Na+-Coupled Nutrient Cotransport Induced Luminal Negative Potential and Claudin-15 Play an Important Role in Paracellular Na+ Recycling in Mouse Small Intestine

 , , and
, , and
Abstract
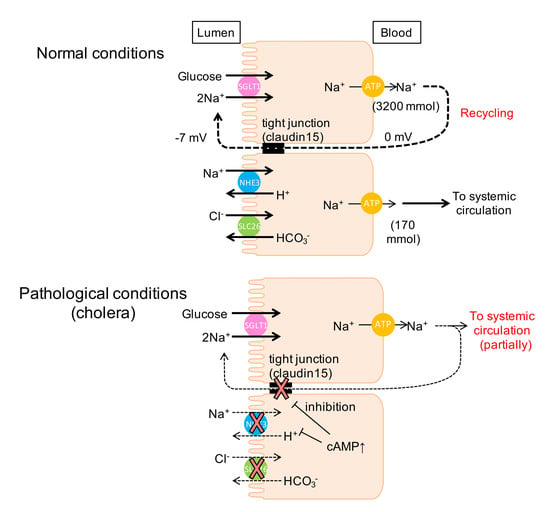
:
1. Introduction
2. Results
2.1. Baseline Na+ Absorption Mechanisms in Wild-Type Mice
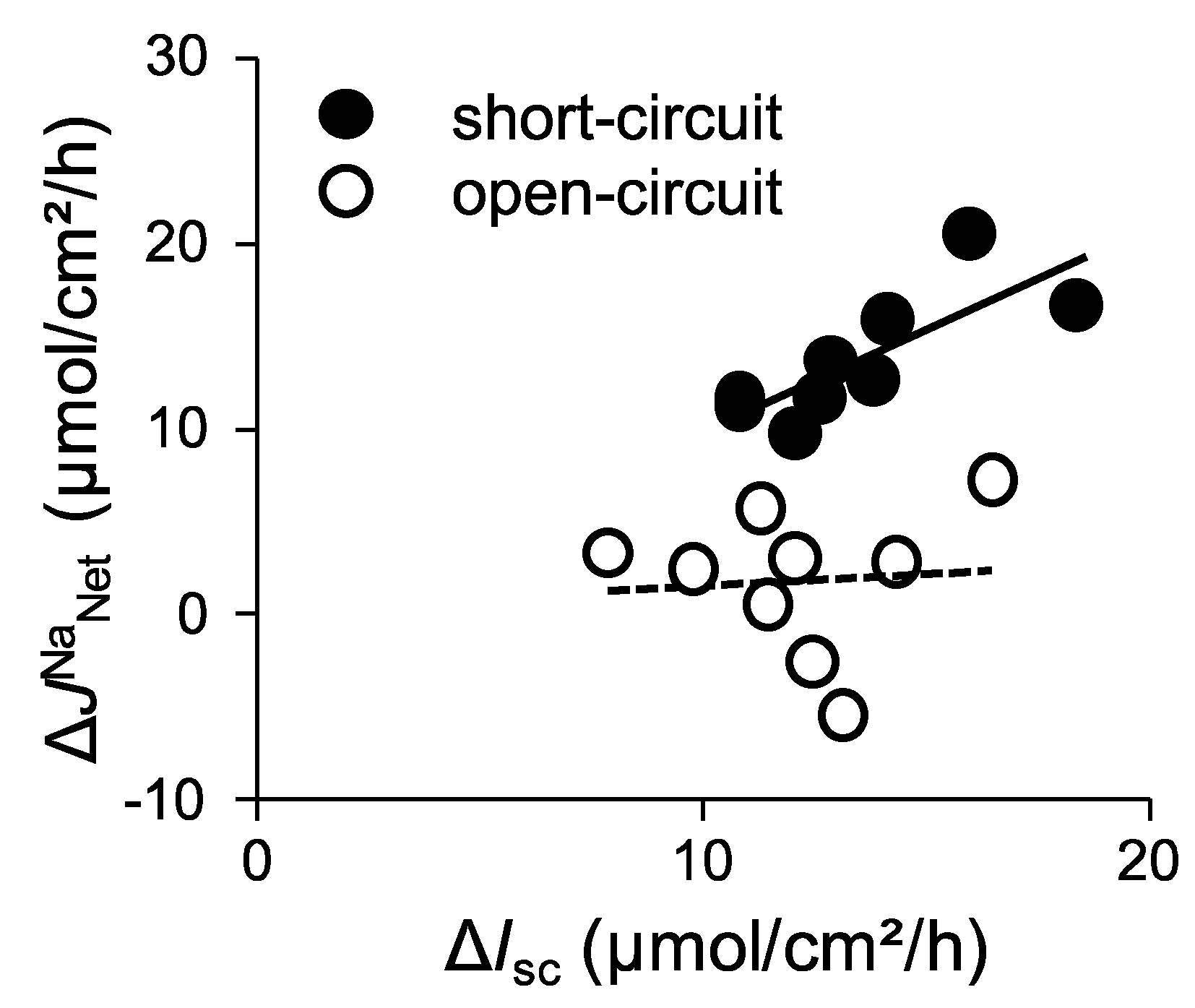
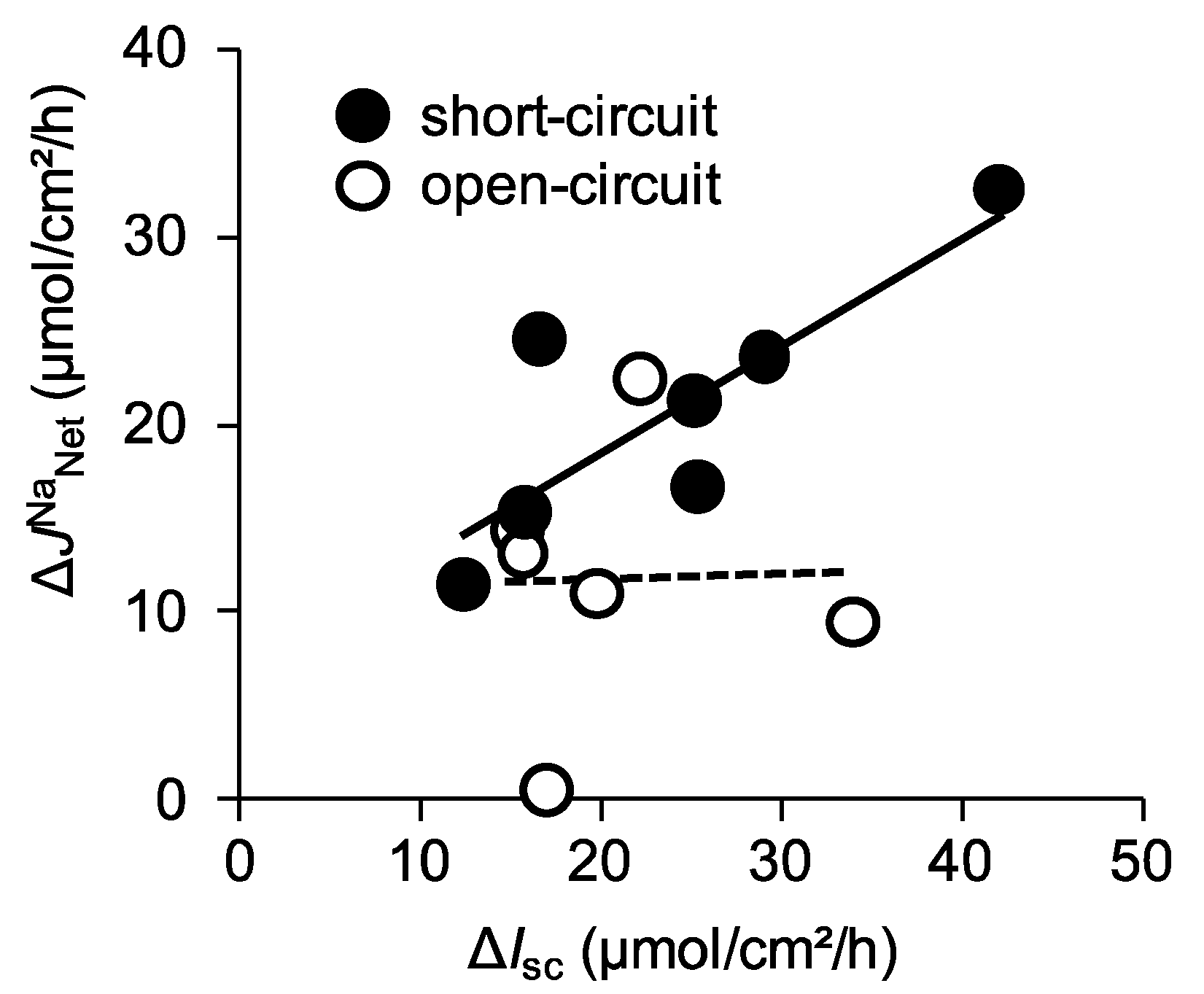
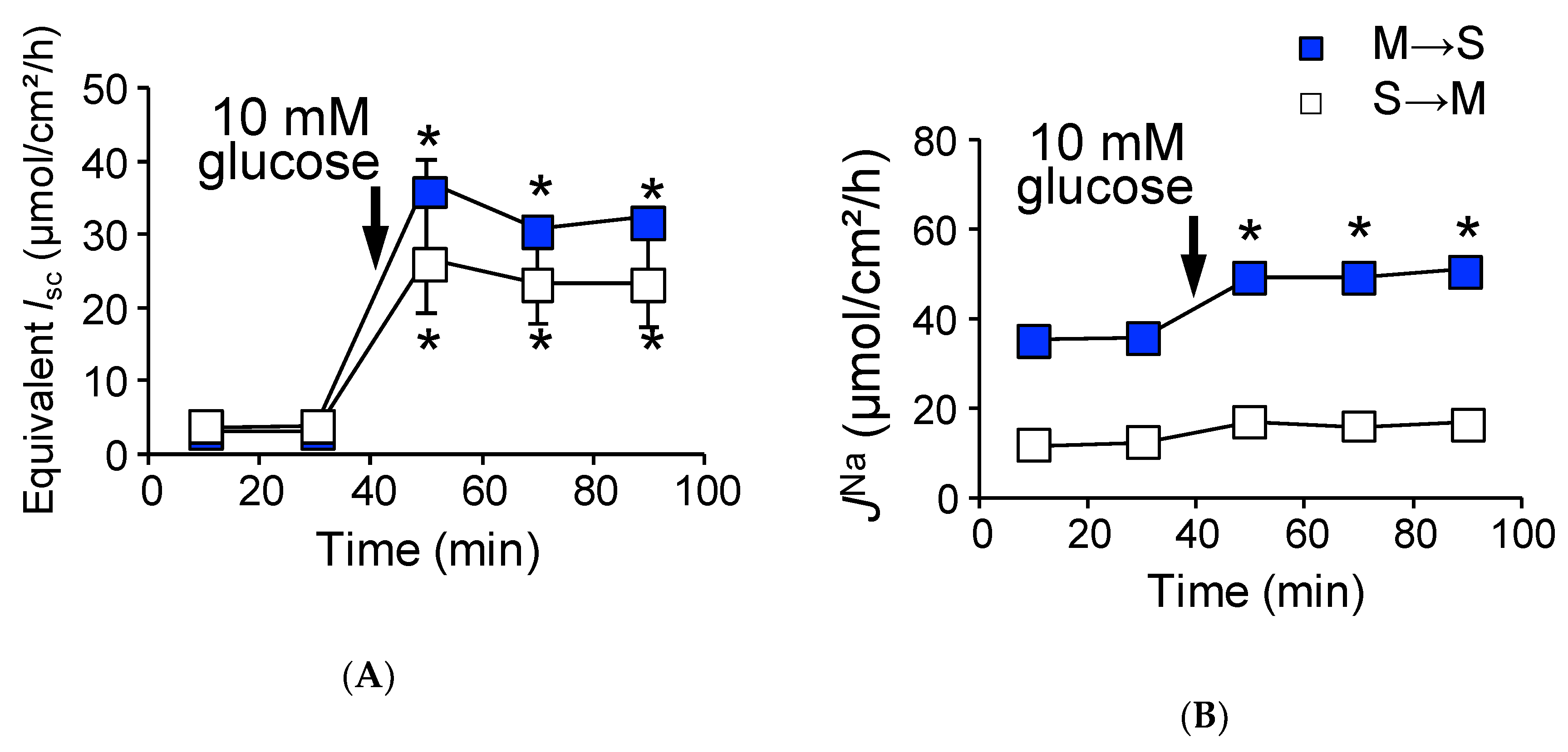
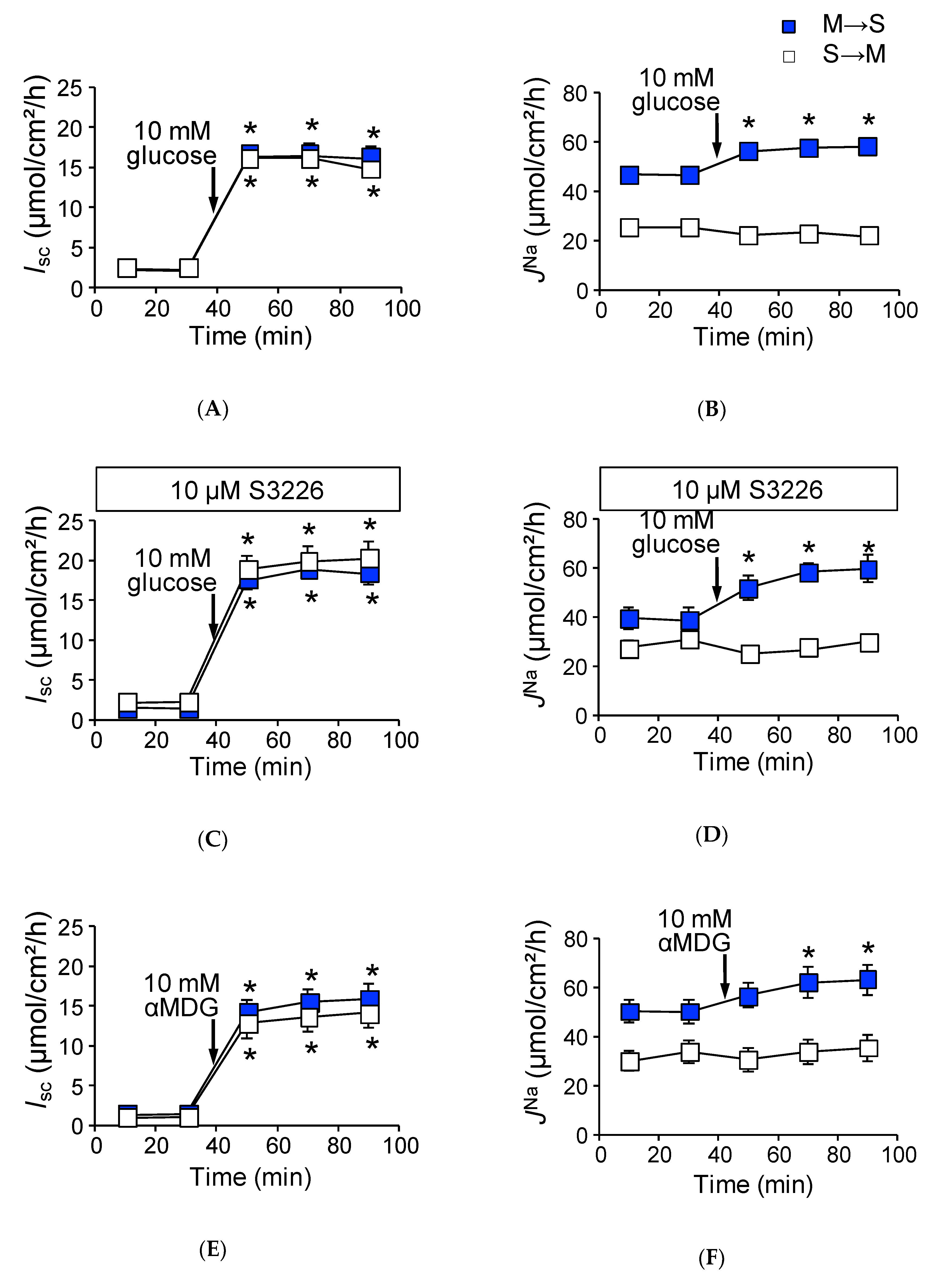
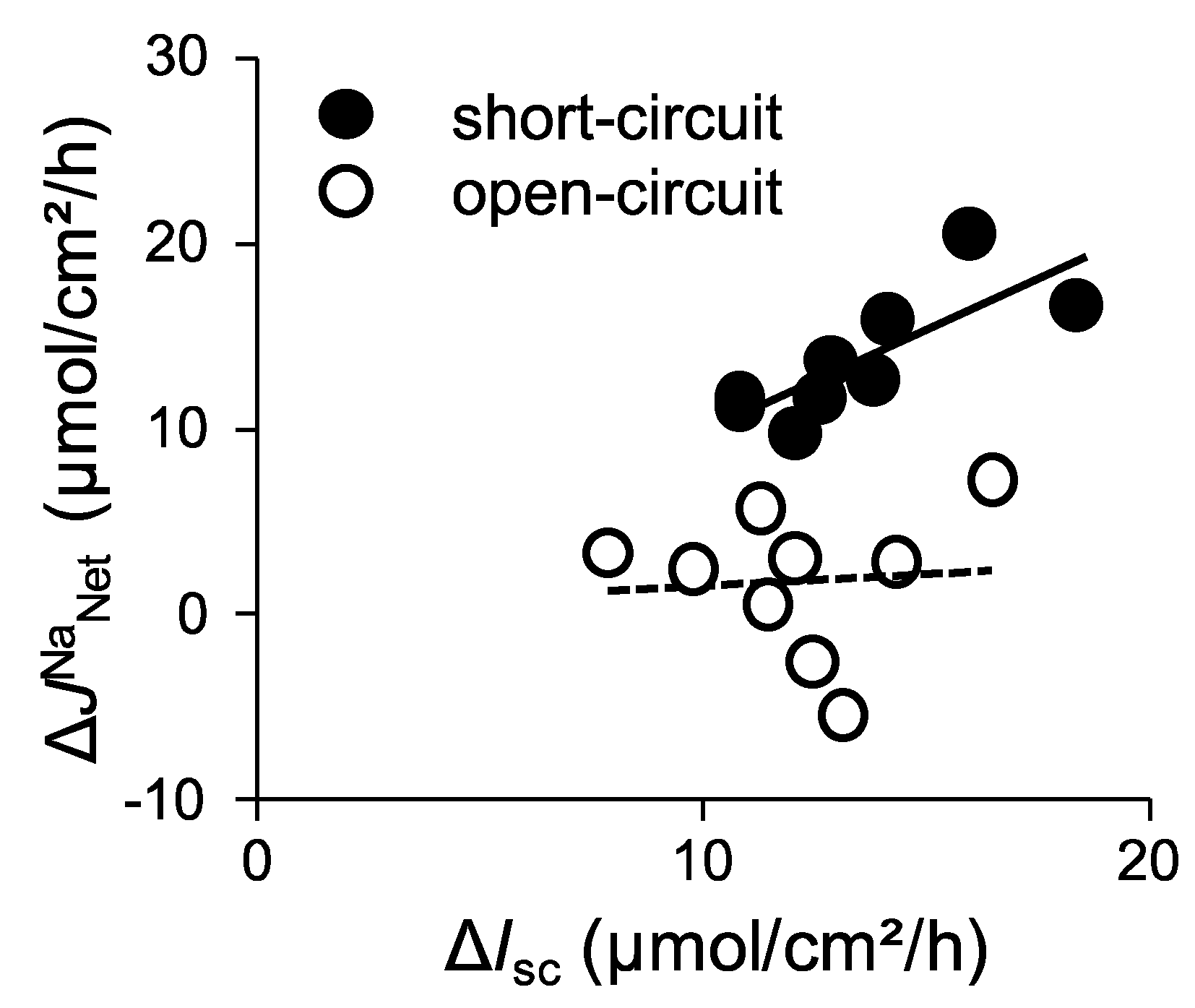
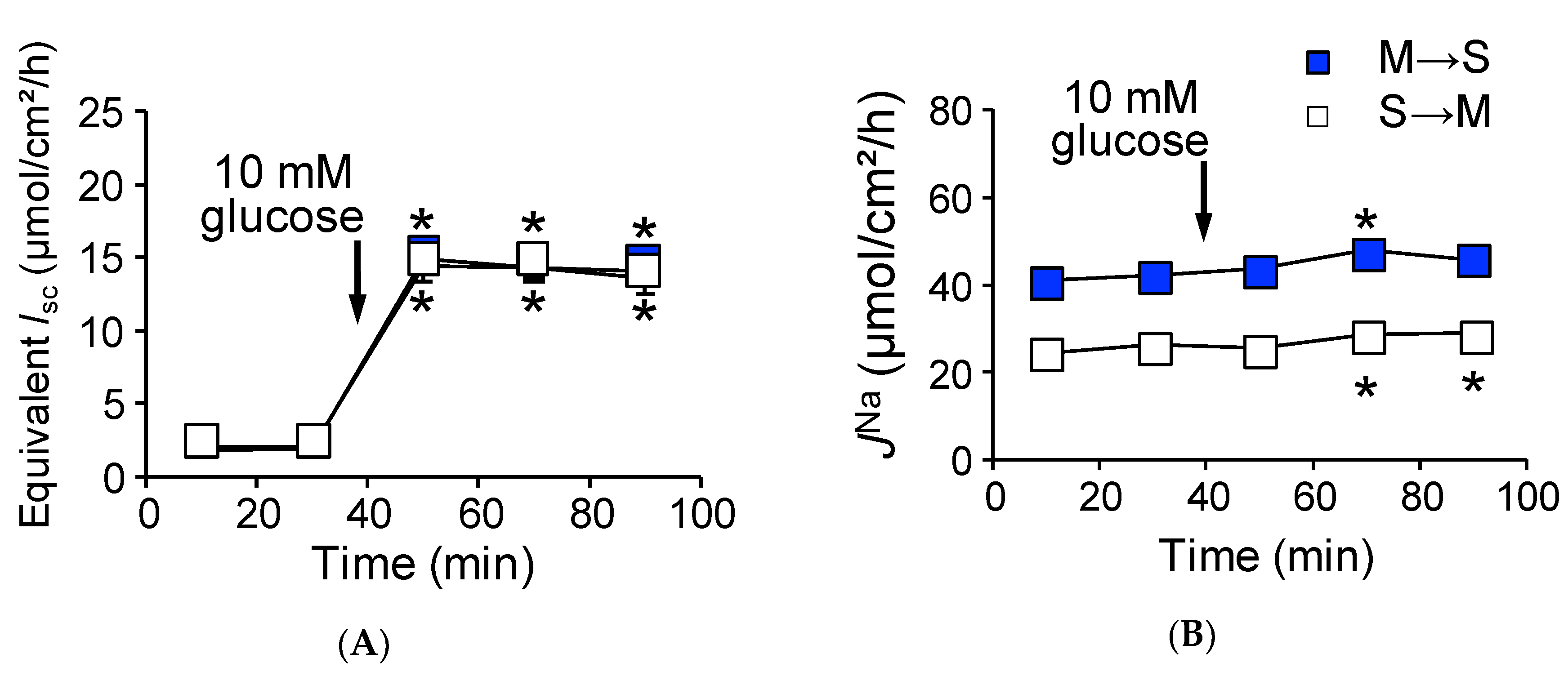
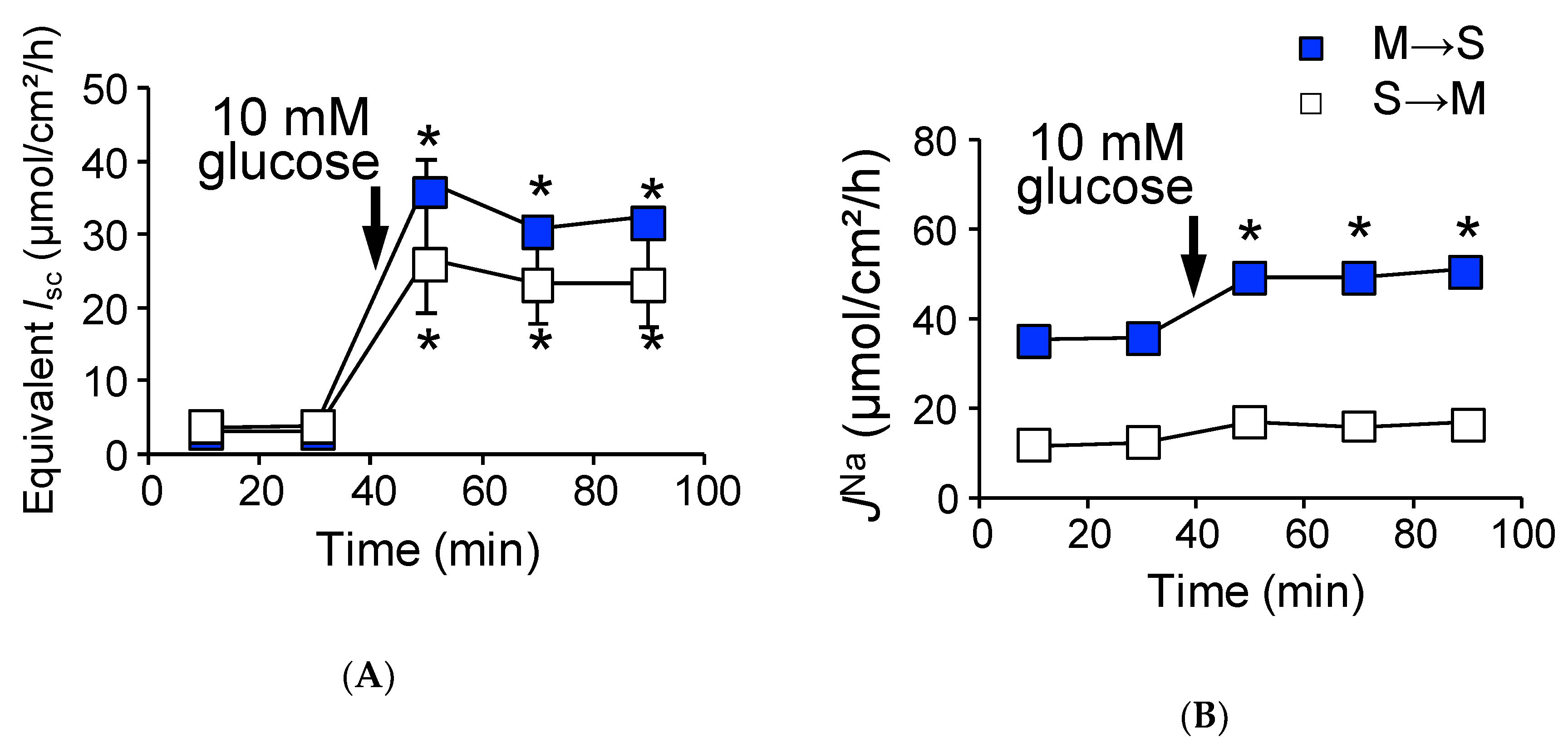
2.2. Activation of SGLT1 Concomitantly Increases Mucosal to Serosal 22Na+ Fluxes under Short-Circuit Conditions
2.3. Activation of SGLT1 does not Increase Mucosal to Serosal 22Na+ Fluxes under Open-Circuit Conditions
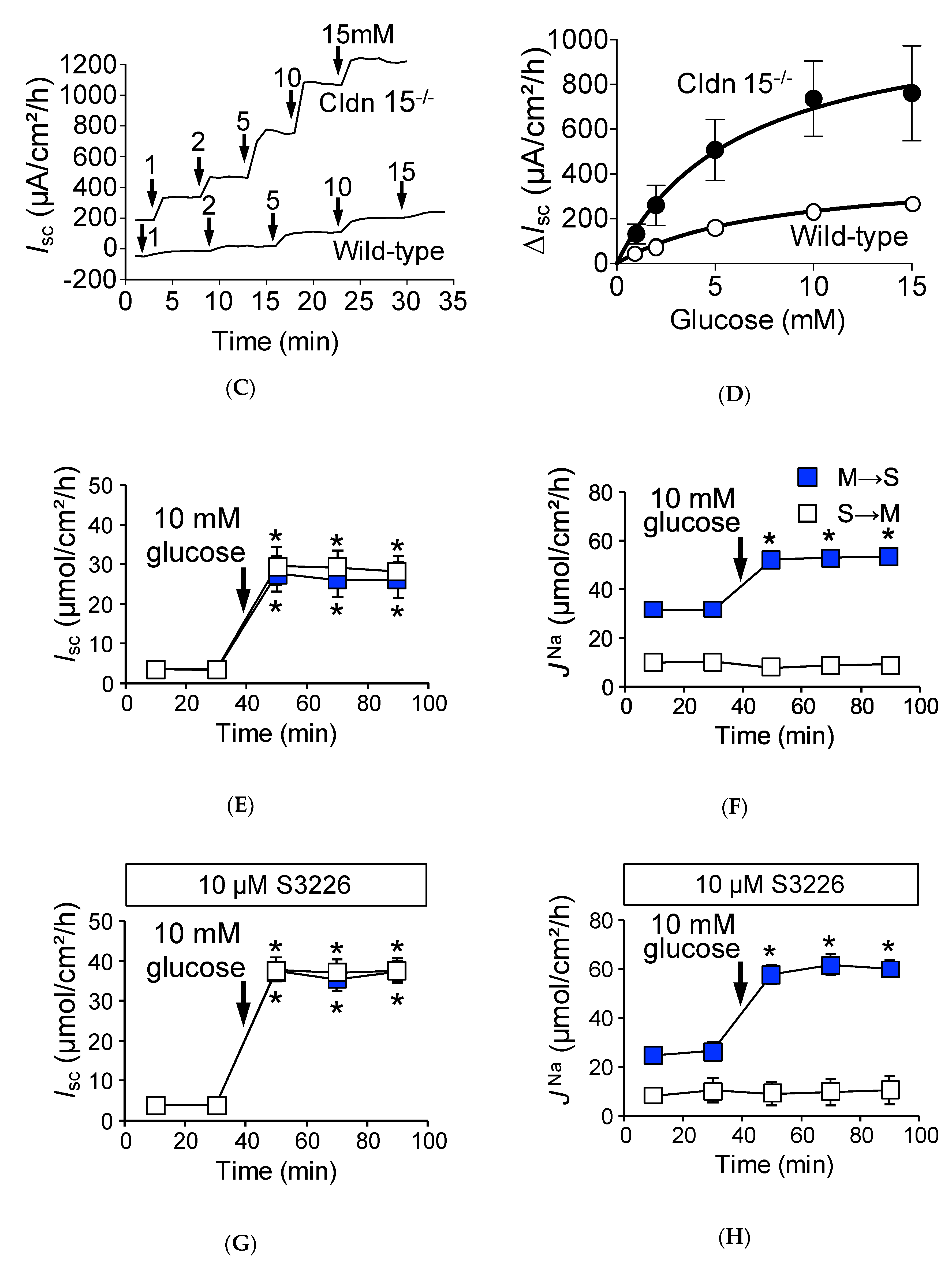
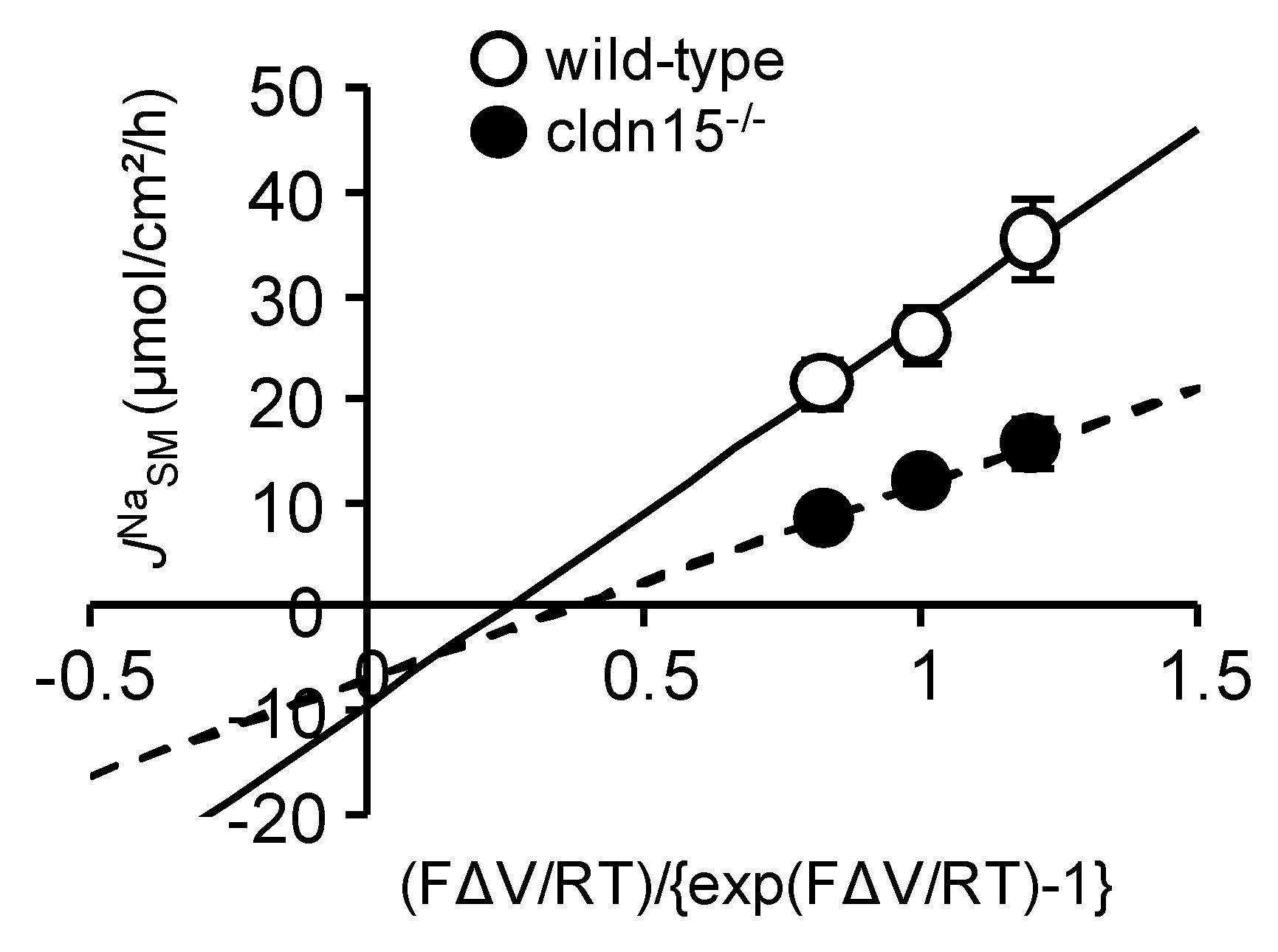
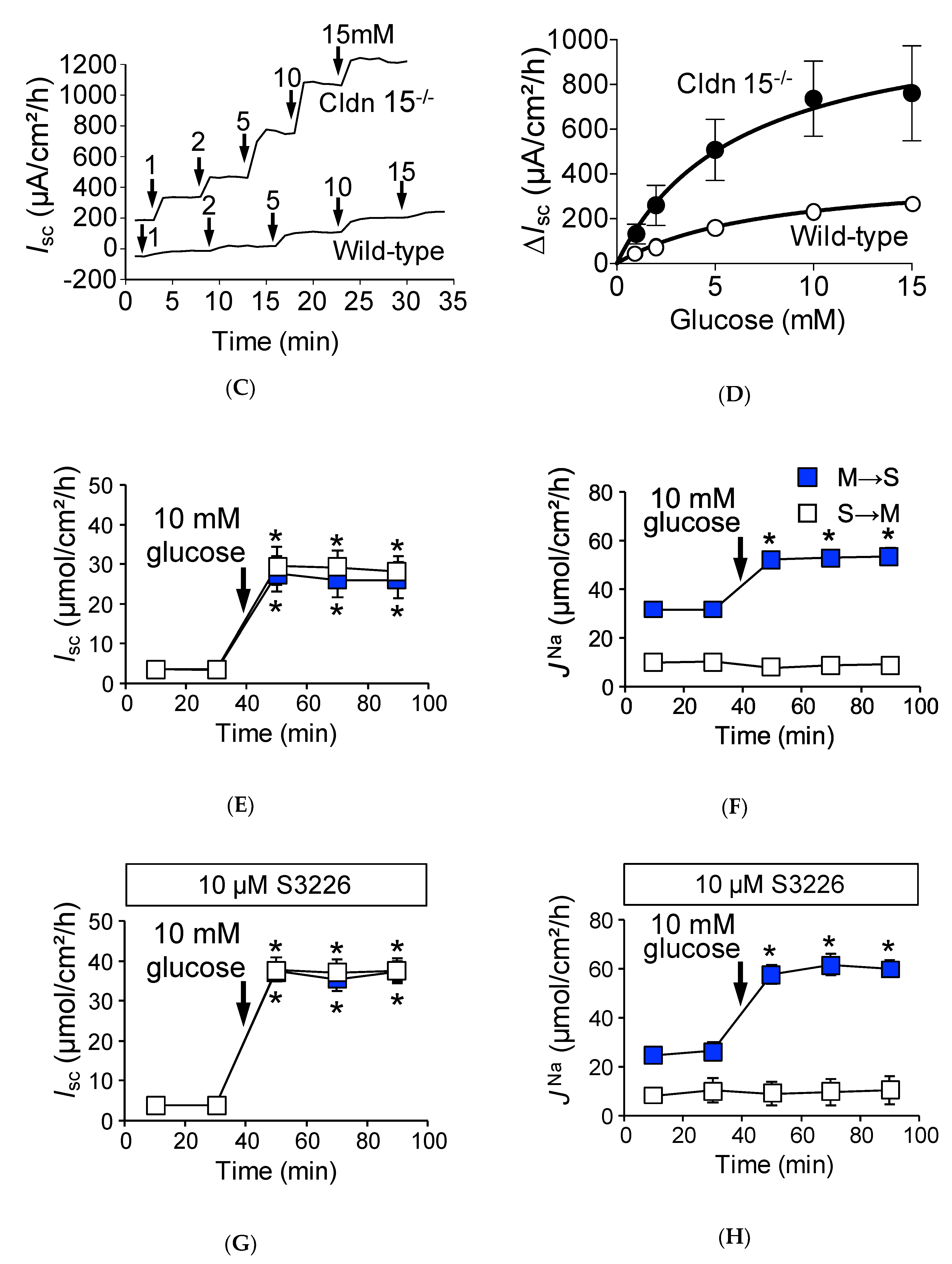
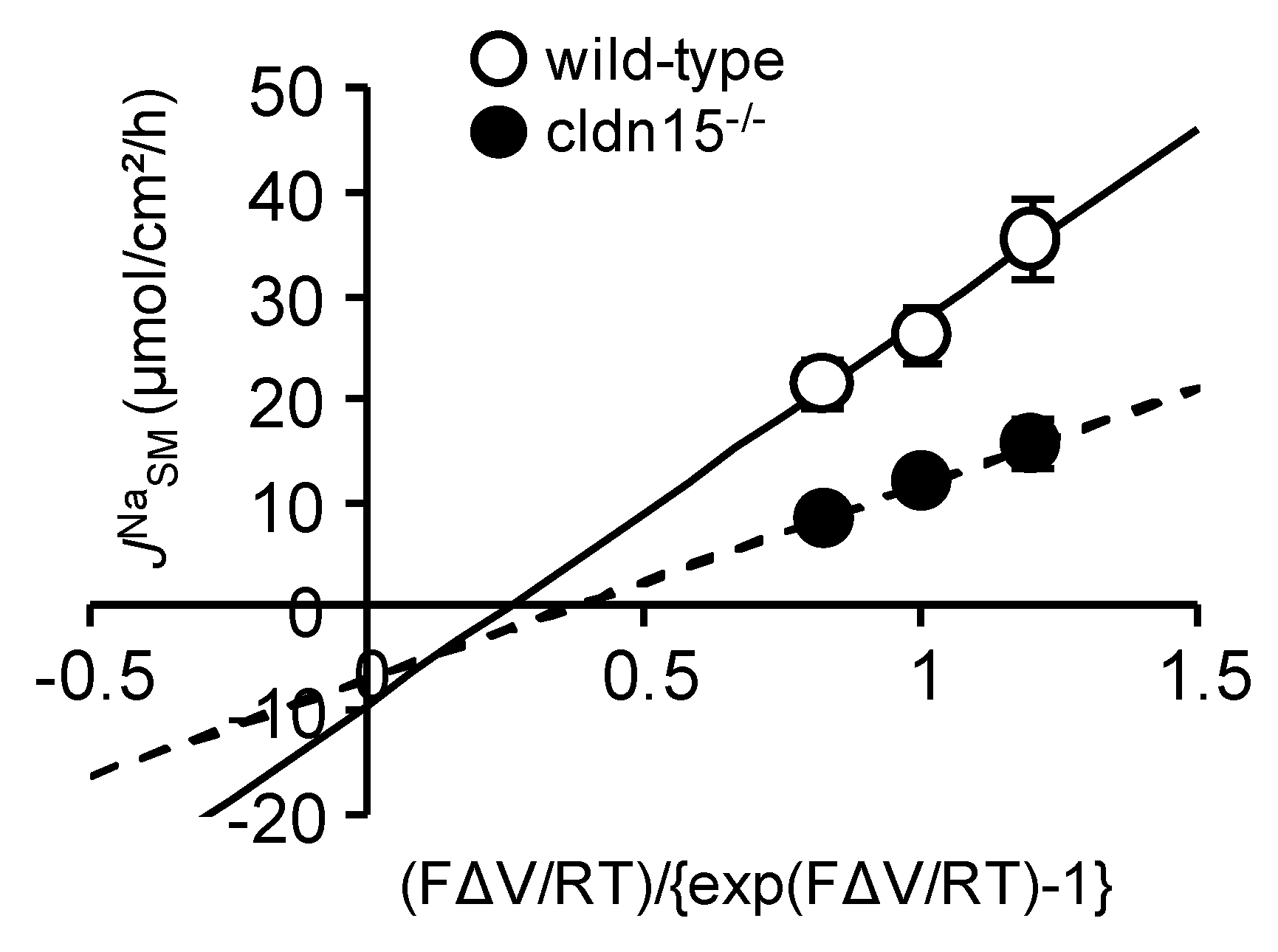
2.4. Baseline Na+ Absorption Mechanisms in Claudin-15 Deficient Mice
2.5. Na+-Dependent Glucose Transporter SGLT1 Is Up-Regulated in Cldn15−/− Mice
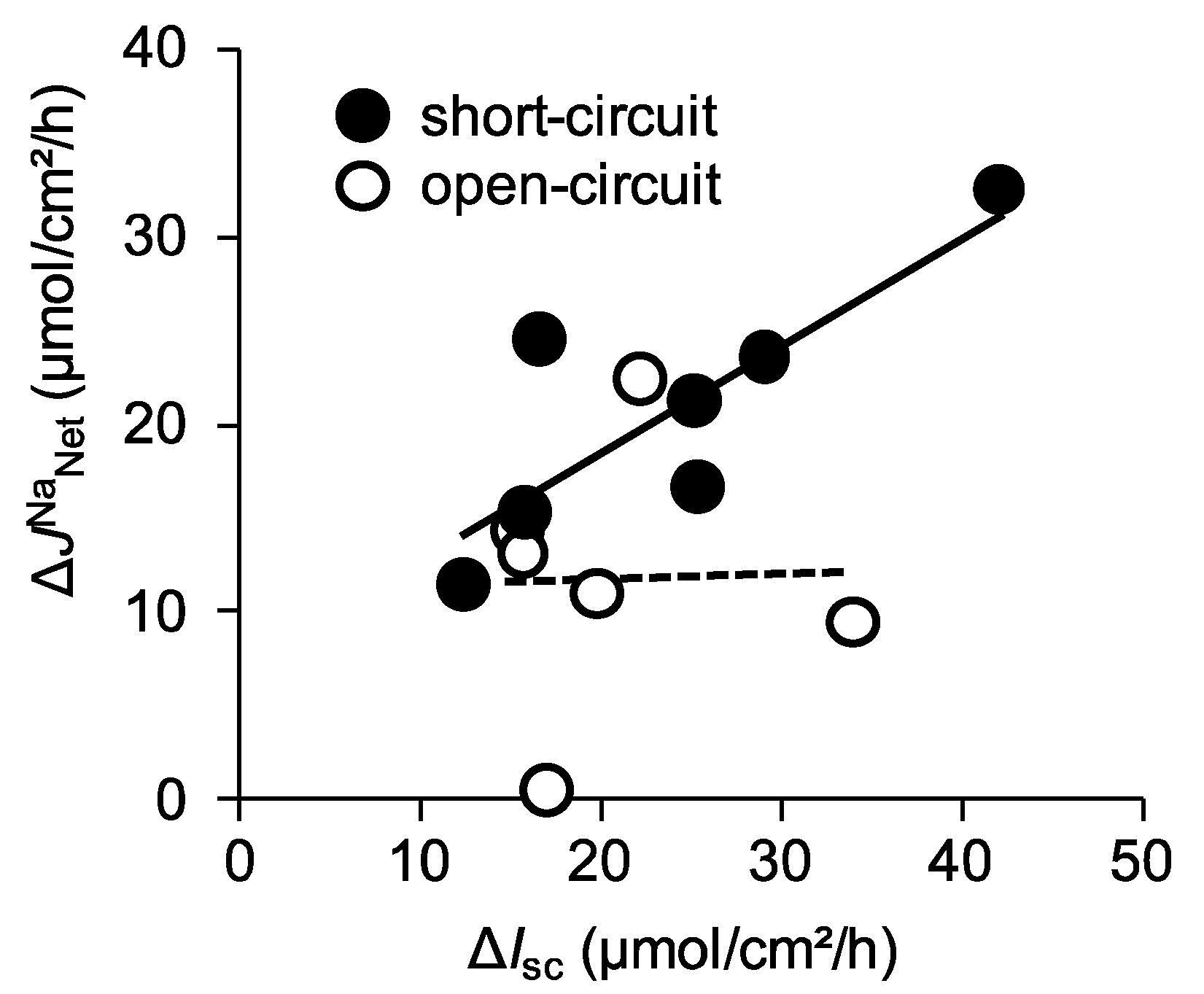
2.6. Absence of Claudin-15 Increases Glucose-Induced Mucosal to Serosal 22Na+ Flux
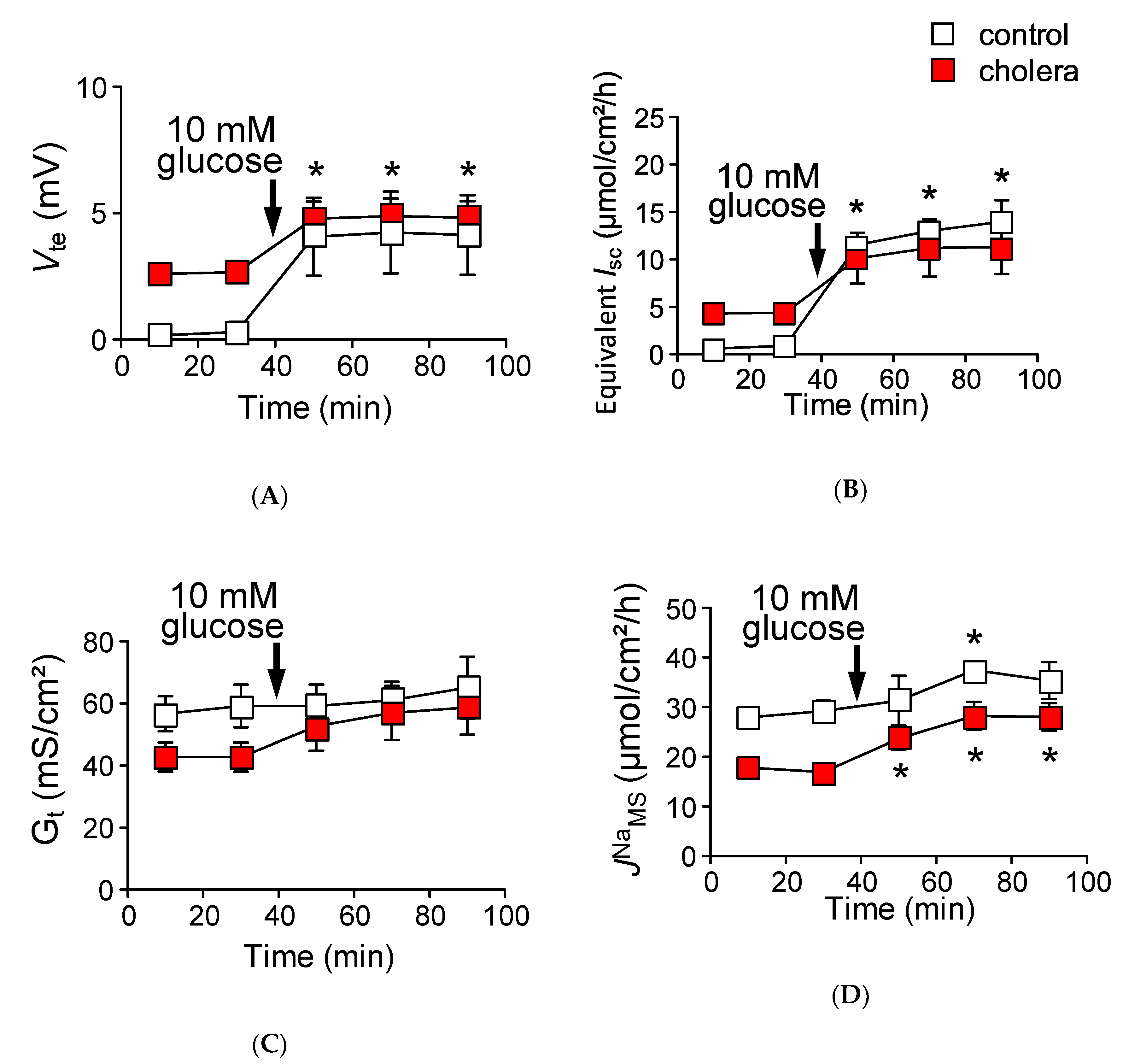
2.7. The Efficiency of Na+ Recycling Systems Is Reduced in a Cholera Toxin-Induced Diarrhea Model
3. Discussion
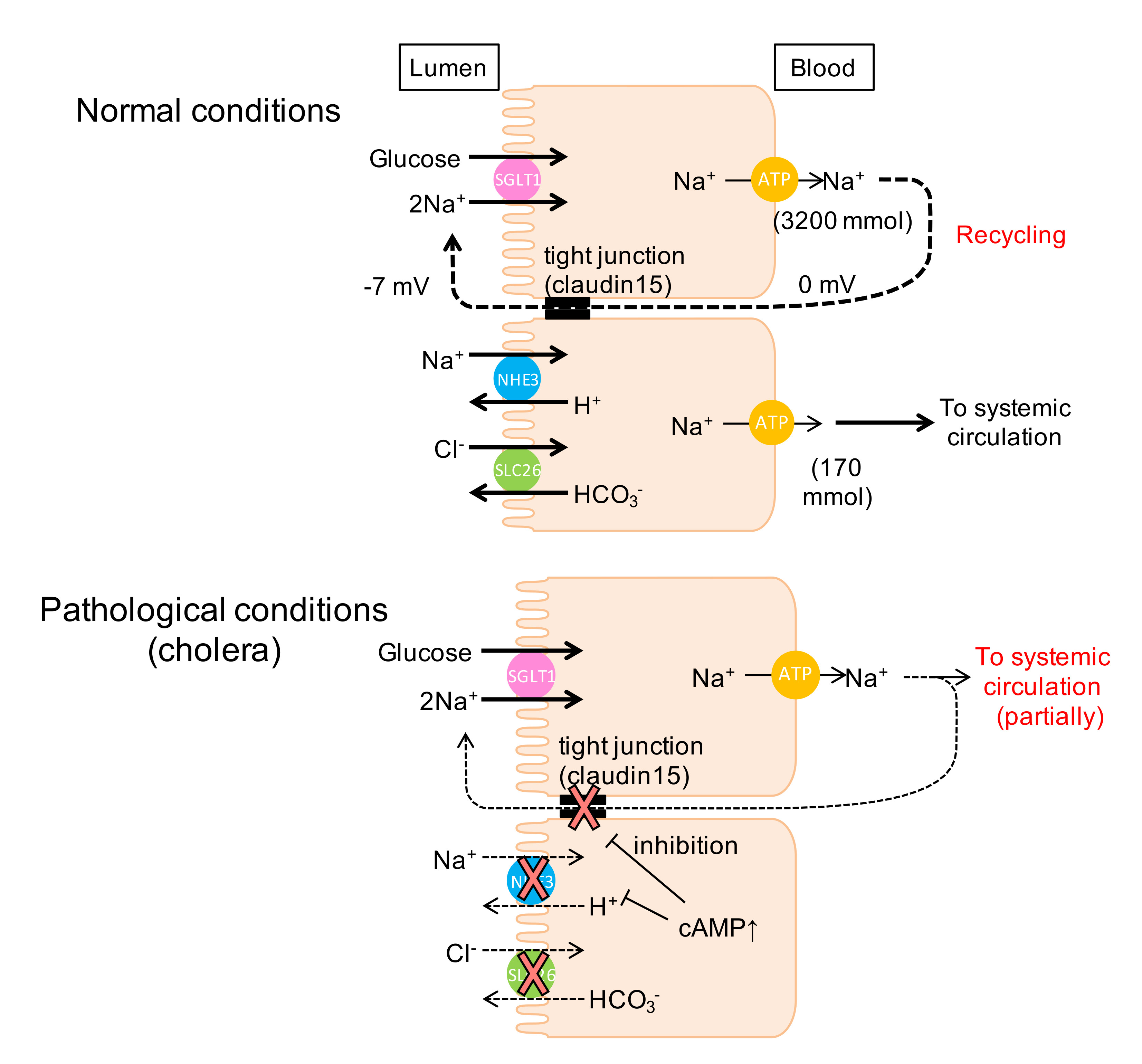
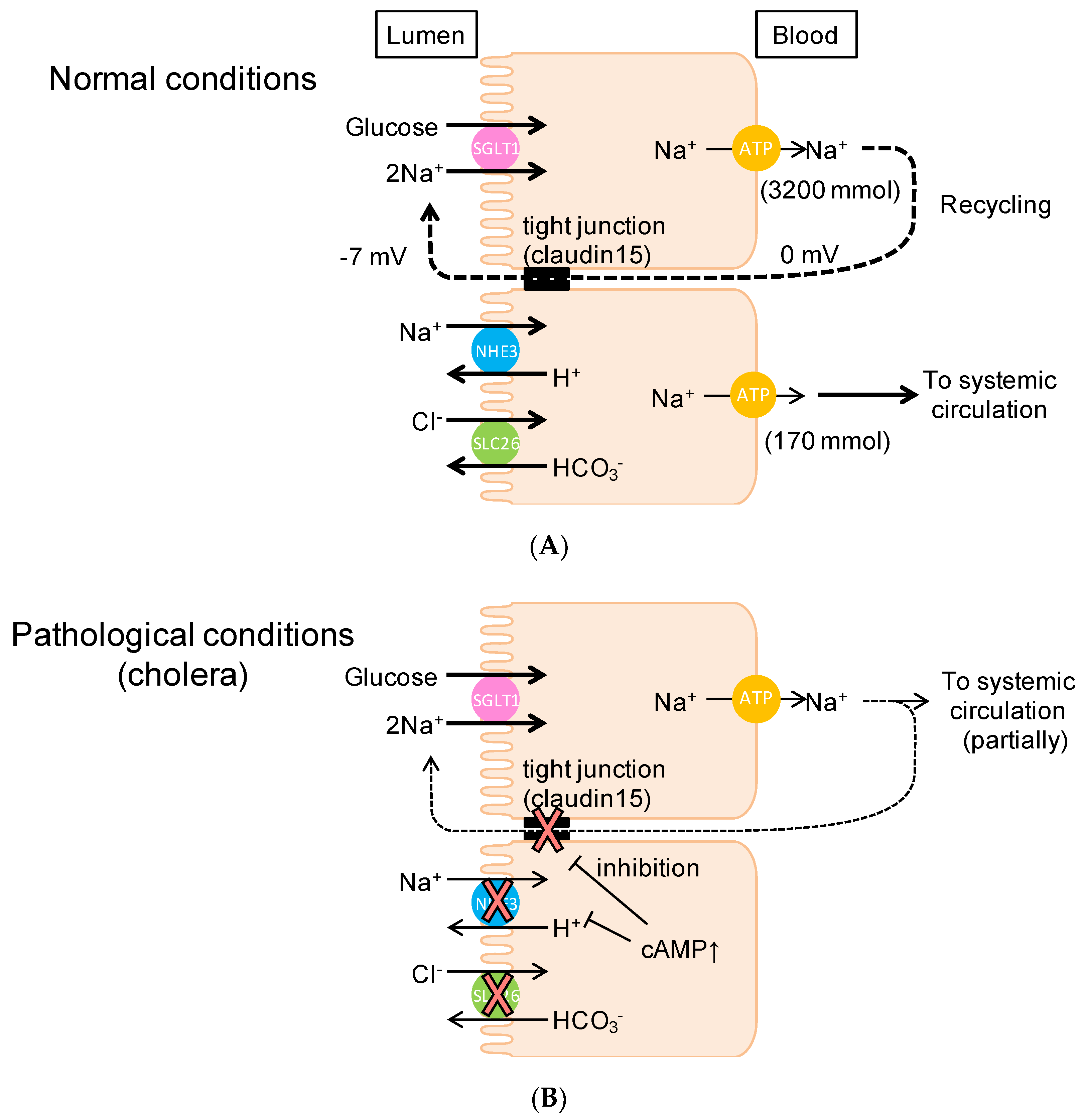
3.1. Intestinal Nutrient Absorption Mechanisms Need a Large Amount of Luminal Na+
3.2. Paracellular Na+ Permeability Is Decreased in Cldn15−/− Mice
3.3. Luminal Negative Potential Is Important for Na+ Recirculation
3.4. Physiological Relevance of the Na+ Recirculation System in the Small Intestine
4. Materials and Methods
4.1. Ethical Approval
4.2. Animals
4.3. Measurement of Electrical Parameters and Unidirectional Fluxes of 22Na+ and 36Cl−
4.4. Cholera Toxin-Induced Diarrhea Model
4.5. Chemicals
4.6. Real-Time Quantitative PCR
4.7. Immunofluorescence
4.8. Statistical and Data Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| αMDG | α-methyl-d-glucose |
| Gt | transmural conductance |
| Isc | short-circuit current |
| J | flux |
| Vte | transepithelial potential difference |
| MS | mucosal to serosal |
| SM | serosal to mucosal |
| NHE3 | Na+/H+ exchanger-3 isoform |
| IBMX | isobutylmethylxanthine |
| Km | Michaelis–Menten constant |
| Vmax | value of the maximum change |
| ORS | oral rehydration solution |
References
- Kato, A.; Romero, M.F. Regulation of Electroneutral NaCl Absorption by the Small Intestine. Annu. Rev. Physiol. 2011, 73, 261–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peerce, B.E.; Wright, E.M. Conformational changes in the intestinal brush border sodium-glucose cotransporter labeled with fluorescein isothiocyanate. Proc. Natl. Acad. Sci. USA 1984, 81, 2223–2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, P.D.; Wang, H.; Huang, W.; Leibach, F.H.; Ganapathy, V. Cloning of a cDNA encoding the human sodium-dependent vitamin transporter mediating the uptake of pantothenate, biotin and lipoate. J. Biol. Chem. 1998, 273, 7501–7506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, S.Y.M.; Muzyka, W.R.; Elliott, J.F.; Cheeseman, C.I.; Young, J.D. Poly(a)+ RNA from the mucosa of rat jejunum induces novel na+-dependent and na+-independent leucine transport activities in in oocytes of xenopus laevis. Mol. Membr. Biol. 1994, 11, 109–118. [Google Scholar] [CrossRef]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef]
- Hediger, M.A.; Coady, M.J.; Ikeda, T.S.; Wright, E.M. Expression cloning and cDNA sequencing of the Na+/glucose co-transporter. Nature 1987, 330, 379–381. [Google Scholar] [CrossRef]
- Mackenzie, B.; Loo, D.D.F.; Wright, E.M. Relationships between Na+/glucose cotransporter (SGLT1) currents and fluxes. J. Membr. Biol. 1998, 162, 101–106. [Google Scholar] [CrossRef]
- Phillips, S.F. Diarrhea: A current view of the pathophysiology. Gastroenterology 1972, 63, 495–518. [Google Scholar] [CrossRef]
- Powell, D.W. Barrier function of epithelia. Am. J. Physiol. 1981, 241, G275–G288. [Google Scholar] [CrossRef]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef]
- Rahner, C.; Mitic, L.L.; Anderson, J.M. Heterogeneity in expression and subcellular localization of claudins 2, 3, 4, and 5 in the rat liver, pancreas, and gut. Gastroenterology 2001, 120, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Rendón-Huerta, E.; Teresa, F.; Teresa, G.M.; Xochitl, G.-S.; Georgina, A.-F.; Veronica, Z.-Z.; Montaño, L.F. Distribution and Expression Pattern of Claudins 6, 7, and 9 in Diffuse- and Intestinal-Type Gastric Adenocarcinomas. J. Gastrointest. Cancer 2010, 41, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Kitano, Y.; Hata, M.; Katsuno, T.; Moriwaki, K.; Sasaki, H.; Hayashi, H.; Suzuki, Y.; Noda, T.; Furuse, M.; et al. Megaintestine in Claudin-15-Deficient Mice. Gastroenterology 2008, 134, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Hayashi, H.; Imasato, M.; Yamazaki, Y.; Hagiwara, A.; Wada, M.; Noda, T.; Watanabe, M.; Suzuki, Y.; Tsukita, S. Loss of claudin-15, but not claudin-2, causes Na+ deficiency and glucose malabsorption in mouse small intestine. Gastroenterology 2011, 140, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Halperin, M.L.; Wolman, S.L.; Greenberg, G.R. Paracellular recirculation of sodium is essential to support nutrient absorption in the gastrointestinal tract: An hypothesis. Clin. Investig. Med. 1986, 9, 209–211. [Google Scholar]
- Binder, H.J.; Brown, I.; Ramakrishna, B.S.; Young, G.P. Oral rehydration therapy in the second decade of the twenty-first century. Curr. Gastroenterol. Rep. 2014, 16, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawenis, L.R.; Stien, X.; Shull, G.E.; Schultheis, P.J.; Woo, A.L.; Walker, N.M.; Clarke, L.L. Intestinal NaCl transport in NHE2 and NHE3 knockout mice. Am. J. Physiol. Liver Physiol. 2002, 282, G776–G784. [Google Scholar] [CrossRef]
- Pappenheimer, J.R.; Reiss, K.Z. Contribution of solvent drag through intercellular junctions to absorption of nutrients by the small intestine of the rat. J. Membr. Biol. 1987, 100, 123–136. [Google Scholar] [CrossRef]
- Lin, R.; Murtazina, R.; Cha, B.; Chakraborty, M.; Sarker, R.; Chen, T.; Lin, Z.; Hogema, B.M.; De Jonge, H.R.; Seidler, U.; et al. d-glucose acts via sodium/glucose cotransporter 1 to increase NHE3 in mouse jejunal brush border by a Na+/H+ exchange regulatory factor 2dependent process. Gastroenterology 2011, 140, 560–571. [Google Scholar] [CrossRef] [Green Version]
- Schwark, J.-R.; Jansen, H.W.; Lang, H.-J.; Krick, W.; Burckhardt, G.; Hropot, M. S3226, a novel inhibitor of Na+/H+ exchanger subtype 3 in various cell types. Pflugers Arch. Eur. J. Physiol. 1998, 436, 797–800. [Google Scholar] [CrossRef]
- Halm, D.R.; Dawson, D.C. Control of potassium transport by turtle colon: Role of membrane potential. Am. J. Physiol. 1984, 247, C26–C32. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, N.; Nakayama, M.; Watanabe, M.; Tajima, H.; Yamauchi, Y.; Ikari, A.; Hayashi, H. Luminal Na+ homeostasis has an important role in intestinal peptide absorption in vivo. Am. J. Physiol. 2018, 315, G799–G809. [Google Scholar] [CrossRef] [PubMed]
- Frizzell, R.A.; Schultz, S.G. Ionic Conductances of Extracellular Shunt Pathway in Rabbit Ileum. J. Gen. Physiol. 1972, 59, 318–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, M. Intestinal ion transport and the pathophysiology of diarrhea Find the latest version: Intestinal ion transport and the pathophysiology of diarrhea. J. Clin. Investig. 2003, 111, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Powell, D.W. Intestinal conductance and permselectivity changes with theophylline and choleragen. Am. J. Physiol. 1974, 227, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Gawenis, L.R.; Boyle, K.T.; Palmer, B.A.; Walker, N.M.; Clarke, L.L. Lateral intercellular space volume as a determinant of CFTR-mediated anion secretion across small intestinal mucosa. Am. J. Physiol. Liver Physiol. 2004, 286, G1015–G1023. [Google Scholar] [CrossRef]
- Smith, D.E.; Clémençon, B.; Hediger, M.A. Proton-coupled oligopeptide transporter family SLC15: Physiological, pharmacological and pathological implications. Mol. Aspects Med. 2013, 34, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, N.; Hempstock, W.; Hayashi, H. The Mode of Action of NHE3 Inhibitors in Intestinal Na + Absorption. Gastroenterol. Med. Res. 2019, 4, 297–299. [Google Scholar]
- Uchida, R.; Iwamoto, K.; Nagayama, S.; Miyajima, A.; Okamoto, H.; Ikuta, N.; Fukumi, H.; Terao, K.; Hirota, T. Effect of γ-cyclodextrin inclusion complex on the absorption of R-α-lipoic acid in rats. Int. J. Mol. Sci. 2015, 16, 10105–10120. [Google Scholar] [CrossRef] [Green Version]
- Gorboulev, V.; Schürmann, A.; Vallon, V.; Kipp, H.; Jaschke, A.; Klessen, D.; Friedrich, A.; Scherneck, S.; Rieg, T.; Cunard, R.; et al. Na+-d-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 2012, 61, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Yu, A.S.L.; Cheng, M.H.; Angelow, S.; Günzel, D.; Kanzawa, S.A.; Schneeberger, E.E.; Fromm, M.; Coalson, R.D. Molecular Basis for Cation Selectivity in Claudin-2–based Paracellular Pores: Identification of an Electrostatic Interaction Site. J. Gen. Physiol. 2009, 133, 111–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, M.; Tamura, A.; Takahashi, N.; Tsukita, S. Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 2013, 144, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, P.J.; Meneton, P.; Riddle, T.M.; Duffy, J.J.; Doetschman, T.; Shull, G.E.; Miller, M.L.; Soleimani, M.; Lorenz, J.N.; Wang, T.; et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat. Genet. 1998, 19, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Schweinfest, C.W.; Spyropoulos, D.D.; Henderson, K.W.; Kim, J.H.; Chapman, J.M.; Barone, S.; Worrell, R.T.; Wang, Z.; Soleimani, M. slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct up-regulation of ion transporters in the colon. J. Biol. Chem. 2006, 281, 37962–37971. [Google Scholar] [CrossRef] [Green Version]
- Duffey, M.E.; Hainau, B.; Ho, S.; Bentzel, C.J. Regulation of epithelial tight junction permeability by cyclic AMP. Nature 1981, 294, 451–453. [Google Scholar] [CrossRef]
- Field, M.; Fromm, D.; al-Awqati, Q.; Greenough, W.B. Effect of cholera enterotoxin on Ion transport across isolated ileal mucosa. J. Clin. Investig. 1972, 51, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Duggan, C.; Fontaine, O.; Pierce, N.F.; Glass, R.I.; Mahalanabis, D.; Alam, N.H.; Bhan, M.K.; Santosham, M. Scientific Rationale for a Change in the Composition of Oral Rehydration Solution. JAMA 2004, 291, 2628–2631. [Google Scholar] [CrossRef]
- Singh, A.K.; Amlal, H.; Haas, P.J.; Dringenberg, U.; Fussell, S.; Barone, S.L.; Engelhardt, R.; Zuo, J.; Seidler, U.; Soleimani, M. Fructose-induced hypertension: Essential role of chloride and fructose absorbing transporters PAT1 and Glut5. Kidney Int. 2008, 74, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Fayad, I.M.; Kamel, M.; Hirschhorn, N.; Abu-Zikry, M. Hypernatraemia surveillance during a national diarrhoeal diseases control project in Egypt. Lancet 1992, 339, 389–393. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| JNa, µmol/cm2/h | Isc, µmol/cm2/h | Gt, mS/cm2 | n | |||
|---|---|---|---|---|---|---|
| M→S | S→M | Net | ||||
| Short-Circuit Conditions | ||||||
| Control | 51.4 ± 2.3 | 24.6 ± 1.7 | 26.9 ± 1.5 | 2.4 ± 0.5 | 58.7±2.2 | 4 |
| S3226 | 38.9 ± 3.4 * | 28.3 ± 1.9 | 10.6 ± 3.9 * | 1.7 ± 0.2 | 54.6±2.8 | 6 |
| Open-Circuit Conditions | ||||||
| Control | 44.2 ± 2.6 N.S. | 22.8 ± 1.6 N.S. | 21.4 ± 3.7 N.S. | 1.8 ± 0.3 N.S. | 55.2±2.4 N.S. | 3 |
| JNa, µmol/cm2/h | Isc, µmol/cm2/h | Gt, mS/cm2 | n | |||
|---|---|---|---|---|---|---|
| M→S | S→M | Net | ||||
| Short-Circuit Conditions | ||||||
| Control | 31.9 ± 1.9 † | 10.4 ± 0.8 † | 21.4 ± 2.4 | 3.3 ± 0.4 † | 17.7 ± 0.7 † | 6 |
| S3226 | 22.3 ± 1.5 * | 5.8 ± 0.5 * | 16.4 ± 1.0 | 3.9 ± 0.1 | 13.6 ± 1.0 * | 4 |
| Open-Circuit Conditions | ||||||
| Control | 35.6 ± 2.4 N.S. | 12.0 ± 0.9 N.S. | 23.7 ± 2.4 | 4.0 ± 0.6 | 19.4 ± 1.4 | 6 |
| JNa, µmol/cm2/h | Isc, µmol/cm2/h | Gt, mS/cm2 | n | |||
|---|---|---|---|---|---|---|
| M→S | S→M | Net | ||||
| Open-Circuit Conditions | ||||||
| Control | 29.2 ± 2.1 | 18.4 ± 3.6 | 10.8 ± 2.8 | −1.0 ± 0.2 | 61.6 ± 9.3 | 3 |
| Cholera | 16.9 ± 1.4* | 17.3 ± 1.4 | −0.3 ± 1.0 * | 3.9± 0.3 * | 39.6 ± 1.1 * | 5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayama, M.; Ishizuka, N.; Hempstock, W.; Ikari, A.; Hayashi, H. Na+-Coupled Nutrient Cotransport Induced Luminal Negative Potential and Claudin-15 Play an Important Role in Paracellular Na+ Recycling in Mouse Small Intestine. Int. J. Mol. Sci. 2020, 21, 376. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020376
Nakayama M, Ishizuka N, Hempstock W, Ikari A, Hayashi H. Na+-Coupled Nutrient Cotransport Induced Luminal Negative Potential and Claudin-15 Play an Important Role in Paracellular Na+ Recycling in Mouse Small Intestine. International Journal of Molecular Sciences. 2020; 21(2):376. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020376
Chicago/Turabian StyleNakayama, Michiko, Noriko Ishizuka, Wendy Hempstock, Akira Ikari, and Hisayoshi Hayashi. 2020. "Na+-Coupled Nutrient Cotransport Induced Luminal Negative Potential and Claudin-15 Play an Important Role in Paracellular Na+ Recycling in Mouse Small Intestine" International Journal of Molecular Sciences 21, no. 2: 376. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020376