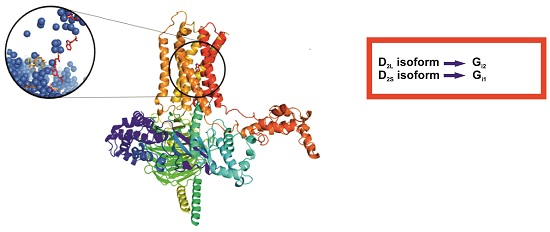
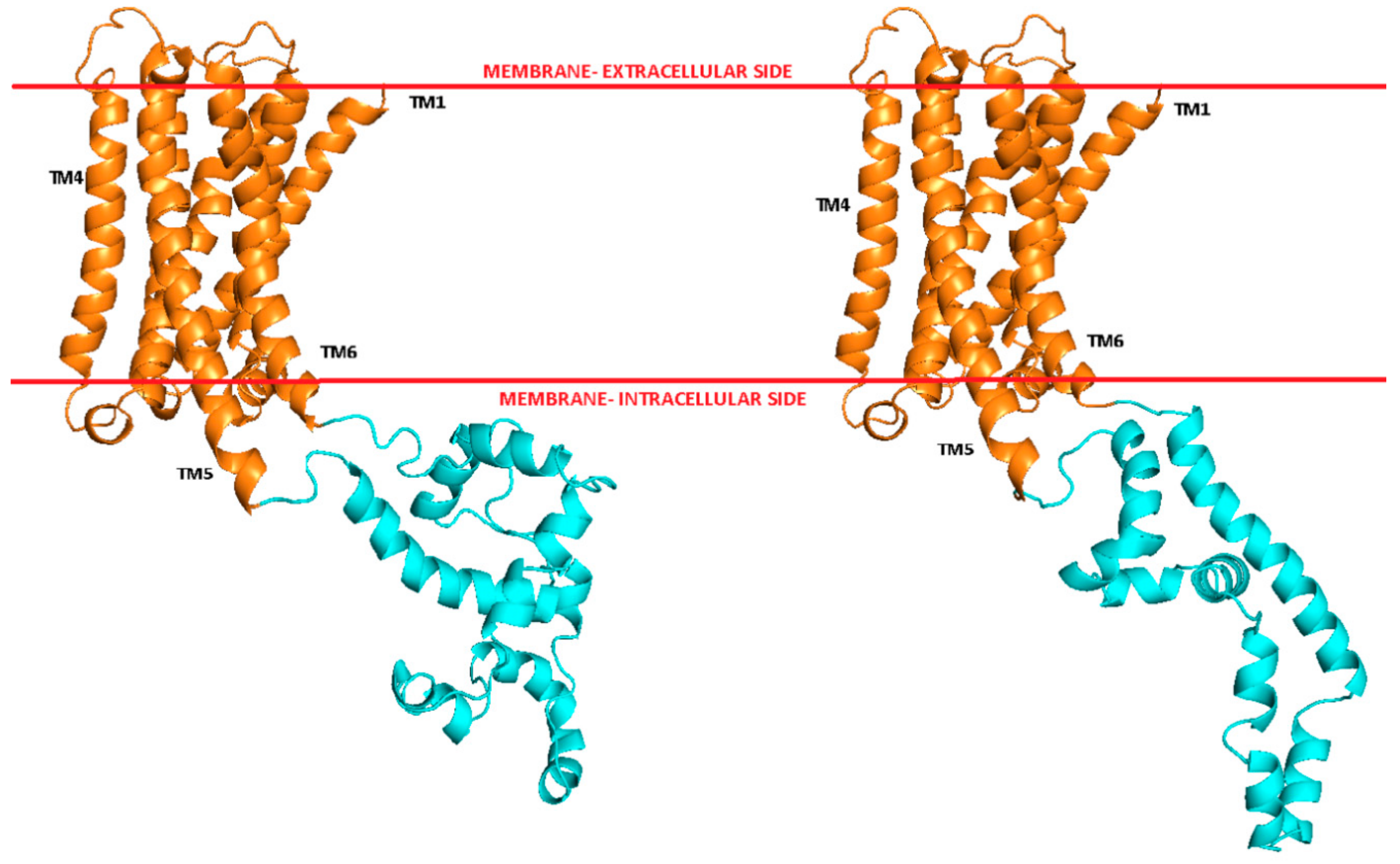
Figure 1.
Models of transmembrane helices, TM, (orange) and ICL3 loop (cyan) of D2L (left) and D2S (right) receptors.
Figure 1.
Models of transmembrane helices, TM, (orange) and ICL3 loop (cyan) of D2L (left) and D2S (right) receptors.
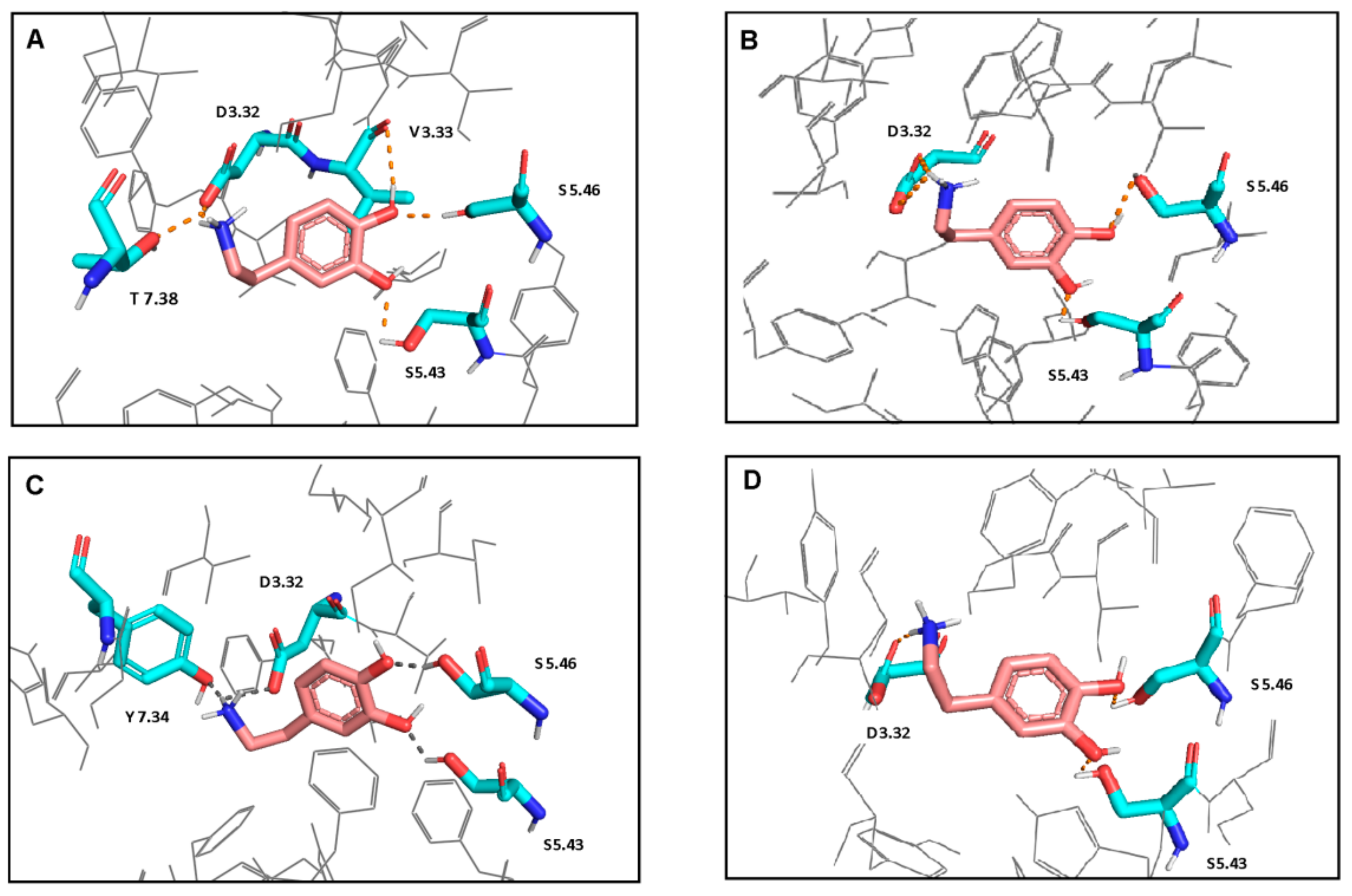
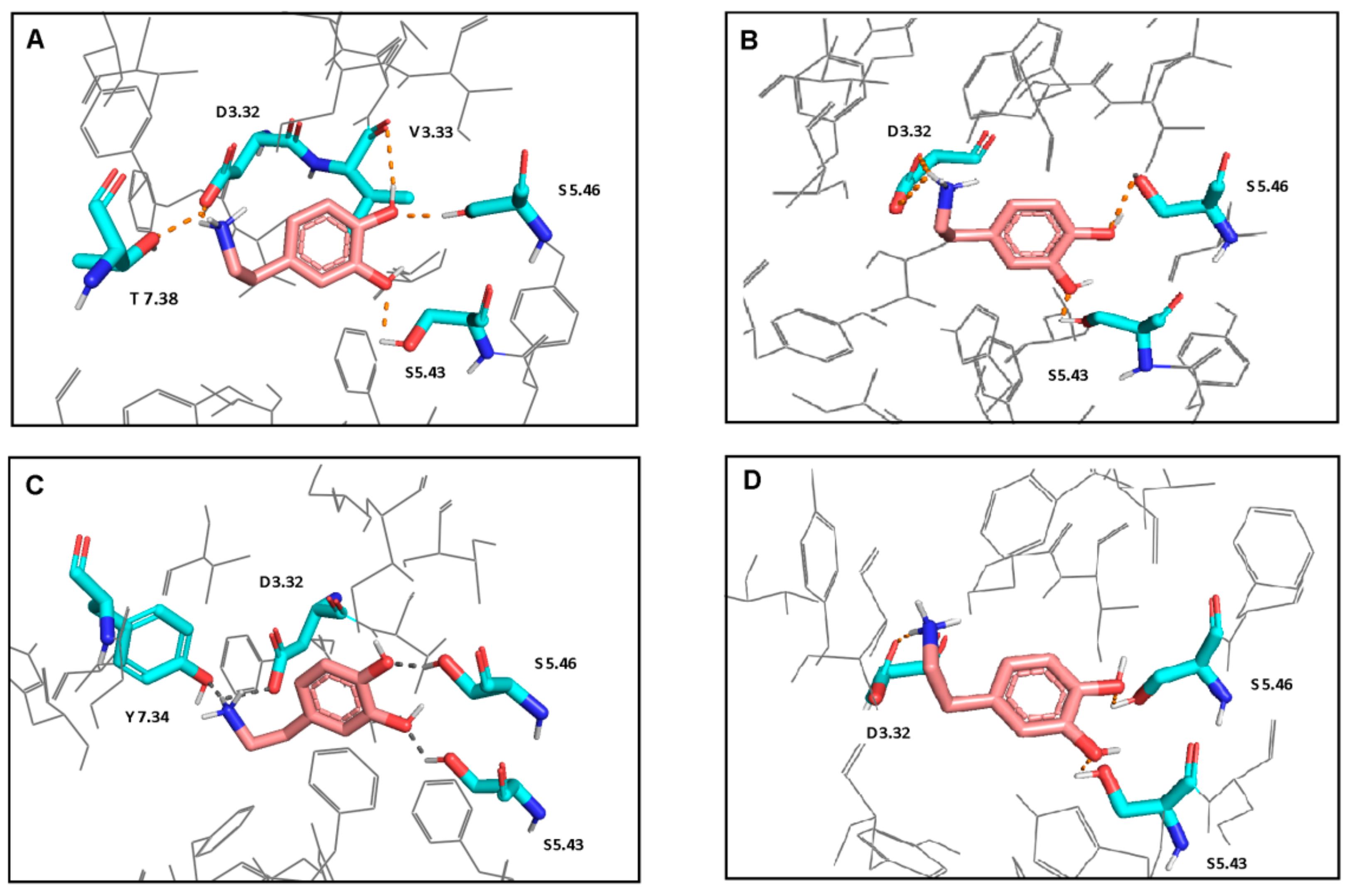
Figure 2.
Interactions of dopamine with L1 (A), L2 (B), S1 (C), and S2 (D) models. Ligand is shown in stick representation with pink carbon atoms. Proteins are colored in gray and shown in wire representation. The interacting residues are shown in stick representation with blue nitrogen, red oxygen and cyan carbon atoms. Hydrogen bonds and salt bridges are shown as the orange dot- lines. Non-polar hydrogen atoms are not shown. Polar hydrogen atoms are shown only for the ligand and the interacting residues.
Figure 2.
Interactions of dopamine with L1 (A), L2 (B), S1 (C), and S2 (D) models. Ligand is shown in stick representation with pink carbon atoms. Proteins are colored in gray and shown in wire representation. The interacting residues are shown in stick representation with blue nitrogen, red oxygen and cyan carbon atoms. Hydrogen bonds and salt bridges are shown as the orange dot- lines. Non-polar hydrogen atoms are not shown. Polar hydrogen atoms are shown only for the ligand and the interacting residues.
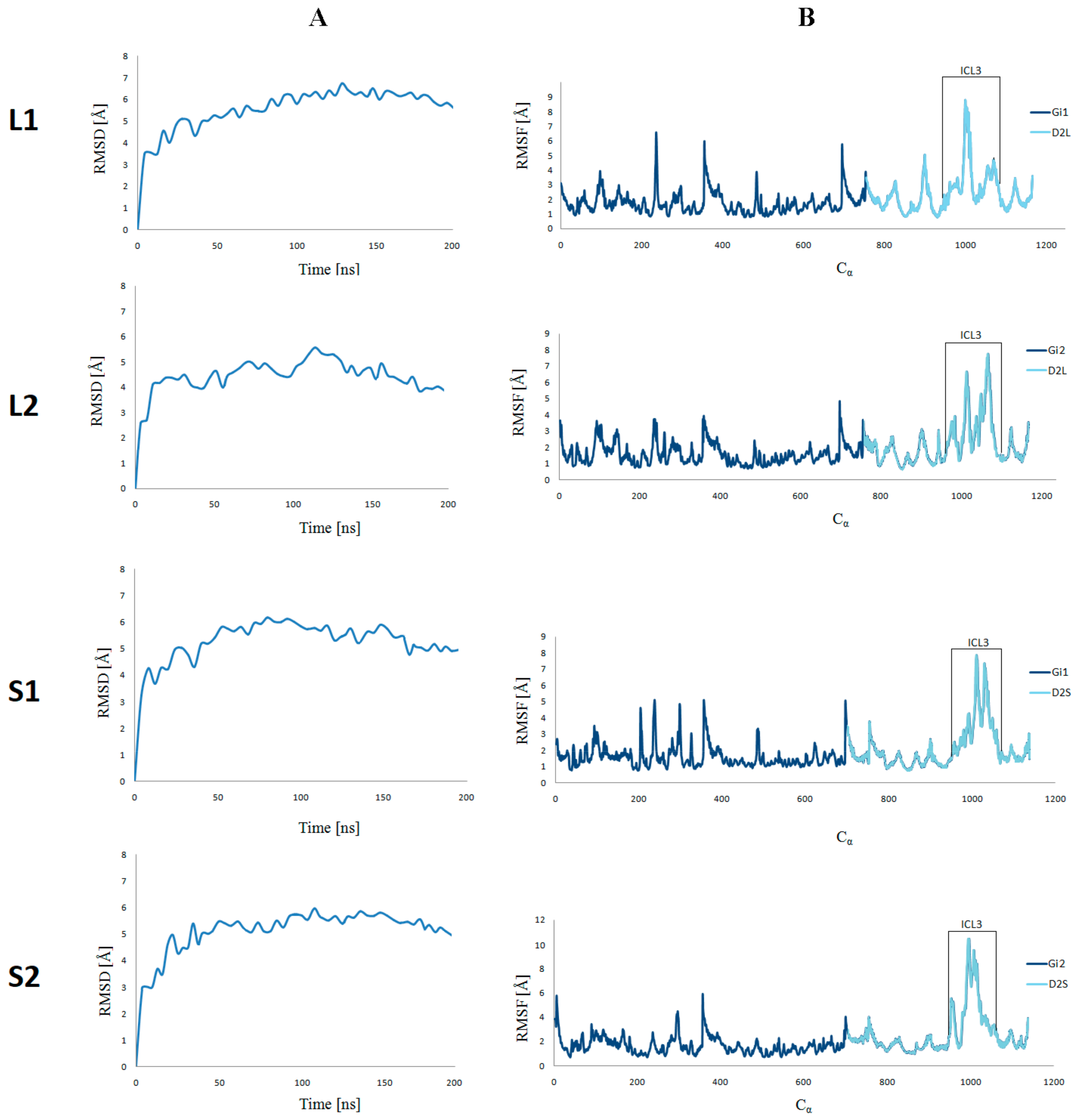
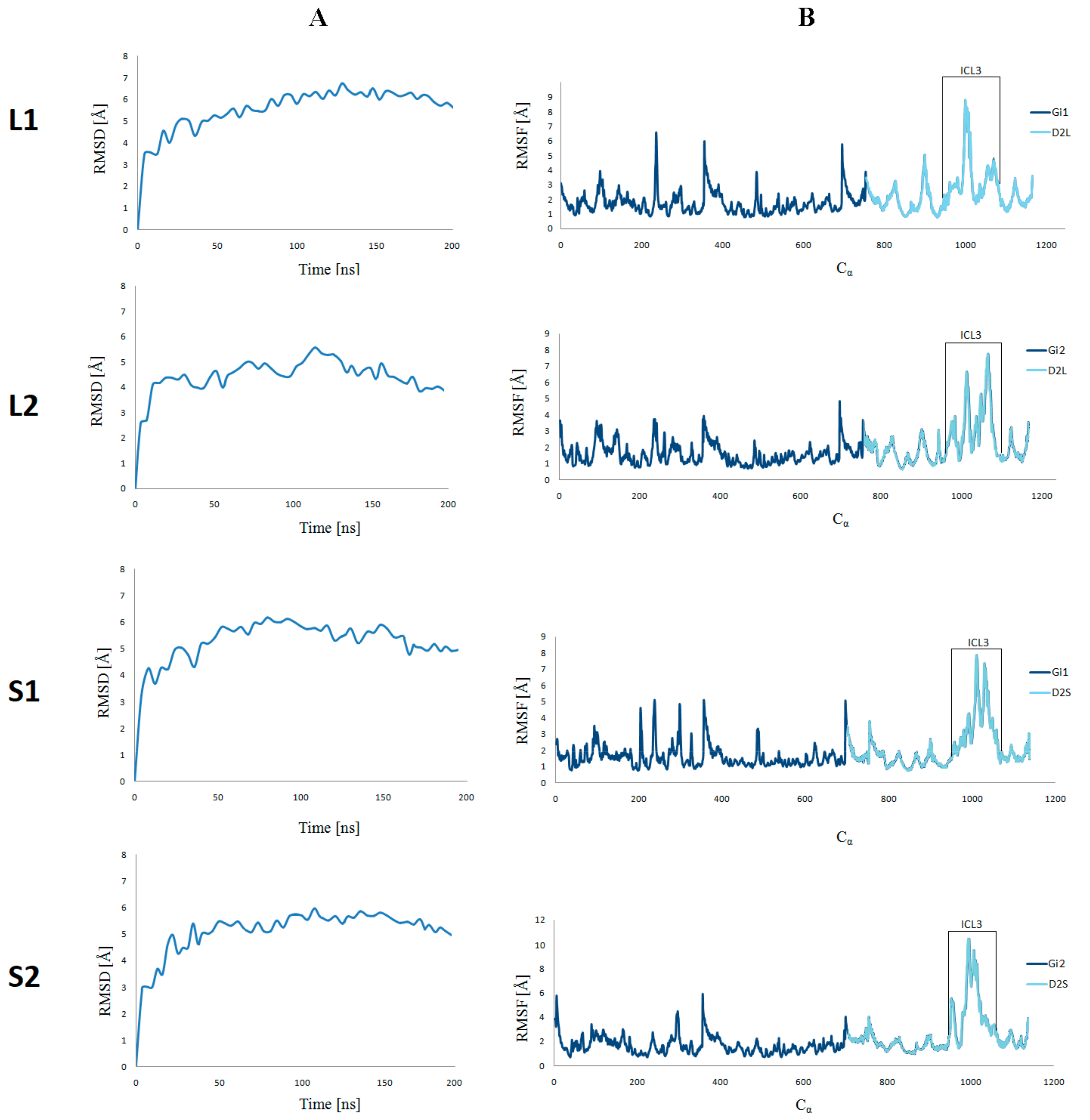
Figure 3.
CαRMSD(A) and CαRMSF (B) of the four selected ligand–receptor complexes against the starting conformations during 200 ns molecular dynamics simulations.
Figure 3.
CαRMSD(A) and CαRMSF (B) of the four selected ligand–receptor complexes against the starting conformations during 200 ns molecular dynamics simulations.
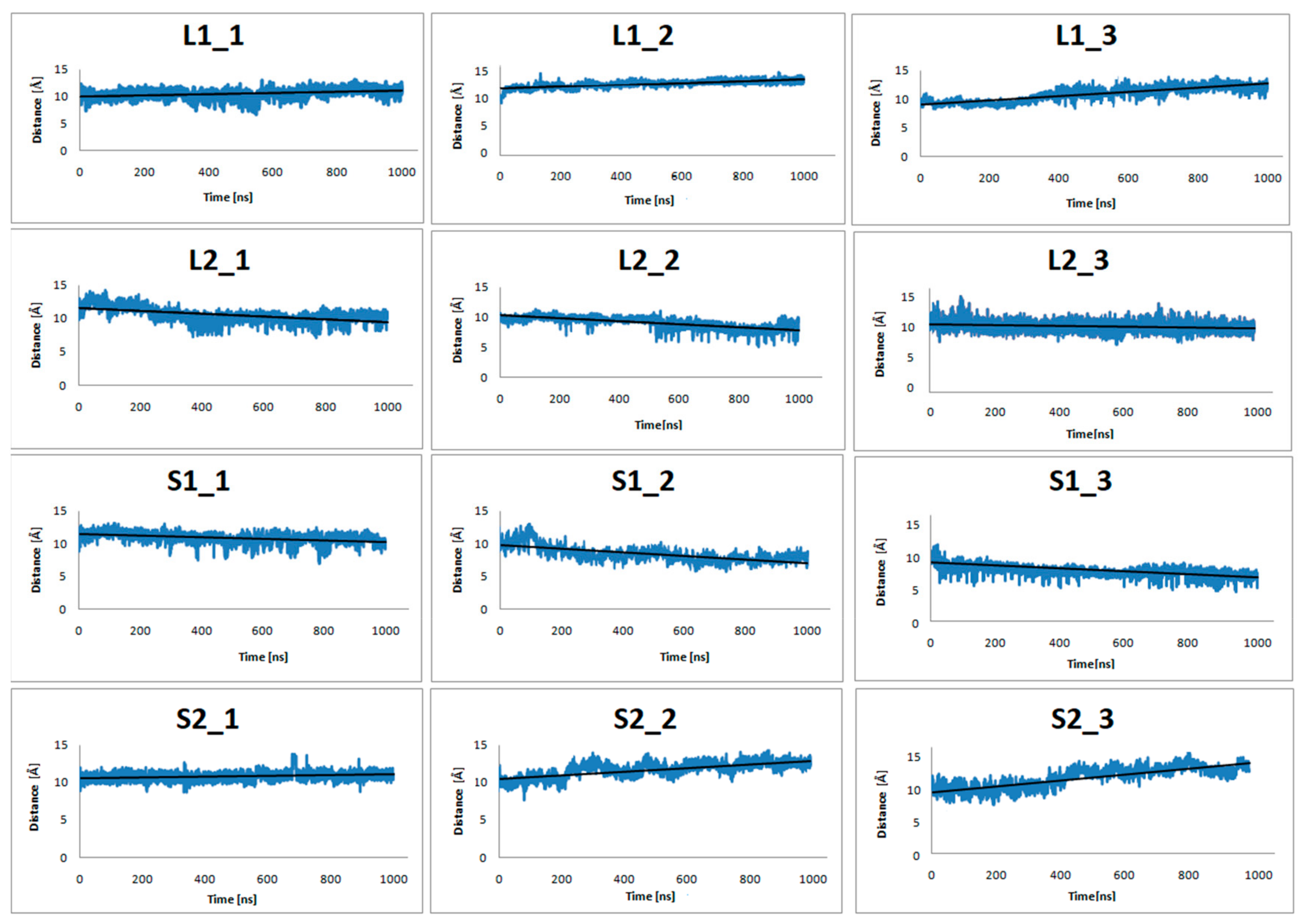
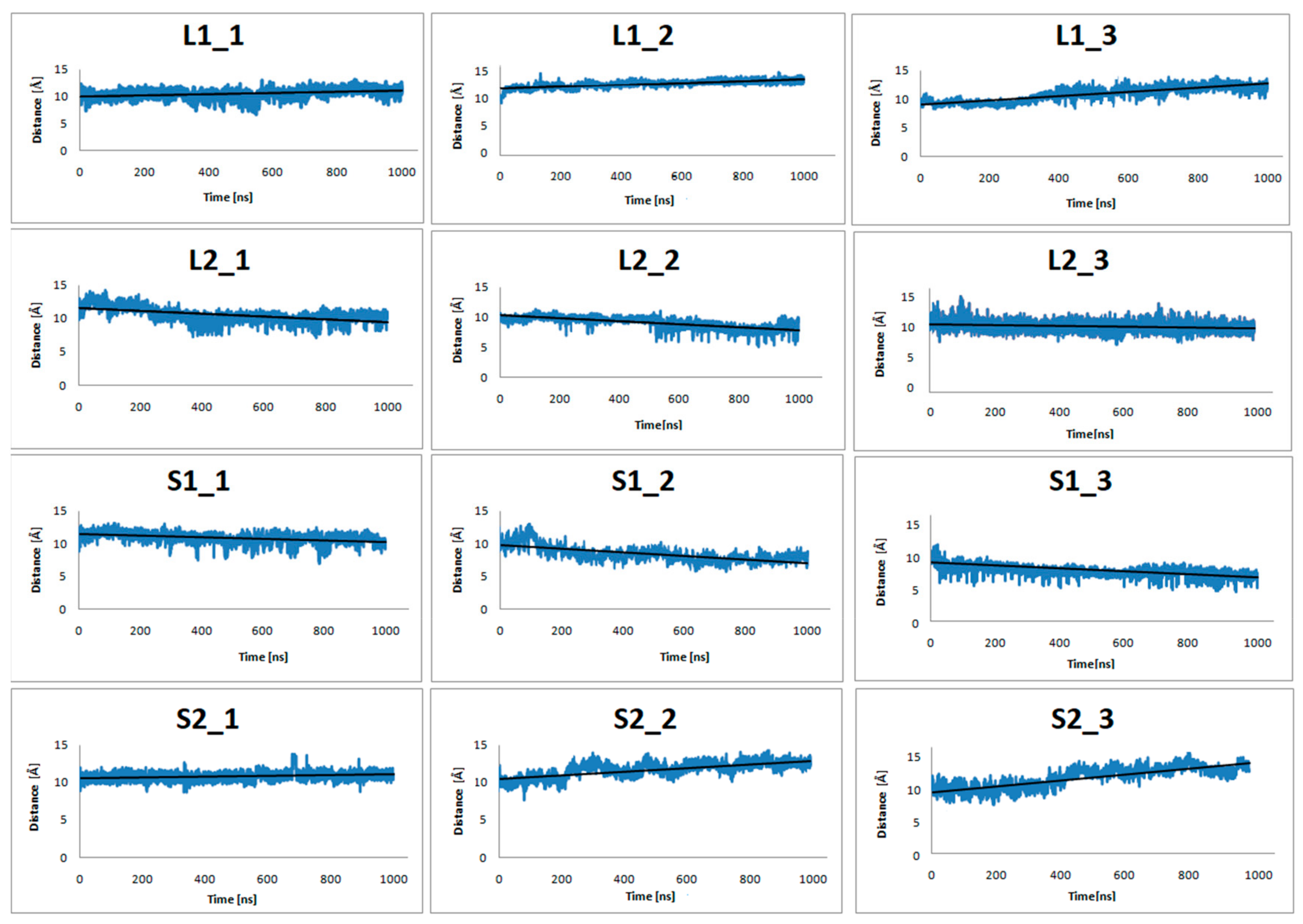
Figure 4.
The distance of center-of-mass (COM) of Y7.53, L2.46, I3.46, T5.54, and V6.40 from the dopamine receptor and D351, C352, G353, L354, and F355from the α5-Gα protein for three replicas of the studied complexes (with dopamine bond) during 1 μs molecular dynamics (MD) simulations.
Figure 4.
The distance of center-of-mass (COM) of Y7.53, L2.46, I3.46, T5.54, and V6.40 from the dopamine receptor and D351, C352, G353, L354, and F355from the α5-Gα protein for three replicas of the studied complexes (with dopamine bond) during 1 μs molecular dynamics (MD) simulations.
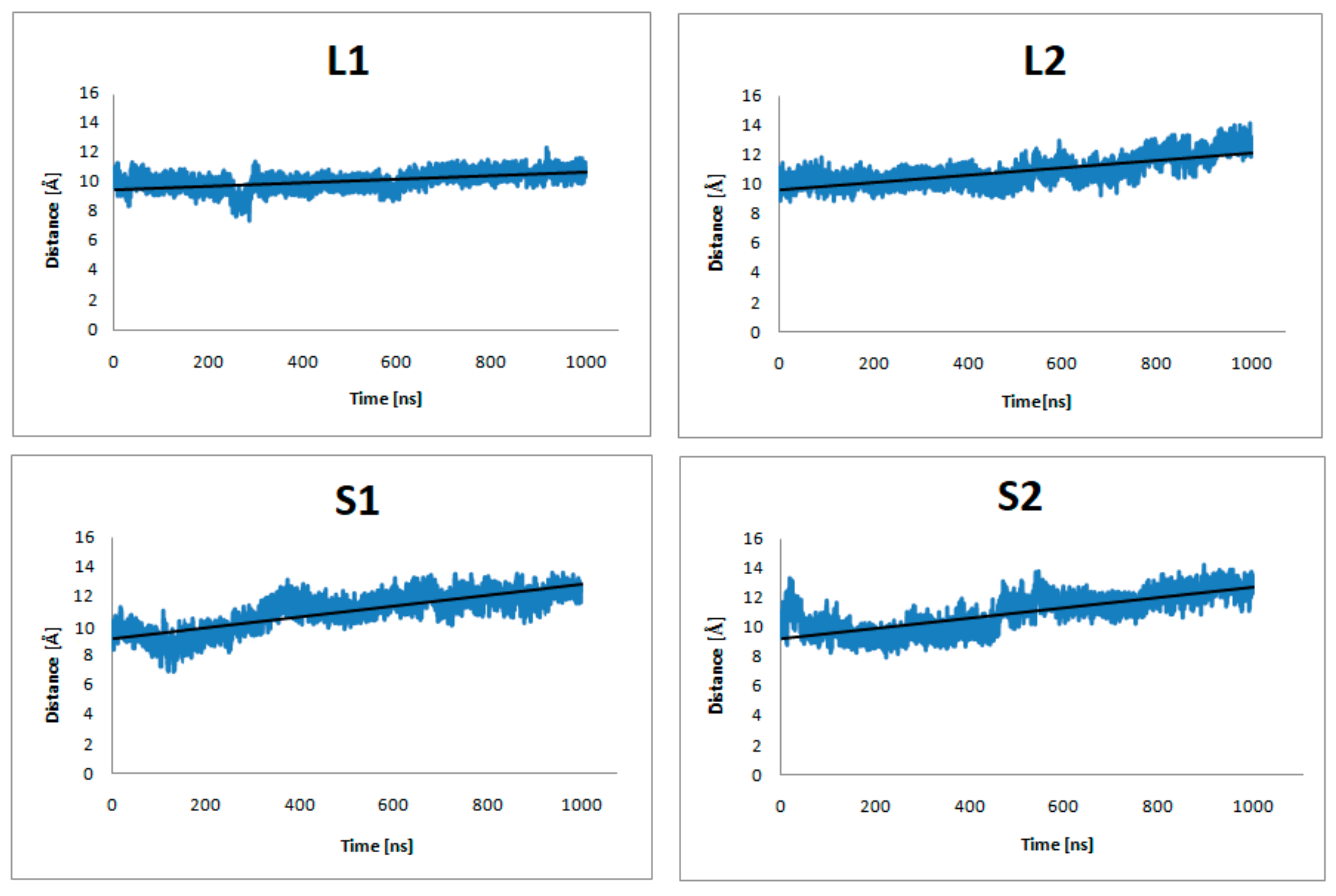
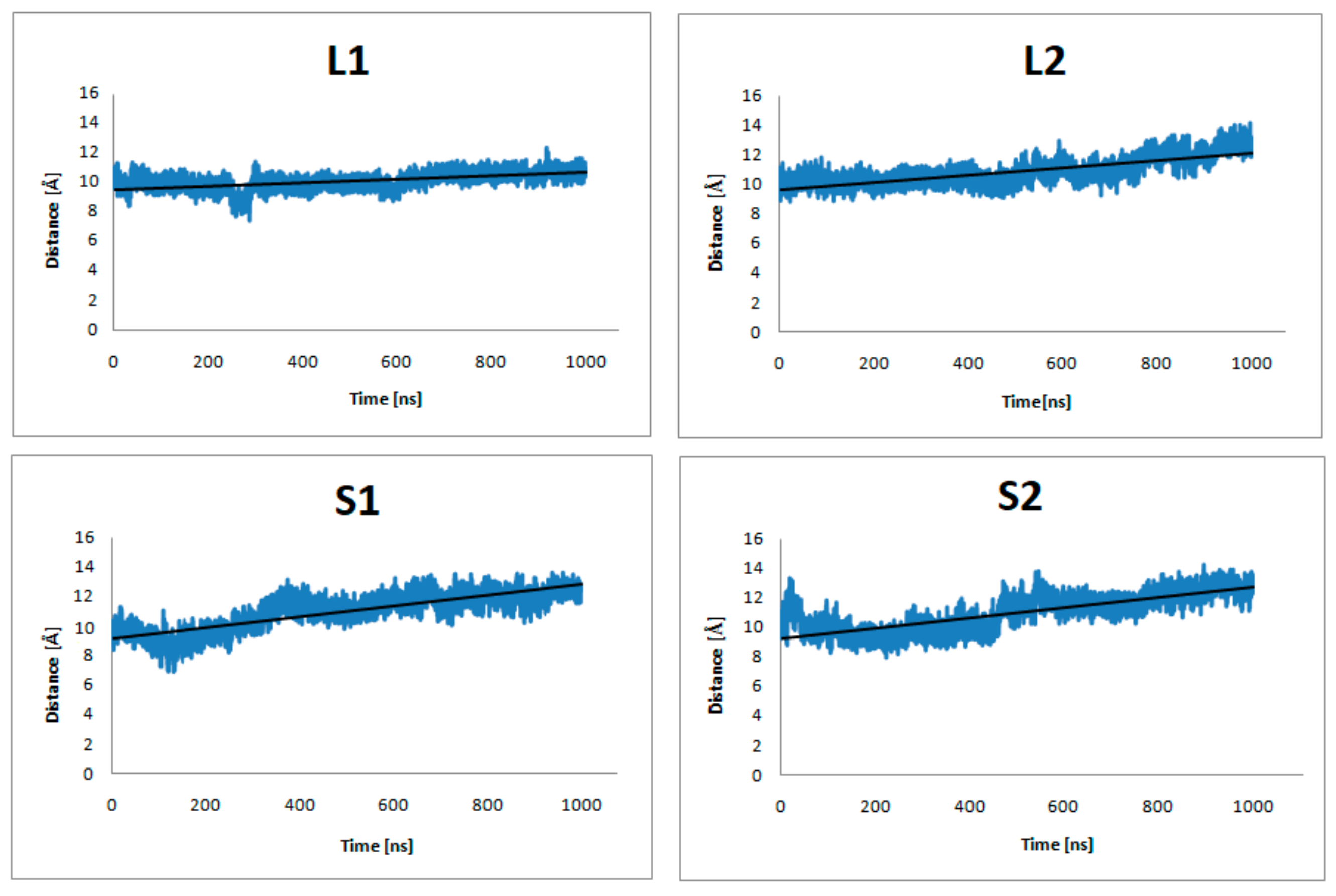
Figure 5.
The distance of center-of-mass (COM) of Y7.53, L2.46, I3.46, T5.54, and V6.40 from the dopamine receptor and D351, C352, G353, L354, and F355 from the α5-Gα protein of the studied complexes (no dopamine bond) during 1 μs MD simulations.
Figure 5.
The distance of center-of-mass (COM) of Y7.53, L2.46, I3.46, T5.54, and V6.40 from the dopamine receptor and D351, C352, G353, L354, and F355 from the α5-Gα protein of the studied complexes (no dopamine bond) during 1 μs MD simulations.
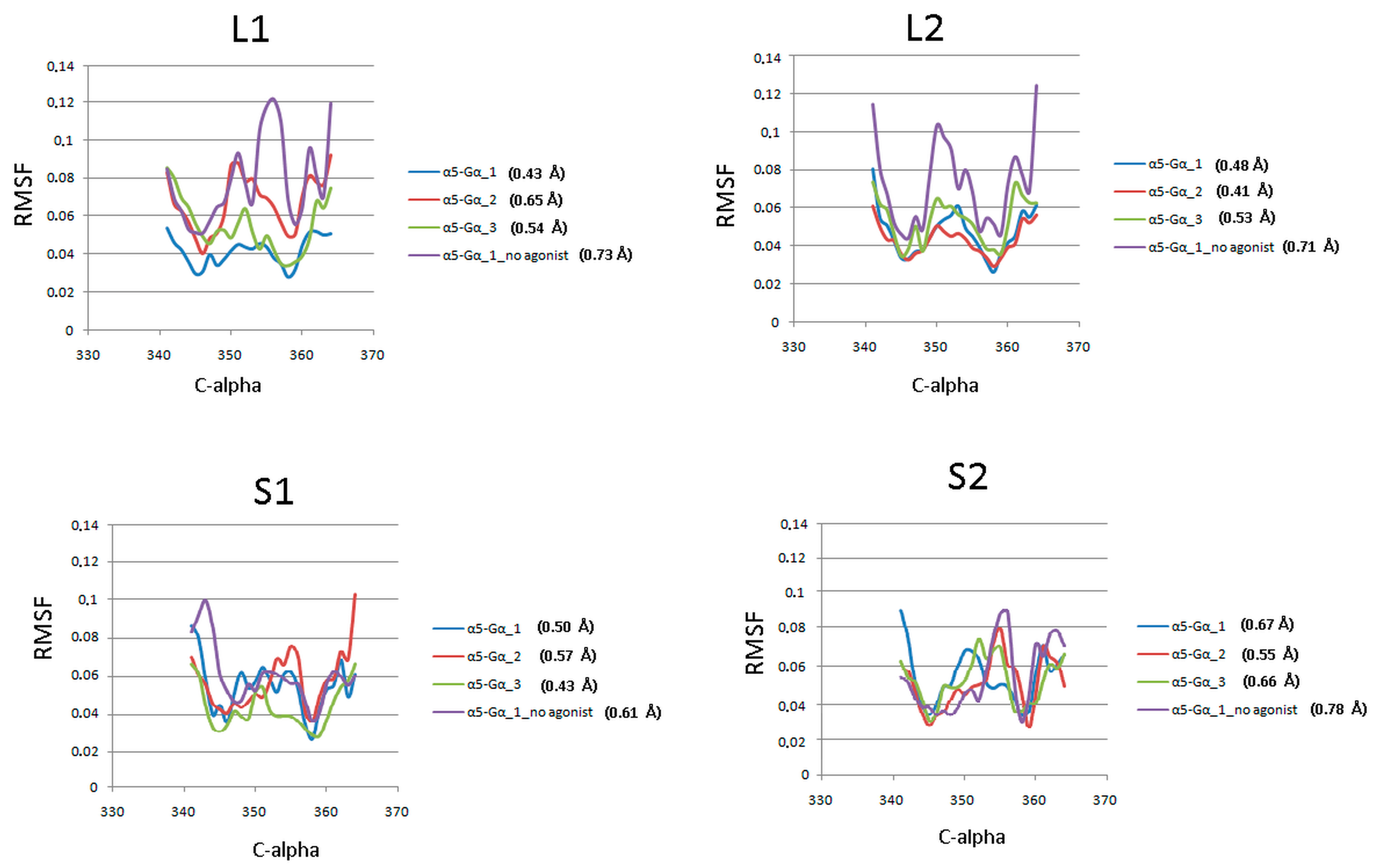
Figure 6.
CαRMSF of the α5-Gα protein against the starting conformations during 1 µs molecular dynamics simulations. Each plot shows scores for three replicas and a receptor without agonist with the average value of RMSF.
Figure 6.
CαRMSF of the α5-Gα protein against the starting conformations during 1 µs molecular dynamics simulations. Each plot shows scores for three replicas and a receptor without agonist with the average value of RMSF.
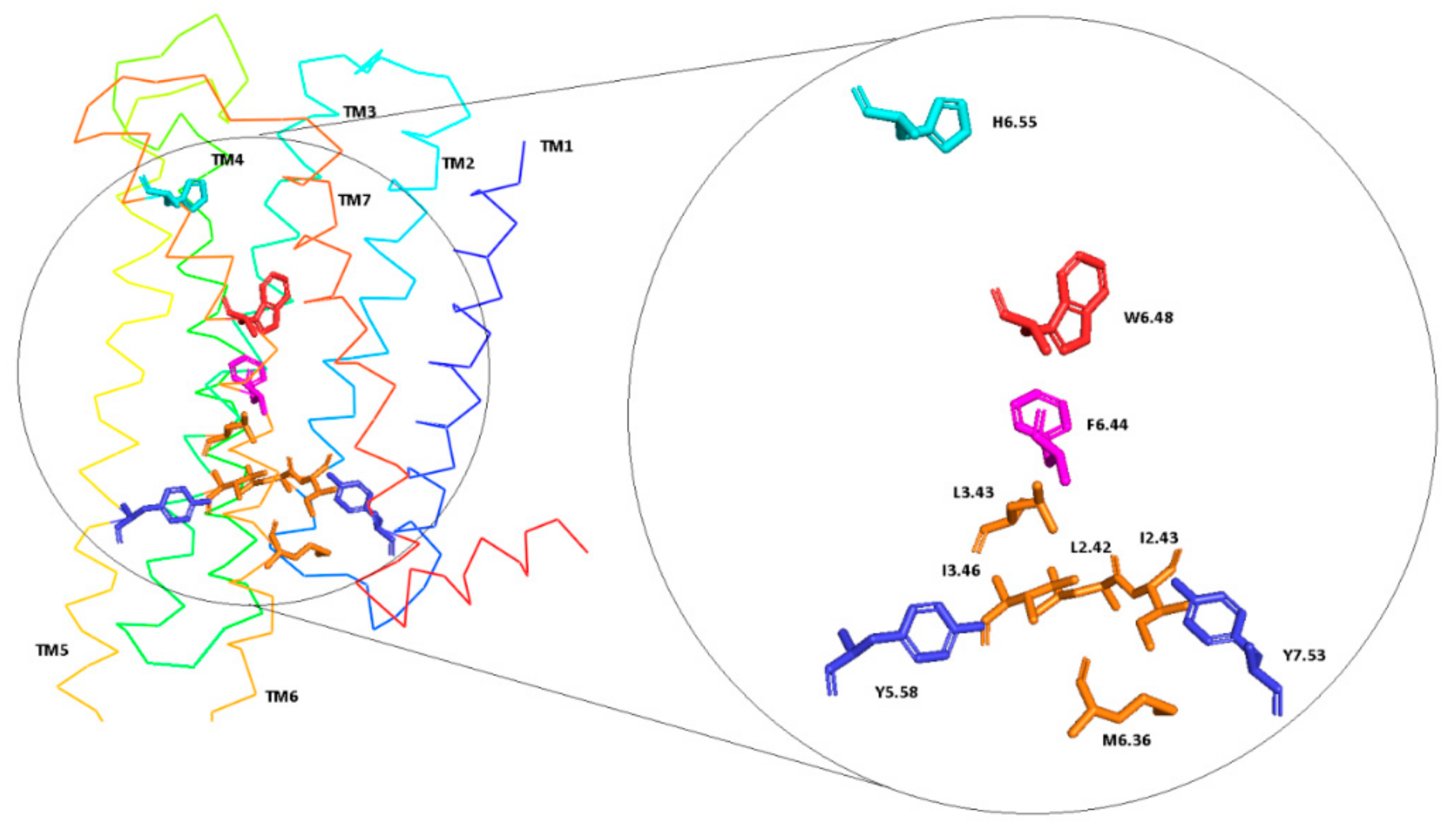
Figure 7.
D2 receptor in inactive conformation with residues involved in molecular switches.
Figure 7.
D2 receptor in inactive conformation with residues involved in molecular switches.
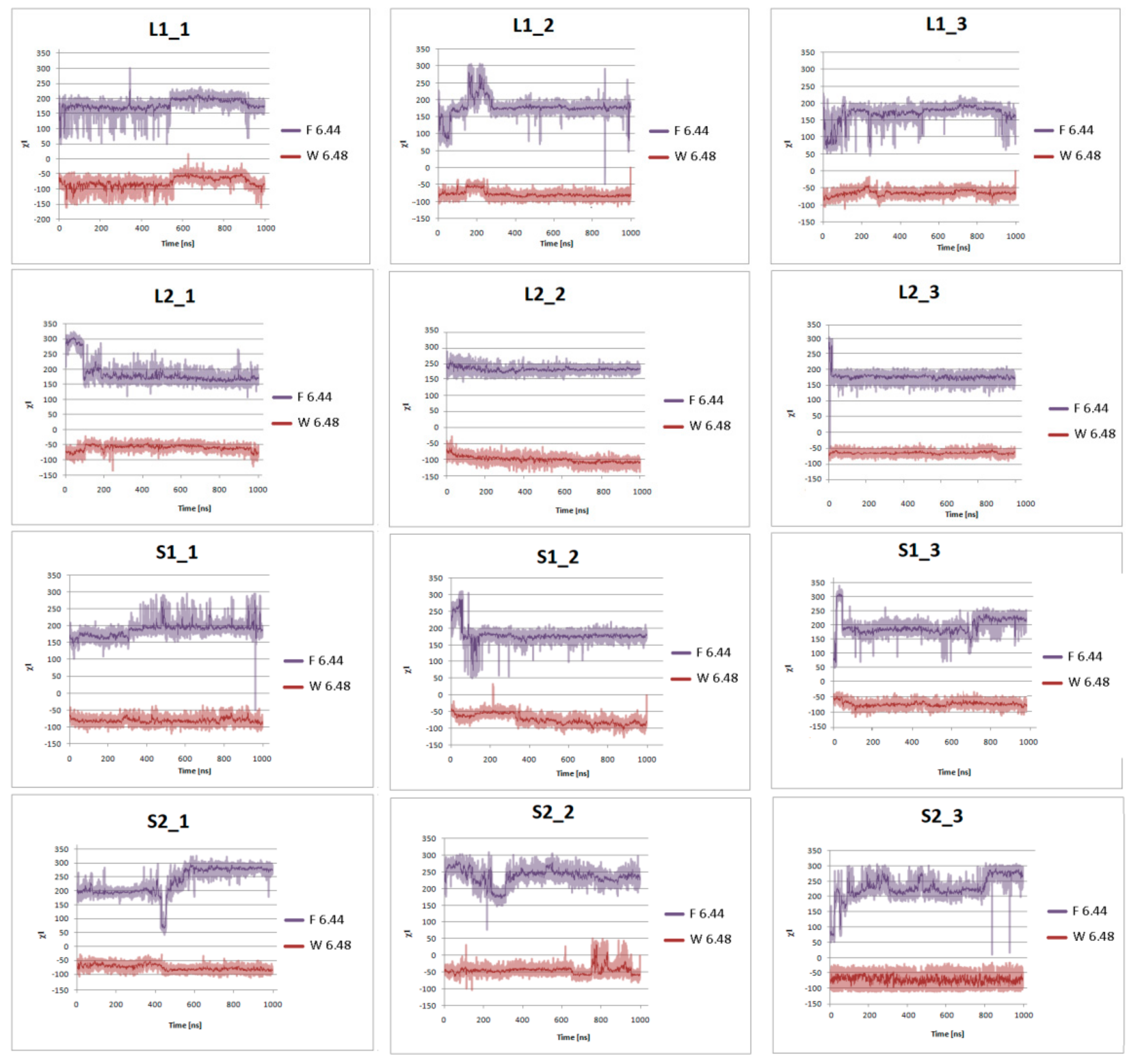
Figure 8.
Side chain torsion angle changes of F6.44 and W6.48 for L1, L2, S1, and S2 receptors in complex with dopamine in three replicas during 1 μs molecular dynamics simulations.
Figure 8.
Side chain torsion angle changes of F6.44 and W6.48 for L1, L2, S1, and S2 receptors in complex with dopamine in three replicas during 1 μs molecular dynamics simulations.
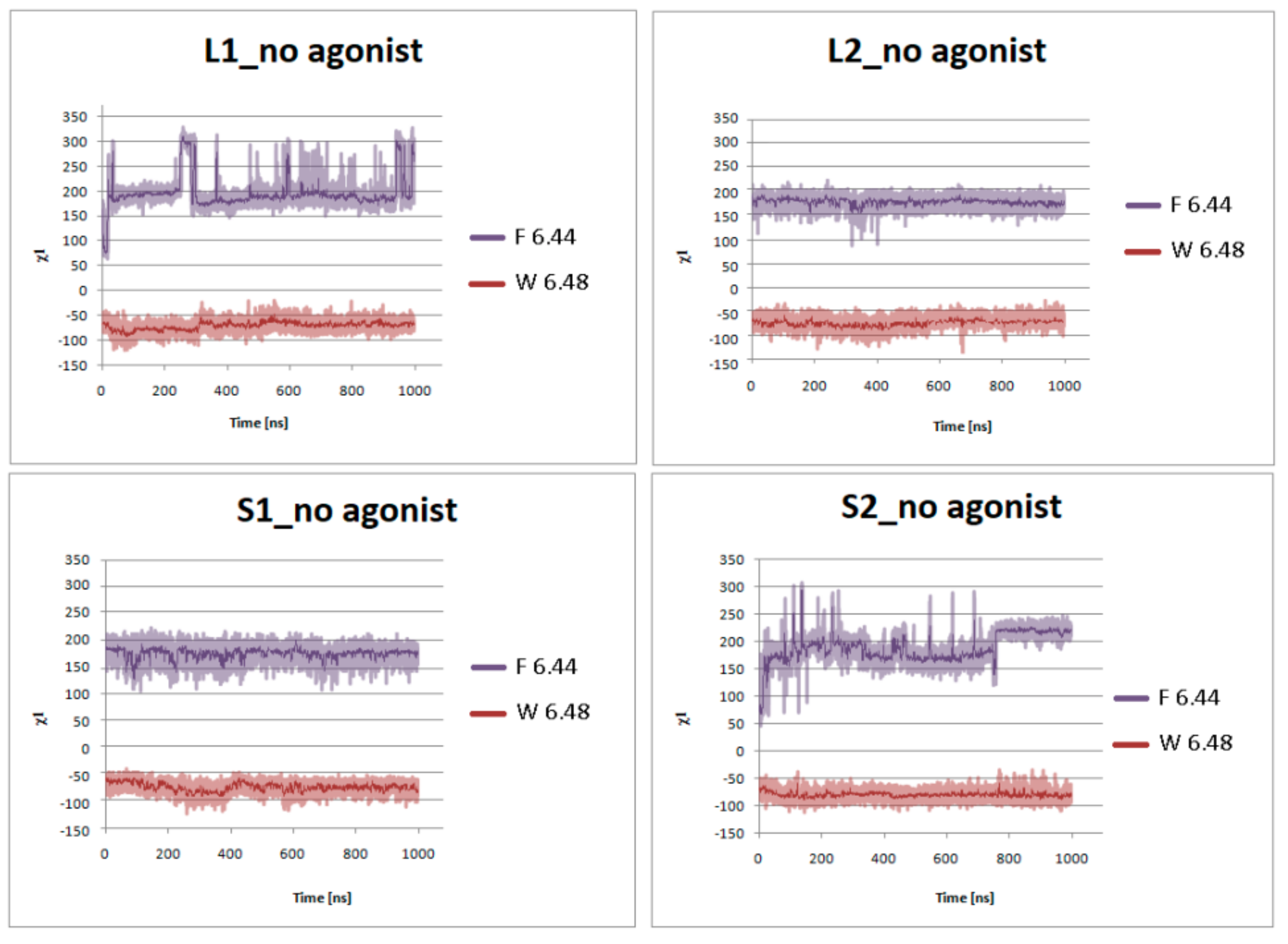
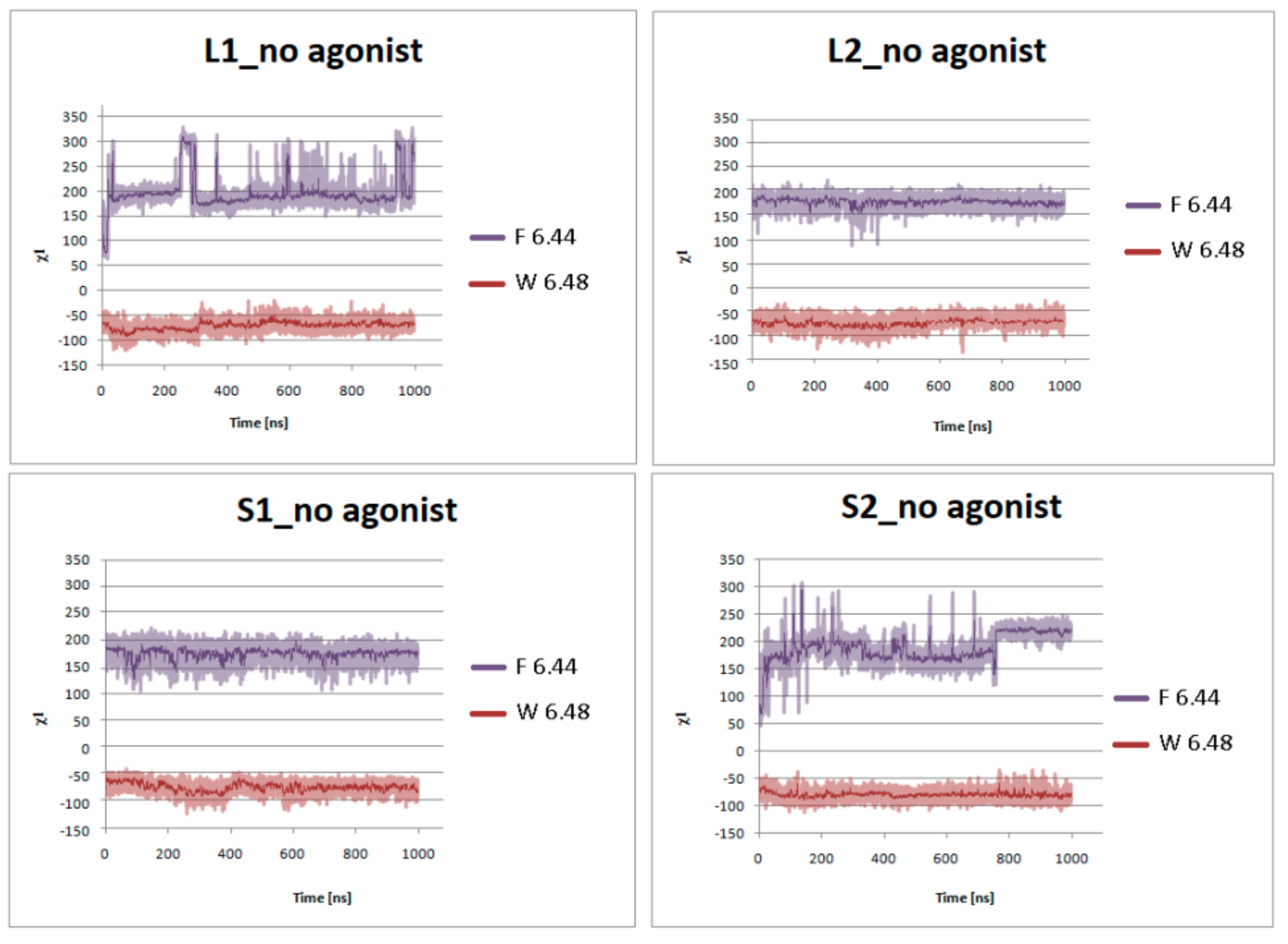
Figure 9.
Side chain torsion angle changes of F6.44 and W6.48 for L1, L2, S1, and S2 receptors without dopamine during 1 μs molecular dynamics simulations.
Figure 9.
Side chain torsion angle changes of F6.44 and W6.48 for L1, L2, S1, and S2 receptors without dopamine during 1 μs molecular dynamics simulations.
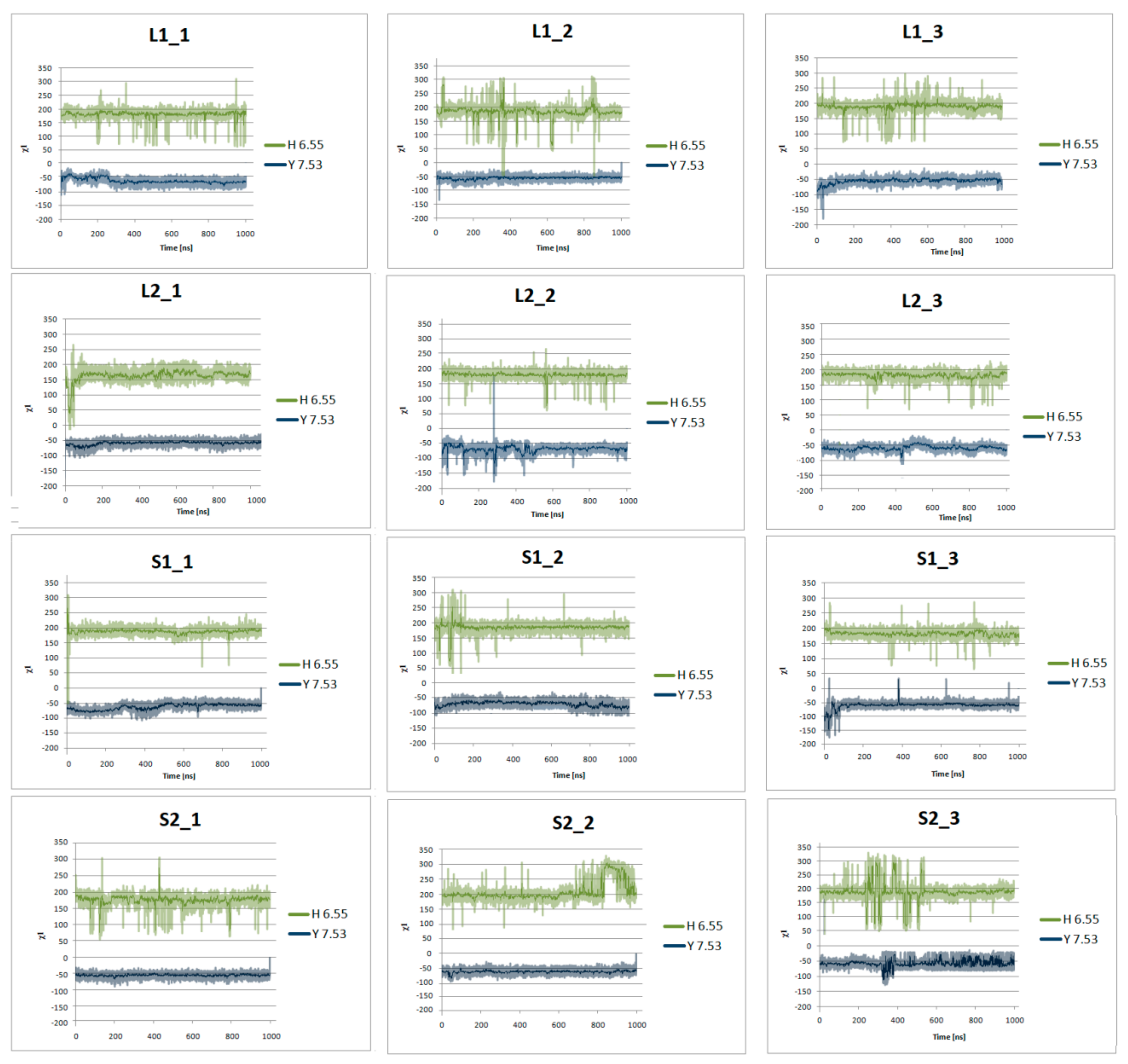
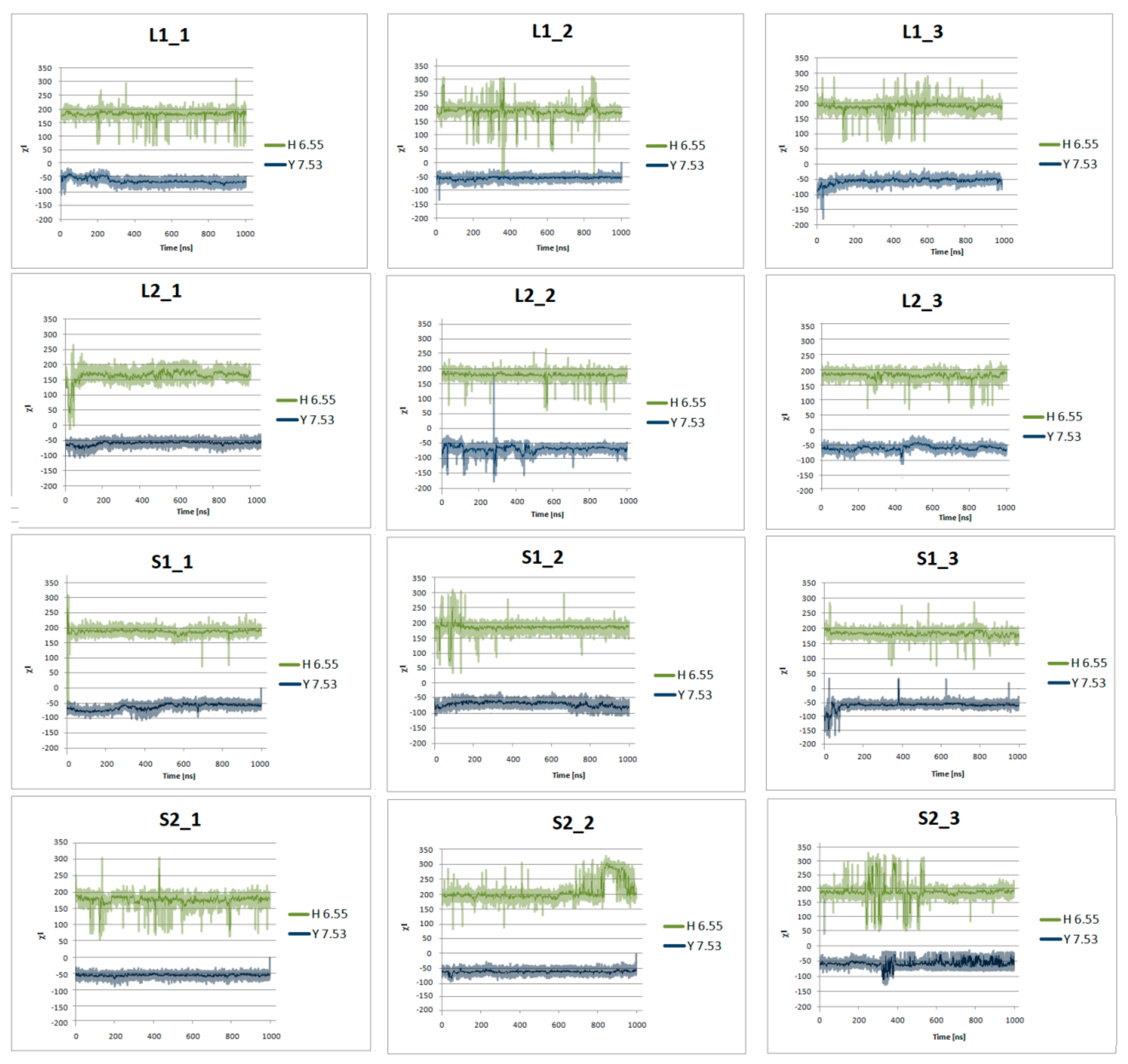
Figure 10.
Side chain torsion angle changes of H6.55 and Y7.53 for L1, L2, S1, and S2 receptors in complex with dopamine in three replicas during 1μs molecular dynamics simulations.
Figure 10.
Side chain torsion angle changes of H6.55 and Y7.53 for L1, L2, S1, and S2 receptors in complex with dopamine in three replicas during 1μs molecular dynamics simulations.
Figure 11.
Side chain torsion angle changes of H6.55 and Y7.53 for L1, L2, S1, and S2 receptors without dopamine during 1 μs molecular dynamics simulations.
Figure 11.
Side chain torsion angle changes of H6.55 and Y7.53 for L1, L2, S1, and S2 receptors without dopamine during 1 μs molecular dynamics simulations.
Figure 12.
Superimposition of TM6 for initial simulation (grey) and TM6 (cyan) after 1 µs MD simulations with conformational state described below.
Figure 12.
Superimposition of TM6 for initial simulation (grey) and TM6 (cyan) after 1 µs MD simulations with conformational state described below.
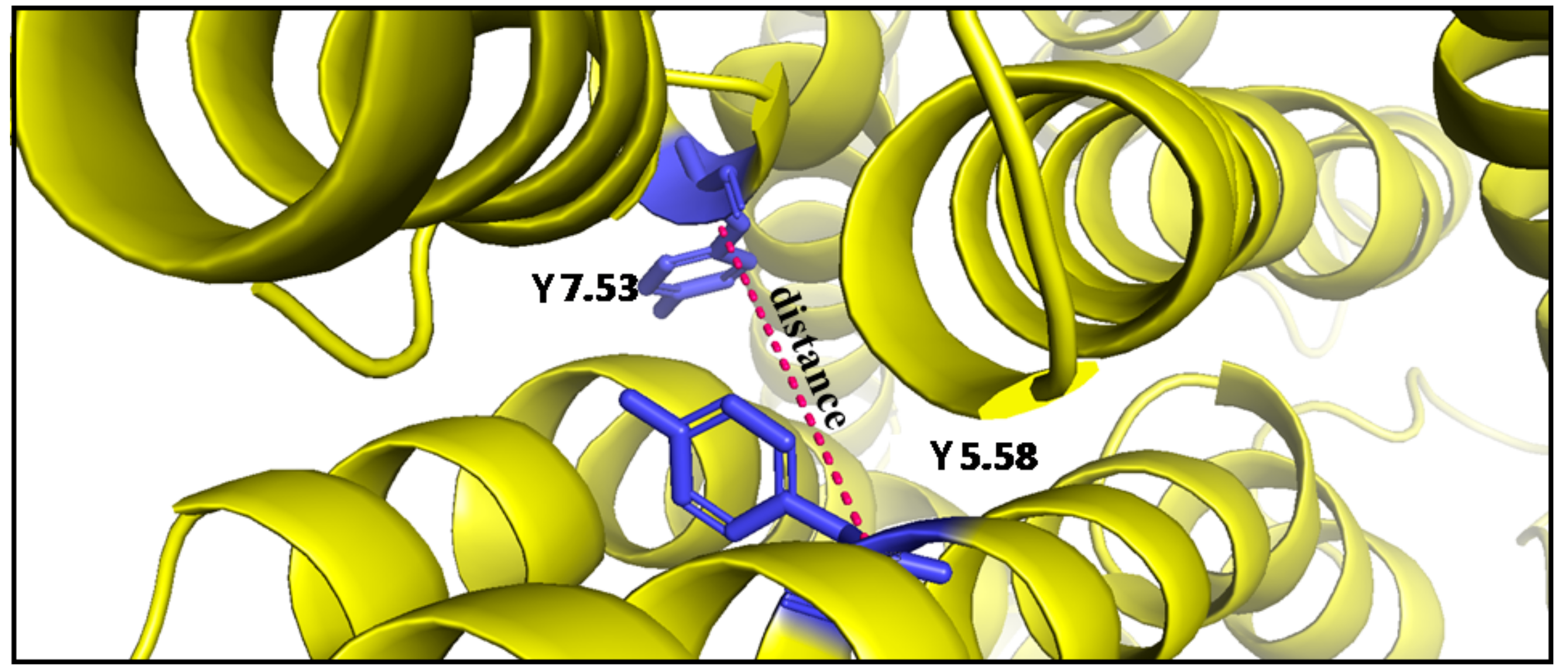
Figure 13.
Distance between TM5 and TM7 (yellow) at the Cα atom of Y5.58 and Cα atom of Y7.53 (blue).
Figure 13.
Distance between TM5 and TM7 (yellow) at the Cα atom of Y5.58 and Cα atom of Y7.53 (blue).
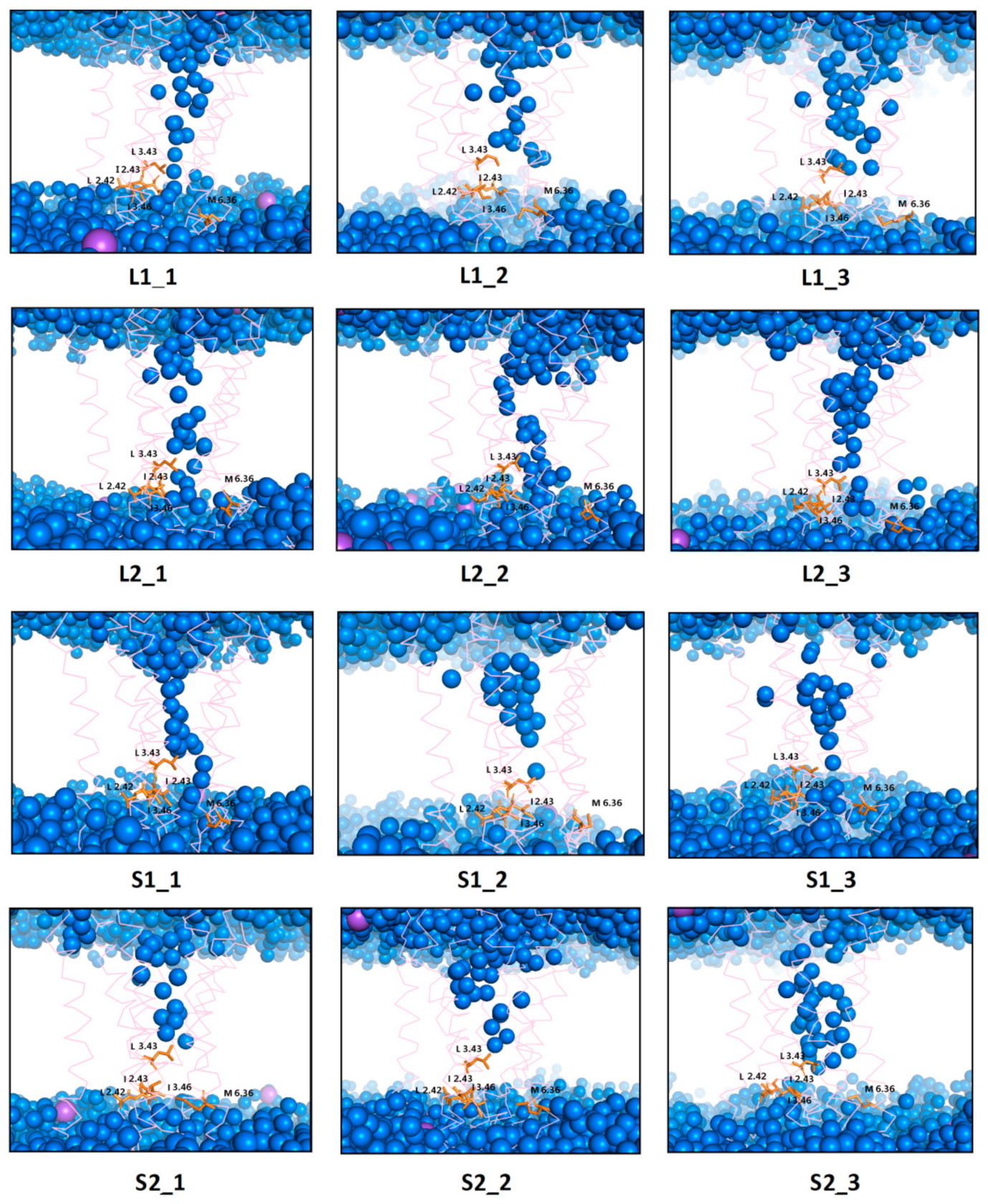
Figure 14.
Internal water channel with marked residues (orange) forming a hydrophobic barrier for receptors with dopamine after 1 µs MD simulations. The helices are marked as pink springs and the sodium ions as a purple sphere.
Figure 14.
Internal water channel with marked residues (orange) forming a hydrophobic barrier for receptors with dopamine after 1 µs MD simulations. The helices are marked as pink springs and the sodium ions as a purple sphere.
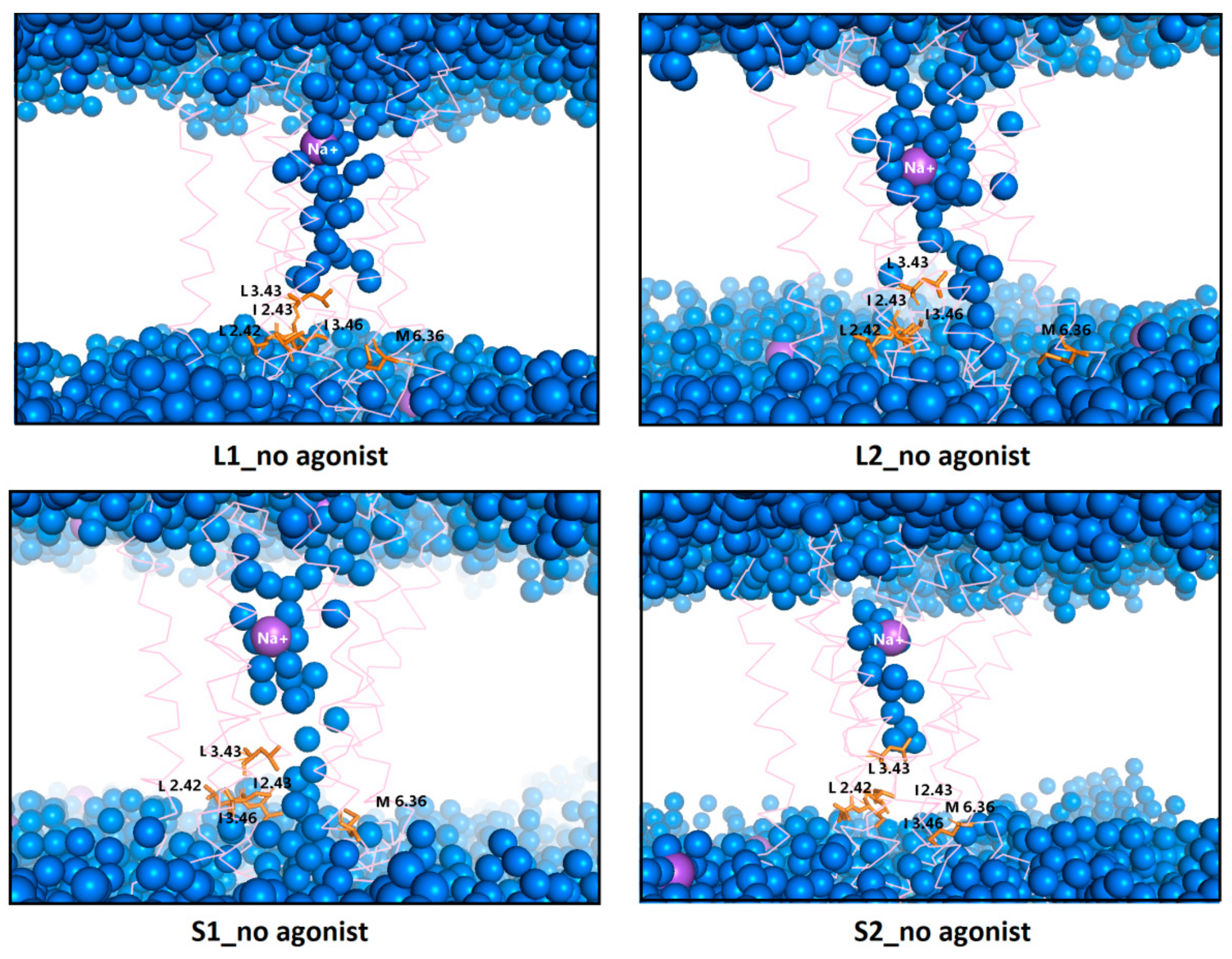
Figure 15.
Internal water channel with marked residues (orange) forming a hydrophobic barrier for receptors without dopamine after 1 µs MD simulations. The helices aremarked as pink springs and the sodium ions as a purple sphere.
Figure 15.
Internal water channel with marked residues (orange) forming a hydrophobic barrier for receptors without dopamine after 1 µs MD simulations. The helices aremarked as pink springs and the sodium ions as a purple sphere.
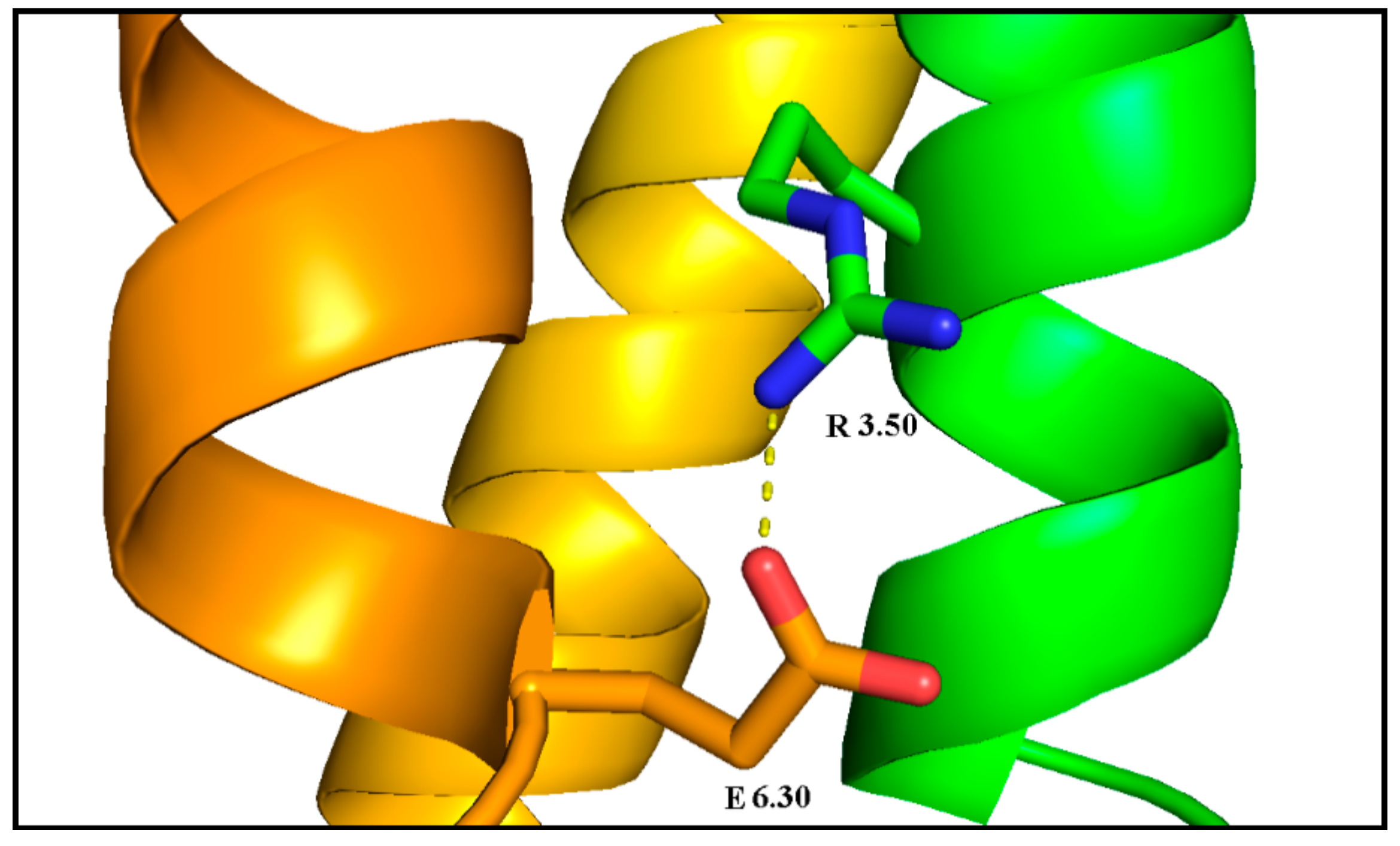
Figure 16.
Ionic lock of inactive the dopamine D2 receptor (PDB ID: 6CM4): the lock between R3.50 (green with blue nitrogen atoms) of the DRY motif and E6.30 (orange with red oxygen atoms).
Figure 16.
Ionic lock of inactive the dopamine D2 receptor (PDB ID: 6CM4): the lock between R3.50 (green with blue nitrogen atoms) of the DRY motif and E6.30 (orange with red oxygen atoms).
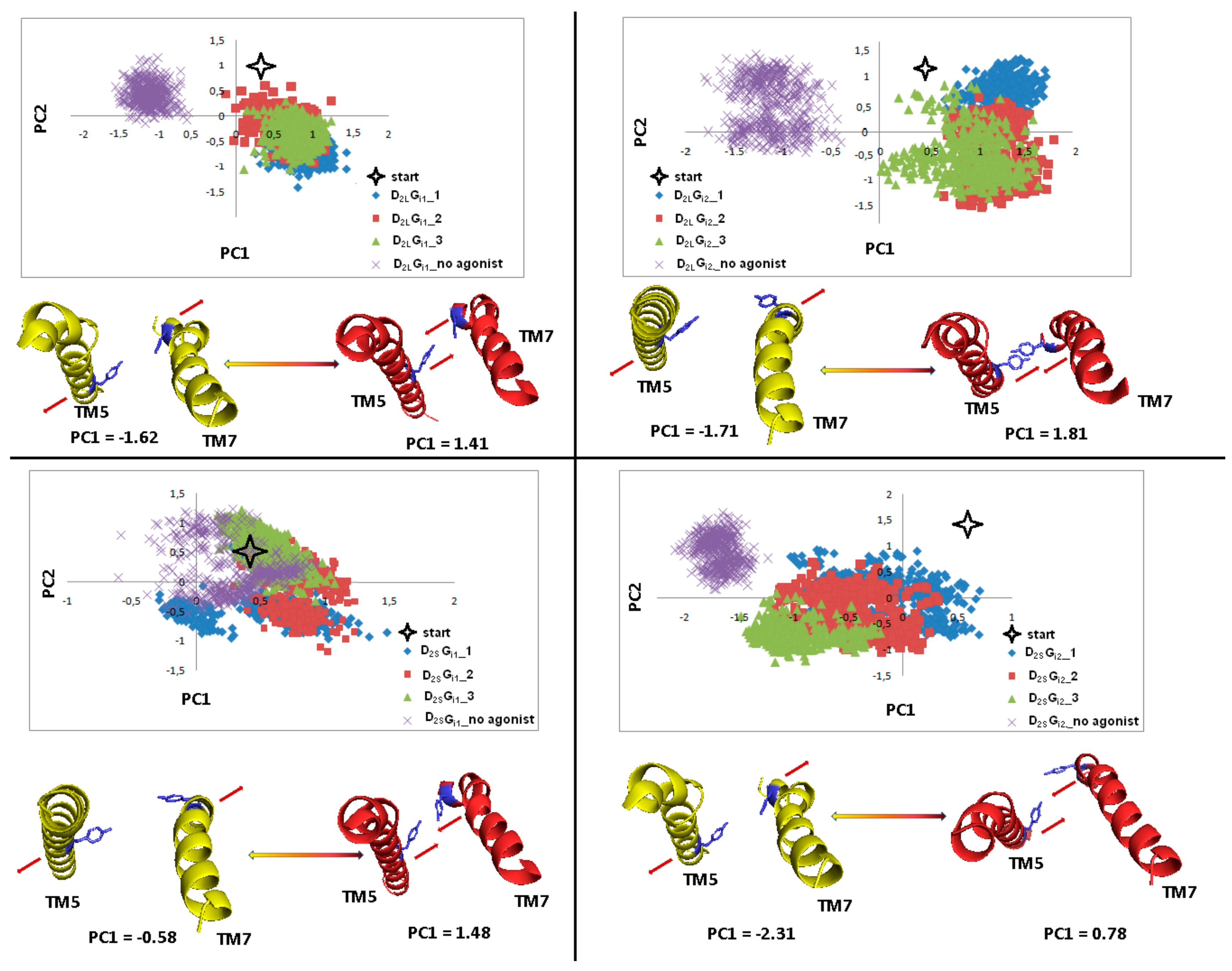
Figure 17.
Motion of the protein in phase space along the first two principal componentsfor TM6 in the last 100 ns of dynamics. The yellow and red cartoons correspond to the PC1 values and show extreme projections along PC1.
Figure 17.
Motion of the protein in phase space along the first two principal componentsfor TM6 in the last 100 ns of dynamics. The yellow and red cartoons correspond to the PC1 values and show extreme projections along PC1.
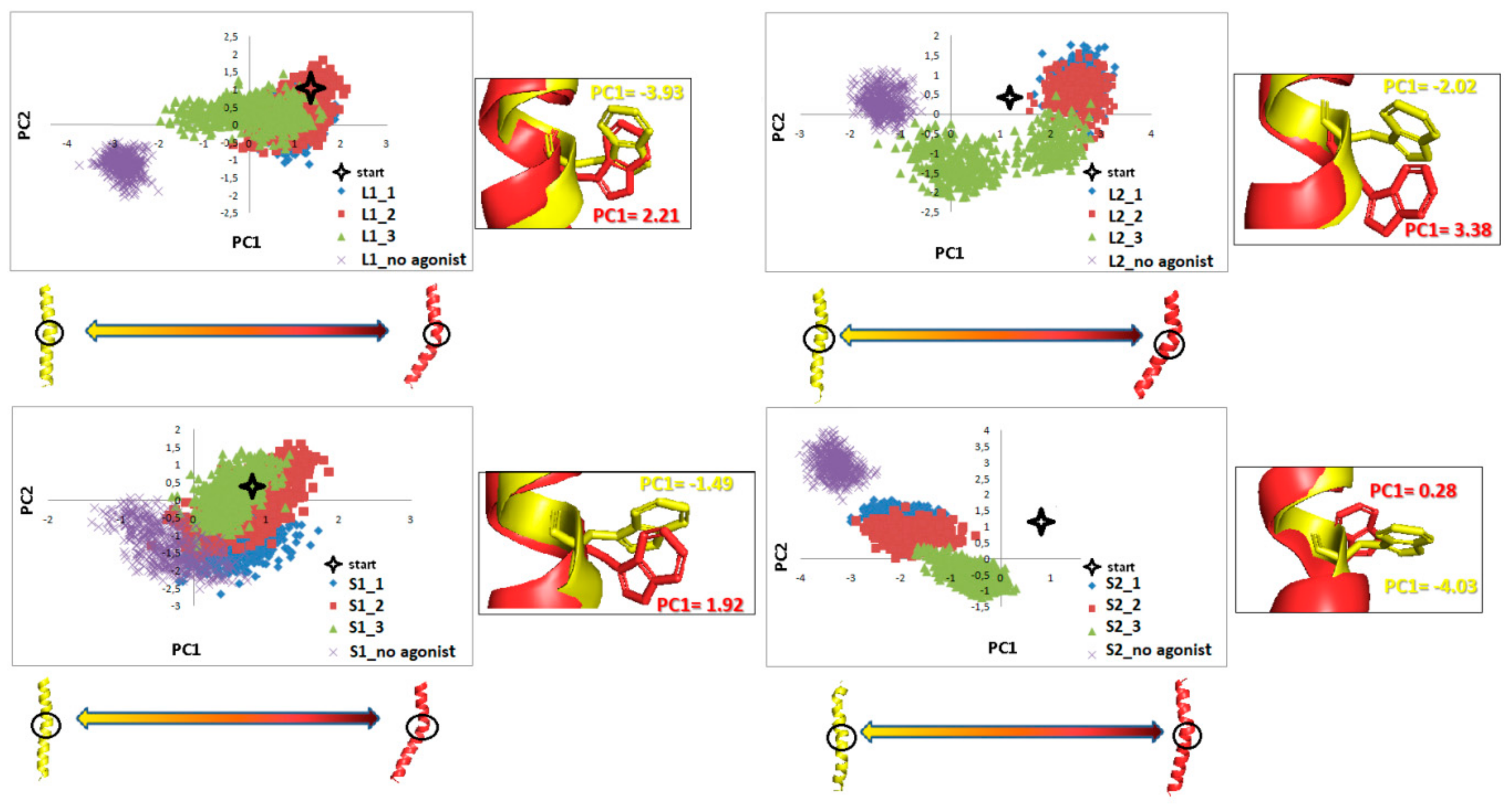
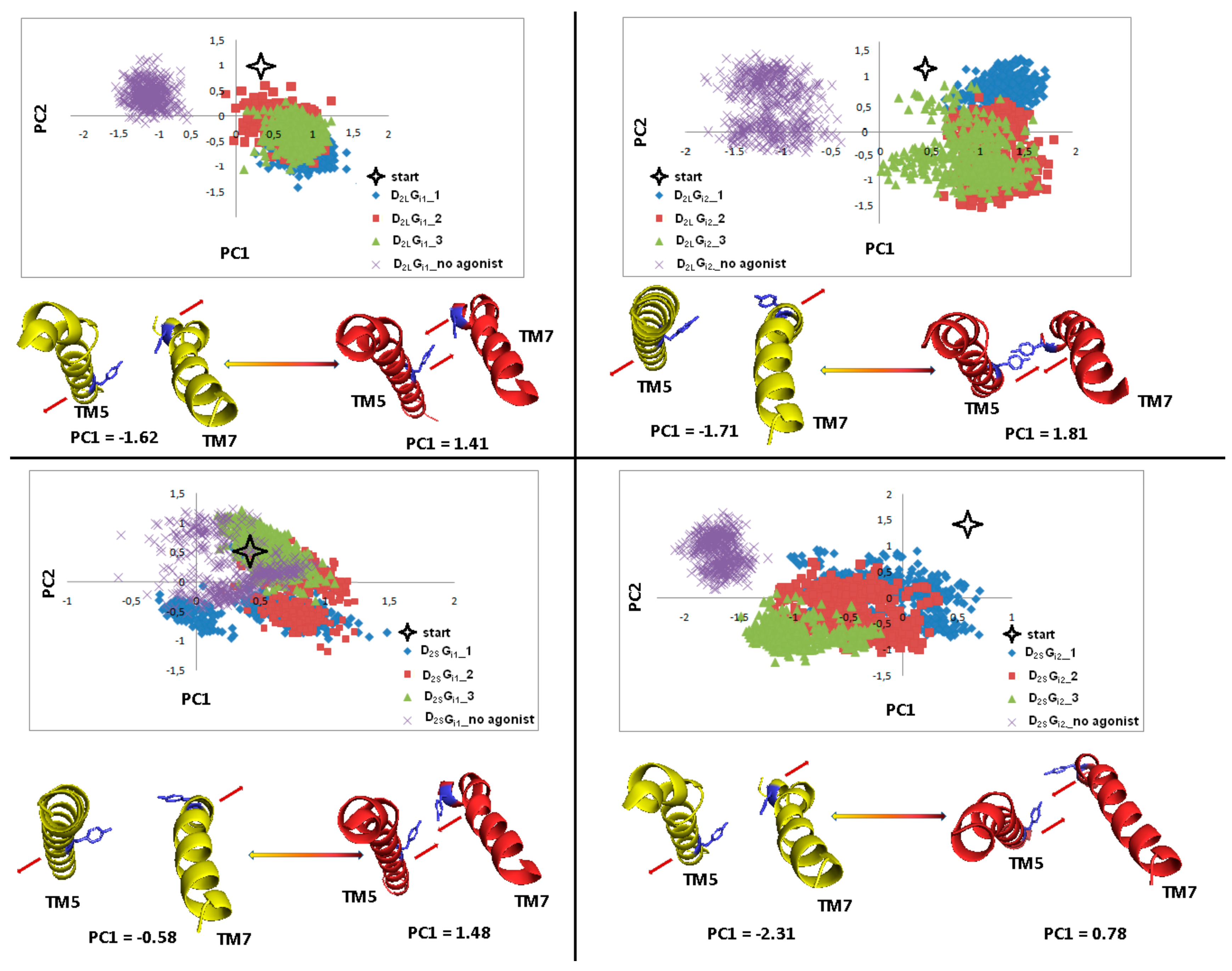
Figure 18.
Motion of the protein in phase space along the first two principal components for TM5 and TM7 in the part including Y5.58 and Y7.53 (blue). The yellow and red cartoons, with the red arrows describing the tendency to the extreme conformations, correspond to the PC1 values and show extreme projections along PC1.
Figure 18.
Motion of the protein in phase space along the first two principal components for TM5 and TM7 in the part including Y5.58 and Y7.53 (blue). The yellow and red cartoons, with the red arrows describing the tendency to the extreme conformations, correspond to the PC1 values and show extreme projections along PC1.
Table 1.
Results of validation of the best-scored receptor-G protein complexes models.
Table 1.
Results of validation of the best-scored receptor-G protein complexes models.
| Web Server | Parameters | Scores |
|---|
| L1 | L2 | S1 | S2 |
|---|
| ProCheck | Favored region (%) | 91.2 | 92.4 | 92.7 | 92.1 | 94.3 | 93.5 | 93.7 | 91.8 | 94.5 | 94.3 | 92.1 | 94.6 | 94.2 | 91.4 | 94.0 | 93.3 |
| Allowed region (%) | 7.1 | 6.9 | 6.2 | 6.8 | 4.6 | 5.0 | 4.3 | 7.8 | 4.3 | 4.2 | 6.7 | 4.4 | 3.4 | 7.1 | 4.7 | 5.5 |
| Outlier region (%) | 1.7 | 0.7 | 1.1 | 1.1 | 1.1 | 1.5 | 2.0 | 0.4 | 1.2 | 1.5 | 1.2 | 1.0 | 2.4 | 1.5 | 1.3 | 1.2 |
| Verify 3D | Averaged 3D–1D score ≥0.2 (%) | 82.4 | 83.1 | 83.2 | 83.4 | 84.2 | 83.4 | 83.9 | 83.4 | 84.7 | 83.2 | 82.9 | 84.3 | 83.3 | 82.4 | 83.9 | 83.4 |
| ERRAT | Overall quality factor | 90.8 | 91.3 | 91.1 | 91.4 | 94.2 | 93.2 | 93.6 | 92.3 | 93.3 | 93.4 | 91.7 | 92.6 | 94.7 | 90.9 | 92.4 | 92.3 |
Table 2.
Values in (kJ/mol) of the average short-range Lennard-Jones potential energy of 1 µs MD simulations for the COM of C-terminal part of α5-Gα: G353, L354, F355 and residues from the DRY-motif of TM3: D3.49, R3.50, from IL2: M140, Y142 and TM6: K6.28, M6.36 for all replicas and receptor without dopamine.
Table 2.
Values in (kJ/mol) of the average short-range Lennard-Jones potential energy of 1 µs MD simulations for the COM of C-terminal part of α5-Gα: G353, L354, F355 and residues from the DRY-motif of TM3: D3.49, R3.50, from IL2: M140, Y142 and TM6: K6.28, M6.36 for all replicas and receptor without dopamine.
| Complex | Replica |
|---|
| 1 | 2 | 3 | No Agonist |
|---|
| L1 | −850.34 kJ/mol | −824.64 kJ/mol | −867.54 kJ/mol | −734.98 kJ/mol |
| L2 | −1134.79 kJ/mol | −914.32 kJ/mol | −1156.62 kJ/mol | −847.72 kJ/mol |
| S1 | −919.73 kJ/mol | −962.38 kJ/mol | −898.54 kJ/mol | −805.22 kJ/mol |
| S2 | −766.54 kJ/mol | −685.39 kJ/mol | −772.47 kJ/mol | −550.27 kJ/mol |
Table 3.
Distance values for receptors between Y5.58 and Y7.53 1 µs MD simulations.
Table 3.
Distance values for receptors between Y5.58 and Y7.53 1 µs MD simulations.
| Complex | Replica |
|---|
| 1 | 2 | 3 | No Agonist |
|---|
| L1 | 13.3 ± 0.4 Å | 14.1 ± 0.3 Å | 14.2 ±0.5 Å | 16.1 ±0.4 Å |
| L2 | 10.1 ± 0.5 Å | 8.5 ± 0.3 Å | 11.7 ± 0.4 Å | 15.1 ± 0.3 Å |
| S1 | 12.3 ± 0.3 Å | 12.6 ± 0.5 Å | 11.4 ± 0.4 Å | 15.6 ±0.3 Å |
| S2 | 18.5 ± 0.3 Å | 16.2 ± 0.3 Å | 17.9 ±0.2 Å | 19.1 ±0.3 Å |
Table 4.
Distance values for receptors between R3.50 and E6.30 after 1 µs MD simulations.
Table 4.
Distance values for receptors between R3.50 and E6.30 after 1 µs MD simulations.
| Complex | Replica |
|---|
| 1 | 2 | 3 | No Agonist |
|---|
| L1 | 18.1 ± 0.2 Å | 18.8 ± 0.2 Å | 19.5 ± 0.4 Å | 18.0 ± 0.3 Å |
| L2 | 21.7 ± 0.4 Å | 22.1 + 0.3 Å | 20.1 ± 0.5 Å | 19.0 + 0.3 Å |
| S1 | 21.9 ± 0.2 Å | 22.0 ± 0.4 Å | 21.9 ± 0.2 Å | 14.7 ± 0.3 Å |
| S2 | 16.8 ± 0.3 Å | 14.4 ± 0.3 Å | 18.8 ± 0.2 Å | 13.6 ± 0.4 Å |
Table 5.
The average distances between the side chain COM of D309, L310, and K312 in Gα1 or K307, D310, and L311 in Gα2 and side chain COM R233 in ICL3 of D2S or D2L during the last 100 ns of the simulations.
Table 5.
The average distances between the side chain COM of D309, L310, and K312 in Gα1 or K307, D310, and L311 in Gα2 and side chain COM R233 in ICL3 of D2S or D2L during the last 100 ns of the simulations.
| Complex | Distance |
|---|
| D309 | L310 | K312 |
|---|
| L1 | 12.1 ±0.2 Å | 13.6 ±0.3 Å | 11.1 ±0.2 Å |
| S1 | 13.1 ±0.3 Å | 11.8 ±0.3 Å | 10.8 ±0.2 Å |
| | K307 | D310 | L311 |
| L2 | 7.5 ±0.3 Å | 5.9 ±0.3 Å | 8.4 ±0.2 Å |
| S2 | 10.0 ±0.4 Å | 8.1 ±0.2 Å | 10.6 ±0.2 Å |
Table 6.
Values of the overlap of the covariance matrices.
Table 6.
Values of the overlap of the covariance matrices.
| Complex | L1 | L2 | S1 | S2 |
|---|
| L1 | - | 0.758 | 0.731 | 0.698 |
| L2 | 0.758 | - | 0.659 | 0.686 |
| S1 | 0.731 | 0.659 | - | 0.761 |
| S2 | 0.698 | 0.686 | 0.761 | - |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}