Recent Advances in Drosophila Models of Charcot-Marie-Tooth Disease

1
Department of Molecular Pathobiology of Brain Disease, Kyoto Prefectural University of Medicine, Kyoto 6028566, Japan
2
Department of Neurology, Kyoto Prefectural University of Medicine, Kyoto 6028566, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(19), 7419; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197419
Submission received: 31 August 2020
/
Revised: 6 October 2020
/
Accepted: 6 October 2020
/
Published: 8 October 2020
(This article belongs to the Special Issue Role of Drosophila in Human Disease Research 2.0)
Abstract
:Charcot-Marie-Tooth disease (CMT) is one of the most common inherited peripheral neuropathies. CMT patients typically show slowly progressive muscle weakness and sensory loss in a distal dominant pattern in childhood. The diagnosis of CMT is based on clinical symptoms, electrophysiological examinations, and genetic testing. Advances in genetic testing technology have revealed the genetic heterogeneity of CMT; more than 100 genes containing the disease causative mutations have been identified. Because a single genetic alteration in CMT leads to progressive neurodegeneration, studies of CMT patients and their respective models revealed the genotype-phenotype relationships of targeted genes. Conventionally, rodents and cell lines have often been used to study the pathogenesis of CMT. Recently, Drosophila has also attracted attention as a CMT model. In this review, we outline the clinical characteristics of CMT, describe the advantages and disadvantages of using Drosophila in CMT studies, and introduce recent advances in CMT research that successfully applied the use of Drosophila, in areas such as molecules associated with mitochondria, endosomes/lysosomes, transfer RNA, axonal transport, and glucose metabolism.
1. Introduction
1.1. Clinical Features of CMT
Charcot-Marie-Tooth disease (CMT) is the most common inherited peripheral neuropathy. The average prevalence of CMT is reported to be about 1 in 2,500 people [1]; however, the CMT prevalence rate varies markedly among epidemiological studies due to a large variety of CMT symptoms [2]. At present, the reported prevalence of CMT among Europeans is about 10–30 per 100,000 people, while that in the East Asia is 5.3–10.8 per 100,000 people [3,4,5,6]. The diagnosis of CMT is based on clinical symptoms, electrophysiological studies, genetic testing, and nerve biopsy [7]. CMT is usually juvenile-onset, and typical symptoms are slow, progressive muscle weakness and sensory disturbance in a distal dominant pattern. Patients show clumsiness, foot deformity (such as pes cavus), and gait disturbance. Some patients show additional symptoms such as hearing loss and scoliosis [8]. Interestingly, there are accumulating case reports of cerebral white matter abnormalities mainly in X-linked CMT type 1, and recently, abnormal diffusion-tensor imaging on brain MRI was shown to correlate with clinical disability in various CMT subgroups, suggesting subclinical central nervous system involvement in addition to clinical peripheral neuropathy [9,10,11]. Most patients develop symptoms in childhood, while there are marked individual differences in the severity and progression rate even in those with the same genetic alteration [12]. A nerve conduction study is performed for patients to estimate the background pathology of CMT. A decreased nerve conduction velocity (NCV) (< 38 m/s) indicates CMT1. The main pathological feature of CMT1 is a destruction of the myelin sheath, which is produced by Schwann cells. On the other hand, the presence of decreased compound muscle and sensory action potentials with normal NCV (>38 m/s) indicates CMT2, whose pathological feature is primary axonal damage. The intermediate NCV (30-45 m/s) is associated with the mixed pathology of damaged myelin (demyelination) and axon (axonopathy) [13]. Genetic testing has made marked progress in recent years, and more than 100 genes containing causative mutations have been identified with the widespread use of next-generation sequencing technologies, revealing significant genetic heterogeneity of CMT [2,14,15] (Figure 1). Previous studies showed that about 60% of CMT patients received a definitive diagnosis by genetic testing, and over 90% of genetically diagnosed patients show an alteration in one of the following four genes: Peripheral Myelin Protein 22-kDa (PMP22), Gap Junction Beta 1 (GJB1), Myelin Protein Zero (MPZ), and Mitofusin 2 (MFN2) [2,16,17]. However, in the Mediterranean area (e.g., southern Italy and eastern Spain), mutations in Ganglioside-induced differentiation-associated protein 1 (GDAP1) are the third most common genetic diagnosis of CMT after PMP22 duplication and mutations in GJB1 [18,19]. No GDAP1 mutation was identified in a German cohort, suggesting that the geographical area affects the genetic distribution [20]. Nerve biopsy, which was previously the key diagnostic step, is being replaced by genetic testing, but it is still important in atypical cases [7]. Typical findings of nerve biopsy in demyelinating-type CMT are onion bulb formation and a thinned myelin sheath, which are the result of repeated de- and remyelination, and these findings are uniformly observed throughout the nerve. In contrast, axonopathy leads to a decreased number of axons and the disappearance of Schwann cells in the absence of demyelination [21].
1.2. Classification of CMT
CMT is clinically divided into subgroups according to the combination of the inheritance pattern and NCV, which helps to estimate the underlying pathology (demyelination or axonopathy) (https://neuromuscular.wustl.edu/) [8] (Table 1). At present, CMT1 is further classified into seven subgroups, from CMT1A to 1G. CMT1A (MIM #118220) is the most frequent subtype of CMT caused by a 1.5-Mb duplication on chromosome 17p11.2 containing the PMP22 gene, present in about half of all CMT patients [16,17,22]. Mutations within MPZ gene are causative for CMT1B (MIM #118200), accounting for around 10% of gene-mutation-identified CMT. CMT1 is the most common subtype of CMT, but CMT1 studies in Drosophila are not practical, because Drosophila does not have a mature myelin sheath like that of humans.
CMT2, which accounts for 20% of genetically diagnosed CMT patients, shows an autosomal dominant inheritance pattern and decreased nerve action potential amplitudes with normal NCV (which indicates axonopathy) [16,17]. Among many genes in which mutations are reported to cause CMT2, research on the following important genes and their functions has markedly progressed using Drosophila models: MFN2, Ganglioside Induced Differentiation Associated Protein 1 (GDAP1), RAB7A, Member RAS Oncogene Family (RAB7), Glycyl-tRNA Synthetase (GARS), Alanyl-tRNA Synthetase (AARS), Methionyl-tRNA Synthetase (MARS), Histidyl-tRNA synthetase (HARS), and Sorbitol dehydrogenase (SORD). The proteins MFN2 and GDAP1 are associated with mitochondrial dynamics, RAB7 is associated with the endosomal function, aminoacyl-tRNA transferase (aaRS), which is encoded by GARS, AARS, MARS and HARS genes, which are associated with accurate protein translation, and the SORD protein is associated with glucose metabolism (also see Section 2, Section 3, Section 4, Section 5 and Section 6).
The term CMT3 was previously used for infant-onset and the severe CMT subtype. However, CMT3 is no longer used because it proved to be a severe form of early-onset CMT1 or CMT4 by genetic analysis. CMT3, which was historically termed Dejerine–Sottas neuropathy, is now known as the component of the hereditary neuropathies with infantile onset [23,24].
CMT4 is a rare autosomal recessive form of demyelinating CMT with a prevalence of about 1% of genetically identified CMT, although one report suggested a prevalence of 30% in a population with a high rate of consanguineous marriage [16,25]. Unlike CMT1, among various genes in which mutations are reported to cause CMT4, Factor-induced gene 4 (FIG4) of CMT4J (MIM #611228) is actively studied using the Drosophila model because FIG4 phospholipid phosphatase has functions including the maintenance of membrane trafficking in both neurons and Schwann cells [26,27,28] (see also Section 3).
All forms of X-linked CMT are designated as CMTX, found in around 10% of all CMT patients [29,30,31,32,33]. CMTX1 (MIM #302800), which is caused by mutations in GJB1 gene, is the most frequent form and accounts for 90% of CMTX. Among CMTX1 patients, the disease severity and NCV pattern exhibit marked individual differences due to the diversity of mutation sites (more than 250 have been reported) [34]. GJB1 encodes Connexin-32 that is expressed on the surface of glial cells and forms gap junctions [35]. Recent reports showed that cerebral white matter lesions are sometimes observed in CMTX1 patients [36]. Overall, Connexin-32 is an interesting molecule to investigate the association between axons and myelin sheaths; however, unfortunately, no fly orthologue of human GJB1 has been identified to date.
1.3. Various CMT Models
Until now, various models such as rodents, the zebrafish, fruit fly, yeast, cell lines and induced pluripotent stem cells (iPSCs) have been developed for CMT modeling [37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57]. Rodent CMT models have advanced understanding of neuropathy; however, regarding MFN2 of CMT2A, which is the most frequent subtype of axonal CMT, it used to be difficult to reproduce the phenotype in rodent due to embryonic lethality [58]. In addition, such models are expensive to maintain and are subject to ethical restrictions. Except for the Cartagena Protocol on Biosafety, there are currently no ethical or social restrictions on experiments with Drosophila (https://bch.cbd.int/protocol). Regarding the iPSC models, it is also an important tool for analyzing tissues not suitable for biopsy, such as nerves, but there is a limitation whereby the results of in vitro experiments are markedly influenced by the culture conditions. Compared with rodents and iPSCs, the strengths of Drosophila as a laboratory animal are as follows: fewer ethical restrictions, a short lifecycle, a large number of genetically homogenous offspring, low maintenance cost and established genetic engineering techniques [59,60]. In the Drosophila CMT model, motor deficit in CMT patients is reproduced by a decline in climbing ability during the adult stage and in crawling ability during the larval stage [61]. The locomotive ability of adult flies is assessed by a negative geotaxis assay, first described as climbing activity [62]. Gently tapping a vial containing flies causes them to drop to the bottom, and flies are recorded as they climb up the wall of the vial. The percentage of flies that reach a certain height within a specified time is measured and statistically analyzed. On the other hand, the crawling ability of larvae is used as a method to measure larval locomotive ability [63]. In the larval crawling assay, larvae in the third instar stage are placed on an agar plate and recorded, and the speed of larval migration and distance are statistically analyzed. Although Drosophila does not have a mature myelin sheath, the basic structure and physiology of the axons are highly conserved between humans and Drosophila, and progress has been made mainly in axonal CMT research. Here, we introduce Drosophila models that aided in elucidating the pathophysiology of CMT.
2. Drosophila CMT Models for Investigating Aberrant Mitochondrial Dynamics
Mitochondria are highly dynamic organelles that are responsible for cell viability. Mitochondria have various functions, such as ATP production, apoptosis, regulation of calcium signaling and reactive oxygen species (ROS) production. Mitochondria continuously repeat fusion and fission to maintain their homeostasis and functions. Mitochondrial fusion promotes diffusion of the matrix content and dilution of oxidized metabolism and damaged mitochondrial DNA, whereas fission is an important process in mitophagy [64]. Mitochondrial fusion and fission are complex processes, and how mitochondrial abnormalities cause disease is not fully understood. However, mutants of following mitochondrial molecules are known to associated with CMT and have been actively studied using Drosophila: MFN2, ganglioside-induced differentiation associated protein 1 (GDAP1), and solute carrier family 25 member 46 (SLC25A46).
2.1. MFN2
MFN2 is a GTPase localized to the outer mitochondrial membrane and forms a dimer during the process of mitochondrial fusion [65]. Mutations of the MFN2 gene are causative for CMT2A2 (MIM #609260), which is the most common genotype of inherited axonal-type neuropathy. A previous study showed that mammalian MFN2 mutations impaired axonal transport of mitochondria [66]. Failure to meet the demand for ATP at the distal axon is one possible cause of neurodegeneration in axonal CMT. Knockdown of Mitochondrial assembly regulatory factor (Marf), the Drosophila homolog of human MFN2, also led to phenotypes as follows: (1) motor dysfunction (reduced climbing ability) that rescued with knock-in of the human wild-type MFN2 but not with MFN2 with R94Q mutation (one of the most common mutations associated with CMT2A), (2) impaired transport of mitochondria to the distal part of the axon and (3) fragmented and clustered mitochondria [67]. These phenotypes could reproduce key symptoms of patients. The Marf knockdown strain also showed fragmented endoplasmic reticulum (ER) cisternae and increased levels of ER stress markers such as X-box binding protein 1 (Xbp1) and binding immunoglobulin protein (BiP), suggesting that ER stress is also involved in the pathogenicity of Marf knockdown [67].
Regarding the relationship between mitochondrial morphology and function, Trevisan et al. reported that the neural function was dependent on the capacity of mitochondrial energy production, but was independent of their morphology and distribution [68]. They reported that the single knockdown of Marf or Optic atrophy gene 1 (Opa1), each encoding a key molecule for mitochondrial fusion, caused mitochondrial fragmentation, impaired transport of mitochondria, impaired ATP production, and increased lethality in adult flies. Interestingly, double knockdown of genes Marf and Dynamin related protein 1 (Drp1), which encoded another GTPase necessary for mitochondrial fission, improved the capacity for ATP production and survival rate but did not rescue aberrant mitochondrial morphology and distribution. On the other hand, double knockdown of Opa1 and Drp1 rescued aberrant mitochondrial morphology and distribution, but it did not improve the capacity for ATP production or viability. These results indicate that neuronal cell viability depends on mitochondrial functions rather than the mitochondrial distribution or morphology. In terms of the relationship between the mitochondrial function and morphology, El Fissi et al. reported the diversity of the impaired mitochondrial function and morphology depending on the mutation site in Marf. Interestingly, all Marf transgenic flies showed reduced climbing ability; flies carrying mutations within the GTPase domain (corresponds to R94Q and T105M in MFN2) of Marf showed unfused and aggregated mitochondria. On the other hand, flies carrying mutations within the helix bundle 1 domain (corresponds to R364W and L76P in MFN2) of Marf showed enhanced mitochondrial fusion and giant mitochondria, rescued by overexpression of the fission factor encoding the gene Drp1 [69]. From these results, not only impaired mitochondrial fusion but also excessive fusion of mitochondria may underlie CMT caused by mutant MFN2, and the diversity of the mutation site in the MFN2 gene and various functional alterations of MFN2 protein caused by each MFN2 mutation may lead to the marked individual differences noted in CMT2A patients.
With respect to the development of treatment, Garrido-Maraver et al. showed that folate metabolism-related gene expression was upregulated in Marf knockdown Drosophila. Although oral folate supplementation did not have a therapeutic effect on this model, overexpression of one gene involved in folate metabolism reduced mortality and ameliorated locomotor deficits in Marf knockdown Drosophila [70]. These results may help to develop new therapeutic targets. Consequently, Marf/MFN2 is one of the most important molecules in elucidating the effects of mitochondrial abnormality on human disease, and it is expected to continue to be actively studied in Drosophila.
2.2. GDAP1
Mutations within the GDAP1 gene are causative for CMT4A (MIM #214400, classified into demyelinating CMT) or CMT2K (MIM #607831, classified into axonal CMT) [71,72]. GDAP1 is a transmembrane protein present in the outer mitochondrial membrane of both neurons and Schwann cells, similar in structure to glutathione S-transferase (GST) but without GST activity [73,74]. GDAP1 is involved in regulation of the mitochondrial morphology and function, but much remains unknown. GDAP1 gene mutations in patients with an autosomal recessive inheritance pattern were reported to impair mitochondrial fission; however, impaired fusion was also observed in the presence of GDAP1 mutations in those with an autosomal dominant inheritance pattern [73,74]. López Del Amo et al. showed that knockdown of dGdap1, which is a homolog of human GDAP1, caused degeneration of the fly’s retina and muscle, and these phenotypes were rescued by human GDAP1 expression [75]. Both knockdown and overexpression of dGdap1 resulted in reduced climbing ability. Knockdown of dGdap1 also resulted in mitochondrial aggregation and large, elongated mitochondria in muscle. On the other hand, overexpression of dGdap1 resulted in a decrease in the size of mitochondria and cluster formation of mitochondria in the retina. Additionally, with both the overexpression and knockdown of dGdap1, early inactivation of the insulin pathway followed by reduced carbohydrate degradation and increased β-oxidation of lipids were observed [75,76]. These dGdap1-based results suggest that impaired energy metabolism due to mitochondrial dysfunction may be closely associated with neurodegeneration.
2.3. SLC25A46
SLC25A46 is a transmembrane protein that exists in the outer mitochondrial membrane and is considered to be involved in mitochondrial fission. Mutations within the SLC25A46 gene are causative for hereditary motor and sensory neuropathy (HMSN) type 6B (MIM #616505), and patients exhibit the autosomal recessive inherited form of axonal neuropathy and optic nerve atrophy [50]. The SLC25A46 knockdown mouse model presented with optic nerve atrophy, axonal degeneration in the peripheral nervous system, giant mitochondria and severe ataxia due to shedding of Purkinje cells in the cerebellum [77]. There are two SLC25A46 paralogues in Drosophila, SLC25A46a [53] and SLC25A46b [54]. Previous reports showed that knockdown of each of SLC25A46a and SLC25A46b in Drosophila similarly led to motor deficit such as reduced crawling and climbing abilities, a shortened synaptic length in the neuromuscular junction (NMJ), decreased ATP production and ROS accumulation [78,79].
It was also reported that human SLC25A46b and Histone deacetylase 1 (HDAC1) interacted with each other based on bioinfomatics analysis, and that Drosophila Histone deacetylase RPD3 (Rpd3), a homolog of human HDAC1, encoded the protein which regulated the acetylation of histone H4K8 in the Drosophila SLC25A46b genomic region. The down-regulation of Rpd3 expression reduced motor deficits and synaptic morphological abnormalities caused by dSCL25A46b knockdown [80]. These findings suggest a novel perspective whereby not only genetic factors, but also epigenetic regulators, may be involved in neurodegeneration due to mitochondrial dysfunction.
3. Drosophila CMT Models for Investigating Membrane Trafficking Defects
3.1. FIG4
Mutations within the FIG4 gene are causative for CMT4J (MIM #611228) and amyotrophic lateral sclerosis (ALS) type 11 (MIM #612577) [81,82]. ALS is a progressive neurodegenerative disease that selectively damages motor neurons, resulting in motor deficits and fatal respiratory muscle paralysis. FIG4 encodes a phosphatase present on the surface of late endosomal membranes and it forms a protein complex with Fab1, which is a lipid kinase, and Vac14. This complex regulates the conversion between phosphatidylinositol 3,5-bisphosphate (PI [3,5] P2) and phosphatidylinositol 3-phosphate (PI3P), with both being lipids making up membranes that are also responsible for signal transduction [83]. However, recent studies of Drosophila revealed a different function of FIG4 other than phosphatase. In Drosophila, dFIG4 is a single homolog for human FIG4. Neuron-specific dFIG4 knockdown Drosophila showed ALS-like symptoms such as reduced motor performance, an abnormal morphology of NMJ in motor neurons, and a shortened lifespan. In addition, interestingly, these flies also showed enlarged lysosomes that were partially rescued by expression of the catalytically inactive dFIG4 mutant protein [84,85]. This suggests that dFIG4 functions independently of enzymatic activity to maintain lysosomal membrane homeostasis.
Furthermore, Muraoka et al. and Shimada et al., using genetic screening, showed that Drosophila long non-coding RNA (lncRNA) interacted genetically with dFIG4 and that knockdown of this lncRNA improved phenotypes including motor deficits such as reduced climbing ability and enlarged lysosomes caused by dFIG4 knockdown [86,87]. Up to now, exome analyses are mainly carried out for the genetic diagnosis of CMT. However, these reports implicate the importance of information on non-protein coding regions in the human genome to elucidate the pathogenesis of CMT. Although the role of these lncRNAs is not yet well-understood, lysosomal membrane metabolism by the FIG4 complex may involve more molecules than previously considered.
3.2. RAB7
Mutations within RAB7 gene are causative for CMT2B (MIM #606071). Rab7 is a small GTPase involved in late endosomal maturation [88,89]. Janssens et al. reported that CMT2B model Drosophila, which was expressing a CMT2B-causing Rab7 mutation in both alleles in all sensory neurons, showed a reduced response to temperature and pain stimuli as well as motor deficit, being similar to the symptoms of CMT2B patients. In the thermotaxis assay, the third instar larvae were placed in the center of a Petri dish with one side warmed to 30℃ and the other maintained at 22℃. Then, less than 10% of control larvae were on 30℃ side, while 17.4% of CMT2B model larvae were on the 30℃ side, suggesting impaired ability of larvae to perceive non-optimal temperatures in this model. In the nociception assay, the authors subjected larvae to touch with a soldering iron maintained at 43℃ as a pain stimulus, which caused a rolling motion in larvae. It took significantly longer to cause a rolling motion in the CMT2B model larvae than in the control, and the proportion of larvae without a rolling motion was also higher in the transgenic CMT2B model larvae. The authors then examined the impact of the CMT2B-causing Rab7 mutation on vesicle trafficking. Analysis of axonal transport in this transgenic CMT2B model larvae showed a reduced pausing time of RAB7-positive vesicles in the axon [90]. This “reduced pausing time of RAB7-positive vesicles” was confirmed and further investigated in a study using the CMT2B model of Xenopus [91]. In this study, a reduced frequency of pausing and an increased velocity of RAB7-positive axonal endosomes were observed in retinal cell axons of Xenopus carrying the CMT2B mutation. They also showed that these axonal endosomes transported the ribonucleoprotein particles, and that the CMT2B mutation of RAB7 reduced the synthesis of axonal mitochondrial outer membrane proteins and altered both the axonal mitochondrial morphology and mitochondrial membrane potential. These results indicate that the RAB7-positive endosome may be the site of local translation in the axon and that such local protein synthesis may be required to maintain the mitochondrial function [91]. These in vivo studies of CMT2B-causing RAB7 mutations will continue to reveal new roles of endosomes and mitochondria in axonal integrity.
4. Drosophila CMT Models for Investigating Mutant Aminoacyl-tRNA Synthetases
Aminoacyl-tRNA synthetases (aaRS) are enzymes that bind specific amino acids to tRNAs in an ATP-dependent manner. To date, mutants of six types of aaRS have been reported to underlie autosomal dominant axonal or intermediate CMT: Glycyl-tRNA synthetase (GARS) mutants for CMT2D (MIM #601472) and distal hereditary motor neuronopathy 5A (MIM #600287) [92]; Tyrosyl-tRNA synthetase (YARS) mutants for Dominant Intermediate-CMT, Type C (MIM #608323) [93]; Alanyl-tRNA synthetase (AARS) mutants for CMT2N (MIM #613287) [94]; Histidyl-tRNA synthetase (HARS) mutants for CMT2W (MIM #616625) [95]; Lysil-tRNA synthetase (KARS) mutants for CMT recessive intermediate B (CMTRIB, MIM #613641) [96]; Methionyl-tRNA synthetase (MARS) mutants for CMT2U (MIM #616280) [97].
Fly models of CMT caused by mutations in aaRS encoding genes reproduced the clinical phenotypes of CMT patients, such as an impaired motor function, axonal degeneration, muscle denervation and synaptic dysfunction [51,98,99,100,101]; however, notably, the development of motor symptoms did not depend on the enzymatic activity of aaRS in these models. Niehues et al. reported that Gars-mutated Drosophila with impaired locomotion showed reduced global protein synthesis in peripheral neurons, and this reduced protein synthesis could not be rescued by overexpression of wild-type Drosophila Gars. It was suggested that reduced translation may be responsible for the CMT-related phenotypes in Drosophila, and that CMT-causing GARS mutations might have a toxic gain-of-function effect on protein translation [100]. Furthermore, Bervoets et al. showed that Drosophila models with CMT-causing Yars mutations exhibited conformational changes in the nuclear YARS protein and over-activation of the transcriptional regulator E2F1. It was suggested that aaRS may have a role as a transcriptional regulator. Regarding the therapeutic approach, both pharmacological prevention of the transfer of mutant YARS protein from the cytoplasm to nucleus and genetic removal of mutant Yars from the nucleus improved motor deficits and neural morphological abnormalities in Drosophila [102]. Other aaRSs are also expected to have some signal-modulating effect, which may offer new therapeutic targets of CMT.
5. Drosophila CMT Models for Investigating Impaired Axonal Transport
Axonal transport is essential to maintain axonal homeostasis. Motor proteins transport various cargos such as proteins and mRNA along cytoskeletal filaments. Three large superfamilies of motor protein have been identified: kinesins, dyneins and myosins [103]. Among them, some mutant members of the kinesin and dynein families can cause hereditary neuropathy: Kinesin family member (KIF) 1Bβ for CMT2A1 (MIM #118210) [104], KIF1A for Hereditary Sensory and autonomic neuropathy IIC (MIM #614213) [105], and Dynein, cytoplasmic 1, heavy chain 1 (DYNC1H1) for CMT2O (MIM #614228) [106].
Regarding KIF1A, Kern et al. reported that Drosophila with the unc-104 (a synonym of KIF1A) hypomorphic mutant showed aberrant apposition of the active zone (AZ) and postsynaptic densities (PSDs) without synaptic retraction, indicating that the protein encoded by unc-104/KIF1A might regulate synapse formation and maturation. The authors reported that unc-104/KIF1A knockdown resulted in about a 10-fold increase in PSDs unopposed by AZ compared with the wild-type [107]. Further, using the same mutant Drosophila, Zhang et al. revealed a decrease in component proteins of synaptic vesicles (such as vesicular glutamate transporter (VGLUT) and cysteine string protein (CSP)) at NMJ and their accumulation in neuronal cell bodies in the presence of unc-104/KIF1A knockdown. They also showed that the level of Rab3 protein, which is KIF1A cargo and regulates the exocytosis of synaptic vesicles, was markedly decreased in unc-104/KIF1A mutant NMJ. Interestingly, the overexpression of RAB3 partially rescued aberrant apposition between pre- and postsynaptic structures without the improvement of axonal transport itself, suggesting that the overproduction of Rab3 might enhance feedback from postsynaptic structures, which was markedly impaired in unc-104/KIF1A mutant NMJ [108].
Recently, it was shown using an in vitro model that the KIF3 complex bound and transported mRNAs and that the specific sequences of mRNAs increased the selectivity and efficiency of transport [109]. Given the clinical characteristics of CMT such as distal dominancy, impaired synapse maturation caused by impaired intracellular transport may be a significant pathogenesis in CMT.
6. Drosophila CMT Model for Investigating Mutant Sorbitol Dehydrogenase
Sorbitol dehydrogenase (SORD) is one of the enzymes involved in the polyol pathway and is ubiquitously expressed in mammalian tissues [110]. In the polyol pathway, glucose is converted to sorbitol by aldose reductase and sorbitol is oxidized to fructose by SORD. Under hyperglycemic conditions, accumulations of sorbitol and fructose due to increased polyol pathway flux may contribute to tissue damage [111,112]. In 2020, Cortese et al. reported that biallelic mutations in SORD caused inherited neuropathies including CMT and distal hereditary motor neuropathy (MIM #618912) [113]. Notably, in that paper, the pathogenic variant of SORD, c.757delG (p.Ala253GlnfsTer27), was shown to be the most frequent cause of the recessive form of inherited neuropathy (frequency of carriers was ~3 per 1,000 individuals). Of 45 patients carrying biallelic mutations in SORD, 98% showed distal dominant lower limb weakness and 43% showed sensory deficits (note: 65% of patients showed a reduced sensory action potential, suggesting asymptomatic sensory nerve impairment), and these patients had serum sorbitol levels more than 100 times higher than those of controls. Patient-derived fibroblasts also showed increased intracellular sorbitol. Furthermore, the pathogenicity of biallelic SORD mutations was validated by SORD deficiency models of Drosophila. Drosophila has two functional SORD proteins that are 90% identical to each other, named Sodh1 and Sodh2 [114]. The authors reported that both Drosophila with neuron-specific knockdown of Sodh1 and Sodh2 and that with homozygous Sodh2 loss-of-function mutation (Sodh2MB01265/MB01265) showed motor deficit and progressive neurodegeneration. In detail, these two Drosophila models showed reduced climbing ability at a late stage (40 days after eclosion (DAE)) and the loss of photoreceptor terminals in the lamina layer of compound eyes, and the extent of this structural abnormality ameliorated at 10 DAE compared with 2 DAE. In addition, Sodh2MB01265/MB01265 mutant flies exhibited a four-fold increase in sorbitol concentrations in brain homogenates compared with controls. The authors concluded that these results recapitulated the characteristics of patients, such as progressive motor deficit, neurodegeneration, and an elevated sorbitol level. Interestingly, the oral administration of inhibitors of aldose reductase, which converts glucose to sorbitol, decreased sorbitol levels in brain homogenates and improved the climbing ability and abnormal structure of photoreceptor terminals at a late stage in Sodh2MB01265/MB01265 mutant flies. Analysis of the Drosophila model, performed concurrently with identification of the novel gene, helped to confirm that SORD mutations were pathogenic rather than simply incidental findings.
7. Conclusions
The highly conserved neural structure between humans and Drosophila facilitates the study of CMT in the Drosophila model. Especially, the motor symptoms of CMT patients are well-reproduced by the climbing and crawling assays of Drosophila. Studies of CMT using Drosophila models only commenced in 2009, and there are still many unstudied genes containing CMT-causing mutations in Drosophila. The study of genetic interactions, in which Drosophila excels, is an area of research that is difficult using rodent models, and unique developments in this area will promote advancements in CMT research. It is our hope that the use of Drosophila will help facilitate such advancements.
Author Contributions
Conceptualization, F.K.-M. and Y.-i.N.; writing—original draft preparation, F.K.-M.; writing—review and editing, Y.-i.N. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by a Grant-in-Aid (19K16924 to F.K-M.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan and by the Research Committee of Charcot-Marie-Tooth Disease (Grant Number: 17929553) of the Japan Agency for Medical Research and Development (AMED).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| CMT | Charcot-Marie-Tooth disease |
| HMSN | Hereditary motor and sensory neuropathy |
| ALS | Amyotrophic lateral sclerosis |
| NMJ AZ PSDs | Neuromuscular junction Active zone Postsynaptic densities |
References
- Skre, H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974, 6, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Pareyson, D.; Saveri, P.; Pisciotta, C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr. Opin. Neurol. 2017, 30, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Barreto, L.C.; Oliveira, F.S.; Nunes, P.S.; de Franca Costa, I.M.; Garcez, C.A.; Goes, G.M.; Neves, E.L.; de Souza Siqueira Quintans, J.; de Souza Araujo, A.A. Epidemiologic Study of Charcot-Marie-Tooth Disease: A Systematic Review. Neuroepidemiology 2016, 46, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Lefter, S.; Hardiman, O.; Ryan, A.M. A population-based epidemiologic study of adult neuromuscular disease in the Republic of Ireland. Neurology 2017, 88, 304–313. [Google Scholar] [CrossRef]
- Lousa, M.; Vazquez-Huarte-Mendicoa, C.; Gutierrez, A.J.; Saavedra, P.; Navarro, B.; Tugores, A. Genetic epidemiology, demographic, and clinical characteristics of Charcot-Marie-tooth disease in the island of Gran Canaria (Spain). J. Peripher. Nerv. Syst. 2019, 24, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Choi, Y.C.; Oh, J.W.; Yi, S.W. Prevalence, Mortality, and Cause of Death in Charcot-Marie-Tooth Disease in Korea: A Nationwide, Population-Based Study. Neuroepidemiology 2020, 54, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Duchesne, M.; Mathis, S.; Richard, L.; Magdelaine, C.; Corcia, P.; Nouioua, S.; Tazir, M.; Magy, L.; Vallat, J.M. Nerve Biopsy Is Still Useful in Some Inherited Neuropathies. J. Neuropathol. Exp. Neurol. 2018, 77, 88–99. [Google Scholar] [CrossRef]
- Pareyson, D.; Marchesi, C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 2009, 8, 654–667. [Google Scholar] [CrossRef]
- Lee, M.; Park, C.H.; Chung, H.K.; Kim, H.J.; Choi, Y.; Yoo, J.H.; Yoon, Y.C.; Hong, Y.B.; Chung, K.W.; Choi, B.O.; et al. Cerebral white matter abnormalities in patients with charcot-marie-tooth disease. Ann. Neurol. 2017, 81, 147–151. [Google Scholar] [CrossRef]
- Lu, Y.Y.; Lyu, H.; Jin, S.Q.; Zuo, Y.H.; Liu, J.; Wang, Z.X.; Zhang, W.; Yuan, Y. Clinical and Genetic Features of Chinese X-linked Charcot-Marie-Tooth Type 1 Disease. Chin. Med. J. (Engl) 2017, 130, 1049–1054. [Google Scholar] [CrossRef]
- Hu, G.; Zhang, L.; Zhang, M.; Yang, C.; Nie, X.; Xiang, F.; Chen, L.; Dong, Z.; Yu, S. Novel gap junction protein beta-1 gene mutation associated with a stroke-like syndrome and central nervous system involvement in patients with X-linked Charcot-Marie-Tooth Type 1: A case report and literature review. Clin. Neurol. Neurosurg. 2019, 180, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Saifi, G.M.; Szigeti, K.; Snipes, G.J.; Garcia, C.A.; Lupski, J.R. Molecular mechanisms, diagnosis, and rational approaches to management of and therapy for Charcot-Marie-Tooth disease and related peripheral neuropathies. J. Investig. Med. 2003, 51, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Yamamoto, M.; Yoshihara, T.; Koike, H.; Nakagawa, M.; Yoshikawa, H.; Ohnishi, A.; Hayasaka, K.; Onodera, O.; Baba, M.; et al. Demyelinating and axonal features of Charcot-Marie-Tooth disease with mutations of myelin-related proteins (PMP22, MPZ and Cx32): A clinicopathological study of 205 Japanese patients. Brain 2003, 126 Pt 1, 134–151. [Google Scholar] [CrossRef] [Green Version]
- Pipis, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Next-generation sequencing in Charcot-Marie-Tooth disease: Opportunities and challenges. Nat. Rev. Neurol 2019, 15, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Rossor, A.M.; Carr, A.S.; Devine, H.; Chandrashekar, H.; Pelayo-Negro, A.L.; Pareyson, D.; Shy, M.E.; Scherer, S.S.; Reilly, M.M. Peripheral neuropathy in complex inherited diseases: An approach to diagnosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 846–863. [Google Scholar] [CrossRef]
- Murphy, S.M.; Laura, M.; Fawcett, K.; Pandraud, A.; Liu, Y.T.; Davidson, G.L.; Rossor, A.M.; Polke, J.M.; Castleman, V.; Manji, H.; et al. Charcot-Marie-Tooth disease: Frequency of genetic subtypes and guidelines for genetic testing. J. Neurol. Neurosurg. Psychiatry 2012, 83, 706–710. [Google Scholar] [CrossRef]
- Saporta, A.S.; Sottile, S.L.; Miller, L.J.; Feely, S.M.; Siskind, C.E.; Shy, M.E. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 2011, 69, 22–33. [Google Scholar] [CrossRef]
- Sivera, R.; Sevilla, T.; Vilchez, J.J.; Martinez-Rubio, D.; Chumillas, M.J.; Vazquez, J.F.; Muelas, N.; Bataller, L.; Millan, J.M.; Palau, F.; et al. Charcot-Marie-Tooth disease: Genetic and clinical spectrum in a Spanish clinical series. Neurology 2013, 81, 1617–1625. [Google Scholar] [CrossRef] [Green Version]
- Manganelli, F.; Tozza, S.; Pisciotta, C.; Bellone, E.; Iodice, R.; Nolano, M.; Geroldi, A.; Capponi, S.; Mandich, P.; Santoro, L. Charcot-Marie-Tooth disease: Frequency of genetic subtypes in a Southern Italy population. J. Peripher. Nerv. Syst. 2014, 19, 292–298. [Google Scholar] [CrossRef]
- Gess, B.; Schirmacher, A.; Boentert, M.; Young, P. Charcot-Marie-Tooth disease: Frequency of genetic subtypes in a German neuromuscular center population. Neuromuscul. Disord. 2013, 23, 647–651. [Google Scholar] [CrossRef]
- Schroder, J.M. Neuropathology of Charcot-Marie-Tooth and related disorders. Neuromol. Med. 2006, 8, 23–42. [Google Scholar] [CrossRef]
- Patel, P.I.; Roa, B.B.; Welcher, A.A.; Schoener-Scott, R.; Trask, B.J.; Pentao, L.; Snipes, G.J.; Garcia, C.A.; Francke, U.; Shooter, E.M.; et al. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992, 1, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Baets, J.; Deconinck, T.; De Vriendt, E.; Zimon, M.; Yperzeele, L.; Van Hoorenbeeck, K.; Peeters, K.; Spiegel, R.; Parman, Y.; Ceulemans, B.; et al. Genetic spectrum of hereditary neuropathies with onset in the first year of life. Brain 2011, 134 Pt 9, 2664–2676. [Google Scholar] [CrossRef]
- Plante-Bordeneuve, V.; Said, G. Dejerine-Sottas disease and hereditary demyelinating polyneuropathy of infancy. Muscle Nerve 2002, 26, 608–621. [Google Scholar] [CrossRef]
- Tazir, M.; Bellatache, M.; Nouioua, S.; Vallat, J.M. Autosomal recessive Charcot-Marie-Tooth disease: From genes to phenotypes. J. Peripher. Nerv. Syst. 2013, 18, 113–129. [Google Scholar] [CrossRef]
- Nicot, A.S.; Laporte, J. Endosomal phosphoinositides and human diseases. Traffic 2008, 9, 1240–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaccari, I.; Carbone, A.; Previtali, S.C.; Mironova, Y.A.; Alberizzi, V.; Noseda, R.; Rivellini, C.; Bianchi, F.; Del Carro, U.; D’Antonio, M.; et al. Loss of Fig4 in both Schwann cells and motor neurons contributes to CMT4J neuropathy. Hum. Mol. Genet. 2015, 24, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; McCollum, M.; Ravi, V.; Arpag, S.; Moiseev, D.; Castoro, R.; Mobley, B.; Burnette, B.; Siskind, C.; Day, J.; et al. Myelin abnormality in Charcot-Marie-Tooth type 4J recapitulates features of acquired demyelination. Ann. Neurol. 2018, 83, 756–770. [Google Scholar] [CrossRef]
- Shy, M.E.; Siskind, C.; Swan, E.R.; Krajewski, K.M.; Doherty, T.; Fuerst, D.R.; Ainsworth, P.J.; Lewis, R.A.; Scherer, S.S.; Hahn, A.F. CMT1X phenotypes represent loss of GJB1 gene function. Neurology 2007, 68, 849–855. [Google Scholar] [CrossRef]
- Ionasescu, V.V.; Trofatter, J.; Haines, J.L.; Summers, A.M.; Ionasescu, R.; Searby, C. X-linked recessive Charcot-Marie-Tooth neuropathy: Clinical and genetic study. Muscle Nerve 1992, 15, 368–373. [Google Scholar] [CrossRef]
- Kanhangad, M.; Cornett, K.; Brewer, M.H.; Nicholson, G.A.; Ryan, M.M.; Smith, R.L.; Subramanian, G.M.; Young, H.K.; Zuchner, S.; Kennerson, M.L.; et al. Unique clinical and neurophysiologic profile of a cohort of children with CMTX3. Neurology 2018, 90, e1706–e1710. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Hong, S.H.; Ki, C.S.; Kim, B.J.; Shim, J.S.; Cho, S.H.; Park, J.H.; Kim, J.W. A novel locus for X-linked recessive CMT with deafness and optic neuropathy maps to Xq21.32-q24. Neurology 2005, 64, 1964–1967. [Google Scholar] [CrossRef] [PubMed]
- Kennerson, M.L.; Yiu, E.M.; Chuang, D.T.; Kidambi, A.; Tso, S.C.; Ly, C.; Chaudhry, R.; Drew, A.P.; Rance, G.; Delatycki, M.B.; et al. A new locus for X-linked dominant Charcot-Marie-Tooth disease (CMTX6) is caused by mutations in the pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) gene. Hum. Mol. Genet. 2013, 22, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Vondracek, P.; Seeman, P.; Hermanova, M.; Fajkusova, L. X-linked Charcot-Marie-Tooth disease: Phenotypic expression of a novel mutation Ile127Ser in the GJB1 (connexin 32) gene. Muscle Nerve 2005, 31, 252–255. [Google Scholar] [CrossRef]
- Bergoffen, J.; Scherer, S.S.; Wang, S.; Scott, M.O.; Bone, L.J.; Paul, D.L.; Chen, K.; Lensch, M.W.; Chance, P.F.; Fischbeck, K.H. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 1993, 262, 2039–2042. [Google Scholar] [CrossRef]
- Paulson, H.L.; Garbern, J.Y.; Hoban, T.F.; Krajewski, K.M.; Lewis, R.A.; Fischbeck, K.H.; Grossman, R.I.; Lenkinski, R.; Kamholz, J.A.; Shy, M.E. Transient central nervous system white matter abnormality in X-linked Charcot-Marie-Tooth disease. Ann. Neurol. 2002, 52, 429–434. [Google Scholar] [CrossRef]
- Hayasaka, K.; Himoro, M.; Sato, W.; Takada, G.; Uyemura, K.; Shimizu, N.; Bird, T.D.; Conneally, P.M.; Chance, P.F. Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat. Genet. 1993, 5, 31–34. [Google Scholar] [CrossRef]
- Martini, R.; Zielasek, J.; Toyka, K.V.; Giese, K.P.; Schachner, M. Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nat. Genet. 1995, 11, 281–286. [Google Scholar] [CrossRef]
- Huxley, C.; Passage, E.; Manson, A.; Putzu, G.; Figarella-Branger, D.; Pellissier, J.F.; Fontes, M. Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum. Mol. Genet. 1996, 5, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Sereda, M.; Griffiths, I.; Puhlhofer, A.; Stewart, H.; Rossner, M.J.; Zimmerman, F.; Magyar, J.P.; Schneider, A.; Hund, E.; Meinck, H.M.; et al. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron 1996, 16, 1049–1060. [Google Scholar] [CrossRef] [Green Version]
- Magyar, J.P.; Martini, R.; Ruelicke, T.; Aguzzi, A.; Adlkofer, K.; Dembic, Z.; Zielasek, J.; Toyka, K.V.; Suter, U. Impaired differentiation of Schwann cells in transgenic mice with increased PMP22 gene dosage. J. Neurosci. 1996, 16, 5351–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzini, P.; Neuberg, D.H.; Schachner, M.; Nelles, E.; Willecke, K.; Zielasek, J.; Toyka, K.V.; Suter, U.; Martini, R. Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J. Neurosci. 1997, 17, 4545–4551. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, C.S.; Sherman, D.L.; Fleetwood-Walker, S.M.; Cottrell, D.F.; Tait, S.; Garry, E.M.; Wallace, V.C.; Ure, J.; Griffiths, I.R.; Smith, A.; et al. Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron 2000, 26, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.M.; Perea, J.; McGuigan, A.; King, R.H.; Muddle, J.R.; Gabreels-Festen, A.A.; Thomas, P.K.; Huxley, C. Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A. J. Anat. 2002, 200, 377–390. [Google Scholar] [CrossRef]
- Grandis, M.; Leandri, M.; Vigo, T.; Cilli, M.; Sereda, M.W.; Gherardi, G.; Benedetti, L.; Mancardi, G.; Abbruzzese, M.; Nave, K.A.; et al. Early abnormalities in sciatic nerve function and structure in a rat model of Charcot-Marie-Tooth type 1A disease. Exp. Neurol. 2004, 190, 213–223. [Google Scholar] [CrossRef]
- Dequen, F.; Filali, M.; Lariviere, R.C.; Perrot, R.; Hisanaga, S.; Julien, J.P. Reversal of neuropathy phenotypes in conditional mouse model of Charcot-Marie-Tooth disease type 2E. Hum. Mol. Genet. 2010, 19, 2616–2629. [Google Scholar] [CrossRef] [Green Version]
- Fledrich, R.; Schlotter-Weigel, B.; Schnizer, T.J.; Wichert, S.P.; Stassart, R.M.; Meyer zu Horste, G.; Klink, A.; Weiss, B.G.; Haag, U.; Walter, M.C.; et al. A rat model of Charcot-Marie-Tooth disease 1A recapitulates disease variability and supplies biomarkers of axonal loss in patients. Brain 2012, 135 Pt 1, 72–87. [Google Scholar] [CrossRef]
- d’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Vanden Berghe, P.; Timmerman, V.; Robberecht, W.; Van Den Bosch, L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef]
- Chapman, A.L.; Bennett, E.J.; Ramesh, T.M.; De Vos, K.J.; Grierson, A.J. Axonal Transport Defects in a Mitofusin 2 Loss of Function Model of Charcot-Marie-Tooth Disease in Zebrafish. PLoS ONE 2013, 8, e67276. [Google Scholar] [CrossRef] [Green Version]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Storkebaum, E.; Leitao-Goncalves, R.; Godenschwege, T.; Nangle, L.; Mejia, M.; Bosmans, I.; Ooms, T.; Jacobs, A.; Van Dijck, P.; Yang, X.L.; et al. Dominant mutations in the tyrosyl-tRNA synthetase gene recapitulate in Drosophila features of human Charcot-Marie-Tooth neuropathy. Proc. Nat. Acad. Sci. USA 2009, 106, 11782–11787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eschenbacher, W.H.; Song, M.; Chen, Y.; Bhandari, P.; Zhao, P.; Jowdy, C.C.; Engelhard, J.T.; Dorn, G.W., 2nd. Two rare human mitofusin 2 mutations alter mitochondrial dynamics and induce retinal and cardiac pathology in Drosophila. PLoS ONE 2012, 7, e44296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saporta, M.A.; Dang, V.; Volfson, D.; Zou, B.; Xie, X.S.; Adebola, A.; Liem, R.K.; Shy, M.; Dimos, J.T. Axonal Charcot-Marie-Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Exp. Neurol. 2015, 263, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohara, R.; Imamura, K.; Morii, F.; Egawa, N.; Tsukita, K.; Enami, T.; Shibukawa, R.; Mizuno, T.; Nakagawa, M.; Inoue, H. Modeling Drug-Induced Neuropathy Using Human iPSCs for Predictive Toxicology. Clin. Pharmacol. Ther. 2017, 101, 754–762. [Google Scholar] [CrossRef]
- Kitani-Morii, F.; Imamura, K.; Kondo, T.; Ohara, R.; Enami, T.; Shibukawa, R.; Yamamoto, T.; Sekiguchi, K.; Toguchida, J.; Mizuno, T.; et al. Analysis of neural crest cells from Charcot-Marie-Tooth disease patients demonstrates disease-relevant molecular signature. Neuroreport 2017, 28, 814–821. [Google Scholar] [CrossRef]
- Juneja, M.; Burns, J.; Saporta, M.A.; Timmerman, V. Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J. Neurol. Neurosurg. Psychiatry 2019, 90, 58–67. [Google Scholar] [CrossRef]
- Rzepnikowska, W.; Kaminska, J.; Kabzinska, D.; Binieda, K.; Kochanski, A. A Yeast-Based Model for Hereditary Motor and Sensory Neuropathies: A Simple System for Complex, Heterogeneous Diseases. Int. J. Mol. Sci. 2020, 21, 4277. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Grunwald, D.J.; Eisen, J.S. Headwaters of the zebrafish—Emergence of a new model vertebrate. Nat. Rev. Genet. 2002, 3, 717–724. [Google Scholar] [CrossRef]
- Venken, K.J.; Bellen, H.J. Emerging technologies for gene manipulation in Drosophila melanogaster. Nat. Rev. Genet. 2005, 6, 167–178. [Google Scholar] [CrossRef]
- Nichols, C.D.; Becnel, J.; Pandey, U.B. Methods to assay Drosophila behavior. J. Vis. Exp 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bourg, E.; Lints, F.A. Hypergravity and aging in Drosophila melanogaster. 4. Climbing activity. Gerontology 1992, 38, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Min, V.A.; Condron, B.G. An assay of behavioral plasticity in Drosophila larvae. J. Neurosci. Methods 2005, 145, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.K.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016, 540, 74–79. [Google Scholar] [CrossRef]
- Misko, A.L.; Sasaki, Y.; Tuck, E.; Milbrandt, J.; Baloh, R.H. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 2012, 32, 4145–4155. [Google Scholar] [CrossRef] [Green Version]
- Debattisti, V.; Pendin, D.; Ziviani, E.; Daga, A.; Scorrano, L. Reduction of endoplasmic reticulum stress attenuates the defects caused by Drosophila mitofusin depletion. J. Cell Biol. 2014, 204, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Trevisan, T.; Pendin, D.; Montagna, A.; Bova, S.; Ghelli, A.M.; Daga, A. Manipulation of Mitochondria Dynamics Reveals Separate Roles for Form and Function in Mitochondria Distribution. Cell Rep. 2018, 23, 1742–1753. [Google Scholar] [CrossRef] [Green Version]
- El Fissi, N.; Rojo, M.; Aouane, A.; Karatas, E.; Poliacikova, G.; David, C.; Royet, J.; Rival, T. Mitofusin gain and loss of function drive pathogenesis in Drosophila models of CMT2A neuropathy. EMBO Rep. 2018, 19, e45241. [Google Scholar] [CrossRef]
- Garrido-Maraver, J.; Celardo, I.; Costa, A.C.; Lehmann, S.; Loh, S.H.Y.; Martins, L.M. Enhancing folic acid metabolism suppresses defects associated with loss of Drosophila mitofusin. Cell Death Dis. 2019, 10, 288. [Google Scholar] [CrossRef] [Green Version]
- Baxter, R.V.; Ben Othmane, K.; Rochelle, J.M.; Stajich, J.E.; Hulette, C.; Dew-Knight, S.; Hentati, F.; Ben Hamida, M.; Bel, S.; Stenger, J.E.; et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 2002, 30, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Bouhouche, A.; Birouk, N.; Azzedine, H.; Benomar, A.; Durosier, G.; Ente, D.; Muriel, M.P.; Ruberg, M.; Slassi, I.; Yahyaoui, M.; et al. Autosomal recessive axonal Charcot-Marie-Tooth disease (ARCMT2): Phenotype-genotype correlations in 13 Moroccan families. Brain 2007, 130 Pt 4, 1062–1075. [Google Scholar] [CrossRef] [Green Version]
- Niemann, A.; Ruegg, M.; La Padula, V.; Schenone, A.; Suter, U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: New implications for Charcot-Marie-Tooth disease. J. Cell Biol. 2005, 170, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Zimon, M.; Baets, J.; Fabrizi, G.M.; Jaakkola, E.; Kabzinska, D.; Pilch, J.; Schindler, A.B.; Cornblath, D.R.; Fischbeck, K.H.; Auer-Grumbach, M.; et al. Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 2011, 77, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Del Amo, V.; Seco-Cervera, M.; Garcia-Gimenez, J.L.; Whitworth, A.J.; Pallardo, F.V.; Galindo, M.I. Mitochondrial defects and neuromuscular degeneration caused by altered expression of Drosophila Gdap1: Implications for the Charcot-Marie-Tooth neuropathy. Hum. Mol. Genet. 2015, 24, 21–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Del Amo, V.; Palomino-Schatzlein, M.; Seco-Cervera, M.; Garcia-Gimenez, J.L.; Pallardo, F.V.; Pineda-Lucena, A.; Galindo, M.I. A Drosophila model of GDAP1 function reveals the involvement of insulin signalling in the mitochondria-dependent neuromuscular degeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 801–809. [Google Scholar] [CrossRef]
- Li, Z.; Peng, Y.; Hufnagel, R.B.; Hu, Y.C.; Zhao, C.; Queme, L.F.; Khuchua, Z.; Driver, A.M.; Dong, F.; Lu, Q.R.; et al. Loss of SLC25A46 causes neurodegeneration by affecting mitochondrial dynamics and energy production in mice. Hum. Mol. Genet. 2017, 26, 3776–3791. [Google Scholar] [CrossRef]
- Suda, K.; Ueoka, I.; Azuma, Y.; Muraoka, Y.; Yoshida, H.; Yamaguchi, M. Novel Drosophila model for mitochondrial diseases by targeting of a solute carrier protein SLC25A46. Brain Res. 2018, 1689, 30–44. [Google Scholar] [CrossRef]
- Ali, M.S.; Suda, K.; Kowada, R.; Ueoka, I.; Yoshida, H.; Yamaguchi, M. Neuron-specific knockdown of solute carrier protein SLC25A46a induces locomotive defects, an abnormal neuron terminal morphology, learning disability, and shortened lifespan. IBRO Rep. 2020, 8, 65–75. [Google Scholar] [CrossRef]
- Suda, K.; Muraoka, Y.; Ortega-Yanez, A.; Yoshida, H.; Kizu, F.; Hochin, T.; Kimura, H.; Yamaguchi, M. Reduction of Rpd3 suppresses defects in locomotive ability and neuronal morphology induced by the knockdown of Drosophila SLC25A46 via an epigenetic pathway. Exp. Cell Res. 2019, 385, 111673. [Google Scholar] [CrossRef]
- Chow, C.Y.; Zhang, Y.; Dowling, J.J.; Jin, N.; Adamska, M.; Shiga, K.; Szigeti, K.; Shy, M.E.; Li, J.; Zhang, X.; et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 2007, 448, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCartney, A.J.; Zhang, Y.; Weisman, L.S. Phosphatidylinositol 3,5-bisphosphate: Low abundance, high significance. Bioessays 2014, 36, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyotani, A.; Azuma, Y.; Yamamoto, I.; Yoshida, H.; Mizuta, I.; Mizuno, T.; Nakagawa, M.; Tokuda, T.; Yamaguchi, M. Knockdown of the Drosophila FIG4 induces deficient locomotive behavior, shortening of motor neuron, axonal targeting aberration, reduction of life span and defects in eye development. Exp. Neurol. 2016, 277, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, R.; Cunningham, K.M.; Zhang, K.; Lloyd, T.E. FIG4 regulates lysosome membrane homeostasis independent of phosphatase function. Hum. Mol. Genet. 2016, 25, 681–692. [Google Scholar] [CrossRef]
- Muraoka, Y.; Nakamura, A.; Tanaka, R.; Suda, K.; Azuma, Y.; Kushimura, Y.; Lo Piccolo, L.; Yoshida, H.; Mizuta, I.; Tokuda, T.; et al. Genetic screening of the genes interacting with Drosophila FIG4 identified a novel link between CMT-causing gene and long noncoding RNAs. Exp. Neurol. 2018, 310, 1–13. [Google Scholar] [CrossRef]
- Shimada, S.; Muraoka, Y.; Ibaraki, K.; Takano-Shimizu-Kouno, T.; Yoshida, H.; Yamaguchi, M. Identification of CR43467 encoding a long non-coding RNA as a novel genetic interactant with dFIG4, a CMT-causing gene. Exp. Cell Res. 2020, 386, 111711. [Google Scholar] [CrossRef]
- Auer-Grumbach, M.; De Jonghe, P.; Wagner, K.; Verhoeven, K.; Hartung, H.P.; Timmerman, V. Phenotype-genotype correlations in a CMT2B family with refined 3q13-q22 locus. Neurology 2000, 55, 1552–1557. [Google Scholar] [CrossRef]
- Langemeyer, L.; Frohlich, F.; Ungermann, C. Rab GTPase Function in Endosome and Lysosome Biogenesis. Trends Cell Biol. 2018, 28, 957–970. [Google Scholar] [CrossRef]
- Janssens, K.; Goethals, S.; Atkinson, D.; Ermanoska, B.; Fransen, E.; Jordanova, A.; Auer-Grumbach, M.; Asselbergh, B.; Timmerman, V. Human Rab7 mutation mimics features of Charcot-Marie-Tooth neuropathy type 2B in Drosophila. Neurobiol. Dis. 2014, 65, 211–219. [Google Scholar] [CrossRef]
- Cioni, J.M.; Lin, J.Q.; Holtermann, A.V.; Koppers, M.; Jakobs, M.A.H.; Azizi, A.; Turner-Bridger, B.; Shigeoka, T.; Franze, K.; Harris, W.A.; et al. Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 2019, 176, 56–72e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonellis, A.; Ellsworth, R.E.; Sambuughin, N.; Puls, I.; Abel, A.; Lee-Lin, S.Q.; Jordanova, A.; Kremensky, I.; Christodoulou, K.; Middleton, L.T.; et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003, 72, 1293–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordanova, A.; Irobi, J.; Thomas, F.P.; Van Dijck, P.; Meerschaert, K.; Dewil, M.; Dierick, I.; Jacobs, A.; De Vriendt, E.; Guergueltcheva, V.; et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006, 38, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latour, P.; Thauvin-Robinet, C.; Baudelet-Mery, C.; Soichot, P.; Cusin, V.; Faivre, L.; Locatelli, M.C.; Mayencon, M.; Sarcey, A.; Broussolle, E.; et al. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2010, 86, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Vester, A.; Velez-Ruiz, G.; McLaughlin, H.M.; Program, N.C.S.; Lupski, J.R.; Talbot, K.; Vance, J.M.; Zuchner, S.; Roda, R.H.; Fischbeck, K.H.; et al. A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum. Mutat. 2013, 34, 191–199. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, H.M.; Sakaguchi, R.; Liu, C.; Igarashi, T.; Pehlivan, D.; Chu, K.; Iyer, R.; Cruz, P.; Cherukuri, P.F.; Hansen, N.F.; et al. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am. J. Hum. Genet. 2010, 87, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, M.; McLaughlin, H.; Houlden, H.; Guo, M.; Yo-Tsen, L.; Hadjivassilious, M.; Speziani, F.; Yang, X.L.; Antonellis, A.; Reilly, M.M.; et al. Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1247–1249. [Google Scholar] [CrossRef] [Green Version]
- Ermanoska, B.; Motley, W.W.; Leitao-Goncalves, R.; Asselbergh, B.; Lee, L.H.; De Rijk, P.; Sleegers, K.; Ooms, T.; Godenschwege, T.A.; Timmerman, V.; et al. CMT-associated mutations in glycyl- and tyrosyl-tRNA synthetases exhibit similar pattern of toxicity and share common genetic modifiers in Drosophila. Neurobiol. Dis. 2014, 68, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Grice, S.J.; Liu, J.L.; Webber, C. Synergistic interactions between Drosophila orthologues of genes spanned by de novo human CNVs support multiple-hit models of autism. PLoS Genet. 2015, 11, e1004998. [Google Scholar] [CrossRef]
- Niehues, S.; Bussmann, J.; Steffes, G.; Erdmann, I.; Kohrer, C.; Sun, L.; Wagner, M.; Schafer, K.; Wang, G.; Koerdt, S.N.; et al. Impaired protein translation in Drosophila models for Charcot-Marie-Tooth neuropathy caused by mutant tRNA synthetases. Nat. Commun. 2015, 6, 7520. [Google Scholar] [CrossRef] [Green Version]
- Bussmann, J.; Storkebaum, E. Molecular pathogenesis of peripheral neuropathies: Insights from Drosophila models. Curr. Opin. Genet. Dev. 2017, 44, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Bervoets, S.; Wei, N.; Erfurth, M.L.; Yusein-Myashkova, S.; Ermanoska, B.; Mateiu, L.; Asselbergh, B.; Blocquel, D.; Kakad, P.; Penserga, T.; et al. Transcriptional dysregulation by a nucleus-localized aminoacyl-tRNA synthetase associated with Charcot-Marie-Tooth neuropathy. Nat. Commun. 2019, 10, 5045. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Tanaka, Y. Kinesin superfamily proteins (KIFs): Various functions and their relevance for important phenomena in life and diseases. Exp. Cell Res. 2015, 334, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Takita, J.; Tanaka, Y.; Setou, M.; Nakagawa, T.; Takeda, S.; Yang, H.W.; Terada, S.; Nakata, T.; Takei, Y.; et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell 2001, 105, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Riviere, J.B.; Ramalingam, S.; Lavastre, V.; Shekarabi, M.; Holbert, S.; Lafontaine, J.; Srour, M.; Merner, N.; Rochefort, D.; Hince, P.; et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am. J. Hum. Genet. 2011, 89, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Weedon, M.N.; Hastings, R.; Caswell, R.; Xie, W.; Paszkiewicz, K.; Antoniadi, T.; Williams, M.; King, C.; Greenhalgh, L.; Newbury-Ecob, R.; et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2011, 89, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Kern, J.V.; Zhang, Y.V.; Kramer, S.; Brenman, J.E.; Rasse, T.M. The kinesin-3, unc-104 regulates dendrite morphogenesis and synaptic development in Drosophila. Genetics 2013, 195, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.V.; Hannan, S.B.; Stapper, Z.A.; Kern, J.V.; Jahn, T.R.; Rasse, T.M. The Drosophila KIF1A Homolog unc-104 Is Important for Site-Specific Synapse Maturation. Front. Cell Neurosci. 2016, 10, 207. [Google Scholar] [CrossRef] [Green Version]
- Baumann, S.; Komissarov, A.; Gili, M.; Ruprecht, V.; Wieser, S.; Maurer, S.P. A reconstituted mammalian APC-kinesin complex selectively transports defined packages of axonal mRNAs. Sci. Adv. 2020, 6, eaaz1588. [Google Scholar] [CrossRef] [Green Version]
- El-Kabbani, O.; Darmanin, C.; Chung, R.P. Sorbitol dehydrogenase: Structure, function and ligand design. Curr. Med. Chem. 2004, 11, 465–476. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Serrano, A.M.; Duarte, J.M.N. Brain Metabolism Alterations in Type 2 Diabetes: What Did We Learn From Diet-Induced Diabetes Models? Front. Neurosci. 2020, 14, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese, A.; Zhu, Y.; Rebelo, A.P.; Negri, S.; Courel, S.; Abreu, L.; Bacon, C.J.; Bai, Y.; Bis-Brewer, D.M.; Bugiardini, E.; et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 2020, 52, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Luque, T.; Hjelmqvist, L.; Marfany, G.; Danielsson, O.; El-Ahmad, M.; Persson, B.; Jornvall, H.; Gonzalez-Duarte, R. Sorbitol dehydrogenase of Drosophila. Gene, protein, and expression data show a two-gene system. J. Biol. Chem. 1998, 273, 34293–34301. [Google Scholar] [CrossRef] [Green Version]
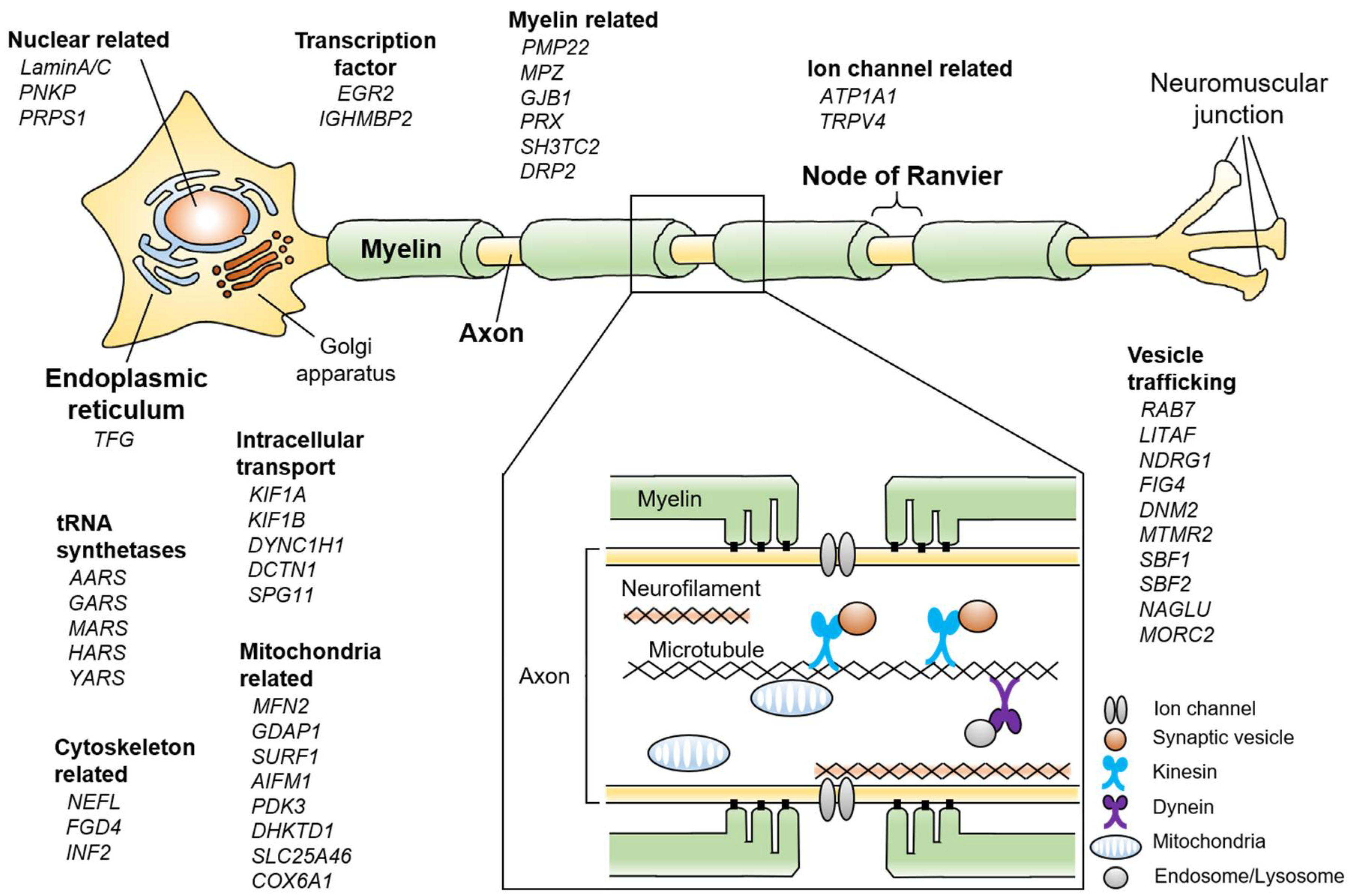
Figure 1.
Schematic summary showing various Charcot-Marie-Tooth disease (CMT)-related genes and pathways in the peripheral nerve. If each gene has multiple functions, the most representative one is described. The enlarged box shows a cross-sectional view of the peripheral nerve.
Figure 1.
Schematic summary showing various Charcot-Marie-Tooth disease (CMT)-related genes and pathways in the peripheral nerve. If each gene has multiple functions, the most representative one is described. The enlarged box shows a cross-sectional view of the peripheral nerve.


{kind=link}
Table 1.
Characteristics of genes in which mutations are reported to cause CMT.
| CMT Subtype | Genomic Locus | Gene Symbol | Biological Functions | Drosophila Homolog | Phenotype MIM |
|---|---|---|---|---|---|
| CMT1 (Demyelinating, autosomal dominant) | |||||
| CMT1A | 17p12 | PMP22 | Myelin protein | - | 118220 |
| CMT1B | 1q23 | MPZ | Myelin protein | - | 118200 |
| CMT1C | 16p13.3 | LITAF | Regulation of endosomal trafficking | CG13510, CG13559, CG32280 | 601098 |
| CMT1D | 10q21 | EGR2 | Transcription factor | sr | 607678 |
| CMT1E | 17p12 | PMP22 | Myelin protein | - | 118300 |
| CMT1F | 8p21 | NEFL | Neurofilament protein | - | 607684 |
| CMT1G | 8q21 | PMP2 | Myelin protein | - | 618279 |
| HNPP | 17p12 | PMP22 | Myelin protein | - | 162500 |
| CMT2 (Axonal, autosomal dominant) | |||||
| CMT2A1 | 1p36 | KIF1B | Intracellular transport | unc-104 | 118210 |
| CMT2A2 | 1p36 | MFN2 | Mitochondrial dynamics | Marf | 609260 |
| CMT2B | 3q21 | RAB7 | Regulation of vesicular transport | Rab7 | 600882 |
| CMT2C | 12q24 | TRPV4 | Regulation of calcium ion influx | nan, iav | 606071 |
| CMT2D | 7p14 | GARS | Protein translation | gars, GlyRS | 601472 |
| CMT2E | 8p21 | NEFL | Neurofilament protein | - | 607684 |
| CMT2F | 7q11 | HSPB1 | Microtubule regulator and chaperon activity | heat shock protein family B member | 606595 |
| CMT2I/J | 1q22 | MPZ | Myelin protein | - | 607736 |
| CMT2K | 8q21 | GDAP1 | Mitochondrial dynamics | dGdap1 | 607831 |
| CMT2L | 12q24 | HSPB8 | Microtubule regulator and chaperon activity | - | 608673 |
| CMT2M | 19q13 | DNM2 | Endocytosis and regulation of cell motility | shi | 606482 |
| CMT2N | 16q22 | AARS | Protein translation | - | 613287 |
| CMT2O | 14q32 | DYNC1H1 | Intracellular transport | dynein heavy chain 64C | 614228 |
| CMT2P | 9q33 | LRSAM1 | E3 ubiquitin ligase | - | 614436 |
| CMT2Q | 10p14 | DHKTD1 | Mitochondrial biogenesis | CG1544 | 615025 |
| CMT2U | 12q13 | MARS | Protein translation | mars, MetRS | 616280 |
| CMT2V | 17q21 | NAGLU | Lysosomal enzyme | CG13397 | 616491 |
| CMT2W | 5q31 | HARS | Protein translation | hars, HisRS | 616625 |
| CMT2Y | 9q13 | VCP | Regulation of autophagy | TER94 | 616687 |
| CMT2Z | 22q12 | MORC2 | Fatty acid metabolism | - | 616688 |
| CMT2DD | 1p13 | ATP1A1 | Ion channel at Ranvier nodes | - | 618036 |
| HMSN-P | 3q12 | TFG | ER vesicle trafficking | - | 604484 |
| CMT2 (Axonal, autosomal recessive) | |||||
| AR-CMT2A | 1q22 | LaminA/C | Nuclear membrane protein | Lam | 605588 |
| AR-CMT2B | 19q13 | PNKP | Regulation of phosphorylation of nucleic acids | CG9601 | 605589 |
| AR-CMT2F | 7q11 | HSPB1 | Microtubule regulator and chaperon activity | heat shock protein family B member | 606595 |
| AR-CMT2K | 8q21 | GDAP1 | Mitochondrial dynamics | dGdap1 | 607831 |
| AR-CMT2P | 9q33 | LRSAM1 | E3 ubiquitin ligase | - | 614436 |
| AR-CMT2R | 4q31 | TRIM2 | E3 ubiquitin ligase | - | 615490 |
| AR-CMT2S | 11q13 | IGHMBP2 | Transcription factor | CG30094 | 616155 |
| AR-CMT2T | 3q25 | MME | Neutral endopeptidase | Nep1, Nep2 | 617017 |
| AR-CMT2X | 15q21 | SPG11 | Membrane associated | CG13531 | 616668 |
| AR-CMT2A2B | 1p36 | MFN2 | Mitochondrial dynamics | Marf | 617087 |
| HMSN6B | 5q22 | SLC25A46 | Mitochondrial dynamics | Slc25A46b | 616505 |
| SCAN3 | 1p32.3 | COA7 | Mitochondrial biogenesis | Coa7 | 618387 |
| HSMN IIC | 2q37 | KIF1A | Intracellular transport | unc-104 | 614213 |
| CMT4 (Demyelinating, Autosomal recessive) | |||||
| CMT4A | 8q13-q21.1 | GDAP1 | Mitochondrial dynamics | dGdap1 | 214400 |
| CMT4B1 | 11q22 | MTMR2 | Regulation of phosphorylation of Phosphatidylinositol | mtm | 601382 |
| CMT4B2 | 11p15 | SBF2 | Signaling pathway | Sbf | 604563 |
| CMT4B3 | 22q13 | SBF1 | Signaling pathway | Sbf | 615284 |
| CMT4C | 5q23-q33 | SH3TC2 | Myelin maturation | - | 601596 |
| CMT4D | 8q24 | NDRG1 | Vesicle transport | MESK2 | 601455 |
| CMT4E | 10q21-q22 | EGR2 | Transcription factor | - | 605253 |
| CMT4F | 19q13 | PRX | Myelin maturation | - | 614895 |
| CMT4G | 10q22 | HK1 | Glucose metabolism | Hex-A | 605285 |
| CMT4H | 12p11.2 | FGD4 | Regulation of actin fibers | - | 609311 |
| CMT4J | 6p21 | FIG4 | Endo-lysosomal trafficking | dFig4 | 611228 |
| CMT4K | 9q34 | SURF1 | Mitochondrial biogenesis | Surf1 | 616684 |
| X-linked CMT | |||||
| Dominant | |||||
| CMTX1 | Xq13 | GJB1 | Gap junction formation | - | 302800 |
| CMTX3 | Xq27 | - | - | - | 302802 |
| Semi-dominant | |||||
| CMTX6 | Xp22 | PDK3 | Mitochondrial biogenesis | Pdk | 300905 |
| Recessive | |||||
| CMTX2 | Xp22.2 | - | - | - | 302801 |
| CMTX4 | Xq26 | AIFM1 | Mitochondrial biogenesis | AIF | 310490 |
| CMTX5 | Xq22 | PRPS1 | Nucleotide biosynthesis | Prps | 311070 |
| CMT (Intermediate NCV, autosomal dominant) | |||||
| CMT-DIA | 10q24 | - | - | - | 606483 |
| CMT-IB | 19p13 | DNM2 | Regulation of cellular proliferation | - | 606482 |
| CMT-DIC | 1p35 | YARS | Protein translation | yars, TyrRS | 608323 |
| CMT-DID | 1q22 | MPZ | Myelin protein | - | 607791 |
| CMT-DIE | 14q32 | INF2 | Regulation of actin fibers | form3 | 614455 |
| CMT-DIF | 3q26 | GNB4 | Signaling pathway | Gβ13F | 615185 |
| CMT-DIG | 8p21 | NEFL | Neurofilament protein | - | 617882 |
| CMT (Intermediate NCV, autosomal recessive) | |||||
| CMT RIA | 8q21.1 | GDAP1 | Mitochondrial dynamics | dGdap1 | 608340 |
| CMT RIB | 16q23 | KARS | Protein translation | kars, LysRS | 613641 |
| CMT RIC | 1p36 | PLEKHG5 | Signaling pathway | CG42674 | 615376 |
| CMT RID | 12q24 | COX6A1 | Mitochondrial biogenesis | levy, COX6AL, CG14077 | 616039 |
| CMTXI | Xq22 | DRP2 | Myelin maturation | - | 300052 |
Abbreviations; CMT: Charcot-Marie-Tooth disease; MIM: Mendelian Inheritance in Man; HNPP: Hereditary neuropathy with liability to pressure palsy; HMSN-P: hereditary motor and sensory neuropathy with proximal dominant involvement; ER: Endoplasmic reticulum; SCAN: spinocerebellar ataxia with axonal neuropathy; NCV: nerve conduction velocity.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kitani-Morii, F.; Noto, Y.-i. Recent Advances in Drosophila Models of Charcot-Marie-Tooth Disease. Int. J. Mol. Sci. 2020, 21, 7419. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197419
AMA Style
Kitani-Morii F, Noto Y-i. Recent Advances in Drosophila Models of Charcot-Marie-Tooth Disease. International Journal of Molecular Sciences. 2020; 21(19):7419. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197419
Chicago/Turabian StyleKitani-Morii, Fukiko, and Yu-ichi Noto. 2020. "Recent Advances in Drosophila Models of Charcot-Marie-Tooth Disease" International Journal of Molecular Sciences 21, no. 19: 7419. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197419
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.