Alleviation of Multidrug Resistance by Flavonoid and Non-Flavonoid Compounds in Breast, Lung, Colorectal and Prostate Cancer

 ,
, 
Abstract
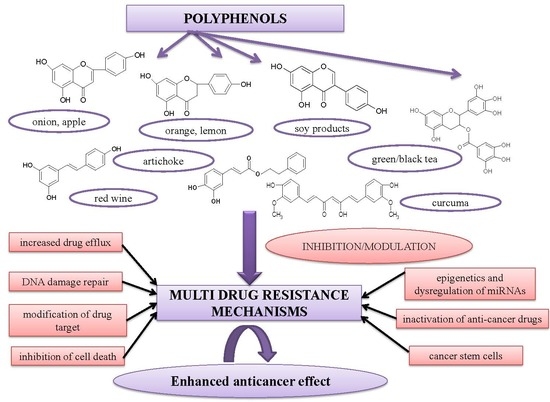
:
1. Introduction
2. Mechanism of Multidrug Resistance in Cancer
2.1. Increase of Drug Efflux
2.2. Detoxification Mechanisms and Inactivation of Anticancer Drugs
2.3. DNA Damage Repair
2.4. Modification of Drug Target
2.5. Inhibition of Cell Death
2.6. Cancer Stem Cells
2.7. Tumor Heterogeneity
2.8. Tumor Microenvironment (TME)
2.9. Epithelial to Mesenchymal Transition (EMT)
2.10. Epigenetic Variations
2.11. Dysregulation of microRNA (miRNAs)
2.12. Modulation of Reactive Oxygen Species (ROS)
3. Role of Polyphenols in MDR
3.1. In Vitro Studies
3.1.1. Flavonoid Compounds
Flavones
Flavonols
Flavanones
Flavan-3-ols
Isoflavones
3.1.2. Non-Flavonoid Compounds
Stilbenes
Lignans
Ellagitannins
Hydroxy-Benzoic Acids
Hydroxy-Cinnamic Acids
Other Compounds
3.1.3. Synergic and Pleiotropic Activity of Polyphenols
3.2. In Vivo and Clinical Studies
3.2.1. Flavonoid Compounds
Flavones and Flavonols
Flavan-3-ols
Isoflavones
3.2.2. Non-Flavonoid Compounds
Stilbenes
Hydroxy-Cinammic Acids
Lignans
Other Compounds
3.2.3. Bioavailability and Toxicity of the Polyphenols
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ↑ | upregulation |
| ↓ | downregulation |
| 5-FU | 5-fluorouracil |
| ABC | ATP-binding cassette transporter proteins |
| ABCB1, ABCG1 | isoforms of ATP-binding cassette transporter proteins |
| ARE | antioxidant response element |
| ABT-263 | small molecule that inhibits Bcl-2; 4-(4-{[2-(4-Chlorophenyl)-5,5-dimethyl-1-cyclohexen-1-yl]methyl}c-1-piperazinyl)-N-[(4-{[(2R)-4-(4-morpholinyl)-1-(phenylsulfanyl)-2-butanyl]amino}-3-[(trifluoromethyl)sulfonyl]phenyl)sulfonyl]benzamide |
| ABT-737 | small molecule that inhibits Bcl-2; 4-{4-[(4′-Chloro-2-biphenylyl)methyl]-1-piperazinyl}-N-[(4-{[(2R)-4-(dimethylamino)-1-(phenylsulfanyl)-2-butanyl]amino}-3-nitrophenyl)sulfonyl]benzamide |
| AKT | protein kinase B |
| ALDH | aldehyde dehydrogenase |
| ALP | autophagy lysosomes systems |
| AMPK | AMP-activated protein kinase |
| APAF1 | apoptotic protease activating factor 1 |
| APR-246 | drug that binds to p53 (restoring p53 function) and depletes glutathione; PRIMA-1, 2-hydroxymethyl-2-methoxymethyl-aza-bicyclo[2.2.2]octan-3-one |
| AR | androgen receptor |
| ATR | serine/threonine protein kinase |
| Axl, Tyro3 | receptors for tyrosine kinase |
| BASE | base excision repair |
| Bax | Bcl-2-associated X protein/Bcl-2-like protein 4 |
| Bcl-2 | B cell lymphoma 2 protein |
| Bcl-XL | B cell lymphoma extra-large protein |
| BCRA1, 2 | breast cancer susceptible genes |
| BCRP | breast cancer resistant protein |
| BER | base excision repair |
| BH | Bcl-2 homology domain |
| BIM | Bcl-2 like protein 11 |
| BNDQ | quercetin and doxorubicin co-encapsulated biotin receptor-targeting nanoparticles |
| BPIS | bound polyphenols of inner shell from foxtail millet bran |
| BRAF | serine/threonine-protein kinase B-Raf |
| C | catechin |
| CAB | carboplatin |
| CAPE | caffeic acid phenethyl ester |
| CAR | constitutive androstane receptor |
| caspase-3, 8, 9 | cysteine aspartic proteases-3, 8, 9 |
| CBZ | cabazitaxel |
| CD44, 24, 133 | cluster of differentiation 44, 24, 133 |
| CDF | difluorinated curcumin |
| CDK 2,4,6 | cyclin-dependent kinases 2,4,6 |
| CDPP | cisplatin |
| CEA | carcioembryonic antigen |
| cFLIP | regulator of caspase-8 activation; cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein |
| CgA | chromogranin |
| Chk1/2 | Check point kinase 1/2 |
| cIAP-1,2 | cellular inhibitor of apoptosis protein 1,2 |
| COMT | catechol-O-methyl transferase |
| COX-2 | ciclo-oxygenase 2 |
| CPT11 | irinotecan |
| CREB-1 | element binding protein-1 |
| CRPC | castration-resistant prostate cancer |
| CSC | cancer stem cells |
| CXCR4 | CXC chemokine receptor type 4 |
| CYP1A1, CYP1B1, CYP19A1, CYP17A1 | isoforms of cytochrome 450 |
| CYP3A4 | cytochrome P450 3A4 |
| DDR | DNA damage response |
| DIABLO | direct IAP-binding protein with Low pI |
| DMBA | 7,12-dimethylbenz[a] anthracene |
| DNA | deoxyribonucleic acid |
| DOC | docetaxel |
| DOX | doxorubicin (adriamycin) |
| DPPT | deoxypodophyllotoxin |
| DR4/5 | pro-apoptotic death receptors |
| EC | epicatechin |
| EGC | epigallocatechin |
| EGCG | epigallocatechingallate |
| EGF | epidermal growth factor |
| EGFR | epithelial growth factor receptor |
| EGFR(T790M) | epithelial growth factor receptor with a mutation that replace threonine by methionine at position 790 |
| EGR-1 | early growth response protein 1 |
| EMT | epithelial-mesenchymal transition |
| ENL | enterolactone |
| ER | estrogen receptors |
| ERα/ERβ | estrogen receptor alpha/estrogen receptor beta |
| ERK 1,2 | extracellular-signal regulated kinase |
| ETS2 | proto-oncogene 2, transcription factor (v-ets, Avian Erythroblastosis Virus E26 Oncogene Homolog 2) |
| EZH2 | enhancer of zeste homolog 2 (histone methyltransferase) |
| FASN | fatty acid synthase |
| FBAP5 | fatty acid-binding protein 5 |
| FGF | fibroblast growth factor |
| FOLFOX | 5-fluorouracil, oxaliplatin, folinic acid |
| GF | gefitinib |
| GRP78 | glucose regulated protein |
| GSH | reduced glutathione |
| GSK3 | glycogen synthase kinase 3 |
| GST | glutathione-S transferase |
| GPX | glutathione peroxidase |
| GTE | green tea extract |
| HER-2, 3 | human epidermal growth factor 2, 3 |
| Her2/neu | receptor tyrosine-proteinkinase erbB-2 |
| HGFR/MET | hepatocyte growth factor receptor |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| hMLH1 | mismatch repair gene of human mutL homolog 1 |
| HNK | honokiol |
| HO-1 | hemeoxygenase 1 |
| HR | homologous recombination |
| HRas | transforming protein p21 |
| IAP | inhibitors of apoptosis proteins |
| ICAM-1 | intercellular adhesion molecule 1 |
| IGF-1R | insulin growth factor receptor |
| IL-6, 8, 17, 18 | interleukin-6, 8, 17, 18 |
| i.p. | intraperitoneal administration |
| i.v | intravenous administration |
| JNK | c-Jun N-terminal kinase |
| Keap 1 | kelch-like ECH-associated protein 1 |
| KRAS | gene identified in Kirsten rat sarcoma |
| MAPK | mitogen activated protein kinase |
| MDM2 | mouse double minute 2 homolog |
| MDR | multidrug resistance |
| Meriva | turmeric/phospholipid formulation |
| MET | tyrosine-proteinkinase |
| MLH1,2 | human mutL homolog 1,2 |
| MMP | mitochondrial membrane potential |
| MMP-2,9 | metalloproteinases 2,9 |
| MMR | mismatch repair |
| MRP1/2 | multidrug resistance associated protein 1/2 |
| mRNA | messenger RNA |
| miRNA | microRNA |
| miR-16, 17, 21, 200c, 17-5p, 892c | microRNA-16, 17, 21, 200c, 17-5p, 892c |
| MSH 1/2 | DNA mismatch repair protein 1/2 |
| mTOR | mammalian target of rapamycin |
| NAD(P)H | reduced nicotinamide adenine dinucleotide phosphate |
| NER | nucleotide excision repair |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGF | nerve growth factor |
| NHEJ | non-homologous end-joining |
| NOX | NADPH oxidases |
| NPs | nanoparticles |
| Nrf2 | erythroid 2-related factor 2 |
| NSCLC | non-small cell lung cancer |
| NSE | neurospecific enolase |
| OX | oxaliplatin |
| p53 | tumor suppressor protein |
| P-gp (MDR1) | P-glycoprotein (multidrug resistance protein 1) |
| PI3K/AKT | phosphoinositide 3-kinase/protein kinase B |
| PKC | proteinkinase C |
| p.o. | oral administration |
| PPARβ/δ | peroxisome proliferator-activated receptor β/δ |
| PPT | podophyllotoxin |
| PS | phospho-sulindac |
| PSA | prostate serum antigen |
| PTEN | phosphatase and tensin homolog |
| PTX | paclitaxel |
| PXR | pregnane X receptor |
| RARs | retinoic acid receptors |
| RES | resveratrol |
| ROS | reactive oxygen species |
| SCC | squamous cell carcinoma |
| SChA | schizandrin A |
| SCLC | small cell lung cancer |
| SECO | secoisolariciresinol |
| Smac | second mitochondria-derived activator of caspase |
| SOD | superoxide-dismutase |
| Src | proto-oncogene tyrosine-protein kinase |
| SRT501 | small molecule, a form of resveratrol designed to target sirtuin 1 protein |
| STAT3 | signal transducer and activator of transcription 3 |
| T-box 3 | T-box transcription factor 3 |
| Tf-PEG-CUR | transferrin-poly(ethylene glycol)-curcumin |
| TGF-β | transforming growth factor |
| TKI | tyrosine kinase inhibitors |
| TME | tumor microenvironment |
| TP53 | gene coding tumor suppressor protein p53 |
| TRAIL | TNF-related apoptosis-inducing ligand |
| UGT | uridine diphospho-glucuronosyltransferase |
| VCR | vincristine |
| VEGF | vascular endothelial growth factor |
| VEGFR2 | vascular endothelial growth factor receptor 2 |
| Wnt/β-catenin | wingless-type MMTV integration site family member (MMTV, mouse mammary tumor virus)/beta-catenin signaling pathway |
| xCT | glutamate cysteine antiporter |
| XIAP | X-linked inhibitor of apoptosis protein |
| YB-1 | Y-box binding protein-1 |
References
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundqvist, A.; Andersson, E.; Ahlberg, I.; Nilbert, M.; Gerdtham, U. Socioeconomic inequalities in breast cancer incidence and mortality in Europe—A systematic review and meta-analysis. Eur. J. Public Health 2016, 26, 804–813. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.N.; Schwab, R.B.; Martinez, M.E. Reproductive risk factors and breast cancer subtypes: A review of the literature. Breast Cancer Res. Treat 2014, 144, 1–10. [Google Scholar] [CrossRef]
- Banin Hirata, B.K.; Oda, J.M.M.; Losi Guembarovski, R.; Ariza, C.B.; de Oliveira, C.E.; Watanabe, M.A.E. Molecular markers for breast cancer: Prediction on tumor behavior. Dis. Markers 2014. [Google Scholar] [CrossRef] [PubMed]
- Kamińska, M.; Ciszewski, T.; Łopacka-Szatan, K.; Miotła, P.; Starosławska, E. Breast cancer risk factors. Prz. Menopauzalny 2015, 14, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samavat, H.; Kurzer, M.S. Estrogen metabolism and breast cancer. Cancer Lett. 2015, 356, 231–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surakasula, A.; Nagarjunapu, G.C.; Raghavaiah, K.V. A comparative study of pre- and post-menopausal breast cancer: Risk factors, presentation, characteristics and management. J. Res. Pharm. Pr. 2014, 3, 12–18. [Google Scholar] [CrossRef]
- Farouk, O.; Ebrahim, M.A.; Senbel, A.; Emarah, Z.; Abozeed, W.; Seisa, M.O.; Mackisack, S.; Jalil, S.A.; Abdelhady, S. Breast cancer characteristics in very young Egyptian women ≤ 35 years. Breast Cancer: Targets Ther. 2016, 8, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, H.L.; Smith, L.; Tomlinson, D.C. Multidrug-resistant breast cancer: Current perspectives. Targets Ther. 2014, 6, 1–13. [Google Scholar]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Welch, H.G.; Prorok, P.C.; O’Malley, A.J.; Kramer, B.S. Breast-cancer tumor size, overdiagnosis, and mammography screening effectiveness. N. Engl. J. Med. 2016, 375, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Ridge, C.A.; McErlean, A.M.; Ginsberg, M.S. Seminars in Interventional Radiology in Epidemiology of Lung Cancer; Thieme Medical Publishers: New York, NY, USA, 2013; Volume 30, pp. 093–098. [Google Scholar]
- Didkowska, J.; Wojciechowska, U.; Mańczuk, M.; Łobaszewski, J. Lung cancer epidemiology: Contemporary and future challenges worldwide. Ann. Transl. Med. 2016, 4, 150. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Zhang, L.-Q.; Huang, J.-F.; Liu, K.; Chuai, Z.-R.; Yang, Z.; Wang, Y.-X.; Shi, D.-C.; Liu, Q.; Huang, Q. BRAF mutations in patients with non-small cell lung cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e101354. [Google Scholar] [CrossRef] [Green Version]
- Gazdar, A.F.; Zhou, C. Lung Cancer in Never-Smokers: A Different Disease in IASLC Thoracic Oncology; Pass, H.I., Ball, D., Scagliotti, G.V., Eds.; Elsevier: Cambridge, MA, USA, 2018; pp. 23–29. [Google Scholar]
- Midha, A.; Dearden, S.; McCormack, R. EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: A systematic review and global map by ethnicity (mutMapII). Am. J. Cancer Res. 2015, 5, 2892–2911. [Google Scholar]
- Øines, M.; Helsingen, L.M.; Bretthauer, M.; Emilsson, L. Epidemiology and risk factors of colorectal polyps. Best Pr. Res. Clin. Gastroenterol. 2017, 31, 419–424. [Google Scholar] [CrossRef]
- Sakai, E.; Nakajima, A.; Kaneda, A. Accumulation of aberrant DNA methylation during colorectal cancer development. World J. Gastroenterol. 2014, 20, 978–987. [Google Scholar] [CrossRef]
- Wong, S.H.; Kwong, T.N.Y.; Wu, C.-Y.; Yu, J. Clinical applications of gut microbiota in cancer biology. Semin. Cancer Biol. 2019, 55, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Schwedhelm, C.; Hoffmann, G.; Knüppel, S.; Laure Preterre, A.; Iqbal, K.; Bechthold, A.; De Henauw, S.; Michels, N.; Devleesschauwer, B. Food groups and risk of colorectal cancer. Int. J. Cancer 2018, 142, 1748–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.S.-H.; Chay, W.Y.; Tan, M.-H.; Chow, K.Y.; Lim, W.-Y. Reproductive factors, obesity and risk of colorectal cancer in a cohort of Asian women. Cancer Epidemiol. 2019, 58, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Ward, H.A.; Jenab, M.; Rothwell, J.A.; Boutron-Ruault, M.-C.; Carbonnel, F.; Kvaskoff, M.; Kaaks, R.; Kühn, T.; Boeing, H.; et al. Heterogeneity of Colorectal Cancer Risk Factors by Anatomical Subsite in 10 European Countries: A Multinational Cohort Study. Clin. Gastroenterol. Hepatol. 2019, 17, 1323–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witold, K.; Anna, K.; Maciej, T.; Jakub, J. Adenomas–Genetic factors in colorectal cancer prevention. Rep. Pr. Oncol. Radiother. 2018, 23, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Tsaur, I.; Heidegger, I.; Kretschmer, A.; Borgmann, H.; Gandaglia, G.; Briganti, A.; de Visschere, P.; Mathieu, R.; Valerio, M.; van den Bergh, R.; et al. Aggressive variants of prostate cancer—Are we ready to apply specific treatment right now? Cancer Treat. Rev. 2019, 75, 20–26. [Google Scholar] [CrossRef]
- Leitzmann, M.F.; Rohrmann, S. Risk factors for the onset of prostatic cancer: Age, location, and behavioral correlates. Clin. Epidemiol. 2012, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- McAllister, M.J.; Underwood, M.A.; Leung, H.Y.; Edwards, J. A review on the interactions between the tumor microenvironment and androgen receptor signaling in prostate cancer. Transl. Res. 2019, 206, 91–106. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.-S.H.; Neal, J.W.; Wakelee, H. Review of the current targeted therapies for non-small-cell lung cancer. World J. Clin. Oncol. 2014, 5, 576–587. [Google Scholar] [CrossRef]
- Eid, S.Y.; El-Readi, M.Z.; Fatani, S.H.; Eldin, E.E.M.N.; Wink, M. Natural products modulate the multifactorial multidrug resistance of cancer. Pharm. 2015, 6, 146–176. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Yang, H.L.; Yang, Y.J.; Wang, L.; Lee, S.C. Overcome cancer cell drug resistance using natural products. Evid. Based Complement. Altern. Med. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabekura, T. Overcoming multidrug resistance in human cancer cells by natural compounds. Toxins 2010, 2, 1207–1224. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Liu, K.; Shen, Q.; Li, Q.; Hao, J.; Han, F.; Jiang, R.W. Reversal of Multidrug Resistance in Cancer by Multi-Functional Flavonoids. Front. Oncol. 2019, 9, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotecha, R.; Takami, A.; Espinoza, J.L. Dietary phytochemicals and cancer chemoprevention: A review of the clinical evidence. Oncotarget 2016, 7, 52517–52529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangney, C.C.; Rasmussen, H.E. Polyphenols, inflammation, and cardiovascular disease. Curr. Atheroscler. Rep. 2013, 15, 324. [Google Scholar] [CrossRef] [PubMed]
- Mrduljaš, N.; Krešić, G.; Bilušić, T. Polyphenols: Food Sources and Health Benefits in Functional Food-Improve Health through Adequate Food; Hueda, M.C., Ed.; IntechOpen: London, UK, 2017; Available online: https://www.intechopen.com/ (accessed on 2 January 2020). [CrossRef] [Green Version]
- Estrela, J.M.; Mena, S.; Obrador, E.; Benlloch, M.; Castellano, G.; Salvador, R.; Dellinger, R.W. Polyphenolic Phytochemicals in Cancer Prevention and Therapy: Bioavailability versus Bioefficacy. J. Med. Chem. 2017, 60, 9413–9436. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, Q.; Li, D.; Zhou, Y.; Zheng, X.; Sun, H.; Yan, J.; Zhang, L.; Lin, Y.; Wang, X. Wogonin enhances antitumor activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo through ROS-mediated downregulation of cFLIPL and IAP proteins. Apoptosis 2013, 18, 618–626. [Google Scholar] [CrossRef]
- Kim, K.; Vance, T.M.; Chun, O.K. Estimated intake and major food sources of flavonoids among US adults: Changes between 1999–2002 and 2007–2010 in NHANES. Eur. J. Nutr. 2016, 55, 833–843. [Google Scholar] [CrossRef]
- Wang, Z.; Li, X.; Wang, D.; Zou, Y.; Qu, X.; He, C.; Deng, Y.; Jin, Y.; Zhou, Y.; Zhou, Y. Concurrently suppressing multidrug resistance and metastasis of breast cancer by co-delivery of paclitaxel and honokiol with pH-sensitive polymeric micelles. Acta Biomater. 2017, 62, 144–156. [Google Scholar] [CrossRef]
- Roy, A.; Ernsting, M.J.; Undzys, E.; Li, S.D. A highly tumor-targeted nanoparticle of podophyllotoxin penetrated tumor core and regressed multidrug resistant tumors. Biomaterials 2015, 52, 335–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, X.; Wang, G.; Cai, Q.; Zheng, X.; Zhang, J.; Chen, Q.; Wu, B.; Zhu, X.; Hao, H.; Zhou, F. A Promising Microtubule Inhibitor Deoxypodophyllotoxin Exhibits Better Efficacy to Multidrug-Resistant Breast Cancer than Paclitaxel via Avoiding Efflux Transport. Drug Metab. Dispos. 2018, 46, 542–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molavi, O.; Narimani, F.; Asiaee, F.; Sharifi, S.; Tarhriz, V.; Shayanfar, A.; Hejazi, M.; Lai, R. Silibinin sensitizes chemo-resistant breast cancer cells to chemotherapy. Pharm. Biol. 2017, 55, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, Y.; De Silva, F.; Krol, E.S.; Alcorn, J. Flaxseed lignans enhance the cytotoxicity of chemotherapeutic agents against breast cancer cell lines MDA-MB-231 and SKBR3. Nutr. Cancer 2018, 70, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Zhang, D.; Chu, X.; Wang, J. Schizandrin A enhances chemosensitivity of colon carcinoma cells to 5-fluorouracil through up-regulation of miR-195. Biomed. Pharm. 2018, 99, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.M.; Munekata, P.E.; Putnik, P.; Kovačević, D.B.; Muchenje, V.; Barba, F.J. Sources, Chemistry, and Biological Potential of Ellagitannins and Ellagic Acid Derivatives. In Studies in Natural Products Chemistry; Ur-Rahman, A., Ed.; Elsevier: Cambridge, MA, USA, 2018; Volume 60, pp. 189–221. [Google Scholar]
- Wei, Y.; Pu, X.; Zhao, L. Preclinical studies for the combination of paclitaxel and curcumin in cancer therapy. Oncol. Rep. 2017, 37, 3159–3166. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-M.; Kao, C.-L.; Tseng, Y.-T.; Lo, Y.-C.; Chen, C.-Y. Ginger phytochemicals inhibit cell growth and modulate drug resistance factors in docetaxel resistant prostate cancer cell. Molcules 2017, 22, 1477. [Google Scholar] [CrossRef]
- Harris, A.L.; Hochhauser, D. Mechanisms of multidrug resistance in cancer treatment. Acta Oncol. 1992, 31, 205–213. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef]
- Linn, S.C.; Giaccone, G. MDR1/P-glycoprotein expression in colorectal cancer. Eur. J. Cancer 1995, 31, 1291–1294. [Google Scholar] [CrossRef]
- Kong, X.B.; Yang, Z.K.; Liang, L.J.; Huang, J.F.; Lin, H.L. Overexpression of P-glycoprotein in hepatocellular carcinoma and its clinical implication. World J. Gastroenterol. 2000, 6, 134–135. [Google Scholar] [CrossRef]
- Clarke, R.; Leonessa, F.; Trock, B. Multidrug resistance/P-glycoprotein and breast cancer: Review and meta-analysis. Semin. Oncol. 2005, 32, S9–S15. [Google Scholar] [CrossRef]
- Triller, N.; Korosec, P.; Kern, I.; Kosnik, M.; Debeljak, A. Multidrug resistance in small cell lung cancer: Expression of P-glycoprotein, multidrug resistance protein 1 and lung resistance protein in chemo-naive patients and in relapsed disease. Lung Cancer 2006, 54, 235–240. [Google Scholar] [CrossRef]
- Sanchez, C.; Mendoza, P.; Contreras, H.R.; Vergara, J.; McCubrey, J.A.; Huidobro, C.; Castellon, E.A. Expression of multidrug resistance proteins in prostate cancer is related with cell sensitivity to chemotherapeutic drugs. Prostate 2009, 69, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Aldonza, M.B.D.; Hong, J.-Y.; Bae, S.Y.; Song, J.; Kim, W.K.; Oh, J.; Shin, Y.; Lee, S.H.; Lee, S.K. Suppression of MAPK signaling and reversal of mTOR-dependent MDR1-associated multidrug resistance by 21α-methylmelianodiol in lung cancer cells. PLoS ONE 2015, 10, e0127841. [Google Scholar] [CrossRef]
- Xu, J.W.; Li, Q.Q.; Tao, L.L.; Cheng, Y.Y.; Yu, J.; Chen, Q.; Liu, X.P.; Xu, Z.D. Involvement of EGFR in the promotion of malignant properties in multidrug resistant breast cancer cells. Int. J. Oncol. 2011, 39, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Fan, Z.Z.; Li, Q. Signal transduction pathways and transcriptional mechanisms of ABCB1/Pgp-mediated multiple drug resistance in human cancer cells. J. Int. Med. Res. 2012, 40, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.X.; Sun, Y.B.; Wang, S.Q.; Duan, L.; Huo, Q.L.; Ren, F.; Li, G.F. Grape seed procyanidin reversal of p-glycoprotein associated multi-drug resistance via down-regulation of NF-kappaB and MAPK/ERK mediated YB-1 activity in A2780/T cells. PLoS ONE 2013, 8, e71071. [Google Scholar] [CrossRef]
- Akiyama, K.; Ohga, N.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Ishikawa, S.; Maishi, N.; Akino, T.; Kondoh, M.; Matsuda, A.; et al. Tumor Endothelial Cells Acquire Drug Resistance by MDR1 Up-Regulation via VEGF Signaling in Tumor Microenvironment. Am. J. Pathol. 2012, 180, 1283–1293. [Google Scholar] [CrossRef]
- Mirzaei, S.A.; Dinmohammadi, F.; Alizadeh, A.; Elahian, F. Inflammatory pathway interactions and cancer multidrug resistance regulation. Life Sci. 2019, 235, 116825. [Google Scholar] [CrossRef]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.M.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Sykes, D.B.; Miller, D.S. Constitutive androstane receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. Mol. Pharm. 2010, 78, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, M.; Robbins, D.; Chen, T. Targeting xenobiotic receptors PXR and CAR in human diseases. Drug Discov. Today 2015, 20, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Alexa-Stratulat, T.; Pešić, M.; Gašparović, A.Č.; Trougakos, I.P.; Riganti, C. What sustains the multidrug resistance phenotype beyond ABC efflux transporters? Looking beyond the tip of the iceberg. Drug Resist. Updates 2019, 46, 100643. [Google Scholar] [CrossRef] [PubMed]
- Sulova, Z.; Macejova, D.; Seres, M.; Sedlak, J.; Brtko, J.; Breier, A. Combined treatment of P-gp-positive L1210/VCR cells by verapamil and all-trans retinoic acid induces down-regulation of P-glycoprotein expression and transport activity. Toxicol. Vitr. 2008, 22, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Abd Ellah, N.H.; Taylor, L.; Ayres, N.; Elmahdy, M.M.; Fetih, G.N.; Jones, H.N.; Ibrahim, E.A.; Pauletti, G.M. NF-kappaB decoy polyplexes decrease P-glycoprotein-mediated multidrug resistance in colorectal cancer cells. Cancer Gene 2016, 23, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Bush, J.A.; Li, G. Cancer chemoresistance: The relationship between p53 and multidrug transporters. Int. J. Cancer 2002, 98, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, G.F.; Yang, J.M.; Vassil, A.; Yang, J.; Bash-Babula, J.; Hait, W.N. Regulation of expression of the multidrug resistance protein MRP1 by p53 in human prostate cancer cells. J. Clin. Investig. 2000, 105, 1261–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavrovskaya, A.A. Cellular mechanisms of multidrug resistance of tumor cells. Biochem. Mosc. 2000, 65, 95–106. [Google Scholar]
- Wang, X.K.; Fu, L.W. Interaction of tyrosine kinase inhibitors with the MDR- related ABC transporter proteins. Curr. Drug Metab. 2010, 11, 618–628. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharm. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef] [Green Version]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Sakurai, Y. Increased accumulation of vincristine and adriamycin in drug-resistant P388 tumor cells following incubation with calcium antagonists and calmodulin inhibitors. Cancer Res. 1982, 42, 4730–4733. [Google Scholar]
- Tsuruo, T.; Iida, H.; Nojiri, M.; Tsukagoshi, S.; Sakurai, Y. Circumvention of vincristine and Adriamycin resistance in vitro and in vivo by calcium influx blockers. Cancer Res. 1983, 43, 2905–2910. [Google Scholar] [PubMed]
- Dalton, W.S.; Grogan, T.M.; Meltzer, P.S.; Scheper, R.J.; Durie, B.G.; Taylor, C.W.; Miller, T.P.; Salmon, S.E. Drug-resistance in multiple myeloma and non-Hodgkin’s lymphoma: Detection of P-glycoprotein and potential circumvention by addition of verapamil to chemotherapy. J. Clin. Oncol. 1989, 7, 415–424. [Google Scholar] [CrossRef]
- To, K.K.; Tomlinson, B. Targeting the ABCG2-overexpressing multidrug resistant (MDR) cancer cells by PPARgamma agonists. Br. J. Pharm. 2013, 170, 1137–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Kosuri, K.V.; Wu, X.; Wang, L.; Villalona-Calero, M.A.; Otterson, G.A. An epigenetic mechanism for capecitabine resistance in mesothelioma. Biochem. Biophys. Res. Commun. 2010, 391, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Novotna, R.; Wsol, V.; Xiong, G.; Maser, E. Inactivation of the anticancer drugs doxorubicin and oracin by aldo-keto reductase (AKR) 1C3. Toxicol. Lett. 2008, 181, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Brozovic, A.; Goncalves, A.C.; Jurkovicova, D.; Line, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates 2019, 46, 100645. [Google Scholar] [CrossRef]
- Tew, K.D. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994, 54, 4313–4320. [Google Scholar] [CrossRef] [Green Version]
- Tew, K.D. Glutathione-Associated enzymes in anticancer drug resistance. Cancer Res. 2016, 76, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starlard-Davenport, A.; Lyn-Cook, B.; Beland, F.A.; Pogribny, I.P. The role of UDP-glucuronosyltransferases and drug transporters in breast cancer drug resistance. Exp. Oncol. 2010, 32, 172–180. [Google Scholar] [PubMed]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar]
- Slyskova, J.; Sabatella, M.; Ribeiro-Silva, C.; Stok, C.; Theil, A.F.; Vermeulen, W.; Lans, H. Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 2018, 46, 9537–9549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Balaguer, F.; Sempere, L.; Xicola, R.M.; Bujanda, L.; et al. The efficacy of adjuvant chemotherapy with 5-fluorouracil in colorectal cancer depends on the mismatch repair status. Eur. J. Cancer 2009, 45, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- O’Hare, T.; Eide, C.A.; Deininger, M.W. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110, 2242–2249. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Tian, Z.; Tang, J.; Yang, Q.; Li, X.; Zhu, J.; Wu, G. Atypical ubiquitin-binding protein SHARPIN promotes breast cancer progression. Biomed. Pharm. 2019, 119, 109414. [Google Scholar] [CrossRef]
- Baguley, B.C. Multiple drug resistance mechanisms in cancer. Mol. Biotechnol. 2010, 46, 308–316. [Google Scholar] [CrossRef]
- Longley, D.; Johnston, P. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Bruyere, C.; Meijer, L. Targeting cyclin-dependent kinases in anti-neoplastic therapy. Curr. Opin. Cell Biol. 2013, 25, 772–779. [Google Scholar] [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxid Med. Cell Longev. 2017. [Google Scholar] [CrossRef] [Green Version]
- Wangpaichitr, M.; Wu, C.; You, M.; Kuo, M.T.; Feun, L.; Lampidis, T.; Savaraj, N. Inhibition of mTOR restores cisplatin sensitivity through down-regulation of growth and anti-apoptotic proteins. Eur. J. Pharm. 2008, 591, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Sewify, E.M.; Afifi, O.A.; Mosad, E.; Zaki, A.H.; El Gammal, S.A. Cyclin D1 amplification in multiple myeloma is associated with multidrug resistance expression. Clin. Lymphoma Myeloma Leuk. 2014, 14, 215–222. [Google Scholar] [CrossRef]
- Gillet, J.P.; Gottesman, M.M. Mechanisms of Multidrug Resistance in Cancer. In Multi-Drug Resistance in Cancer (Methods in Molecular Biology); Zhou, J., Ed.; Springer: New York, NY, USA, 2010; Volume 596, pp. 47–76. [Google Scholar]
- O’Brien, S.M.; Cunningham, C.C.; Golenkov, A.K.; Turkina, A.G.; Novick, S.C.; Rai, K.R. Phase I to II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in patients with advanced chronic lymphocytic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 7697–7702. [Google Scholar] [CrossRef]
- O’Brien, S.; Moore, J.O.; Boyd, T.E.; Larratt, L.M.; Skotnicki, A.; Koziner, B.; Chanan-Khan, A.A.; Seymour, J.F.; Bociek, R.G.; Pavletic, S.; et al. Randomized phase III trial of fludarabine plus cyclophosphamide with or without oblimersen sodium (Bcl-2 antisense) in patients with relapsed or refractory chronic lymphocytic leukemia. J. Clin. Oncol. 2007, 25, 1114–1120. [Google Scholar] [CrossRef]
- Soderquist, R.S.; Eastman, A. BCL2 Inhibitors as Anticancer Drugs: A Plethora of Misleading BH3 Mimetics. Mol. Cancer 2016, 15, 2011–2017. [Google Scholar] [CrossRef] [Green Version]
- Bedikian, A.Y.; Millward, M.; Pehamberger, H.; Conry, R.; Gore, M.; Trefzer, U.; Pavlick, A.C.; DeConti, R.; Hersh, E.M.; Hersey, P.; et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: The Oblimersen Melanoma Study Group. J. Clin. Oncol. 2006, 24, 4738–4745. [Google Scholar] [CrossRef]
- Ng, K.P.; Hillmer, A.M.; Chuah, C.T.; Juan, W.C.; Ko, T.K.; Teo, A.S.; Ariyaratne, P.N.; Takahashi, N.; Sawada, K.; Fei, Y.; et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med. 2012, 18, 521–528. [Google Scholar] [CrossRef]
- Merino, D.; Lalaoui, N.; Morizot, A.; Solary, E.; Micheau, O. TRAIL in cancer therapy: Present and future challenges. Expert Opin. Ther. Targets 2007, 11, 1299–1314. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.S. TRAIL as a target in anti-cancer therapy. Cancer Lett. 2009, 285, 1–5. [Google Scholar] [CrossRef]
- Hetschko, H.; Voss, V.; Seifert, V.; Prehn, J.H.; Kogel, D. Upregulation of DR5 by proteasome inhibitors potently sensitizes glioma cells to TRAIL-induced apoptosis. FEBS J. 2008, 275, 1925–1936. [Google Scholar] [CrossRef]
- Hunter, T.B.; Manimala, N.J.; Luddy, K.A.; Catlin, T.; Antonia, S.J. Paclitaxel and TRAIL synergize to kill paclitaxel-resistant small cell lung cancer cells through a caspase-independent mechanism mediated through AIF. Anticancer Res. 2011, 31, 3193–3204. [Google Scholar]
- Zhang, Y.Q.; Tang, X.Q.; Sun, L.; Dong, L.; Qin, Y.; Liu, H.Q.; Xia, H.; Cao, J.G. Rosiglitazone enhances fluorouracil-induced apoptosis of HT-29 cells by activating peroxisome proliferator-activated receptor gamma. World J. Gastroenterol. 2007, 13, 1534–1540. [Google Scholar] [CrossRef] [Green Version]
- Moitra, K.; Lou, H.; Dean, M. Multidrug efflux pumps and cancer stem cells: Insights into multidrug resistance and therapeutic development. Clin. Pharm. 2011, 89, 491–502. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 5416923. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [Green Version]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Schmidt, F.; Efferth, T. Tumor Heterogeneity, Single-Cell Sequencing, and Drug Resistance. Pharmaceuticals 2016, 9, 33. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Kwak, E.L.; Ahronian, L.G.; Siravegna, G.; Mussolin, B.; Borger, D.R.; Godfrey, J.T.; Jessop, N.A.; Clark, J.W.; Blaszkowsky, L.S.; Ryan, D.P.; et al. Molecular Heterogeneity and Receptor Coamplification Drive Resistance to Targeted Therapy in MET-Amplified Esophagogastric Cancer. Cancer Discov. 2015, 5, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [Green Version]
- Sergina, N.V.; Rausch, M.; Wang, D.; Blair, J.; Hann, B.; Shokat, K.M.; Moasser, M.M. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 2007, 445, 437–441. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Huang, S.; Kruser, T.J.; Nechrebecki, M.M.; Armstrong, E.A.; Benavente, S.; Gondi, V.; Hsu, K.T.; Harari, P.M. Mechanisms of acquired resistance to cetuximab: Role of HER (ErbB) family members. Oncogene 2008, 27, 3944–3956. [Google Scholar] [CrossRef] [Green Version]
- Riggins, R.B.; Schrecengost, R.S.; Guerrero, M.S.; Bouton, A.H. Pathways to tamoxifen resistance. Cancer Lett. 2007, 256, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Medeiros Alencar, V.H.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet Lond. Engl. 2013, 381, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Van Veggel, B.; de Langen, A.J.; Hashemi, S.M.S.; Monkhorst, K.; Heideman, D.A.M.; Thunnissen, E.; Smit, E.F. Afatinib and Cetuximab in Four Patients with EGFR Exon 20 Insertion-Positive Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1222–1226. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Sledge, G.W.; Neuberg, D.; Bernardo, P.; Ingle, J.N.; Martino, S.; Rowinsky, E.K.; Wood, W.C. Phase III trial of doxorubicin, paclitaxel, and the combination of doxorubicin and paclitaxel as front-line chemotherapy for metastatic breast cancer: An intergroup trial (E1193). J. Clin. Oncol. 2003, 21, 588–592. [Google Scholar] [CrossRef]
- Del Re, M.; Bordi, P.; Rofi, E.; Restante, G.; Valleggi, S.; Minari, R.; Crucitta, S.; Arrigoni, E.; Chella, A.; Morganti, R.; et al. The amount of activating EGFR mutations in circulating cell-free DNA is a marker to monitor osimertinib response. Br. J. Cancer 2018, 119, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Milane, L.; Ganesh, S.; Shah, S.; Duan, Z.F.; Amiji, M. Multi-modal strategies for overcoming tumor drug resistance: Hypoxia, the Warburg effect, stem cells, and multifunctional nanotechnology. J. Control Release 2011, 155, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Videira, M.; Reis, R.L.; Brito, M.A. Deconstructing breast cancer cell biology and the mechanisms of multidrug resistance. Biochim. Biophys. Acta 2014, 1846, 312–325. [Google Scholar] [CrossRef] [PubMed]
- McMillin, D.W.; Negri, J.M.; Mitsiades, C.S. The role of tumour-stromal interactions in modifying drug response: Challenges and opportunities. Nat. Rev. Drug Discov. 2013, 12, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shee, K.; Yang, W.; Hinds, J.W.; Hampsch, R.A.; Varn, F.S.; Traphagen, N.A.; Patel, K.; Cheng, C.; Jenkins, N.P.; Kettenbach, A.N.; et al. Therapeutically targeting tumor microenvironment-mediated drug resistance in estrogen receptor-positive breast cancer. J. Exp. Med. 2018, 215, 895–910. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Zhi, Q.; Wang, Y.; Li, Z.; Zhou, J.; Huang, J. Hypoxia Induces Multidrug Resistance via Enhancement of Epidermal Growth Factor-Like Domain 7 Expression in Non-Small Lung Cancer Cells. Chemotherapy 2017, 62, 172–180. [Google Scholar] [CrossRef]
- Faria, M.; Shepherd, P.; Pan, Y.; Chatterjee, S.S.; Navone, N.; Gustafsson, J.A.; Strom, A. The estrogen receptor variants beta2 and beta5 induce stem cell characteristics and chemotherapy resistance in prostate cancer through activation of hypoxic signaling. Oncotarget 2018, 9, 36273–36288. [Google Scholar] [CrossRef]
- Jahanban-Esfahlan, R.; de la Guardia, M.; Ahmadi, D.; Yousefi, B. Modulating tumor hypoxia by nanomedicine for effective cancer therapy. J. Cell Physiol. 2018, 233, 2019–2031. [Google Scholar] [CrossRef]
- Chen, J.; Ding, Z.; Peng, Y.; Pan, F.; Li, J.; Zou, L.; Zhang, Y.; Liang, H. HIF-1α inhibition reverses multidrug resistance in colon cancer cells via downregulation of MDR1/P-glycoprotein. PLoS ONE 2014, 9, e98882. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.; Friedl, P. Cancer invasion and resistance: Interconnected processes of disease progression and therapy failure. Trends Mol. Med. 2012, 18, 13–26. [Google Scholar] [CrossRef]
- Tezcan, O.; Ojha, T.; Storm, G.; Kiessling, F.; Lammers, T. Targeting cellular and microenvironmental multidrug resistance. Expert Opin. Drug Deliv. 2016, 13, 1199–1202. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Liu, Y.; Zhang, J.-T. A new mechanism of drug resistance in breast cancer cells: Fatty acid synthase overexpression-mediated palmitate overproduction. Mol. Cancer 2008, 7, 263–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Fayi, M.S.; Gou, X.; Forootan, S.S.; Al-Jameel, W.; Bao, Z.; Rudland, P.R.; Cornford, P.A.; Hussain, S.A.; Ke, Y. The increased expression of fatty acid-binding protein 9 in prostate cancer and its prognostic significance. Oncotarget 2016, 7, 82783–82797. [Google Scholar] [PubMed]
- Wu, X.; Qin, L.; Fako, V.; Zhang, J.T. Molecular mechanisms of fatty acid synthase (FASN)-mediated resistance to anti-cancer treatments. Adv. Biol. Regul. 2014, 54, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Bauerschlag, D.O.; Maass, N.; Leonhardt, P.; Verburg, F.A.; Pecks, U.; Zeppernick, F.; Morgenroth, A.; Mottaghy, F.M.; Tolba, R.; Meinhold-Heerlein, I.; et al. Fatty acid synthase overexpression: Target for therapy and reversal of chemoresistance in ovarian cancer. J. Transl. Med. 2015, 13, 146. [Google Scholar] [CrossRef] [Green Version]
- Plava, J.; Cihova, M.; Burikova, M.; Matuskova, M.; Kucerova, L.; Miklikova, S. Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Mol Cancer 2019, 18, 67. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Butera, G.; Pacchiana, R.; Donadelli, M. Autocrine mechanisms of cancer chemoresistance. Semin. Cell Dev. Biol. 2018, 78, 3–12. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, B.C.; Fujii, T.; Dorfman, J.D.; Goodwin, J.M.; Zhu, A.X.; Lanuti, M.; Tanabe, K.K. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008, 68, 2391–2399. [Google Scholar] [CrossRef] [Green Version]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molcules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofides, A.; Karantanos, T.; Bardhan, K.; Boussiotis, V.A. Epigenetic regulation of cancer biology and anti-tumor immunity by EZH2. Oncotarget 2016, 7, 85624–85640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.W.; Gwak, S.Y.; Shim, G.A.; Liu, L.; Lim, Y.C.; Kim, J.M.; Jung, M.G.; Koo, B.S. EZH2 is associated with poor prognosis in head-and-neck squamous cell carcinoma via regulating the epithelial-to-mesenchymal transition and chemosensitivity. Oral Oncol. 2016, 52, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726. [Google Scholar] [CrossRef]
- An, X.; Sarmiento, C.; Tan, T.; Zhu, H. Regulation of multidrug resistance by microRNAs in anti-cancer therapy. Acta Pharm. Sinb. 2017, 7, 38–51. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.-Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.-H.; Chen, Z.-S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updates 2018, 41, 1–25. [Google Scholar] [CrossRef]
- Cen, J.; Zhang, L.; Liu, F.; Zhang, F.; Ji, B.S. Long-Term Alteration of Reactive Oxygen Species Led to Multidrug Resistance in MCF-7 Cells. Oxid Med. Cell Longev. 2016, 2016, 7053451. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Tang, Y.; Zhou, H.; Liu, Y.; Huang, J.; Li, L.; Liu, W.; Feng, Y.; Zhou, Y.; Chen, T.; et al. STAT3 mediates multidrug resistance of Burkitt lymphoma cells by promoting antioxidant feedback. Biochem. Biophys. Res. Commun. 2017, 488, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, S.; Turkekul, K.; Serttas, R.; Erdogan, Z. The natural flavonoid apigenin sensitizes human CD44(+) prostate cancer stem cells to cisplatin therapy. Biomed. Pharm. 2017, 88, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.S.; Satelli, A.; Moridani, M.; Jenkins, M.; Rao, U.S. Luteolin induces apoptosis in multidrug resistant cancer cells without affecting the drug transporter function: Involvement of cell line-specific apoptotic mechanisms. Int. J. Cancer 2012, 130, 2703–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Wang, H.; Fan, L.; Wu, X.; Xin, A.; Ren, H.; Wang, X.J. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic. Biol. Med. 2011, 50, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Sabzichi, M.; Hamishehkar, H.; Ramezani, F.; Sharifi, S.; Tabasinezhad, M.; Pirouzpanah, M.; Ghanbari, P.; Samadi, N. Luteolin-loaded phytosomes sensitize human breast carcinoma MDA-MB 231 cells to doxorubicin by suppressing Nrf2 mediated signalling. Asian Pac. J. Cancer Prev. 2014, 15, 5311–5316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zhao, Q.; Wang, B.; Yuan, S.; Wang, X.; Li, K. Quercetin reversed MDR in breast cancer cells through down-regulating P-gp expression and eliminating cancer stem cells mediated by YB-1 nuclear translocation. Phytother Res. 2018, 32, 1530–1536. [Google Scholar] [CrossRef]
- Chieli, E.; Romiti, N.; Rodeiro, I.; Garrido, G. In vitro effects of Mangifera indica and polyphenols derived on ABCB1/P-glycoprotein activity. Food Chem. Toxicol. 2009, 47, 2703–2710. [Google Scholar] [CrossRef]
- Taur, J.S.; Rodriguez-Proteau, R. Effects of dietary flavonoids on the transport of cimetidine via P-glycoprotein and cationic transporters in Caco-2 and LLC-PK1 cell models. Xenobiotica 2008, 38, 1536–1550. [Google Scholar] [CrossRef]
- Chung, S.Y.; Sung, M.K.; Kim, N.H.; Jang, J.O.; Go, E.J.; Lee, H.J. Inhibition of P-glycoprotein by natural products in human breast cancer cells. Arch. Pharm. Res. 2005, 28, 823–828. [Google Scholar] [CrossRef]
- Jeng, L.B.; Kumar Velmurugan, B.; Chen, M.C.; Hsu, H.H.; Ho, T.J.; Day, C.H.; Lin, Y.M.; Padma, V.V.; Tu, C.C.; Huang, C.Y. Fisetin mediated apoptotic cell death in parental and Oxaliplatin/irinotecan resistant colorectal cancer cells in vitro and in vivo. J. Cell Physiol. 2018, 233, 7134–7142. [Google Scholar] [CrossRef] [PubMed]
- Febriansah, R.; Putri, D.D.; Sarmoko; Nurulita, N.A.; Meiyanto, E.; Nugroho, A.E. Hesperidin as a preventive resistance agent in MCF-7 breast cancer cells line resistance to doxorubicin. Asian Pac. J. Trop. Biomed. 2014, 4, 228–233. [Google Scholar] [CrossRef] [Green Version]
- El-Readi, M.Z.; Hamdan, D.; Farrag, N.; El-Shazly, A.; Wink, M. Inhibition of P-glycoprotein activity by limonin and other secondary metabolites from Citrus species in human colon and leukaemia cell lines. Eur. J. Pharm. 2010, 626, 139–145. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Zupko, I.; Chang, F.R.; Hunyadi, A.; Wu, C.C.; Weng, T.S.; Wang, H.C. Dietary flavonoid derivatives enhance chemotherapeutic effect by inhibiting the DNA damage response pathway. Toxicol. Appl. Pharm. 2016, 311, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Knop, J.; Misaka, S.; Singer, K.; Hoier, E.; Muller, F.; Glaeser, H.; Konig, J.; Fromm, M.F. Inhibitory Effects of Green Tea and (-)-Epigallocatechin Gallate on Transport by OATP1B1, OATP1B3, OCT1, OCT2, MATE1, MATE2-K and P-Glycoprotein. PLoS ONE 2015, 10, e0139370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jodoin, J.; Demeule, M.; Beliveau, R. Inhibition of the multidrug resistance P-glycoprotein activity by green tea polyphenols. Biochim. Biophys. Acta 2002, 1542, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Esmaeili, M.A. Combination of siRNA-directed gene silencing with epigallocatechin-3-gallate (EGCG) reverses drug resistance in human breast cancer cells. J. Chem. Biol. 2016, 9, 41–52. [Google Scholar] [CrossRef] [Green Version]
- La, X.; Zhang, L.; Li, Z.; Li, H.; Yang, Y. (-)-Epigallocatechin Gallate (EGCG) Enhances the Sensitivity of Colorectal Cancer Cells to 5-FU by Inhibiting GRP78/NF-κB/miR-155–5p/MDR1 Pathway. J. Agric. Food Chem. 2019, 67, 2510–2518. [Google Scholar] [CrossRef]
- Wang, P.; Henning, S.M.; Heber, D.; Vadgama, J.V. Sensitization to docetaxel in prostate cancer cells by green tea and quercetin. J. Nutr. Biochem. 2015, 26, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.C.; Lee, C. Reversal of Cisplatin resistance by epigallocatechin gallate is mediated by downregulation of axl and tyro 3 expression in human lung cancer cells. Korean J. Physiol. Pharm. 2014, 18, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.P.; Wang, G.; Zhao, Z.B.; Wang, Q.; Shi, Y. Synergistic cytotoxic effect of genistein and doxorubicin on drug-resistant human breast cancer MCF-7/Adr cells. Oncol. Rep. 2014, 32, 1647–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ahmed, F.; Ali, S.; Philip, P.A.; Kucuk, O.; Sarkar, F.H. Inactivation of nuclear factor κB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res. 2005, 65, 6934–6942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigalli, J.P.; Scholz, P.N.; Tocchetti, G.N.; Ruiz, M.L.; Weiss, J. The phytoestrogens daidzein and equol inhibit the drug transporter BCRP/ABCG2 in breast cancer cells: Potential chemosensitizing effect. Eur. J. Nutr. 2019, 58, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Alayev, A.; Berger, S.M.; Kramer, M.Y.; Schwartz, N.S.; Holz, M.K. The combination of rapamycin and resveratrol blocks autophagy and induces apoptosis in breast cancer cells. J. Cell Biochem. 2015, 116, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Wu, X.-N.; Chen, J.; Wang, W.-X.; Lu, Z. Resveratrol reverses multidrug resistance in human breast cancer doxorubicin-resistant cells. Expther. Med. 2014, 7, 1611–1616. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Hoti, S.; Prasad, N. Resveratrol modulates expression of ABC transporters in non-small lung cancer cells: Molecular docking and gene expression studies. J. Cancer Sci. 2014, 6, 497–504. [Google Scholar] [CrossRef]
- Khaleel, S.A.; Al-Abd, A.M.; Ali, A.A.; Abdel-Naim, A.B. Didox and resveratrol sensitize colorectal cancer cells to doxorubicin via activating apoptosis and ameliorating P-glycoprotein activity. Sci. Rep. 2016, 6, 36855. [Google Scholar] [CrossRef]
- Zhu, Y.; He, W.; Gao, X.; Li, B.; Mei, C.; Xu, R.; Chen, H. Resveratrol overcomes gefitinib resistance by increasing the intracellular gefitinib concentration and triggering apoptosis, autophagy and senescence in PC9/G NSCLC cells. Sci. Rep. 2015, 5, 17730. [Google Scholar] [CrossRef] [Green Version]
- Vinod, B.S.; Nair, H.H.; Vijayakurup, V.; Shabna, A.; Shah, S.; Krishna, A.; Pillai, K.S.; Thankachan, S.; Anto, R.J. Resveratrol chemosensitizes HER-2-overexpressing breast cancer cells to docetaxel chemoresistance by inhibiting docetaxel-mediated activation of HER-2–Akt axis. Cell Death Discov. 2015, 1, 15061. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Subramaniam, D.; Ramalingam, S.; Dhar, A.; Postier, R.G.; Umar, S.; Zhang, Y.; Anant, S. Honokiol radiosensitizes colorectal cancer cells: Enhanced activity in cells with mismatch repair defects. Am. J. Physiol. Gastrointest Liver Physiol. 2011, 301, G929–G937. [Google Scholar] [CrossRef]
- Li, Y.; Revalde, J.; Paxton, J.W. The effects of dietary and herbal phytochemicals on drug transporters. Adv. Drug Deliv. Rev. 2017, 116, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.-Y.; Chung, Y.-C.; Hou, Y.-C.; Tsai, Y.-W.; Chen, C.-H.; Chang, H.-P.; Chou, J.-L.; Hsu, C.-P. Effects of ellagic acid on chemosensitivity to 5-fluorouracil in colorectal carcinoma cells. Anticancer Res. 2012, 32, 4413–4418. [Google Scholar] [PubMed]
- Berdowska, I.; Zieliński, B.; Saczko, J.; Sopel, M.; Gamian, A.; Fecka, I. Modulatory impact of selected ellagitannins on the viability of human breast cancer cells. J. Funct. Foods 2018, 42, 122–128. [Google Scholar] [CrossRef]
- Wang, R.; Ma, L.; Weng, D.; Yao, J.; Liu, X.; Jin, F. Gallic acid induces apoptosis and enhances the anticancer effects of cisplatin in human small cell lung cancer H446 cell line via the ROS-dependent mitochondrial apoptotic pathway. Oncol. Rep. 2016, 35, 3075–3083. [Google Scholar] [CrossRef] [PubMed]
- Nowakowska, A.; Tarasiuk, J. Comparative effects of selected plant polyphenols, gallic acid and epigallocatechin gallate, on matrix metalloproteinases activity in multidrug resistant MCF7/DOX breast cancer cells. Acta Biochim. Pol. 2016, 63, 571–575. [Google Scholar] [CrossRef] [Green Version]
- Phan, A.N.H.; Hua, T.N.M.; Kim, M.-K.; Vo, V.T.A.; Choi, J.-W.; Kim, H.-W.; Rho, J.K.; Kim, K.W.; Jeong, Y. Gallic acid inhibition of Src-Stat3 signaling overcomes acquired resistance to EGF receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer. Oncotarget 2016, 7, 54702–54713. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Shan, S.; Li, H.; Shi, J.; Zhang, X.; Li, Z. Reversal effects of bound polyphenol from foxtail millet bran on multidrug resistance in human HCT-8/Fu colorectal cancer cell. J. Agric. Food Chem. 2018, 66, 5190–5199. [Google Scholar] [CrossRef]
- Omene, C.O.; Wu, J.; Frenkel, K. Caffeic Acid Phenethyl Ester (CAPE) derived from propolis, a honeybee product, inhibits growth of breast cancer stem cells. Investig. New Drugs 2012, 30, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Khoram, N.M.; Bigdeli, B.; Nikoofar, A.; Goliaei, B. Caffeic acid phenethyl ester increases radiosensitivity of estrogen receptor-positive and-negative breast cancer cells by prolonging radiation-induced DNA damage. J. Breast Cancer 2016, 19, 18–25. [Google Scholar] [CrossRef]
- Ozturk, G.; Ginis, Z.; Akyol, S.; Erden, G.; Gurel, A.; Akyol, O. The anticancer mechanism of caffeic acid phenethyl ester (CAPE): Review of melanomas, lung and prostate cancers. Eur. Rev. Med. Pharm. Sci. 2012, 16, 2064–2068. [Google Scholar]
- Sonoki, H.; Tanimae, A.; Furuta, T.; Endo, S.; Matsunaga, T.; Ichihara, K.; Ikari, A. Caffeic acid phenethyl ester down-regulates claudin-2 expression at the transcriptional and post-translational levels and enhances chemosensitivity to doxorubicin in lung adenocarcinoma A549 cells. J. Nutr. Biochem. 2018, 56, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zeng, F.; Xu, L.; Zhou, J.; Liu, X.; Le, H. Anticancer effects of cinnamic acid in lung adenocarcinoma cell line h1299-derived stem-like cells. Oncol. Res. 2012, 20, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.-D.; Qin, Y.; Yang, C.; Li, L. Effect of curcumin on human colon cancer multidrug resistance in vitro and in vivo. Clinics 2013, 68, 694–701. [Google Scholar] [CrossRef]
- Lu, Y.; Wei, C.; Xi, Z. Curcumin suppresses proliferation and invasion in non-small cell lung cancer by modulation of MTA1-mediated Wnt/β-catenin pathway. In Vitro Cell. Dev. Biol. Anim. 2014, 50, 840–850. [Google Scholar] [CrossRef] [PubMed]
- De Porras, V.R.; Bystrup, S.; Martínez-Cardús, A.; Pluvinet, R.; Sumoy, L.; Howells, L.; James, M.I.; Iwuji, C.; Manzano, J.L.; Layos, L. Curcumin mediates oxaliplatin-acquired resistance reversion in colorectal cancer cell lines through modulation of CXC-Chemokine/NF-κB signalling pathway. Sci. Rep. 2016, 6, 24675. [Google Scholar] [CrossRef] [Green Version]
- Vinod, B.S.; Antony, J.; Nair, H.H.; Puliyappadamba, V.T.; Saikia, M.; Shyam Narayanan, S.; Bevin, A.; John Anto, R. Mechanistic evaluation of the signaling events regulating curcumin-mediated chemosensitization of breast cancer cells to 5-fluorouracil. Cell Death Discov. 2013, 4, e505. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Yu, Y.; Padhye, S.B.; Sarkar, F.H.; Majumdar, A.P. Difluorinated-curcumin (CDF) restores PTEN expression in colon cancer cells by down-regulating miR-21. PLoS ONE 2013, 8, e68543. [Google Scholar] [CrossRef]
- Shen, J.; Chen, Y.-J.; Jia, Y.-W.; Zhao, W.-Y.; Chen, G.-H.; Liu, D.-F.; Chen, Y.-Y.; Zhang, C.; Liu, X.P. Reverse effect of curcumin on CDDP-induced drug-resistance via Keap1/p62-Nrf2 signaling in A549/CDDP cell. Asian Pac. J. Trop. Med. 2017, 10, 1190–1196. [Google Scholar] [CrossRef]
- Gu, Y.; Li, J.; Li, Y.; Song, L.; Li, D.; Peng, L.; Wan, Y.; Hua, S. Nanomicelles loaded with doxorubicin and curcumin for alleviating multidrug resistance in lung cancer. Int. J. Nanomed. 2016, 11, 5757–5770. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.X.; Zhao, Y.L.; Li, Y.; Miao, Q.; Li, Z.-K.; Ren, X.L.; Song, L.Q.; Yin, H.; Zhang, J. Curcumin reverses cis-platin resistance and promotes human lung adenocarcinoma A549/DDP cell apoptosis through HIF-1α and caspase-3 mechanisms. Phytomedicine 2012, 19, 779–787. [Google Scholar] [CrossRef]
- Jiang, M.; Huang, O.; Zhang, X.; Xie, Z.; Shen, A.; Liu, H.; Geng, M.; Shen, K. Curcumin induces cell death and restores tamoxifen sensitivity in the antiestrogen-resistant breast cancer cell lines MCF-7/LCC2 and MCF-7/LCC9. Molcules 2013, 18, 701–720. [Google Scholar] [CrossRef] [Green Version]
- Thulasiraman, P.; McAndrews, D.J.; Mohiudddin, I.Q. Curcumin restores sensitivity to retinoic acid in triple negative breast cancer cells. BMC Cancer 2014, 14, 724. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Chen, R.; Zhong, Z.; Shi, Z.; Chen, M.; Wang, Y. Epigallocatechin-3-gallate potentiates the effect of curcumin in inducing growth inhibition and apoptosis of resistant breast cancer cells. Am. J. Chin. Med. 2014, 42, 1279–1300. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; Sobh, M.A.; Eid, H.M.; Salem, A.; Elbelasi, H.H.; El-Naggar, M.H.; AbdelBar, F.M.; Sheashaa, H.; Sobh, M.A.; Badria, F.A. Gingerol-derivatives: Emerging new therapy against human drug-resistant MCF-7. Tumor Biol. 2014, 35, 9941–9948. [Google Scholar] [CrossRef] [PubMed]
- Boumendjel, A.; Di Pietro, A.; Dumontet, C.; Barron, D. Recent advances in the discovery of flavonoids and analogs with high-affinity binding to P-glycoprotein responsible for cancer cell multidrug resistance. Med. Res. Rev. 2002, 22, 512–529. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.Y.; Li, H.B.; Sui, Z.Q.; Corke, H. Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (EGCG): An updated review. Crit. Rev. Food Sci. Nutr. 2018, 58, 924–941. [Google Scholar] [CrossRef]
- Rauf, A.; Imran, M.; Butt, M.S.; Nadeem, M.; Peters, D.G.; Mubarak, M.S. Resveratrol as an anti-cancer agent: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1428–1447. [Google Scholar] [CrossRef]
- Hussain, S.A.; Sulaiman, A.A.; Balch, C.; Chauhan, H.; Alhadidi, Q.M.; Tiwari, A.K. Natural Polyphenols in Cancer Chemoresistance. Nutr. Cancer 2016, 68, 879–891. [Google Scholar] [CrossRef]
- Czerwonka, A.; Maciolek, U.; Kalafut, J.; Mendyk, E.; Kuzniar, A.; Rzeski, W. Anticancer effects of sodium and potassium quercetin-5′-sulfonates through inhibition of proliferation, induction of apoptosis, and cell cycle arrest in the HT-29 human adenocarcinoma cell line. Bioorg. Chem. 2019, 94, 103426. [Google Scholar] [CrossRef]
- Scambia, G.; Ranelletti, F.O.; Panici, P.B.; De Vincenzo, R.; Bonanno, G.; Ferrandina, G.; Piantelli, M.; Bussa, S.; Rumi, C.; Cianfriglia, M.; et al. Quercetin potentiates the effect of adriamycin in a multidrug-resistant MCF-7 human breast-cancer cell line: P-glycoprotein as a possible target. Cancer Chemother. Pharm. 1994, 34, 459–464. [Google Scholar] [CrossRef]
- He, W.T.; Zhu, Y.H.; Zhang, T.; Abulimiti, P.; Zeng, F.Y.; Zhang, L.P.; Luo, L.J.; Xie, X.M.; Zhang, H.L. Curcumin Reverses 5-Fluorouracil Resistance by Promoting Human Colon Cancer HCT-8/5-FU Cell Apoptosis and Down-regulating Heat Shock Protein 27 and P-Glycoprotein. Chin. J. Integr. Med. 2019, 25, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhao, B.; Xiong, P.; Wang, C.; Zhang, J.; Tian, X.; Huang, Y. Curcumin Induces Autophagy via Inhibition of Yes-Associated Protein (YAP) in Human Colon Cancer Cells. Med. Sci. Monit. 2018, 24, 7035–7042. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Yang, Y.; Wang, G.; Chen, X.; Ju, Y. Curcumin attenuates resistance to irinotecan via induction of apoptosis of cancer stem cells in chemoresistant colon cancer cells. Int. J. Oncol. 2018, 53, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Fu, L.; Huang, J.; Dai, Y.; Wang, B.; Xu, G.; Wu, L.; Zhou, H. Curcumin reverses doxorubicin resistance via inhibition the efflux function of ABCB4 in doxorubicinresistant breast cancer cells. Mol. Med. Rep. 2019, 19, 5162–5168. [Google Scholar]
- Hu, C.; Li, M.; Guo, T.; Wang, S.; Huang, W.; Yang, K.; Liao, Z.; Wang, J.; Zhang, F.; Wang, H. Anti-metastasis activity of curcumin against breast cancer via the inhibition of stem cell-like properties and EMT. Phytomedicine 2019, 58, 152740. [Google Scholar] [CrossRef]
- Zhao, W.; Zhou, X.; Qi, G.; Guo, Y. Curcumin suppressed the prostate cancer by inhibiting JNK pathways via epigenetic regulation. J. Biochem. Mol. Toxicol. 2018, 32, e22049. [Google Scholar] [CrossRef]
- Lin, W.; Luo, J.; Sun, Y.; Lin, C.; Li, G.; Niu, Y.; Chang, C. ASC-J9((R)) suppresses prostate cancer cell invasion via altering the sumoylation-phosphorylation of STAT3. Cancer Lett. 2018, 425, 21–30. [Google Scholar] [CrossRef]
- Chen, Q.Y.; Lu, G.H.; Wu, Y.Q.; Zheng, Y.; Xu, K.; Wu, L.J.; Jiang, Z.Y.; Feng, R.; Zhou, J.Y. Curcumin induces mitochondria pathway mediated cell apoptosis in A549 lung adenocarcinoma cells. Oncol. Rep. 2010, 23, 1285–1292. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Debatin, K.M. Sensitization for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by the chemopreventive agent resveratrol. Cancer Res. 2004, 64, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.N.; Lin, V.C.; Rau, K.M.; Shieh, P.C.; Kuo, D.H.; Shieh, J.C.; Chen, W.J.; Tsai, S.C.; Way, T.D. Resveratrol modulates tumor cell proliferation and protein translation via SIRT1-dependent AMPK activation. J. Agric. Food Chem. 2010, 58, 1584–1592. [Google Scholar] [CrossRef]
- Lecumberri, E.; Dupertuis, Y.M.; Miralbell, R.; Pichard, C. Green tea polyphenol epigallocatechin-3-gallate (EGCG) as adjuvant in cancer therapy. Clin. Nutr. 2013, 32, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.C.; Choi, J.S.; Li, X. Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int. J. Pharm. 2006, 313, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Liu, C.; Chen, C.; Yu, X.; Chen, G.; Shi, Y.; Qin, F.; Ou, J.; Qiu, K.; Li, G. Quercetin and doxorubicin co-encapsulated biotin receptor-targeting nanoparticles for minimizing drug resistance in breast cancer. Oncotarget 2016, 7, 32184–32199. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Cao, X.; Li, N.; Zhang, Y.; Lan, L.; Zhou, Y.; Pan, X.; Shen, L.; Yin, Z.; Luo, L. Luteolin is effective in the non-small cell lung cancer model with L 858 R/T 790 M EGF receptor mutation and erlotinib resistance. Br. J. Pharm. 2014, 171, 2842–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, T.; Wang, J.; Yin, Y.; Hua, H.; Jing, J.; Sun, X.; Li, M.; Zhang, Y.; Jiang, Y. (-)-Epigallocatechin gallate sensitizes breast cancer cells to paclitaxel in a murine model of breast carcinoma. Breast Cancer Res. 2010, 12, R8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Rahman, S.S.A.; Shehab, G.; Nashaat, H. Epigallocatechin-3-Gallate: The prospective targeting of cancer stem cells and preventing metastasis of chemically-induced mammary cancer in rats. Am. J. Med. Sci. 2017, 354, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yan, L.; Wang, L.; Tai, W.; Wang, W.; Yang, C. Genistein enhances the effect of cisplatin on the inhibition of non-small cell lung cancer A549 cell growth in vitro and in vivo. Oncol. Lett. 2014, 8, 2806–2810. [Google Scholar] [CrossRef]
- Zhu, H.; Cheng, H.; Ren, Y.; Liu, Z.G.; Zhang, Y.F.; De Luo, B. Synergistic inhibitory effects by the combination of gefitinib and genistein on NSCLC with acquired drug-resistance in vitro and in vivo. Mol. Biol. Rep. 2012, 39, 4971–4979. [Google Scholar] [CrossRef]
- Meng, J.; Guo, F.; Xu, H.; Liang, W.; Wang, C.; Yang, X.D. Combination therapy using co-encapsulated resveratrol and paclitaxel in liposomes for drug resistance reversal in breast cancer cells in vivo. Sci. Rep. 2016, 6, 22390. [Google Scholar] [CrossRef]
- Yang, S.; Li, W.; Sun, H.; Wu, B.; Ji, F.; Sun, T.; Chang, H.; Shen, P.; Wang, Y.; Zhou, D. Resveratrol elicits anti-colorectal cancer effect by activating miR-34c-KITLG in vitro and in vivo. BMC Cancer 2015, 15, 969. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Bao, P.; Qi, H.; You, H. Resveratrol down-regulates survivin and induces apoptosis in human multidrug-resistant SPC-A-1/CDDP cells. Oncol. Rep. 2010, 23, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Omene, C.; Karkoszka, J.; Bosland, M.; Eckard, J.; Klein, C.B.; Frenkel, K. Caffeic acid phenethyl ester (CAPE), derived from a honeybee product propolis, exhibits a diversity of anti-tumor effects in pre-clinical models of human breast cancer. Cancer Lett. 2011, 308, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Lou, S.; Zhao, Z.; Dezort, M.; Lohneis, T.; Zhang, C. Multifunctional Nanosystem for Targeted and Controlled Delivery of Multiple Chemotherapeutic Agents for the Treatment of Drug-Resistant Breast Cancer. ACS Omega 2018, 3, 9210–9219. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Diagaradjane, P.; Anand, P.; Kuzhuvelil, H.B.; Deorukhkar, A.; Gelovani, J.; Guha, S.; Krishnan, S.; Aggarwal, B.B. Curcumin sensitizes human colorectal cancer to capecitabine by modulation of cyclin D1, COX-2, MMP-9, VEGF and CXCR4 expression in an orthotopic mouse model. Int. J. Cancer 2009, 125, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Neerati, P.; Sudhakar, Y.A.; Kanwar, J.R. Curcumin regulates colon cancer by inhibiting P-glycoprotein in in-situ cancerous colon perfusion rat model. J. Cancer Sci. 2013, 5, 313–319. [Google Scholar]
- Howells, L.M.; Sale, S.; Sriramareddy, S.N.; Irving, G.R.; Jones, D.J.; Ottley, C.J.; Pearson, D.G.; Mann, C.D.; Manson, M.M.; Berry, D.P. Curcumin ameliorates oxaliplatin-induced chemoresistance in HCT116 colorectal cancer cells in vitro and in vivo. Int. J. Cancer 2011, 129, 476–486. [Google Scholar] [CrossRef]
- Yan, J.; Wang, Y.; Zhang, X.; Liu, S.; Tian, C.; Wang, H. Targeted nanomedicine for prostate cancer therapy: Docetaxel and curcumin co-encapsulated lipid–polymer hybrid nanoparticles for the enhanced anti-tumor activity in vitro and in vivo. Drug Deliv. 2016, 23, 1757–1762. [Google Scholar] [CrossRef]
- Pramanik, D.; Campbell, N.R.; Das, S.; Gupta, S.; Chenna, V.; Bisht, S.; Sysa-Shah, P.; Bedja, D.; Karikari, C.; Steenbergen, C.; et al. A composite polymer nanoparticle overcomes multidrug resistance and ameliorates doxorubicin-associated cardiomyopathy. Oncotarget 2012, 3, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhang, Z.; Hill, D.L.; Wang, H.; Zhang, R. Curcumin, a dietary component, has anticancer, chemosensitization, and radiosensitization effects by down-regulating the MDM2 oncogene through the PI3K/mTOR/ETS2 pathway. Cancer Res. 2007, 67, 1988–1996. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.-W.; Wong, C.C.; Mattheolabakis, G.; Xie, G.; Huang, L.; Rigas, B. Curcumin enhances the lung cancer chemopreventive efficacy of phospho-sulindac by improving its pharmacokinetics. Int. J. Oncol. 2013, 43, 895–902. [Google Scholar] [CrossRef] [Green Version]
- Cui, T.; Zhang, S.; Sun, H. Co-delivery of doxorubicin and pH-sensitive curcumin prodrug by transferrin-targeted nanoparticles for breast cancer treatment. Oncol. Rep. 2017, 37, 1253–1260. [Google Scholar] [CrossRef]
- Mahammedi, H.; Planchat, E.; Pouget, M.; Durando, X.; Cure, H.; Guy, L.; Van-Praagh, I.; Savareux, L.; Atger, M.; Bayet-Robert, M.; et al. The New Combination Docetaxel, Prednisone and Curcumin in Patients with Castration-Resistant Prostate Cancer: A Pilot Phase II Study. Oncology 2016, 90, 69–78. [Google Scholar] [CrossRef]
- Bayet-Robert, M.; Kwiatkowski, F.; Leheurteur, M.; Gachon, F.; Planchat, E.; Abrial, C.; Mouret-Reynier, M.A.; Durando, X.; Barthomeuf, C.; Chollet, P. Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol. 2010, 9, 8–14. [Google Scholar] [CrossRef] [Green Version]
- D’Archivio, M.; Filesi, C.; Vari, R.; Scazzocchio, B.; Masella, R. Bioavailability of the polyphenols: Status and controversies. Int. J. Mol. Sci. 2010, 11, 1321–1342. [Google Scholar] [CrossRef]
- Tresserra-Rimbau, A.; Lamuela-Raventos, R.M.; Moreno, J.J.; Polyphenols, food and pharma. Current knowledge and directions for future research. Biochem. Pharm. 2018, 156, 186–195. [Google Scholar] [CrossRef]
- Velderrain-Rodriguez, G.R.; Palafox-Carlos, H.; Wall-Medrano, A.; Ayala-Zavala, J.F.; Chen, C.Y.; Robles-Sanchez, M.; Astiazaran-Garcia, H.; Alvarez-Parrilla, E.; Gonzalez-Aguilar, G.A. Phenolic compounds: Their journey after intake. Food Funct. 2014, 5, 189–197. [Google Scholar] [CrossRef]
- Azrad, M.; Vollmer, R.T.; Madden, J.; Dewhirst, M.; Polascik, T.J.; Snyder, D.C.; Ruffin, M.T.; Moul, J.W.; Brenner, D.E.; Demark-Wahnefried, W. Flaxseed-derived enterolactone is inversely associated with tumor cell proliferation in men with localized prostate cancer. J. Med. Food 2013, 16, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Sarrias, A.; Tome-Carneiro, J.; Bellesia, A.; Tomas-Barberan, F.A.; Espin, J.C. The ellagic acid-derived gut microbiota metabolite, urolithin A, potentiates the anticancer effects of 5-fluorouracil chemotherapy on human colon cancer cells. Food Funct. 2015, 6, 1460–1469. [Google Scholar] [CrossRef]
- Alam, M.N.; Almoyad, M.; Huq, F. Polyphenols in Colorectal Cancer: Current State of Knowledge including Clinical Trials and Molecular Mechanism of Action. Biomed. Res. Int. 2018. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.R.; Scott, E.; Brown, V.A.; Gescher, A.J.; Steward, W.P.; Brown, K. Clinical trials of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 161–169. [Google Scholar] [CrossRef]
- Vinod, B.S.; Maliekal, T.T.; Anto, R.J. Phytochemicals as chemosensitizers: From molecular mechanism to clinical significance. Antioxid. Redox Signal. 2013, 18, 1307–1348. [Google Scholar] [CrossRef]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse effects of cancer chemotherapy: Anything new to improve tolerance and reduce sequelae? Front. Pharm. 2018, 9, 245. [Google Scholar] [CrossRef]
- James, M.I.; Iwuji, C.; Irving, G.; Karmokar, A.; Higgins, J.A.; Griffin-Teal, N.; Thomas, A.; Greaves, P.; Cai, H.; Patel, S.R.; et al. Curcumin inhibits cancer stem cell phenotypes in ex vivo models of colorectal liver metastases, and is clinically safe and tolerable in combination with FOLFOX chemotherapy. Cancer Lett. 2015, 364, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Gorelick, D.A. Pharmacokinetic strategies for treatment of drug overdose and addiction. Future Med. Chem. 2012, 4, 227–243. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Krstin, S.; Wink, M. Modulation of multidrug resistant in cancer cells by EGCG, tannic acid and curcumin. Phytomedicine 2018, 50, 213–222. [Google Scholar] [CrossRef]
- Hu, F.; Wei, F.; Wang, Y.; Wu, B.; Fang, Y.; Xiong, B. EGCG synergizes the therapeutic effect of cisplatin and oxaliplatin through autophagic pathway in human colorectal cancer cells. J. Pharm. Sci. 2015, 128, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Schonthal, A.H. Adverse effects of concentrated green tea extracts. Mol. Nutr. Food Res. 2011, 55, 874–885. [Google Scholar] [CrossRef]
- Pisters, K.M.; Newman, R.A.; Coldman, B.; Shin, D.M.; Khuri, F.R.; Hong, W.K.; Glisson, B.S.; Lee, J.S. Phase I trial of oral green tea extract in adult patients with solid tumors. J. Clin. Oncol. 2001, 19, 1830–1838. [Google Scholar] [CrossRef]
- Ullmann, U.; Haller, J.; Decourt, J.D.; Girault, J.; Spitzer, V.; Weber, P. Plasma-kinetic characteristics of purified and isolated green tea catechin epigallocatechin gallate (EGCG) after 10 days repeated dosing in healthy volunteers. Int. J. Vitam. Nutr. Res. 2004, 74, 269–278. [Google Scholar] [CrossRef]
- Popat, R.; Plesner, T.; Davies, F.; Cook, G.; Cook, M.; Elliott, P.; Jacobson, E.; Gumbleton, T.; Oakervee, H.; Cavenagh, J. A phase 2 study of SRT501 (resveratrol) with bortezomib for patients with relapsed and or refractory multiple myeloma. Br. J. Haematol. 2013, 160, 714–717. [Google Scholar] [CrossRef]
- Singh, C.K.; Ndiaye, M.A.; Ahmad, N. Resveratrol and cancer: Challenges for clinical translation. Biochim. Et. Biophys. Acta 2015, 1852, 1178–1185. [Google Scholar] [CrossRef] [Green Version]
- Navarro, V.J.; Bonkovsky, H.L.; Hwang, S.I.; Vega, M.; Barnhart, H.; Serrano, J. Catechins in dietary supplements and hepatotoxicity. Dig. Dis. Sci. 2013, 58, 2682–2690. [Google Scholar] [CrossRef] [Green Version]
- Bonkovsky, H.L. Hepatotoxicity associated with supplements containing Chinese green tea (Camellia sinensis). Ann. Intern. Med. 2006, 144, 68–71. [Google Scholar] [CrossRef]
- Mereles, D.; Hunstein, W. Epigallocatechin-3-gallate (EGCG) for clinical trials: More pitfalls than promises? Int. J. Mol. Sci. 2011, 12, 5592–5603. [Google Scholar] [CrossRef] [Green Version]
- Granja, A.; Pinheiro, M.; Reis, S. Epigallocatechin gallate nanodelivery systems for cancer therapy. Nutrients 2016, 8, 307. [Google Scholar] [CrossRef]
- Tomeh, M.A.; Hadianamrei, R.; Zhao, X. A Review of Curcumin and Its Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2019, 20, 1033. [Google Scholar] [CrossRef] [Green Version]


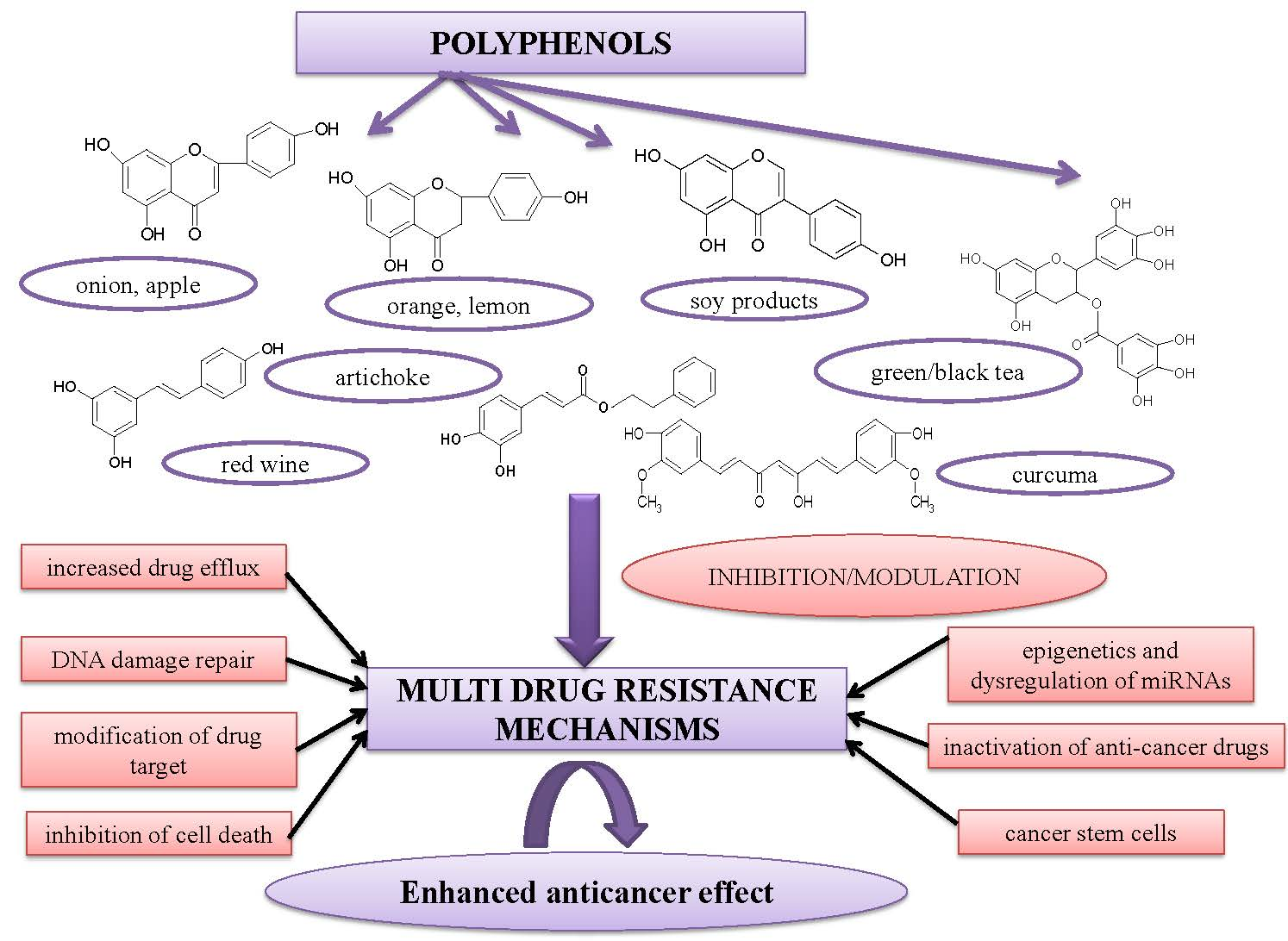{kind=link}
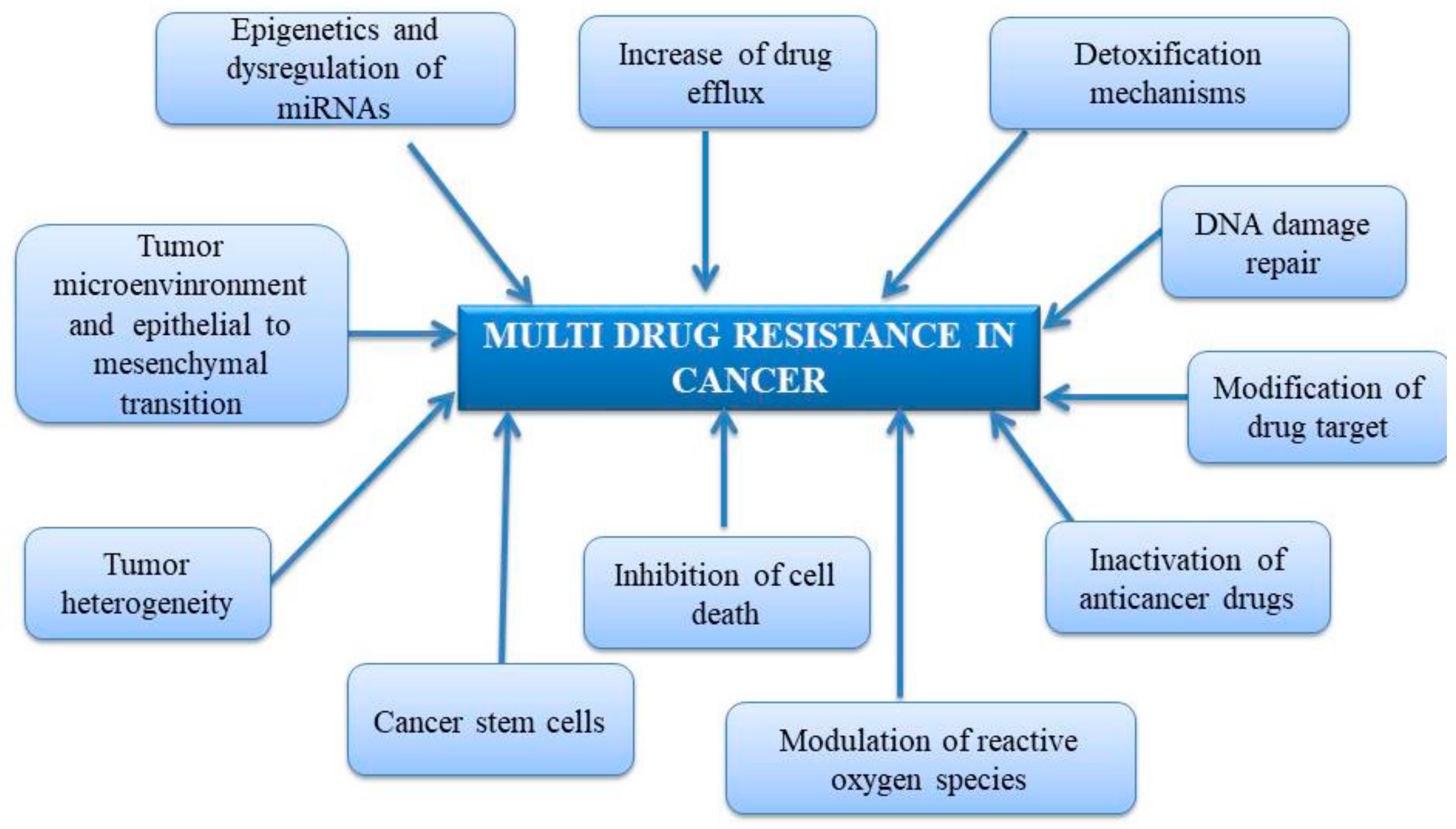{kind=link}
| Phenolic Compounds | Chemical Structure | Representative Compounds | Sources | Reference |
|---|---|---|---|---|
| Flavonoid Compounds | ||||
| Flavones |  | apigenin (R1–OH, R2–H, R3–H) luteolin (R1–OH, R2–OH, R3–H) wogonin (R1–H, R2–H, R3–OCH3) | oranges, lemons, apricots, apples, black currants, bananas, potatoes, spinach, onions, lettuce, parsley, celery, beans, tomatoes, roots of Scutellaria baicalensis Georgi | [37,38,40,41] |
| Flavonols |  | quercetin (R1–OH, R2–OH, R3–H, R4–OH, R5–OH) fisetin (R1–OH, R2–H, R3–OH, R4–OH, R5–H) | ||
| Flavanones |  | naringenin (R1–H, R2–OH) hesperitin (R1–OCH3, R2–OH) | oranges, grapefruits, lemons | [40] |
| Flavan-3-ols |  | catechin (C), epicatechin (EC) (R1–OH, R2–OH, R3–H, R4–H) epigallocatechin (EGC) (R1– OH, R2–OH, R3–OH, R4–H) epigallocatechingallate (EGCG) (R1–OH, R2–OH, R3–OH, R4—  ) ) | green/black tea, grapes, cherries, apricots, peaches | [38,40] |
| Isoflavones |  | genistein (R1–OH, R2–OH, R3–OH) daidzein (R1–OH, R2–H, R3–OH) | soy flour, soy paste (natto, cheonggukang), soy bean (roasted) | [38] |
| Non-Flavonoid Compounds | ||||
| Hydroxy-benzoic acids |  | gallic acid (R1–OH, R2–OH, R3–OH, R4–OH) | blackcurrants, strawberries, raspberries, kiwi, cherry, plums, spinach, broccoli | [40,42] |
| Hydroxy-cinnamic acids |  | caffeic acid (R1–H, R2–OH, R3–OH) ferulic acid (R1–H, R2–OH, R3–OCH3) cinnamic acid (R1–H, R2–H, R3–H) | plums, apples, eggplants, potatoes, wheat, rice, oat, kiwi | [40] |
 | caffeic acid phenethyl ester (CAPE) | artichoke, oregano, thyme, basil, coffee, mushrooms | [40] | |
| Lignans |  | honokiol | bark, root, seeds, leaves of Magnolia sp. | [43] |
 | podophyllotoxin (R–OH) deoxypodophyllotoxin (R–H) | rhizome of American mayapple (Podophyllum peltatum L.) roots of Anthriscus sylvestris L. (Hoffm.) | [44,45] | |
 | silybin (silibinin) | fruits of milk twistle (Silybum marianum L.) Gaerth | [46] | |
 | secoisolariciresinol | flaxseeds | [47] | |
 | schizandrin A | fruits of Schisandra chinensis | [48] | |
| Ellagitannins |  | ellagic acid | raspberries, strawberries, pomegranate black currants, blackberries | [49] |
 | sanguiin-H6 | raspberries | [49] | |
| Stilbenes |  | resveratrol | grapes, mulberries | [40] |
| Other Compounds |  | curcumin | Curcuma roots | [50] |
 | gingerol | fresh/dried ginger rhizomes | [51] | |
| Compound | Type of Cancer | Cell Line | Treatment/Duration | Mechanisms of Overcoming MDR | Reference |
|---|---|---|---|---|---|
| Flavonoid Compounds | |||||
| Apigenin | Prostate | CD44+ PC3 cells | 15 μM apigenin + 7.5 μM CDPP, 48 h | ↓ Bcl-2, ↓ sharpin, ↓ survivin, ↑ caspase 8, ↑ APAF-1, ↑ p53 mRNA, ↓ NF-κB, ↑ p21, ↓ CDK-2, ↓ CDK-4, ↓ CDK-6 | [188] |
| Wogonin | Lung | A549 cell line | 10 μM wagonin + TRAIL (5–20 ng/mL), 24 h | ↑ apoptosis, ↓ cFLIPL, ↓ XIAP, ↓ cIAP-1, ↓ IAP-2 | [41] |
| Luteolin | Breast | ABCG2 expressing MCF-7 cells mitoxantrone resistant | 12.5–100 μM luteolin + 1 μM mitoxantrone, 4 h | ↑ ROS, ↑ DNA damage, ↓ NF-κB ↓ cIAP-1, ↓ survivin, ↓ XIAP ↑ ATR-CHk2-p53 | [189] |
| Breast | MDA-MB 231 cells DOX resistant | 5–20 μM luteolin + 0.08–20 mM DOX, 24 h | ↓ Nrf2 | [191] | |
| Lung | A549 cells | Pre-treatment (24 h) with 5 μM luteolin before DOX (0–3 μg/mL), OX (0–100 μM), bleomycin (0–100 μM), 48 h | ↓ Nrf2 | [190] | |
| Quercetin | Breast | DOX resistant MCF-7 cells | 2.5 μg/mL DOX, 0.5 μg/mL PTX, 0.5 μg/mL VCR + 0.5. μg/ml quercetin - 24 h | ↓ P-gp, ↓ YB-1 nuclear protein translocation, ↓ BCSCs phenotype CD44+/CD24−/low, ↑ apoptosis, cell cycle arrest | [192] |
| Colorectal | VCR resistant Caco-2 cells | 0.5–200 μM quercetin, 24 h | ↓ P-gp | [193] | |
| Colorectal | Caco-2 cells | 20 μM cimetidine + 100 μM quercetin, 4 h | ↓ P-gp | [194] | |
| Fisetin | Colorectal | OX-resistant LoVo cells CPT11-resistant LoVo cells | 0 μM, 40 μM, 80 μM fisetin, 24 h | ↑ apoptosis, ↑ cytochrome C release, ↓ IGF-1R and AKT phosphorylation levels | [196] |
| Naringenin | Breast | Daunomycin resistant MCF-7 cells | 9 × 10−8 M– 7.2 × 10−5 M daunomycin + 50 μM naringenin, 72 h | ↓ P-gp | [195] |
| Hesperitin glycoside (hesperidin) | Breast | MCF-7 DOX resistant cells | 0.5–3.5 μM/L hesperidin + 35–233 nM/L DOX, 24 h | ↓ P-gp | [197] |
| Colorectal | Coco-2 cells overexpressing P-gp | 32 μM hesperidin, 24 h | ↓ P-gp | [198] | |
| Catechin | Breast | MDA-MDB-231 CDPP resistant cells | 5, 10, 20, 40 μM C + 10 μM CDPP, 6 h | ↓ ATR-Chk1 pathway | [199] |
| EGCG | Breast | Tamoxifen-resistant MCF-7 | Nrf2-RNA transfection, 48 h + 50/100 μM EGCG, 24 h | ↓ Nrf2 signaling pathway | [202] |
| Colorectal | HCT-116 DLD1 cells | 50 μM EGCG + 0–30 μM 5-FU, 24 h | ↓ GRP78/ NF-κB/miR-155-5p/MDR1 pathway | [203] | |
| Prostate | PC3, LAPC4 cells | 40 μM EGCG + 5 μM quercetin + 5 nM DOC, 24/48 h | ↓ CD44+/CD24− cells, ↓ MRP1, ↓ PI3K/AKT/ STAT3 | [204] | |
| Lung | A549/H460 CDPP resistant cells | 80 μM EGCG + 0–30 μM CDPP, 24 h | ↓ Axl, Tyro3 | [205] | |
| Genistein | Breast | MCF-7 DOX resistant cells | 0–120 μmol/L genistein + 0.7–70 μM DOX, 48 h | ↓ HER 2/neu, ↑ apoptosis | [206] |
| Prostate Lung | PC-3 cells H460 cells | pre-treatment with 15–30 μmol/L genistein, 24 h 1–2 nM DOC/100 nM/L cisplatin, 48 h | ↑ apoptosis, ↓ NF-κB | [207] | |
| Daidzein | Breast | MCF-7/ MDA-MB 231 cells | pre-treatment with 10 μM daidzein, 24 h before administration of 0–10 mM DOX/ mitoxantrone | ↓ MRP1/2,↓BCRP | [208] |
| NON-FLAVONOID COMPOUNDS | |||||
| Resveratrol | Breast | MCF-7 cells | 100 µM RES + 20 nM rapamycin, 24 h | ↓ mTOR, ↓ AKT, ↑ autophagy | [209] |
| Breast | DOX resistant MCF-7 | 4–16 µM RES + 4–64 µM DOX, 24 h | ↓ P-gp | [210] | |
| Breast | SK-BR-3, MCF7, MDA-MB-231, T47D cells | 15 µM RES + 1 nM DOC | ↓ HER2-AKT axis | [214] | |
| Lung | NCI-H460 cells | 0–20 µg/mL RES + 0–10 µg/mL PTX, 24 h | ↓ P-gp, MRP2, BCRP | [211] | |
| Lung | GF resistant NSCLC- PC9 | 1–20 µM GF + 5–160 µM RES | ↑ apoptosis, ↑ senescence | [213] | |
| Colorectal | HCT 116, HT-29 cells | 0.3 µM DOX + 100 µM RES | ↓ P-gp, ↑ Bax, cell cycle arrest | [212] | |
| Honokiol | Breast | MCF-7/DOX, MDA-MB-231 | 200 µL polymeric micelles with 1 mg PTX + 0.5 mg/L HNK, 24/36 h | ↓ P-gp, ↑ plasma fluidity | [43] |
| Colorectal | HCT-116 cells | 0–50 μM HNK + 0–5 Gy γ-radiation, 24/48 h | ↑ apoptosis, ↓ cyclin A1, D1 | [215] | |
| Secoisolarici resinol | Breast | MDA-MB-231, SKBR3 cells | 25–50 µM SECO, 25–50 µM ENL, 20 nM DOX, 1 nM DOC, 1000 nM CAB, 72 h | ↓ FAS | [47] |
| Schizandrin A | Colorectal | 5-FU resistant HCT116, SW-480 | 0–8 µM 5-FU + 0–40 µM SchA, 48 h | ↑ mir-195 | [48] |
| Silybin | Breast | MDA-MB 435 DOX resistant cell line MCF-7 PTX resistant cell line | 200–600 μM silybin + 0–35 μg/mL DOX/250 nM PTX, 24 h | ↓ STAT3, ERK, AKT | [46] |
| Gallic acid | Lung | SCLC H446 cells | 2–12 µg/mL gallic acid + 3.12–50 µg/mL CDPP | ↑ apoptosis, MMP disruption ↑ Bax, ↑ APAF1, ↑ p53, ↑ DIABLO, ↓ XIAP | [219] |
| Breast | MCF-7/DOX cells MCF-7/DOX500 | 30–120 µM gallic acid + 5–20 µM EGCG, 24 h | ↓ MMP-2/ MMP-9 | [220] | |
| Lung | HCC827, H1650, H1975, H358, H1666 cells TKI resistant | 20–100 µM gallic acid + 0.1–5 µM GF, 5 days | ↓ Src-STAT3, ↑ apoptosis | [221] | |
| Cinnamic acid | Lung | Chemoresistant H1299-derived stem-like cells | 1–32 mM cinnamic acid; 4 mM cinnamic acid + 4–32 µM PTX/ 4–32 μg/mL CDPP, 24 h | ↑ differentiation into CD33 negative cells; ↓chemoresistance to cisplatin and PTX | [227] |
| Caffeic acid/ ferulic acid | Colorectal | HCT-8 cells | Pre-treatment - 0.5–1 mg/mL BPIS (12 h) before 1000–6000 µM 5-FU, 50–400 µM OX, 25–125 µM VCR | ↓ P-gp, MRP1, BCRP | [222] |
| Caffeic acid phenethyl ester (CAPE) | Breast | MDA-MB-231 cells | 10–40 µM CAPE, 4.5 days | ↓ CD44 cells, ↓ progenitor formation | [223] |
| Breast | MDA-MB-231, T47D cells | Pretreatment with 1 µM CAPE (72 h) before irradiation (2–8 Gy) | ↑ DNA damage | [224] | |
| Lung | A549 cells | 10, 50 µM CAPE 10 µM DOX, 24 h | ↑ chemosensitivity to DOX, ↓ claudin -2 | [226] | |
| Ellagic acid | Colorectal | SW480, Colo 320DM, HT-29 cells | 5–25 µM 5-FU + 2–25 µM ellagic acid | ↑ Bax/Bcl-2 ratio, ↑ caspase-3 ↓ mitochondrial potential | [217] |
| Sanguiin-H6 | Breast | DOX resistant MCF-7 | 0–313 µM sanguiin-H6, 48 h | ↓ ABC transporters | [218] |
| Non-Flavonoid Compounds | |||||
| Curcumin | Colorectal | OX-resistant HTOXAR3, LoVOXAR3 DLDOXAR3 | 5–10 μM curcumin + 10–30 μM OX, – 24 h | ↓NF-κB signaling cascade, ↓ CXCL8, CXCL1, CXCL2 | [230] |
| Colorectal | VCR resistant HCT8/VCR | 6.25–100 μM curcumin + 0.5 μg/l VCR, 48 h | ↓ P-gp | [228] | |
| Colorectal | 5-FU and OX resistant HCT-116, SW-620 | 100 nM CDF | ↓ miR-21 | [232] | |
| Lung | A549-CDPP resistant | 20 μg/mL CDDP + 10 μM curcumin, 24 h | ↓ autophagy, ↓ Nrf2 activation | [233] | |
| Lung | A549/DOX cells, P-gp overexpressing DOX resistant overexpressing | Nanomicelles with 1–30 μg/mL DOX + curcumin (1.6 times concentration of DOX), 72 h | ↑ sensitivity to DOX, ↑ cellular uptake | [234] | |
| Lung | CDPP resistant A549 cells | 5–20 μM curcumin + 1.5 μg/mL CDPP | ↑ apoptosis, ↓ HIF-1α | [235] | |
| Breast | Tamoxifen resistant MCF-7/LCC2, MCF-7/LCC9 | 30 μM curcumin, 24 h | ↓ mTOR, ↓ EZH2 | [236] | |
| Breast | MCF-7, MDA-MB-231, SK-BR-3 cells | 10 μM curcumin 6 h before 5-FU (10 μM) | ↓ NF-κB signaling cascade | [231] | |
| Breast | DOX resistant MCF-7 cells | 0–20 mM curcumin + 0–4 mΜ EGCG | ↓ Bcl-2, ↓ survivin, ↑ caspase 7, 9 | [238] | |
| Breast | MDA-MB-231, MDA-MB-468, SK-BR-3, MCF-7 cells | 30 μM curcumin and/or 1 μM trans retinoic acid, 48 h | ↑ sensitivity to retinoic acid ↓ FBAP5, PPARβ/δ | [237] | |
| Gingerol | Prostate | DOC resistant PC3 | 100 µM 6-gingerol + 100 µM 10-gingerol | ↓ MRP1, ↓GST | [51] |
| Breast | cyclophosphamide, 5-5-FU, DOX resistant MCF-7 | 50–250 µM 6-gingerol | ↓ Wnt/β-catenin, ↓ GSK3 | [239] | |
| Compound | Type of Cancer | Model System | Doses and Duration of Administration | Mechanisms of Overcoming MDR | Reference |
|---|---|---|---|---|---|
| Flavonoid Compounds | |||||
| Quercetin | Breast | Female Sprague–Dawley rats | 1.5, 7.5, 10 mg/kg quercetin p.o. + 10 mg/kg tamoxifen p.o. | ↓ P-gp, ↓ MRP2, ↓ BCPR, ↓ CYP3A4 | [257] |
| Breast | Xenograft BALB/c nude mouse model for MCF-7 DOX resistant cells | 5 mg/kg BNDQ i.v. 20 days, every three days | ↓ P-gp | [258] | |
| Wogonin | Lung | Xenograft mouse model for A549 cells | 3 mg/kg TRAIL i.p. + 100 mg/kg wogonin i.p. 3 times/week, 28 days | ↑ ROS, ↑ apoptosis, ↓ cFLIPL, ↓ XIAP, ↓ cIAP-1, ↓ IAP-2 | [41] |
| Fisetin | Colorectal | Xenograft nude mouse model for Lovo OX/irinotecan resistant cells | 400 mg/kg/day fisetin and 800 mg/kg/day fisetin p.o., 4 weeks | ↑ apoptosis, ↑ cytochrome C release, ↓ IGF1R/AKT, ↓ tumor volumes | [196] |
| Luteolin | Lung | Xenograft BALB/c nude mouse model for NCI-H1975 erlotinib resistant cells | 10/30 mg/kg/day luteolin i.p. + 100 mg/kg/day erlotinib i.p. + 2 mg/kg/day CDPP i.p., 15 days | ↓ tumor volumes, ↓ EGFR, ↓ PI3K/AKT mTOR ↑ apoptosis | [259] |
| Genistein | Lung | Xenograft mouse models for A549 cells | 5 mg/kg CDPP i.p., day one + 800 μg/kg genistein p.o., 5 days, 5 mg/kg CDPP i.p. day one + 500 μg/kg genistein p.o., 4 days, every 7 days for 21 days | ↓ tumor volumes, ↓ PI3/AKT | [262] |
| Lung | Xenograft BALB/c mouse models for H1975 cells | 50 mg/kg GF p.o. + 100 mg/kg genistein p.o., 5 weeks | ↓ EGFR, ↓ mTOR, ↑ caspase -3 | [263] | |
| EGCG | Breast | Xenograft BALB/c mouse models for breast 4T1 cancer cells | EGCG 30 mg/kg/day i.v. + PTX 10 mg/kg i.v., every two days, 24 days | ↑ apoptosis, ↓ GRP78, ↓ JNK phosphorylation | [260] |
| Breast | Female Sprague–Dawley rats treated with DMBA | 5 mg/kg PTX i.p. + 10 mg/kg EGCG i.p., twice/week, 4 weeks | ↓ CD44 cells, ↓ VEGF, ↓ MMP-2, ↑ caspase-3 | [261] | |
| Non-Flavonoid Compounds | |||||
| Resveratrol | Breast | Xenograft BALB/c mouse model for MCF-7/Adr resistant cells | Liposomes with 8 mg/kg PTX + 20 mg/kg RES i.v., every two days, 14 days | ↑ cellular uptake of PTX, ↓ P-gp | [264] |
| Colorectal | Xenograft BALB/c nude mouse model for HCT-116 cells | 100 mg/kg RES + 10 mg/kg OX i.v. every day, 14 days | ↑ miR-34c | [265] | |
| Lung | Xenograft BALB/c mouse model (females) for SPC-A-1/CDDP cells | 1 g/kg/day RES p.o., 3 g/kg/day RES p.o., 28 days | ↓ survivin, ↑ apoptosis (caspase 3) | [266] | |
| Caffeic acid phenethyl exter (CAPE) | Breast | Xenograft Ncr-nu/nu mouse models for MCF-7, MDA-MB-213 cells | 10, 50, 250 nmol/mouse CAPE p.o., every day, 60 days | ↓ NF-κB, ↓ EGFR, IFGR, ↓ MDR1 | [267] |
| Podophyllotoxin (PPT) | Breast and prostate | Xenograft BALB/c and NOD-SCID mouse models EMT6/AR1 (breast), PC3 (prostate) cells | 12 mg/kg DOC i.v., every 4 days, 8 days; 5 mg/kg CBZ i.v., every 4 days, 8 days; 180 mg/kg PPT NPs i.v. every 4 days, 8 days | ↓ P-gp, ↑ cellular uptake of chemotherapeutic agents | [44] |
| Deoxypodophyllotoxin (DPPT) | Breast | Xenograft mouse model MCF-7 DOX resistant cells | 1.25 mg/kg DPPT i.v. + 12.5 mg/kg PTX i.v. every 3 days, 10 days | efflux transport | [45] |
| Silybin | Breast | Xenograft mouse model (females) for MDA-MB-231 cells | 1.5 mg/kg nanosystems − 75 μg/mg DOX + 120 μg/mg PTX + 90 μg/mg silybin i.v. every 4 days, 30 days | P-gp | [268] |
| Curcumin | Colorectal | HCT-116 cells in orthotopic mouse model | 1 g/kg curcumin by gavage, daily + 60 mg/kg capecitabine by gavage, twice weekly, 4 weeks | ↓ NF-κB, ↓MMP-2, ↓ CXCR4, ↓ COX-2, ↓ ICAM-1, ↓VEGF | [269] |
| Colorectal | Swiss albino rats with N-Nitroso N-methyl urea–induced carcinogenesis | Pre-treatment with curcumin 50 mg/kg p.o. for one week before administration of irinotecan 30 μg/mL i.v. | ↓ P-gp, ↑ sensitivity of cancer cells to irinotecan | [270] | |
| Colorectal | Xenograft mouse model (6–8 weeks, females) for HCT-116 cells | 1.13% Meriva (equivalent to 0.2% curcuminoids) p.o. + 7.5 mg/kg OX i.v. daily, 21 days | ↓ cancer stem cells, ↓ DNA damage repair | [271] | |
| Prostate | Xenograft BALB/c mouse model for PC3 cells | NPs with 5 mg/kg DOC + 10 mg/kg curcumin i.v. daily, 21 days | ↑ intracellular accumulation of DOC | [272] | |
| Prostate | Xenograft nu/nu mouse models (males, 5–6 weeks old) for PC-3A cells | NP with 6 mg/kg DOX + 24 mg/kg curcumin i.v. twice every three days, 4 weeks | ↓ MDR, MRP | [273] | |
| Prostate | Xenograft mouse model for PC3 cells (nude mice) | 5 mg/kg curcumin p.o. daily, 4 weeks + 160 mg/kg gemcitabine i.p. every 7 days, 21 days + 3 Gy radiation days 4, 6, 10 for 21 days | ↓ MDM2 | [274] | |
| Lung | Xenograft mouse model for A549 cells | 200 mg/kg/day PS + 500 mg/kg/day curcumin p.o., 36 days | ↑ pharmacokinetics ↑ accumulation in cancer tissue, ↓ P-gp, ↓ MRP1/2 | [275] | |
| Breast | Xenograft BALB/c mouse model (6–8 weeks) for MCF-7 cell lines | NPs with Tf-PEG-CUR/DOX—50 mg/kg CUR/DOX i.v. once/week, 7 weeks | ↑ cellular uptake of DOX | [276] | |
| Prostate | CRPC patients, non-randomized open-label phase II trial (n = 30) | 75 mg/m2 DOC i.v. day 1 every 21 days for 6 cycles + 8 mg dexamethasone p.o. 12 h, 3 h and 1 h before DOC administration + 5 mg prednisone p.o. twice/day starting on day 1 + 6000 mg curcumin p.o. 7 days in each cycle | ↓ PSA (50% of patients), ↓ NSE (30% of patients), suggested mechanisms: ↓ NF-κB, ↓ AR, ↓ VEGFR, ↓ MDR1B | [277] | |
| Breast | Advanced-metastatic breast cancer patients, single institution open-label phase I trials (n = 13) | 100 mg/m2 DOC i.v. day 1 of each 3 weeks cycle for 6 cycles + 450 mg curcumin p.o. 7 days consecutive for each cycle + 50 mg methylprednisolone 2 days before and after chemotherapy | ↓ CEA, ↓ VEGF suggested mechanisms: ↓ P-gp | [278] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costea, T.; Vlad, O.C.; Miclea, L.-C.; Ganea, C.; Szöllősi, J.; Mocanu, M.-M. Alleviation of Multidrug Resistance by Flavonoid and Non-Flavonoid Compounds in Breast, Lung, Colorectal and Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 401. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020401
Costea T, Vlad OC, Miclea L-C, Ganea C, Szöllősi J, Mocanu M-M. Alleviation of Multidrug Resistance by Flavonoid and Non-Flavonoid Compounds in Breast, Lung, Colorectal and Prostate Cancer. International Journal of Molecular Sciences. 2020; 21(2):401. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020401
Chicago/Turabian StyleCostea, Teodora, Oana Cezara Vlad, Luminita-Claudia Miclea, Constanta Ganea, János Szöllősi, and Maria-Magdalena Mocanu. 2020. "Alleviation of Multidrug Resistance by Flavonoid and Non-Flavonoid Compounds in Breast, Lung, Colorectal and Prostate Cancer" International Journal of Molecular Sciences 21, no. 2: 401. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020401