; Valence, S.; Desguerre, I.; Marie Lepage, J.; Boddaert, N.; Line Jacquemont, M.; et al. Expanding the spectrum of TUBA1A-related cortical dysgenesis to Polymicrogyria. Eur. J. Hum. Genet. 2013, 21, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Okumura, A.; Hayashi, M.; Tsurui, H.; Yamakawa, Y.; Abe, S.; Kudo, T.; Suzuki, R.; Shimizu, T.; Shimojima, K.; Yamamoto, T. Lissencephaly with marked ventricular dilation, agenesis of corpus callosum, and cerebellar hypoplasia caused by TUBA1A mutation. Brain Dev. 2013, 35, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Aiken, J.; Moore, J.K.; Bates, E.A. TUBA1A mutations identified in lissencephaly patients dominantly disrupt neuronal migration and impair dynein activity. Hum. Mol. Genet. 2019, 28, 1227–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanchi, F.; Corso, V.; Scannapieco, P.; Ievolella, C.; Negrisolo, E.; Tiso, N.; Lanfranchi, G.; Valle, G. TUBA8: A new tissue-specific isoform of alpha-tubulin that is highly conserved in human and mouse. Biochem. Biophys. Res. Commun. 2000, 270, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, M.R.; Morrison, E.; Sirey, T.; Molnar, Z.; Hayward, B.E.; Carr, I.M.; Springell, K.; Woods, C.G.; Ahmed, M.; Hattingh, L.; et al. Mutation of the variant alpha-tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am. J. Hum. Genet. 2009, 85, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Howes, S.C.; Alushin, G.M.; Shida, T.; Nachury, M.V.; Nogales, E. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol. Biol. Cell 2014, 25, 257–266. [Google Scholar] [CrossRef]
- Skoge, R.H.; Ziegler, M. SIRT2 inactivation reveals a subset of hyperacetylated perinuclear microtubules inaccessible to HDAC6. J. Cell Sci. 2016, 129, 2972–2982. [Google Scholar] [CrossRef] [Green Version]
- Magiera, M.M.; Singh, P.; Gadadhar, S.; Janke, C. Tubulin posttranslational modifications and emerging links to human disease. Cell 2018, 173, 1323–1327. [Google Scholar] [CrossRef] [Green Version]
- Szyk, A.; Deaconescu, A.M.; Spector, J.; Goodman, B.; Valenstein, M.L.; Ziolkowska, N.E.; Kormendi, V.; Grigorieff, N.; Roll-Mecak, A. Molecular basis for age-dependent microtubule acetylation by tubulin acetyltransferase. Cell 2014, 157, 1405–1415. [Google Scholar] [CrossRef] [Green Version]
- Skultetyova, L.; Ustinova, K.; Kutil, Z.; Novakova, Z.; Pavlicek, J.; Mikesova, J.; Trapl, D.; Baranova, P.; Havlinova, B.; Hubalek, M.; et al. Human histone deacetylase 6 shows strong preference for tubulin dimers over assembled microtubules. Sci. Rep. 2017, 7, 11547. [Google Scholar] [CrossRef] [Green Version]
- Coombes, C.; Yamamoto, A.; McClellan, M.; Reid, T.A.; Plooster, M.; Luxton, G.W.; Alper, J.; Howard, J.; Gardner, M.K. Mechanism of microtubule lumen entry for the alpha-tubulin acetyltransferase enzyme alphaTAT1. Proc. Natl. Acad. Sci. USA 2016, 113, E7176–E7184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ly, N.; Elkhatib, N.; Bresteau, E.; Piétrement, O.; Khaled, M.; Magiera, M.M.; Janke, C.; Le Cam, E.; Rutenberg, A.D.; Montagnac, G. αTAT1 controls longitudinal spreading of acetylation marks from open microtubules extremities. Sci. Rep. 2016, 6, 35624. [Google Scholar] [CrossRef] [PubMed]
- Janke, C.; Montagnac, G. Causes and Consequences of Microtubule Acetylation. Curr. Biol. 2017, 27, R1287–R1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portran, D.; Schaedel, L.; Xu, Z.; Théry, M.; Nachury, M.V. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat. Cell Biol. 2017, 19, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.S.; Davis, A.A.; Culver, D.G.; Glass, J.D. WldS mice are resistant to paclitaxel (taxol) neuropathy. Ann. Neurol. 2002, 52, 442–447. [Google Scholar] [CrossRef]
- Suzuki, K.; Koike, T. Mammalian Sir2-related protein (SIRT) 2-mediated modulation of resistance to axonal degeneration in slow Wallerian degeneration mice: A crucial role of tubulin deacetylation. Neuroscience 2007, 147, 599–612. [Google Scholar] [CrossRef]
- Ahmad, F.J.; Pienkowski, T.P.; Baas, P.W. Regional differences in microtubule dynamics in the axon. J. Neurosci. 1993, 13, 856–866. [Google Scholar] [CrossRef]
- Song, Y.; Brady, S.T. Post-translational modifications of tubulin: Pathways to functional diversity of microtubules. Trends Cell Biol. 2015, 25, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [Green Version]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelières, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef]
- Bulinski, J.C. Microtubule modification: Acetylation speeds anterograde traffic flow. Curr. Biol. 2007, 17, R18–R20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balabanian, L.; Berger, C.L.; Hendricks, A.G. Acetylated microtubules are preferentially bundled leading to enhanced kinesin-1 motility. Biophys. J. 2017, 113, 1551–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrieux, A.; Aubry, L.; Boscheron, C. CAP-Gly proteins contribute to microtubule-dependent trafficking via interactions with the C-terminal aromatic residue of α-tubulin. Small GTPases 2019, 10, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wloga, D.; Joachimiak, E.; Fabczak, H. Tubulin post-translational modifications and microtubule dynamics. Int. J. Mol. Sci. 2017, 18, 2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, I.; Garnham, C.P.; Roll-Mecak, A. Writing and reading the tubulin code. J. Biol. Chem. 2015, 290, 17163–17172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, Y.; Kurabe, N.; Ikegami, K.; Tsutsumi, K.; Konishi, Y.; Kaplan, O.I.; Kunitomo, H.; Iino, Y.; Blacque, O.E.; Setou, M. Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs). J. Biol. Chem. 2010, 285, 22936–22941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogowski, K.; van Dijk, J.; Magiera, M.M.; Bosc, C.; Deloulme, J.C.; Bosson, A.; Peris, L.; Gold, N.D.; Lacroix, B.; Bosch Grau, M. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 2010, 143, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Tort, O.; Tanco, S.; Rocha, C.; Bièche, I.; Seixas, C.; Bosc, C.; Andrieux, A.; Moutin, M.J.; Avilés, F.X.; Lorenzo, J. The cytosolic carboxypeptidases CCP2 and CCP3 catalyze posttranslational removal of acidic amino acids. Mol. Biol. Cell. 2014, 25, 3017–3027. [Google Scholar] [CrossRef]
- Gundersen, G.G.; Kalnoski, M.H.; Bulinski, J.C. Distinct populations of microtubules: Tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell 1984, 38, 779–789. [Google Scholar] [CrossRef]
- Geuens, G.; Gundersen, G.G.; Nuydens, R.; Cornelissen, F.; Bulinski, J.C.; DeBrabander, M. Ultrastructural colocalization of tyrosinated and detyrosinated alpha-tubulin in interphase and mitotic cells. J. Cell Biol. 1986, 103, 883–1893. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.; Li, Y.; Black, M.M. Composite microtubules of the axon: Quantitative analysis of tyrosinated and acetylated tubulin along individual axonal microtubules. J. Cell Sci. 1993, 104, 339–352. [Google Scholar] [PubMed]
- Nogales, E. Structural insights into microtubule function. Annu. Rev. Biochem. 2000, 69, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Devambez, I.; van Dijk, J.; Benlefki, S.; Layalle, S.; Grau, Y.; Rogowski, K.; Parmentier, M.L.; Soustelle, L. Identification of DmTTLL5 as a major tubulin glutamylase in the drosophila nervous system. Sci. Rep. 2017, 7, 16254. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, J.; Miro, J.; Strub, J.M.; Lacroix, B.; van Dorsselaer, A.; Edde, B.; Janke, C. Polyglutamylation is a post-translational modification with a broad range of substrates. J. Biol. Chem. 2008, 283, 3915–3922. [Google Scholar] [CrossRef] [Green Version]
- Van der Laan, S.; Dubra, G.; Rogowski, K. Tubulin glutamylation: A skeleton key for neurodegenerative diseases. Neural Regen. Res. 2019, 14, 1899–1900. [Google Scholar] [PubMed]
- Lacroix, B.; van Dijk, J.; Gold, N.D.; Guizetti, J.; Aldrian-Herrada, G.; Rogowski, K.; Gerlich, D.W.; Janke, C. Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J. Cell Biol. 2010, 189, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Valenstein, M.L.; Roll-Mecak, A. Graded control of microtubule severing by tubulin glutamylation. Cell 2016, 164, 911–921. [Google Scholar] [CrossRef] [Green Version]
- Boucher, D.; Larcher, J.C.; Gros, F.; Denoulet, P. Polyglutamylation of tubulin as a progressive regulator of in vitro interactions between the microtubule-associated protein Tau and tubulin. Biochemistry 1994, 33, 12471–12477. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Boucher, D.; Lazereg, S.; Pedrotti, B.; Islam, K.; Denoulet, P.; Larcher, J.C. Differential binding regulation of microtubule-associated proteins MAP1A, MAP1B, and MAP2 by tubulin polyglutamylation. J. Biol. Chem. 2001, 276, 12839–12848. [Google Scholar] [CrossRef] [Green Version]
- Short, B. Polyglutamylation makes the cut. J. Cell Biol. 2010, 189, 920. [Google Scholar] [CrossRef] [Green Version]
- Audebert, S.; Koulakoff, A.; Berwald-Netter, Y.; Gros, F.; Denoulet, P.; Eddé, B. Developmental regulation of polyglutamylated alpha- and beta-tubulin in mouse brain neurons. J. Cell Sci. 1994, 107, 2313–2322. [Google Scholar] [PubMed]
- Ikegami, K.; Mukai, M.; Tsuchida, J.; Heier, R.L.; Macgregor, G.R.; Setou, M. TTLL7 is a mammalian beta-tubulin polyglutamylase required for growth of MAP2-positive neurites. J. Biol. Chem. 2006, 281, 30707–30716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, A.; Rechtsteiner, A.; Saxton, W.M. Two kinesins drive anterograde neuropeptide transport. Mol. Biol. Cell 2017, 28, 3542–3553. [Google Scholar] [CrossRef] [PubMed]
- Van de Willige, D.; Hoogenraad, C.C.; Akhmanova, A. Microtubule plus-end tracking proteins in neuronal development. Cell Mol. Life Sci. 2016, 73, 2053–2077. [Google Scholar] [CrossRef] [Green Version]
- Akhmanova, A.; Steinmetz, M.O. Microtubule minus-end regulation at a glance. J. Cell Sci. 2019, 132, jcs227850. [Google Scholar] [CrossRef] [Green Version]
- Kollman, J.M.; Merdes, A.; Mourey, L.; Agard, D.A. Microtubule nucleation by γ-tubulin complexes. Nat. Rev. Mol. Cell Biol. 2011, 12, 709–721. [Google Scholar] [CrossRef]
- Kodani, A.; Salome Sirerol-Piquer, M.; Seol, A.; Garcia-Verdugo, J.M.; Reiter, J.F. Kif3a interacts with Dynactin subunit p150 Glued to organize centriole subdistal appendages. EMBO J. 2013, 32, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, H.; Sakayori, N.; Takahashi, M.; Osumi, N. Ninein is essential for the maintenance of the cortical progenitor character by anchoring the centrosome to microtubules. Biol. Open 2013, 2, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Gupton, S.L. Building blocks of functioning brain: Cytoskeletal dynamics in neuronal development. Int. Rev. Cell Mol. Biol. 2016, 322, 183–245. [Google Scholar]
- Matus, A. Microtubule-associated proteins and neuronal morphogenesis. J. Cell Sci. Suppl. 1991, 15, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, S.; Harada, A.; Hirokawa, N. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci. Lett. 2000, 279, 129–132. [Google Scholar] [CrossRef]
- Dawson, H.N.; Ferreira, A.; Eyster, M.V.; Ghoshal, N.; Binder, L.I.; Vitek, M.P. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 2001, 114, 1179–1187. [Google Scholar] [PubMed]
- Andrieux, A.; Salin, P.A.; Vernet, M.; Kujala, P.; Baratier, J.; Gory-Faure, S.; Bosc, C.; Pointu, H.; Proietto, D.; Schweitzer, A.; et al. The suppression of brain cold-stable microtubules in mice induces synaptic defects associated with neuroleptic-sensitive behavioral disorders. Genes Dev. 2002, 16, 2350–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khuchua, Z.; Wozniak, D.F.; Bardgett, M.E.; Yue, Z.; McDonald, M.; Boero, J.; Hartman, R.E.; Sims, H.; Strauss, A.W. Deletion of the N-terminus of murine map2 by gene targeting disrupts hippocampal ca1 neuron architecture and alters contextual memory. Neuroscience 2003, 119, 101–111. [Google Scholar] [CrossRef]
- Benoist, M.; Palenzuela, R.; Rozas, C.; Rojas, P.; Tortosa, E.; Morales, B.; González-Billault, C.; Ávila, J.; Esteban, J.A. MAP1B-dependent Rac activation is required for AMPA receptor endocytosis during long-term depression. EMBO J. 2013, 32, 2287–2299. [Google Scholar] [CrossRef] [Green Version]
- Takei, Y.; Kikkawa, Y.S.; Atapour, N.; Hensch, T.K.; Hirokawa, N. Defects in synaptic plasticity, reduced NMDA-receptor transport, and instability of postsynaptic density proteins in mice lacking microtubule-associated protein 1A. J. Neurosci. 2015, 35, 15539–15554. [Google Scholar] [CrossRef] [Green Version]
- Melková, K.; Zapletal, V.; Jansen, S.; Nomilner, E.; Zachrdla, M.; Hritz, J.; Nováček, J.; Zweckstetter, M.; Jensen, M.R.; Blackledge, M. Functionally specific binding regions of microtubule-associated protein 2c exhibit distinct conformations and dynamics. J. Biol. Chem. 2018, 293, 13297–13309. [Google Scholar] [CrossRef] [Green Version]
- Melková, K.; Zapletal, V.; Narasimhan, S.; Jansen, S.; Hritz, J.; Škrabana, R.; Zweckstetter, M.; Ringkjøbing Jensen, M.; Blackledge, M.; Žídek, L. Structure and functions of microtubule associated proteins tau and MAP2c: Similarities and differences. Biomolecules 2019, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [Green Version]
- Garner, C.C.; Brugg, B.; Matus, A. A 70-kilodalton microtubule-associated protein (MAP2c), related to MAP2. J. Neurochem. 1988, 50, 609–615. [Google Scholar] [CrossRef]
- Tucker, R.P.; Binder, L.I.; Viereck, C.; Hemmings, B.A.; Matus, A.I. The sequential appearance of low- and high-molecular-weight forms of MAP2 in the developing cerebellum. J. Neurosci. 1988, 8, 4503–4512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Crowther, R.A.; Garner, C.C. Molecular characterization of microtubule-associated proteins tau and MAP2. Trends Neurosci. 1991, 14, 193–199. [Google Scholar] [CrossRef]
- Przyborski, S.A.; Cambray-Deakin, M.A. Developmental regulation of MAP2 variants during neuronal differentiation in vitro. Brain Res. Dev. Brain Res. 1995, 89, 187–201. [Google Scholar] [CrossRef]
- Hartel, R.; Matus, A. Cytoskeletal maturation in cultured hippocampal slices. Neuroscience 1997, 78, 1–5. [Google Scholar]
- Ferhat, L.; Represa, A.; Ferhat, W.; Ben-Ari, Y.; Khrestchatisky, M. MAP2d mRNA is expressed in identified neuronal populations in the developing and adult rat brain and its subcellular distribution differs from that of MAP2b in hippocampal neurones. Eur. J. Neurosci. 1998, 10, 161–171. [Google Scholar] [CrossRef]
- Bullmann, T.; Härtig, W.; Holzer, M.; Arendt, T. Expression of the embryonal isoform (0N/3R) of the microtubule-associated protein tau in the adult rat central nervous system. J. Comp. Neurol. 2010, 518, 2538–2553. [Google Scholar] [CrossRef]
- Liu, C.; Götz, J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS ONE 2013, 8, e84849. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Spillantini, M.G.; Potier, M.C.; Ulrich, J.; Crowther, R.A. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [CrossRef]
- Kosik, K.S.; Orecchio, L.D.; Bakalis, S.; Neve, R.L. Developmentally regulated expression of specific tau sequences. Neuron 1989, 2, 1389–1397. [Google Scholar] [CrossRef]
- Al-Bassam, J.; Ozer, R.S.; Safer, D.; Halpain, S.; Milligan, R.A. MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J. Cell Biol. 2002, 157, 1187–1196. [Google Scholar] [CrossRef]
- Heidary, G.; Fortini, M.E. Identification and characterization of the Drosophila tau homolog. Mech. Dev. 2001, 108, 171–178. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Kutter, S.; Eichner, T.; Deaconescu, A.M.; Kern, D. Regulation of Microtubule Assembly by Tau and not by Pin1. Mol. Biol. 2016, 428, 1742–1759. [Google Scholar] [CrossRef]
- Panda, D.; Samuel, J.C.; Massie, M.; Feinstein, S.C.; Wilson, L. Differential regulation of microtubule dynamics by three- and four-repeat tau: Implications for the onset of neurodegenerative disease. Proc. Natl. Acad. Sci. USA 2003, 100, 9548–9553. [Google Scholar] [CrossRef] [Green Version]
- Panda, D.; Miller, H.P.; Wilson, L. Rapid treadmilling of brain microtubules free of microtubule-associated proteins in vitro and its suppression by tau. Proc. Natl. Acad. Sci. USA 1999, 96, 12459–12464. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Kanai, Y.; Cowan, N.J.; Hirokawa, N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 1992, 360, 674–677. [Google Scholar] [CrossRef]
- Rosenberg, K.J.; Ross, J.L.; Feinstein, H.E.; Feinstein, S.C.; Israelachvili, J. Complementary dimerization of microtubule-associated tau protein: Implications for microtubule bundling and tau-mediated pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 7445–7450. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.C.; Raviv, U.; Miller, H.P.; Gaylord, M.R.; Kiris, E.; Ventimiglia, D.; Needleman, D.J.; Kim, M.W.; Wilson, L.; Feinstein, S.C.; et al. Human microtubule-associated-protein tau regulates the number of protofilaments in microtubules: A synchrotron x-ray scattering study. Biophys. J. 2009, 97, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Hirokawa, N.; Funakoshi, T.; Sato-Harada, R.; Kanai, Y. Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J. Cell Biol. 1996, 132, 667–679. [Google Scholar] [CrossRef]
- Shiomura, Y.; Hirokawa, N. Colocalization of microtubule-associated protein 1A and microtubule-associated protein 2 on neuronal microtubules in situ revealed with double-label immunoelectron microscopy. J. Cell Biol. 1987, 104, 1575–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirokawa, N.; Hisanaga, S.; Shiomura, Y. MAP2 is a component of crossbridges between microtubules and neurofilaments in the neuronal cytoskeleton: Quick-freeze, deep-etch immunoelectron microscopy and reconstitution studies. J. Neurosci. 1988, 8, 2769–2779. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, R.J.; Williams, R.C. Microtubule-associated protein 2 alters the dynamic properties of microtubule assembly and disassembly. J. Biol. Chem. 1993, 268, 9847–9855. [Google Scholar]
- Yamauchi, P.S.; Flynn, G.C.; Marsh, R.L.; Purich, D.L. Reduction in microtubule dynamics in vitro by brain microtubule-associated proteins and by a microtubule-associated protein-2 s repeated sequence analogue. J. Neurochem. 1993, 60, 817–826. [Google Scholar] [CrossRef]
- Vandecandelaere, A.; Pedrotti, B.; Utton, M.A.; Calvert, R.A.; Bayley, P.M. Differences in the regulation of microtubule dynamics by microtubule-associated proteins MAP1B and MAP2. Cell Motil. Cytoskelet. 1996, 35, 134–146. [Google Scholar] [CrossRef]
- Kalcheva, N.; Rockwood, J.M.; Kress, Y.; Steiner, A.; Shafit-Zagardo, B. Molecular and functional characteristics of MAP-2a: Ability of MAP-2a versus MAP-2b to induce stable microtubules in COS cells. Cell Motil. Cytoskelet. 1998, 40, 272–285. [Google Scholar] [CrossRef]
- Dinsmore, J.H.; Solomon, F. Inhibition of MAP2 expression affects both morphological and cell division phenotypes of neuronal differentiation. Cell 1991, 64, 817–826. [Google Scholar] [CrossRef]
- Teng, J.; Takei, Y.; Harada, A.; Nakata, T.; Chen, J.; Hirokawa, N. Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J. Cell Biol. 2001, 155, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Harada, A.; Teng, J.; Takei, Y.; Oguchi, K.; Hirokawa, N. MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J. Cell Biol. 2002, 158, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Simons, C.; Wolf, N.I.; McNeil, N.; Caldovic, L.; Devaney, J.M.; Takanohashi, A.; Crawford, J.; Ru, K.; Grimmond, S.M.; Miller, D.; et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am. J. Hum. Genet. 2013, 92, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Tonduti, D.; Aiello, C.; Renaldo, F.; Dorboz, I.; Saaman, S.; Rodriguez, D.; Fettah, H.; Elmaleh, M.; Biancheri, R.; Barresi, S.; et al. TUBB4A-related hypomyelinating leukodystrophy: New insights from a series of 12 patients. Eur. J. Paediatr. Neurol. 2016, 20, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Niwa, S.; Takahashi, H.; Hirokawa, N. β-Tubulin mutations that cause severe neuropathies disrupt axonal transport. EMBO J. 2013, 32, 1352–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, I.; Pae, C.I.; Hallows, J.L. The cell cycle and human neurodegenerative disease. Prog. Cell Cycle Res. 2003, 5, 31–41. [Google Scholar] [PubMed]
- Thomas, P.; Fenech, M. Chromosome 17 and 21 aneuploidy in buccal cells is increased with ageing and in Alzheimer’s disease. Mutagenesis 2008, 23, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Faggioli, F.; Vijg, J.; Montagna, C. Chromosomal aneuploidy in the aging brain. Mech. Ageing Dev. 2011, 132, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Granic, A.; Potter, H. Mitotic spindle defects and chromosome mis-segregation induced by LDL/cholesterol-implications for Niemann-Pick C1, Alzheimer’s disease, and atherosclerosis. PLoS ONE 2013, 8, e60718. [Google Scholar] [CrossRef] [Green Version]
- Oromendia, A.B.; Amon, A. Aneuploidy: Implications for protein homeostasis and disease. Dis. Models Mech. 2014, 7, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Arendt, T.; Brückner, M.K.; Mosch, B.; Lösche, A. Selective cell death of hyperploid neurons in Alzheimer’s disease. Am. J. Pathol. 2010, 177, 15–20. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Iourov, I.Y. X chromosome aneuploidy in the Alzheimer’s disease brain. Mol. Cytogenet. 2014, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Blumkin, L.; Halevy, A.; Ben-Ami-Raichman, D.; Dahari, D.; Haviv, A.; Sarit, C.; Lev, D.; van der Knaap, M.S.; Lerman-Sagie, T.; Leshinsky-Silver, E. Expansion of the spectrum of TUBB4A-related disorders: A new phenotype associated with a novel mutation in the TUBB4A gene. Neurogenetics 2014, 15, 115. [Google Scholar]
- Arai-Ichinoi, N.; Uematsu, M.; Sato, R.; Suzuki, T.; Kudo, H.; Kikuchi, A.; Hino-Fukuyo, N.; Matsumoto, M.; Igarashi, K.; Haginoya, K.; et al. Genetic heterogeneity in 26 infants with a hypomyelinating leukodystrophy. Hum. Genet. 2016, 135, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Osaka, H.; Shiina, M.; Sasaki, M.; Takanashi, J.; Haginoya, K.; Wada, T.; Morimoto, M.; Ando, N.; Ikuta, Y.; et al. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology 2014, 82, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Purnell, S.M.; Bleyl, S.B.; Bonkowsky, J.L. Clinical exome sequencing identifies a novel TUBB4A mutation in a child with static hypomyelinating leukodystrophy. Pediatr. Neurol. 2014, 50, 608–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyal, K.M.; Michaud, J.; van der Knaap, M.S.; Bugiani, M.; Venkateswaran, S. Severe TUBB4A-related hypomyelination with atrophy of the basal ganglia and cerebellum: Novel neuropathological findings. J. Neuropathol. Exp. Neurol. 2019, 78, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Li, D.; Wu, Y.; Zhang, Q.; Gu, Q.; Xie, H.; Ji, T.; Wang, H.; Zhao, L.; Zhao, H. Hypomyelinating disorders in China: The clinical and genetic heterogeneity in 119 patients. PLoS ONE 2018, 13, e0188869. [Google Scholar] [CrossRef]
- Carvalho, D.; Santos, S.; Martins, B.; Marques, F.P. TUBB4A novel mutation reinforces the genotype-phenotype correlation of hypomyelination with atrophy of the basal ganglia and cerebellum. Brain 2015, 138, e327. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, C.; Poretti, A.; Cohen, J.; Hamosh, A.; Naidu, S. Novel TUBB4A mutations and expansion of the neuroimaging phenotype of hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC). Am. J. Med. Genet. A 2014, 164, 1802–1807. [Google Scholar] [CrossRef]
- Curiel, J.; Rodríguez Bey, G.; Takanohashi, A.; Bugiani, M.; Fu, X.; Wolf, N.I.; Nmezi, B.; Schiffmann, R.; Bugaighis, M.; Pierson, T.; et al. TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes. Hum. Mol. Genet. 2017, 26, 4506–4518. [Google Scholar] [CrossRef] [Green Version]
- Sase, S.; Almad, A.A.; Boecker, C.A.; Guedes-Dias, P.; Li, J.J.; Takanohashi, A.; Patel, A.; McCaffrey, T.; Patel, H.; Sirdeshpande, D.; et al. TUBB4A mutations result in both glial and neuronal degeneration in an H-ABC leukodystrophy mouse model. eLife 2020, 9, e52986. [Google Scholar] [CrossRef]
- Vulinovic, F.; Krajka, V.; Hausrat, T.J.; Seibler, P.; Alvarez-Fischer, D.; Madoev, H.; Park, J.S.; Kumar, K.R.; Sue, C.M.; Lohmann, K.; et al. Motor protein binding and mitochondrial transport are altered by pathogenic TUBB4A variants. Hum. Mutat. 2019, 40, 2444. [Google Scholar] [CrossRef]
- Yen, T.J.; Machlin, P.S.; Cleveland, D.W. Autoregulated instability of beta-tubulin mRNAs by recognition of the nascent amino terminus of beta-tubulin. Nature 1988, 334, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Itakaoka, M.; Seki, Y.; Morimoto, T.; Homma, K.; Miyamoto, Y.; Yamauchi, J. Dystonia-4 (DYT4)-associated TUBB4A mutants exhibit disorganized microtubule networks and inhibit neuronal process growth. Biochem. Biophys. Res. Commun. 2018, 495, 346–352. [Google Scholar] [CrossRef] [PubMed]
- D’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Vanden Berghe, P.; Timmerman, V.; Robberecht, W.; Van Den Bosch, L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Taes, I.; Timmers, M.; Hersmus, N.; Bento-Abreu, A.; Van Den Bosch, L.; Van Damme, P.; Auwerx, J.; Robberecht, W. Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum. Mol. Genet. 2013, 22, 1783–1790. [Google Scholar] [CrossRef]
- Godena, V.K.; Brookes-Hocking, N.; Moller, A.; Shaw, G.; Oswald, M.; Sancho, R.M.; Miller, C.C.; Whitworth, A.J.; De Vos, K.J. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 2014, 5, 5245. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.W.; Li, L.; You, L.; Yang, X.J. Mice lacking α-tubulin acetyltransferase 1 are viable but display α-tubulin acetylation deficiency and dentate gyrus distortion. J. Biol. Chem. 2016, 291, 25279. [Google Scholar] [CrossRef] [Green Version]
- Hempen, B.; Brion, J.P. Reduction of acetylated alpha-tubulin immunoreactivity in neurofibrillary tangle-bearing neurons in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 964–972. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Dolan, P.J.; Johnson, G.V. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schlüter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef]
- Simões-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Zhao, K.; Wu, J.; Xu, Z.; Jin, S.; Zhang, Y.Q. HDAC6 mutations rescue human tau-induced microtubule defects in Drosophila. Proc. Natl. Acad. Sci. USA 2013, 110, 4604–4609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, S.; Sato, S.; Fukuda, T.; Tada, N.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of Parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice. Neurobiol. Dis. 2020, 136, 104717. [Google Scholar] [CrossRef] [PubMed]
- Cartelli, D.; Amadeo, A.; Calogero, A.M.; Casagrande, F.V.M.; De Gregorio, C.; Gioria, M.; Kuzumaki, N.; Costa, I.; Sassone, J.; Ciammola, A.; et al. Parkin absence accelerates microtubule aging in dopaminergic neurons. Neurobiol. Aging 2018, 61, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Berezniuk, I.; Sironi, J.; Callaway, M.B.; Castro, L.M.; Hirata, I.Y.; Ferro, E.S.; Fricker, L.D. CCP1/Nna1 functions in protein turnover in mouse brain: Implications for cell death in Purkinje cell degeneration mice. FASEB J. 2010, 24, 1813–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magiera, M.M.; Bodakuntla, S.; Žiak, J.; Lacomme, S.; Marques Sousa, P.; Leboucher, S.; Hausrat, T.J.; Bosc, C.; Andrieux, A.; Kneussel, M.; et al. Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J. 2018, 37, e100440. [Google Scholar] [CrossRef]
- Gilmore-Hall, S.; Kuo, J.; Ward, J.M.; Zahra, R.; Morrison, R.S.; Perkins, G.; La Spada, A.R. CCP1 promotes mitochondrial fusion and motility to prevent Purkinje cell neuron loss in pcd mice. J. Cell Biol. 2019, 218, 206–219. [Google Scholar] [CrossRef] [Green Version]
- Bodakuntla, S.; Schnitzler, A.; Villablanca, C.; Gonzalez-Billault, C.; Bieche, I.; Janke, C.; Magiera, M.M. Tubulin polyglutamylation is a general traffic-control mechanism in hippocampal neurons. J. Cell Sci. 2020, 133, jcs241802. [Google Scholar] [CrossRef]
- Shashi, V.; Magiera, M.M.; Klein, D.; Zaki, M.; Schoch, K.; Rudnik-Schöneborn, S.; Norman, A.; Lopes Abath Neto, O.; Dusl, M.; Yuan, X. Loss of tubulin deglutamylase CCP1 causes infantile-onset neurodegeneration. EMBO J. 2018, 37, e100540. [Google Scholar] [CrossRef]
- Vu, H.T.; Akatsu, H.; Setou, M.; Ikegami, K. Increase in α-tubulin modifications in the neuronal processes of hippocampal neurons in both kainic acid-induced epileptic seizure and Alzheimer’s disease. Sci. Rep. 2017, 7, 40205. [Google Scholar] [CrossRef] [Green Version]
- Hoffner, G.; Kahlem, P.; Djian, P. Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with beta-tubulin: Relevance to Huntington’s disease. J. Cell Sci. 2002, 115, 941–948. [Google Scholar]
- Zhang, J.; Dong, X.P. Dysfunction of microtubule-associated proteins of MAP2/tau family in Prion disease. Prion 2012, 6, 334–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, L.; Wetzel, A.; Grannó, S.; Heaton, G.; Harvey, K. Back to the tubule: Microtubule dynamics in Parkinson’s disease. Cell Mol. Life Sci. 2017, 74, 409–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.Z.; Liu, F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog. Neurobiol. 2008, 85, 148–175. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 1963, 197, 192–193. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 1993, 10, 1089–1099. [Google Scholar] [CrossRef]
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef] [Green Version]
- Moon, M.; Hong, H.S.; Nam, D.W.; Baik, S.H.; Song, H.; Kook, S.Y.; Kim, Y.S.; Lee, J.; Mook-Jung, I. Intracellular amyloid-β accumulation in calcium-binding protein-deficient neurons leads to amyloid-β plaque formation in animal model of Alzheimer’s disease. J. Alzheimers Dis. 2012, 29, 615–628. [Google Scholar] [CrossRef]
- Fath, T.; Eidenmüller, J.; Brandt, R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer’s disease. J. Neurosci. 2002, 22, 9733–9741. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 2209. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, M.; Dawson, H.N.; Binder, L.I.; Vitek, M.P.; Ferreira, A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 6364–6369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zempel, H.; Luedtke, J.; Kumar, Y.; Biernat, J.; Dawson, H.; Mandelkow, E.; Mandelkow, E.M. Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013, 32, 2920–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, D.; Goode, B.L.; Feinstein, S.C.; Wilson, L. Kinetic stabilization of microtubule dynamics at steady state by tau and microtubule-binding domains of tau. Biochemistry 1995, 34, 11117–11127. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X. Alzheimer disease therapeutics: Focus on the disease and not just plaques and tangles. Biochem. Pharmacol. 2014, 88, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.D.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Mocanu, M.M.; Nissen, A.; Eckermann, K.; Khlistunova, I.; Biernat, J.; Drexler, D.; Petrova, O.; Schönig, K.; Bujard, H.; Mandelkow, E.; et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J. Neurosci. 2008, 28, 737–748. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.X.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K. Dysregulation of protein phosphorylation/dephosphorylation in Alzheimer’s disease: A therapeutic target. J. Biomed. Biotechnol. 2006, 2006, 31825. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, T.; Higuchi, M.; Zhang, B.; Yoshiyama, Y.; Hong, M.; Trojanowski, J.Q.; Lee, V.M. Attenuated neurodegenerative disease phenotype in tau transgenic mouse lacking neurofilaments. J. Neurosci. 2001, 21, 6026–6035. [Google Scholar] [CrossRef] [Green Version]
- Tanemura, K.; Murayama, M.; Akagi, T.; Hashikawa, T.; Tominaga, T.; Ichikawa, M.; Yamaguchi, H.; Takashima, A. Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J. Neurosci. 2002, 22, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gong, B.; Liu, S.; Fa, M.; Ninan, I.; Staniszewski, A.; Arancio, O. Synaptic fatigue is more pronounced in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2005, 2, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Barten, D.M.; Fanara, P.; Andorfer, C.; Hoque, N.; Wong, P.Y.; Husted, K.H.; Cadelina, G.W.; Decarr, L.B.; Yang, L.; Liu, V.; et al. Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027. J. Neurosci. 2012, 32, 7137–7145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cash, A.D.; Aliev, G.; Siedlak, S.L.; Nunomura, A.; Fujioka, H.; Zhu, X.; Raina, A.K.; Vinters, H.V.; Tabaton, M.; Johnson, A.B. Microtubule reduction in Alzheimer’s disease and aging is independent of tau filament formation. Am. J. Pathol. 2003, 162, 1623–1627. [Google Scholar] [CrossRef]
- Zhang, F.; Su, B.; Wang, C.; Siedlak, S.L.; Mondragon-Rodriguez, S.; Lee, H.G.; Wang, X.; Perry, G.; Zhu, X. Posttranslational modifications of α-tubulin in alzheimer disease. Transl. Neurodegener. 2015, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef]
- Bonifati, V. Genetics of Parkinson’s disease–State of the art, 2013. Parkinsonism Relat. Disord. 2014, 1, S23–S28. [Google Scholar] [CrossRef]
- Ren, Y.; Zhao, J.; Feng, J. Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J. Neurosci. 2003, 23, 3316–3324. [Google Scholar] [CrossRef] [Green Version]
- Law, B.M.; Spain, V.A.; Leinster, V.H.; Chia, R.; Beilina, A.; Cho, H.J.; Taymans, J.M.; Urban, M.K.; Sancho, R.M.; Ramírez, M.B.; et al. A direct interaction between leucine-rich repeat kinase 2 and specific β-tubulin isoforms regulates tubulin acetylation. J. Biol. Chem. 2014, 289, 895–908. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Jiang, Q.; Zhao, J.; Ren, Y.; Sutton, M.D.; Feng, J. Parkin stabilizes microtubules through strong binding mediated by three independent domains. J. Biol. Chem. 2005, 280, 17154–17162. [Google Scholar] [CrossRef] [Green Version]
- Cartelli, D.; Aliverti, A.; Barbiroli, A.; Santambrogio, C.; Ragg, E.M.; Casagrande, F.V.; Cantele, F.; Beltramone, S.; Marangon, J.; De Gregorio, C.; et al. α-synuclein is a novel microtubule dynamase. Sci. Rep. 2016, 6, 33289. [Google Scholar] [CrossRef] [Green Version]
- Carnwath, T.; Mohammed, R.; Tsiang, D. The direct and indirect effects of α-synuclein on microtubule stability in the pathogenesis of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2018, 14, 1685–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.F.; Lockhart, P.J.; Farrer, M.J. Parkin genetics: One model for Parkinson’s disease. Hum. Mol. Genet. 2004, 1, R127–R133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimura, H.; Hattori, N.; Kubo, S.I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Cartelli, D.; Goldwurm, S.; Casagrande, F.; Pezzoli, G.; Cappelletti, G. Microtubule destabilization is shared by genetic and idiopathic Parkinson’s disease patient fibroblasts. PLoS ONE 2012, 7, e37467. [Google Scholar] [CrossRef]
- Ren, Y.; Jiang, H.; Yang, F.; Nakaso, K.; Feng, J. Parkin protects dopaminergic neurons against microtubule-depolymerizing toxins by attenuating microtubule-associated protein kinase activation. J. Biol. Chem. 2009, 284, 4009–4017. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Jiang, H.; Hu, Z.; Fan, K.; Wang, J.; Janoschka, S.; Wang, X.; Ge, S.; Feng, J. Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells 2015, 33, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monfrini, E.; Di Fonzo, A. Leucine-rich repeat kinase (LRRK2) genetics and Parkinson’s disease. Adv. Neurobiol. 2017, 14, 3–30. [Google Scholar]
- Gillardon, F. Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability—A point of convergence in parkinsonian neurodegeneration? J. Neurochem. 2009, 110, 1514–1522. [Google Scholar] [CrossRef]
- Gandhi, P.N.; Wang, X.; Zhu, X.; Chen, S.G.; Wilson-Delfosse, A.L. The Roc domain of leucine-rich repeat kinase 2 is sufficient for interaction with microtubules. Neurosci. Res. 2008, 86, 1711–1720. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, D.; Dowman, J.; Hammond, R.; Leete, T.; Inoue, K.; Abeliovich, A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 2006, 52, 587–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, F.; Yabata, T.; Ohta, E.; Maekawa, T.; Shimada, N.; Suzuki, M.; Maruyama, H.; Ichikawa, T.; Obata, F. LRRK2 phosphorylates tubulin-associated tau but not the free molecule: LRRK2-mediated regulation of the tau-tubulin association and neurite outgrowth. PLoS ONE 2012, 7, e30834. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.M.; Covy, J.P.; Melrose, H.L.; Rousseau, L.; Watkinson, R.; Knight, J.; Miles, S.; Farrer, M.J.; Dickson, D.W.; Giasson, B.I.; et al. LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathol. 2013, 126, 809–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hope, A.D.; Myhre, R.; Kachergus, J.; Lincoln, S.; Bisceglio, G.; Hulihan, M.; Farrer, M.J. Alpha-synuclein missense and multiplication mutations in autosomal dominant Parkinson’s disease. Neurosci. Lett. 2004, 367, 97–100. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Alim, M.A.; Ma, Q.L.; Takeda, K.; Aizawa, T.; Matsubara, M.; Nakamura, M.; Asada, A.; Saito, T.; Kaji, H.; Yoshii, M.; et al. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. Alzheimers Dis. 2004, 6, 435–442. [Google Scholar] [CrossRef]
- Chen, L.; Jin, J.; Davis, J.; Zhou, Y.; Wang, Y.; Liu, J.; Lockhart, P.J.; Zhang, J. Oligomeric alpha-synuclein inhibits tubulin polymerization. Biochem. Biophys. Res. Commun. 2007, 356, 548–553. [Google Scholar] [CrossRef]
- Cartelli, D.; Cappelletti, G. Microtubule destabilization paves the way to Parkinson’s disease. Mol. Neurobiol. 2017, 54, 6762–6774. [Google Scholar] [CrossRef]
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. USA 2005, 102, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Carroll, J.; Trojanowski, J.Q.; Yao, Y.; Iba, M.; Potuzak, J.S.; Hogan, A.M.; Xie, S.X.; Ballatore, C.; Smith, A.B., III; et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci. 2012, 32, 3601–3611. [Google Scholar] [CrossRef]
- Cartelli, D.; Casagrande, F.; Busceti, C.L.; Bucci, D.; Molinaro, G.; Traficante, A.; Passarella, D.; Giavini, E.; Pezzoli, G.; Battaglia, G.; et al. Microtubule alterations occur early in experimental parkinsonism and the microtubule stabilizer epothilone D is neuroprotective. Sci. Rep. 2013, 3, 1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Wali, G.; Sutharsan, R.; Bellette, B.; Crane, D.I.; Sue, C.M.; Mackay-Sim, A. Low dose tubulin-binding drugs rescue peroxisome trafficking deficit in patient-derived stem cells in Hereditary Spastic Paraplegia. Biol. Open 2014, 3, 494–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Yao, Y. A brain-penetrant triazolopyrimidine enhances microtubule-stability, reduces axonal dysfunction and decreases tau pathology in a mouse tauopathy model. Mol. Neurodegener. 2018, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fanara, P.; Banerjee, J.; Hueck, R.V.; Harper, M.R.; Awada, M.; Turner, H.; Husted, K.H.; Brandt, R.; Hellerstein, M.K. Stabilization of hyperdynamic microtubules is neuroprotective in amyotrophic lateral sclerosis. J. Biol. Chem. 2007, 282, 23465–23472. [Google Scholar] [CrossRef] [Green Version]
- Rotstein, M.; Bassan, H.; Kariv, N.; Speiser, Z.; Harel, S.; Gozes, I. NAP enhances neurodevelopment of newborn apolipoprotein E-deficient mice subjected to hypoxia. J. Pharmacol. Exp. Ther. 2006, 319, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Shiryaev, N.; Jouroukhin, Y.; Giladi, E.; Polyzoidou, E.; Grigoriadis, N.C.; Rosenmann, H.; Gozes, I. NAP protects memory, increases soluble tau and reduces tau hyperphosphorylation in a tauopathy model. Neurobiol. Dis. 2009, 34, 381–388. [Google Scholar] [CrossRef]
- Gozes, I.; Stewart, A.; Morimoto, B.; Fox, A.; Sutherland, K.; Schmeche, D. Addressing Alzheimer’s disease tangles: From NAP to AL-108. Curr. Alzheimer Res. 2009, 6, 455–460. [Google Scholar] [CrossRef]
- Morimoto, B.H.; Schmechel, D.; Hirman, J.; Blackwell, A.; Keith, J.; Gold, M.; AL-108-211 Study Investigators. A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2013, 35, 325–336. [Google Scholar] [CrossRef]
- Hanson, K.; Tian, N.; Vickers, J.C.; King, A.E. The HDAC6 inhibitor trichostatin A acetylates microtubules and protects axons from excitotoxin-induced degeneration in a compartmented culture model. Front. Neurosci. 2018, 12, 872. [Google Scholar] [CrossRef]
- Selenica, M.L.; Benner, L.; Housley, S.B.; Manchec, B.; Lee, D.C.; Nash, K.R.; Kalin, J.; Bergman, J.A.; Kozikowski, A.; Gordon, M.N.; et al. Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimers Res. Ther. 2014, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wales, P.; Quinti, L.; Zuo, F.; Moniot, S.; Herisson, F.; Rauf, N.A.; Wang, H.; Silverman, R.B.; Ayata, C.; et al. The sirtuin-2 inhibitor AK7 is neuroprotective in models of Parkinson’s disease but not amyotrophic lateral sclerosis and cerebral ischemia. PLoS ONE 2015, 10, e0116919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, V.; Quinti, L.; Kim, J.; Vollor, L.; Narayanan, K.L.; Edgerly, C.; Cipicchio, P.M.; Lauver, M.A.; Choi, S.H.; Silverman, R.B.; et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2012, 2, 1492–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef] [Green Version]
- Nogales, E.; Wolf, S.G.; Khan, I.A.; Ludueña, R.F.; Downing, K.H. Structure of tubulin at 6.5 A and location of the taxol-binding site. Nature 1995, 375, 424–427. [Google Scholar] [CrossRef]
- Nogales, E.; Wolf, S.G.; Downing, K.H. Structure of the alpha beta tubulin dimer by electron crystallography. Nature 1998, 393, 191. [Google Scholar] [CrossRef] [Green Version]
- Varidaki, A.; Hong, Y.; Coffey, E.T. Repositioning microtubule stabilizing drugs for brain disorders. Front. Cell. Neurosci. 2018, 12, 226. [Google Scholar] [CrossRef]
- Brunden, K.R.; Zhang, B.; Carroll, J.; Yao, Y.; Potuzak, J.S.; Hogan, A.M.; Iba, M.; James, M.J.; Xie, S.X.; Ballatore, C.; et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 2010, 30, 13861–13866. [Google Scholar] [CrossRef] [Green Version]
- Sáez-Calvo, G.; Sharma, A.; Balaguer, F.A.; Barasoain, I.; Rodríguez-Salarichs, J.; Olieric, N.; Muñoz-Hernández, H.; Berbís, M.Á.; Wendeborn, S.; Peñalva, M.A.; et al. Triazolopyrimidines are microtubule-stabilizing agents that bind the vinca inhibitor site of tubulin. Cell Chem. Biol. 2017, 24, 737–750.e6. [Google Scholar] [CrossRef] [Green Version]
- Gozes, I. Activity-dependent neuroprotective protein: From gene to drug candidate. Pharmacol. Ther. 2007, 114, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I.; Divinski, I. NAP, a neuroprotective drug candidate in clinical trials, stimulates microtubule assembly in the living cell. Curr. Alzheimer Res. 2007, 4, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Yenjerla, M.; LaPointe, N.E. The neuroprotective peptide NAP does not directly affect polymerization or dynamics of reconstituted neural microtubules. J. Alzheimers Dis. 2010, 19, 1377–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, Y.; Gray, A.J.; Hirata-Fukae, C.; Minami, S.S.; Waterhouse, E.G.; Mattson, M.P.; LaFerla, F.M.; Gozes, I.; Aisen, P.S. Intranasal NAP administration reduces accumulation of amyloid peptide and tau hyperphosphorylation in a transgenic mouse model of Alzheimer’s disease at early pathological stage. J. Mol. Neurosci. 2007, 31, 165–170. [Google Scholar]
- Gozes, I.; Giladi, E.; Pinhasov, A.; Bardea, A.; Brenneman, D.E. Activity-dependent neurotrophic factor: Intranasal administration of femtomolar-acting peptides improve performance in a water maze. J. Pharmacol. Exp. Ther. 2000, 293, 1091–1098. [Google Scholar]
- Magen, I.; Ostritsky, R.; Richter, F.; Zhu, C.; Fleming, S.M.; Lemesre, V.; Stewart, A.J.; Morimoto, B.H.; Gozes, I.; Chesselet, M.F. Intranasal NAP (davunetide) decreases tau hyperphosphorylation and moderately improves behavioral deficits in mice overexpressing α-synuclein. Pharmacol. Res. Perspect. 2014, 2, e00065. [Google Scholar] [CrossRef]
- Leker, R.R.; Teichner, A.; Grigoriadis, N.; Ovadia, H.; Brenneman, D.E.; Fridkin, M.; Giladi, E.; Romano, J.; Gozes, I. NAP, a femtomolar-acting peptide, protects the brain against ischemic injury by reducing apoptotic death. Stroke 2002, 33, 1085–1092. [Google Scholar] [CrossRef] [Green Version]
- Spong, C.Y.; Abebe, D.T.; Gozes, I.; Brenneman, D.E.; Hill, J.M. Prevention of fetal demise and growth restriction in a mouse model of fetal alcohol syndrome. J. Pharmacol. Exp. Ther. 2001, 297, 774–779. [Google Scholar]
- Quraishe, S.; Cowan, C.M. NAP (davunetide) rescues neuronal dysfunction in a Drosophila model of tauopathy. Mol. Psychiatry 2013, 18, 834–842. [Google Scholar] [CrossRef] [Green Version]
- Ivashko-Pachima, Y.; Sayas, C.L.; Malishkevich, A.; Gozes, I. ADNP/NAP dramatically increase microtubule end-binding protein-Tau interaction: A novel avenue for protection against tauopathy. Mol. Psychiatry 2017, 22, 1335–1344. [Google Scholar] [CrossRef]
- Pandey, U.B.; Batlevi, Y.; Baehrecke, E.H.; Taylor, J.P. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy 2007, 3, 643–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002, 21, 6820–6831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, M.; Kudo, N.; Kosono, S.; Ito, A. Chemical and structural biology of protein lysine deacetylases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 297–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Microtubule organization in neurons. Microtubule (MT) organization is tightly regulated in the different neuronal compartments. In axon, MTs form stable, polarized bundles with uniform polarity orientation, exposing their plus minus ends away from the cell body. In proximal dendrites, MTs are organized in antiparallel bundles oriented with their plus ends pointing away or toward the soma. In the growth cone, MTs adopt four characteristic distributions: splayed, captured at the cortical matrix, looped, and bundled. At the top, MT structure (slide view and end view) is shown.
Figure 1.
Microtubule organization in neurons. Microtubule (MT) organization is tightly regulated in the different neuronal compartments. In axon, MTs form stable, polarized bundles with uniform polarity orientation, exposing their plus minus ends away from the cell body. In proximal dendrites, MTs are organized in antiparallel bundles oriented with their plus ends pointing away or toward the soma. In the growth cone, MTs adopt four characteristic distributions: splayed, captured at the cortical matrix, looped, and bundled. At the top, MT structure (slide view and end view) is shown.

Figure 2.
The distribution of tubulin post-translational modifications (PTMs) differs in neuronal compartments and during neuronal differentiation. In young neurons, polyglutamylation and detyrosination are elevated in the growing axon. Acetylation is localized in the cell body connecting neurite outgrowth, while tyrosination is located in distal region of axons contiguous with the growth cone. In mature neurons, acetylation increases in the proximal site of the axon and presynaptic regions. Detyrosinated and Δ2 tubulin levels are relatively high in differentiated dendrites and axons.
Figure 2.
The distribution of tubulin post-translational modifications (PTMs) differs in neuronal compartments and during neuronal differentiation. In young neurons, polyglutamylation and detyrosination are elevated in the growing axon. Acetylation is localized in the cell body connecting neurite outgrowth, while tyrosination is located in distal region of axons contiguous with the growth cone. In mature neurons, acetylation increases in the proximal site of the axon and presynaptic regions. Detyrosinated and Δ2 tubulin levels are relatively high in differentiated dendrites and axons.


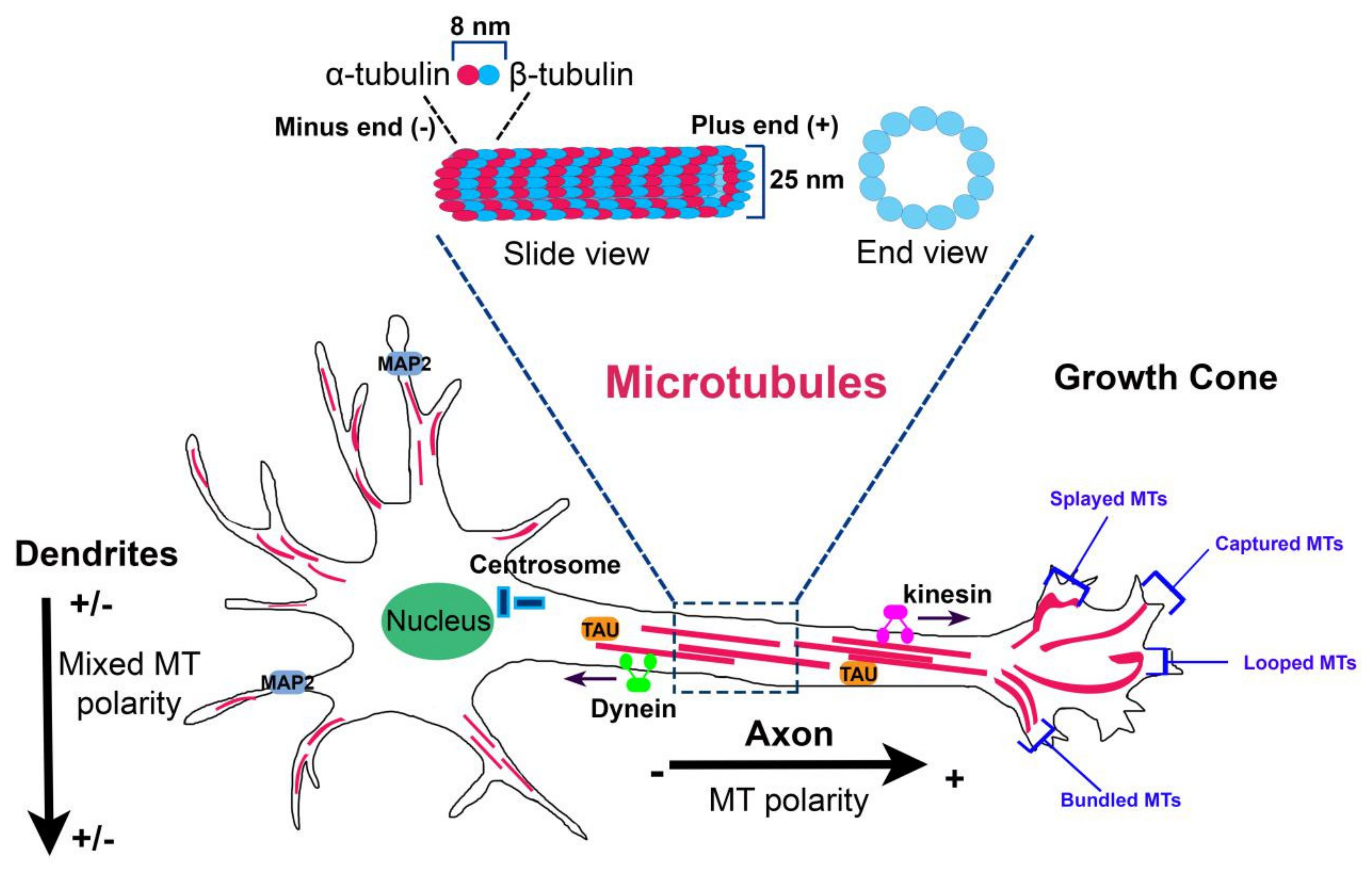{kind=link}
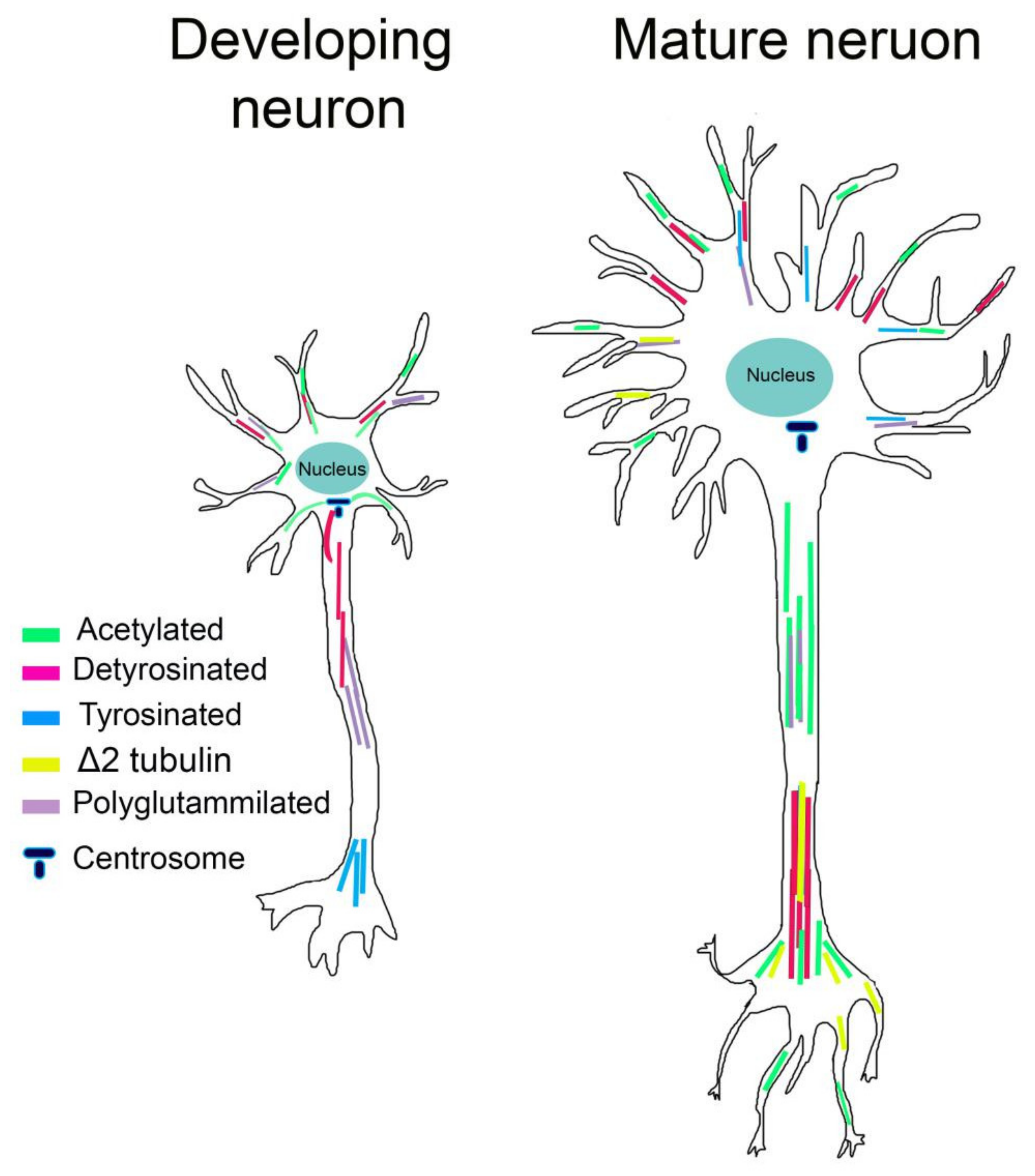{kind=link}
Table 1.
List of tubulin mutations causing neurodegenerative tubulinopathies.
| Gene | Nucleotide Change | Amino Acid Change | Phenotype | References |
|---|---|---|---|---|
| TUBB3 | c.1249G > A | D417N | Polyneuropathy without CFEOM3 | [71,72] |
| TUBB3 | c.1249G > C | D417H | CFEOM3 | [71] |
| TUBB3 | c.1228G > A | E410K | CFEOM3 | [71] |
| TUBB2A | c.1249G > A | D417N | Progressive spastic paraplegia, polyneuropathy and ataxia | [94] |
| TUBA4A | c.128G > T | G43V | FALS | [95] |
| TUBA4A | c.958C > T | R320C | FALS | [95] |
| TUBA4A | c.643C > T | R215C | FALS | [95] |
| TUBA4A | c.959G > A | R320H | FALS | [95] |
| TUBA4A | c.1147G > A | A383T | FALS | [95] |
| TUBA4A | c.1221G > A | W407X | FALS | [95] |
| TUBB4A | c.4C > T | R2W | H-ABC | [98] |
| TUBB4A | c.5G > A | R2G | H-ABC | [98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213] |
| TUBB4A | c.467G > T | R156L | H-ABC | [214] |
| TUBB4A | c.533C > G | T178R | H-ABC | [213] |
| TUBB4A | c.533C > T | T178M | H-ABC | [202] |
| TUBB4A | c.533C > G | T178R | Severe H-ABC | [215] |
| TUBB4A | c.538G > A | V180M | H-ABC | [216] |
| TUBB4A | c.730G > A | G244S | H-ABC | [98,217] |
| TUBB4A | c.731G > T | G244V | H-ABC | [98,202] |
| TUBB4A | c.731G > A | G244D | H-ABC | [202] |
| TUBB4A | c.745G > A | D249N | H-ABC | [98,201,202,213,218] |
| TUBB4A | c.785G > A | R262H | H-ABC | [213,216,218] |
| TUBB4A | c.968T > G | M323R | H-ABC | [98] |
| TUBB4A | c.974G > T | W325L | H-ABC | [216] |
| TUBB4A | c.1054G > A | A352T | H-ABC | [98] |
| TUBB4A | c.1061G > A | C354Y | H-ABC | [98] |
| TUBB4A | c.1099T > A/C | F367I | H-ABC | [98] |
| TUBB4A | c.1162A > G | M388V | H-ABC | [98,213] |
| TUBB4A | c.1163T > C | M388T | H-ABC | [98,202] |
| TUBB4A | c.1164G > A | M388I | H-ABC | [98,217] |
| TUBB4A | c.1181T > G | F394C | H-ABC | [217] |
| TUBB4A | c.1228G > A | E410K | H-ABC | [211,213] |
| TUBB4A | c.544C > A | P182T | H-ABC | [202] |
| TUBB4A | c.568C > T | H190Y | Congenital-onset spastic paraplegia and mild hypomyelination | [101,102] |
| TUBB4A | c.1064A > T | D353V | Early-onset progressive spastic paraplegia | [103] |
| TUBB4A | c.4C > G | R2G | DYT4 | [96,97] |
| TUBB4A | c.811G > A | A271T | DYT4 | [97] |
Table 2.
List of most relevant MT-targeting compounds.
| Drug | Target | Function | Effects | Model | References |
|---|---|---|---|---|---|
| MT-Stabilizing Compounds | |||||
| Paclitaxel | MTs | Stabilizes MTs | Restores axonal transport in spinal axons and reduces the motor phenotype, increases the levels of stable detyrosinated tubulin as well as the numbers of MTs in the ventral root axons | PrP T44 mouse | [290] |
| Epo D | MTs | Stabilizes MTs | Reverses behavioral and cognitive deficits, clears tau pathology and curbs neuron loss | Young and old PS19 mouse | [291] |
| Epo D | MTs | Stabilizes MTs | Attenuates nigrostriatal degeneration | MPTP-treated mice | [292] |
| Epo D | MTs | Stabilizes MTs | Restores the levels of acetylated α-tubulin, restores peroxisome speeds, and ameliorates patient cell sensitivity to hydrogen peroxide | iPSCs derived from patients affected by hereditary spastic paraplegia | [293] |
| Epo D | MTs | Stabilizes MTs | Not reported | Clinical trial | NCT 01492374 NCT 01966666 |
| CNDR-51657 | MTs | Stabilizes MTs | Improves MT density in hippocampal neurons and reduces axonal dystrophy with resulting reduction in tau pathology | PS19 mice | [294] |
| Noscapine | MTs | Stabilizes MTs | Attenuates the deficit of axonal transport and improves MT stability | Mutant SOD1 mice | [295] |
| NAP | May involve MTs | May prevent MTs disruption | Improves short-term memory | ApoE knockout mice | [296] |
| NAP | May involve MTs | May prevent MTs disruption | Increases soluble tau and reduces tau hyperphosphorylation at Ser202 | DM-tau mice | [297] |
| NAP | May involve MTs | May prevent MTs disruption | Improves the individual memory tasks | AD patients | NCT 01056965 [298,299] |
| Tubulin PTMs targeting compounds | |||||
| TSA | HDAC6 (not selective) | Increases tubulin acetylation | Restores fly locomotor behavior and increases tubulin acetylation improving axonal transport | LRRK2 drosophila model of PD | [226] |
| TSA | HDAC6 (not selective) | Increases tubulin acetylation | Preventing axon fragmentation and axonal loss | Mouse cortical neurons expose to kainic acid | [300] |
| TSA | HDAC6 (not selective) | Increases tubulin acetylation | Increases vesicular transport of brain-derived neurotrophic factor by increasing acetylation of α-tubulin | Striatal precursor cells of HD model | [131] |
| TSA TubA | HDAC6 | Increases tubulin acetylation | Corrects mitochondria axonal transport defects | HSPB1 mice | [216] |
| TubA | HDAC6 | Increases tubulin acetylation | Reverts the behavioral defects such as memory impairment and hyperactivity | rTg4510 mice | [301] |
| AGK2 | Sirt2 | Increases tubulin acetylation | Prevents dopamine depletion in disease models of PD | Drosophila model of PD | [302] |
| AK7 | Sirt2 | Increases tubulin acetylation | Prevents dopaminergic neuron loss in vivo in MPTP-treated mice, improves motor function, extends survival, and reduces brain atrophy in HD mice | MPTP mice, R6/2 mice, and HD knock-in mice | [303,304] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sferra, A.; Nicita, F.; Bertini, E. Microtubule Dysfunction: A Common Feature of Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 7354. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197354
AMA Style
Sferra A, Nicita F, Bertini E. Microtubule Dysfunction: A Common Feature of Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(19):7354. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197354
Chicago/Turabian StyleSferra, Antonella, Francesco Nicita, and Enrico Bertini. 2020. "Microtubule Dysfunction: A Common Feature of Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 19: 7354. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197354
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.