Crosstalk between NLRP12 and JNK during Hepatocellular Carcinoma


Department of Pathology, UT Southwestern Medical Center, Dallas, TX 75390, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(2), 496; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020496
Submission received: 19 December 2019
/
Revised: 31 December 2019
/
Accepted: 8 January 2020
/
Published: 13 January 2020
(This article belongs to the Special Issue c-Jun N-terminal kinases: New Insights for Old Cell Signaling Pathways)
{kind=link}
Abstract
:Hepatocellular carcinoma (HCC), a leading cause of cancer-related death, is initiated and promoted by chronic inflammation. Inflammatory mediators are transcriptionally regulated by several inflammatory signaling pathways, including nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK). cJun N-terminal kinase (JNK), a member of the MAPK family, plays a central role in HCC pathogenesis. Pathogen-associated molecular patterns (PAMPs) activate JNK and other MAPK upon recognition by toll-like receptors (TLRs). Apart from TLRs, PAMPs are sensed by several other pattern recognition receptors, including cytosolic NOD-like receptors (NLRs). In a recent study, we demonstrated that the NLR member NLRP12 plays a critical role in suppressing HCC via negative regulation of the JNK pathway. This article briefly reviews the crosstalk between NLRP12 and JNK that occurs during HCC.
1. Introduction
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer [1,2]. With only a 18% survival rate in 5 years, HCC ranked as the second highest death-causing cancer, after pancreatic cancer [3]. Mutations in various oncogenes, including CTCNB1, WNT, AXIN, TP53, CCND1, and CDKN2A are commonly found in HCC [4,5]. The primary cause for sporadic mutations and neoplastic transitions of parenchymal cells is chronic injury and inflammation induced by hepatitis B and C virus (HBV and HCV) infections, chronic alcohol consumption, and drug toxicity [2,6]. Irrespective of etiologies, inflammation plays a central role in the induction and promotion of HCC. For example, inflammatory mediators cause DNA damage, induce mutations, trigger cell death, and promote proliferation of neoplastic hepatocytes [7,8].
The major pathways regulating inflammation in the liver include nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) [8,9,10,11,12,13]. These inflammatory pathways are activated by pathogen-associated molecular patterns (PAMPs), danger-associated molecular patterns (DAMPs), cytokines, growth factors, and stress. Among diverse stimuli, PAMPs are the most potent activator of NF-κB and MAPK pathways. Because of its close anatomical connection with the intestine, the liver is constantly exposed to gut microbiota-derived PAMPs, suggesting that PAMPs constitute a critical player in inflammatory responses and HCC pathogenesis as well [14]. Clinical evidence showing increased endotoxins in patients with chronic liver disorders further underscores the link between chronic liver inflammation and gut-derived PAMPs [15,16,17,18,19,20]. PAMPs are sensed by pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLR), AIM2-like receptors (ALR), and several other cytosolic receptors for nucleic acids [21,22]. Involvement of these PRRs in the pathogenesis of HCC is increasingly evident [23,24,25,26,27]. We recently investigated the role of NLRP12, an NLR member, in HCC pathogenesis [28]. This study demonstrated that Nlrp12-/- mice are highly susceptible to hepatocellular carcinoma [28]. Increased HCC pathogenesis of Nlrp12-/- mice was associated with increased activation of the c-Jun N-terminal kinase (JNK) signaling pathway. Here, we will review our findings focusing on the link between NLRP12 and JNK in the regulation of HCC.
2. JNK in HCC Pathogenesis
The Ras/Raf/MAPK pathway is one of the major pathways associated with HCC [11,29]. Three major MAPK pathways are ERK, P38, and JNK. JNK is activated by dual phosphorylation of the tripeptide motif (Thr-Pro-Tyr) by upstream MAPK kinases MKK4 and MKK7, which are activated by a cascade of kinase signaling [30]. PAMPs activate the JNK pathway following recognition by TLRs. In addition, cytokines (e.g., TNFα, IL-1α), growth factors, mitochondrial stress, and environmental stress can activate JNK [30,31]. Activated JNK phosphorylates AP-1 family transcription factors, such as cJun, cFos, JunB, JunD, ATF2, and non-AP-1 transcription factors, including p53, cMyc, FOXO4, STAT1, and STAT3 [30,32]. These transcription factors regulate a myriad of genes involved in inflammation, proliferation, cell death, and oncogenesis.
Since the JNK pathway plays a central role in maintaining homeostasis in the liver, its dysregulation is associated with inflammatory liver disorders, including steatosis, fibrosis, cirrhosis, and HCC [29,33,34]. JNK induces and activates cJun and cMyc, oncogenic transcription factors that are highly expressed in the HCC tissue [35,36,37,38,39,40]. As such, deletion of cJun in hepatocytes dramatically reduced the number and size of liver tumors induced by diethylnitrosamine (DEN), a hepatotoxin and carcinogen, in mice [41]. Reduced tumor burden in cJun-deficient mice was associated with higher expression of tumor suppressors p53 and p21 [39,41]. Consistently, JNK deficiency or its inhibition abrogates proliferation of tumor cells in the liver [31,34,42]. Compelling evidence suggest that JNK plays an essential role in hepatocyte proliferation and liver regeneration [43,44]. JNK promotes proliferation of hepatocytes via induction of molecules involved in cell cycle progression, such as Cyclin D1 and cMyc, as well as suppression of p53 and p21, which induce apoptosis and inhibit proliferation [39,44,45,46].
Because of its tumor-inducing potential, activation of JNK in the liver needs to be maintained tightly, and such a regulation involves other inflammatory pathways and molecules. Mice deficient in IκB kinase β (IKKβ) in hepatocytes developed increased HCC following the administration of DEN [8]. This finding was unexpected, since transcription factor NF-κB, which is activated by IKKβ, is the major regulator of inflammation. However, further investigation revealed that a higher tumor burden in hepatocyte-specific IKKβ-deficient mice was associated with increased activation of JNK [8]. Similarly, embryonic fibroblasts and fetal hematopoietic cells from p38α-deficient mice showed hyper-proliferation resulting from increased activation of JNK [47,48]. Also, mice deficient in p38α in hepatocytes exhibited increased HCC pathogenesis, which was accompanied by higher activation of JNK [7,48]. All these provide clear evidence that JNK cross-talks with other inflammatory pathways, including NF-κB and p38.
Among three isoforms of JNK (JNK1, JNK2, and JNK3), liver expresses JNK1 and JNK2. However, DEN-induced HCC development was significantly reduced in Jnk1-/- mice, but not Jnk2-/- mice [13,47]. Furthermore, human HCC tissue exhibited increased JNK1 activation, while JNK2 remained similar to normal liver, suggesting that JNK1 is the main player in HCC pathogenesis [13]. In agreement with this, inhibition of JNK with SP600125, a chemical inhibitor, was seen to suppress DEN-induced HCC development [40]. While it is overwhelmingly supported that JNK activation promotes cellular proliferation, animal studies documented an intriguing phenomenon that JNK-mediated HCC pathogenesis is associated with increased apoptosis [31]. Given that proliferation and apoptosis are two opposite biological processes, and tumorigenesis is considered a defect in cell death, how increased apoptosis promotes tumorigenesis is intriguing. These opposing processes in HCC pathogenesis was explained by the fact that hepatocyte death triggers compensatory proliferation of surviving hepatocytes, leading to the induction and promotion of HCC [7]. JNK-mediated hepatocyte death is accompanied by the release of proinflammatory mediators, such as IL-1α, TNFα, and reactive oxygen species (ROS), which stimulate resident macrophages (Kupffer cells), infiltrated macrophages, and dendritic cells to express proinflammatory cytokines, such as IL-6, IL-α, IL-β, and TNFα [7]. These cytokines, along with other growth factors released by myeloid cells, promote the proliferation of neoplastic hepatocytes [10,13,24,49,50,51].
Although most in vitro and in vivo studies depicted JNK as a promotor for HCC, it is surprising that mice double-deficient in Jnk1 and Jnk2 in hepatocytes are susceptible to HCC [52]. In contrast, hematopoietic-specific deletion of Jnk1 and Jnk2 protects animals against HCC [52]. These apparently conflicting observations were explained by the fact that the complete absence of JNK leads to hepatocyte death, promoting hyperactivation of JNK in Kupffer and other myeloid cells and, in turn, induction of inflammatory molecules, which trigger compensatory proliferation of transformed hepatocytes [52]. Considering the crosstalk between JNK and other inflammatory signaling pathways, it is intriguing whether NF-κB, ERK, and P38 become hyperactivated in hepatocytes deficient in both JNK1 and JNK2. Such hyperactivation of other inflammatory pathways in the absence of JNK may promote HCC.
3. NOD-Like Receptors
The innate immune response against pathogens is initiated with germline-encoded PPRs [53,54,55]. Among several families of PRRs, TLRs and NLRs are most discussed [56]. While TLRs are located on the cell surface and the endosomal compartment, NLRs are present in the cytosol [55,57]. NLRs recognize both PAMPs and DAPMs, such as ATP, uric acid, hyaluronan, and other host-derived cellular and metabolic products [58]. NLR-mediated recognition of PAMPs and DAMPs results in the activation of downstream signal transduction pathways, leading to either induction or suppression of inflammatory responses [58,59,60]. Structurally, NLRs share the typical tripartite structural domain organization, including an N-terminal effector domain, a central nucleotide-binding NATCH domain, and a C-terminal LLR domain responsible for the recognition of pattern molecules [22,59]. At least 22 NLR members have been identified in humans, and they are divided into four subfamilies based on their N-terminal effector domains, including: (1) NLRA—contains an acidic transactivation domain; (2) NLRB—having a baculoviral inhibitory repeat-like (BIR) domain; (3) NLRC—possessing caspase activation and recruitment domain (CARD); and (4) NLRP—pyrin domain containing NLRs. Interestingly, in contrast to TLRs, NLRs play diverse roles, including activation of NF-κB and MAPK pathways, formation of the inflammasome, suppression of inflammatory signaling pathways, and transcriptional regulation of genes [22,61]. Notably, NLRs often exhibit a cell type-specific function, and several NLRs share overlapping functions. Because of their key roles in innate immunity and inflammation, NLRs have been implicated in infectious, auto-immune, and inflammatory disorders including cancer [61,62,63,64].
4. NLRP12 and Inflammatory Disorders
NLRP12 belongs to the NLRP subfamily of NLRs. Like other NLRP family proteins, NLRP12 is composed of an N-terminal pyrin domain (PYD), central nucleotide binding domain (NBD), and a C-terminal leucine-rich repeat (LRR). NLRP12 is abundantly expressed in myeloid cell lineage, including macrophages, dendritic cells, monocytes, and neutrophils [65,66]. An initial study found that NLRP12 interacts with ASC in vitro, predicting its role in inflammasome activation [67]. However, the role of NLRP12 in the activation of the inflammasome has not been observed in diverse pathophysiological contexts, except infection caused by Yersinia pestis [68]. Most other studies described NLRP12 as a negative regulator of inflammatory responses [66,69,70,71]. Missense mutations in NLRP12 have been identified in patients with atopic dermatitis and periodic fever syndrome [72,73,74]. Mice deficient in Nlrp12 are highly susceptible to chemically induced colitis and colorectal tumorigenesis [66,69]. Increased inflammation and tumorigenesis of Nlrp12-/- mice are associated with higher inflammatory responses and activation of NF-κB and ERK pathways [66,69]. Supporting this in vivo observation, bone marrow-derived macrophages and dendritic cells of Nlrp12-/- mice are hyper-responsive to TLR ligands, such as LPS, Pam3, and Poly I:C [66,69]. Cytokines, chemokines, and other inflammatory mediators contribute to host defense against bacterial infection. Consistently, higher inflammatory responses in Nlrp12-/- mice during Salmonella Typhimurium infection helped resolve the infection [70]. A recent study demonstrated that NLRP12 dampens antiviral immune responses; however, such a regulation involved the RIG-I pathway but not NF-κB and MAPK [75], suggesting that NLRP12 may regulate inflammatory response and host immunity in multiple ways.
While most studies found NLRP12 to inhibit NF-κB and MAPK pathways in myeloid cells, increasing evidence suggests that NLRP12 regulates these pathways in other cell types as well. T cells of Nlrp12-/- mice are highly responsive to antigen immunization. Thus, Nlrp12-/- mice develop atypical experimental autoimmune encephalomyelitis (EAE) due to increased production of Th2 cytokine IL-4 into the central nervous system [71]. Transfer of naïve Nlrp12-/- T cells into immunodeficient Rag-/- mice also elicited exacerbated colitis [71]. The role of NLRP12 in attenuating these inflammatory disorders was explained by the fact that NLRP12 downregulates NF-κB and ERK activation in T cells [71]. Deficiency of NLRP12 also promotes proliferation of osteocytes, hepatocytes, and microglia [28,76,77]. In the intestine, NLRP12-mediated regulation of inflammatory responses may shape gut microbiota composition [78,79]. Recent studies suggest that altered gut microbiota predisposes susceptibility to colitis and obesity in Nlrp12-/- mice [78,79]. While these observations are interesting, gut microbiota composition can be modulated by other factors, including the animal facility environment, geographical location of the laboratory, mouse handling practices, diet, and so forth. Thus, gut microbiota-dependent disease phenotypes of Nlrp12-/- mice should be validated by independent studies from other laboratories.
5. NLRP12 Suppresses Hepatocellular Carcinoma
Given that chronic inflammation is a major driver for HCC and NLRP12 negatively regulates inflammatory responses, it is intriguing whether NLRP12 plays any role in HCC. In a recent study, we investigated the role of NLRP12 in HCC using mouse models in which tumors were induced by DEN or DEN plus carbon tetrachloride. In both experimental settings, Nlrp12-/- mice developed significantly higher numbers of and larger tumors compared to WT mice [28]. This observation suggests that NLRP12 plays a protective role against HCC. The cancer genomics database suggests that NLRP12 is altered in about 2% of HCC patients [28]. Although NLRP12 is not a major cancer suppressor gene, its expression and activation status may regulate HCC pathogenesis.
Increased HCC pathology in Nlrp12-/- mice was associated with higher expressions of the HCC marker Afp, inflammatory cytokines, and chemokines, including IL-6, TNFα, Cxcl1, Cxcl2, and Ccl2, protooncogene cJun, cMyc, and Cyclin D1, and reduced expression of p21 [28]. IL-6 and TNFα are critical players in HCC pathogenesis with their functions in cellular proliferation and cell death [7,8,80,81,82]. These two cytokines were found to be elevated in the liver of HBV infected patients, further supporting their association in HCC pathogenesis [83,84]. In addition to these pro-inflammatory cytokines, chemokines that recruit macrophages and other myeloid cells in the tumor microenvironment play important roles in HCC [7,8,82]. Inflammatory mediators produced by Kupffer cells and other immune cells contribute to the development of steatosis, fibrosis, and cirrhosis in the liver [6,85,86]. Higher steatosis and fibrosis in DEN-treated Nlrp12-/- mouse livers, therefore, reflect an overall hyperinflammatory response [28]. Notably, inflammatory and proliferative molecules were not dysregulated in healthy Nlrp12-/- livers [28], indicating that NLRP12 suppresses those tumor-promoting mediators in the context of liver injury.
6. NLRP12 Negatively Regulates JNK Activation in the Hepatocyte
As discussed above, inflammatory signaling pathways, including NF-κB, ERK, P38, JNK, and STAT3, regulate inflammatory responses and tumorigenesis. Since NLRP12 has been shown to downregulate the activation of NF-κB and ERK, these pathways were expected to be hyperactivated in Nlrp12-/- livers. Interestingly, Nlrp12-/- HCC showed higher JNK activation, but not NF-κB and ERK [28]. This observation suggests that NLRP12 regulates different inflammatory pathways in a cell type-specific manner. Indeed, higher activation of JNK was seen only in Nlrp12-/- hepatocytes; there was no major difference in JNK activation in Kupffer cells and hepatic stellate cells isolated from wild-type and Nlrp12-/- mouse HCC [28]. The hepatocyte intrinsic function of NLRP12 in regulating JNK was confirmed by in vitro biochemical assays. Primary hepatocytes from healthy Nlrp12-/- mice exhibited increased activation of JNK and expression of cJun, cMyc, and Ccnd1 upon stimulation with LPS and other TLR ligands, e.g., Pam3 and PGN [28]. Knockdown of NLRP12 in the human HCC cell-line HepG2 provided similar results [28]. Corroborating with these data, JNK activation and expression of JNK downstream molecules were markedly reduced upon overexpression of NLRP12 in HepG2 cells [28]. Overall, these studies strongly imply that NLRP12 is a critical negative regulator of the JNK pathway in the liver.
7. NLRP12 Regulates Hepatocyte Proliferation via JNK
A unique feature of the liver is its regeneration capacity. Liver that is subjected to partial hepatectomy regains its complete mass within 7 to 10 days [39,44]. Liver injury or cytotoxicity leads to rapid proliferation of surviving hepatocytes to maintain liver homeostasis [7,87,88]. While compensatory proliferation of hepatocytes is essential for liver homeostasis, this process also triggers HCC during chronic inflammation and injury, or in the presence of mutagens. In agreement with this paradigm, we observed increased apoptosis as well as proliferation in Nlrp12-/- liver upon DEN-induced HCC induction [28]. Notably, healthy WT and Nlrp12-/- livers did not show any difference in cell death and proliferation [28]. The role of NLRP12 in hepatic cell death was examined in vitro using primary hepatocytes from WT and Nlrp12-/- mice. The proliferation rate of hepatocytes was significantly higher in Nlrp12-/- mice, although no major difference in cell death was observed between WT and Nlrp12-/- hepatocytes [28]. Notably, hepatocyte proliferation was increased in the presence of LPS, further supporting the concept that inflammatory pathways enhance proliferation of hepatocytes [28]. Thus, higher apoptotic death of hepatocytes following administration of DEN in the Nlrp12-/- liver was due to extrinsic factors, such as higher abundance of reactive oxygen and nitrogen species, cytotoxic cytokines (e.g., TNFα), and proapoptotic molecules in the tumor microenvironment. Higher proliferation of Nlrp12-/- hepatocytes was associated with increased expression of pro-proliferative molecules, including cMyc, cJun, and Ccnd1. The JNK inhibitor dramatically reduced proliferation rate of hepatocytes and expression of cJun, cMyc, and Ccnd1, suggesting the key role of JNK in the hyperproliferative nature of Nlrp12-/- hepatocytes [28].
8. NLRP12 Suppresses Gut Microbiota-Mediated Activation of JNK in the Liver
JNK can be activated by PAMPs, as well as host-derived factors, such as TNFα, ROS, and stress [30,82,89,90]. However, microbial pattern molecules, such as LPS, MDP, and PGN, are more potent as activators of NF-κB and MAPK pathways compared to endogenous stimuli. Increasing evidence suggests that gut-derived microbial products play pivotal roles in HCC pathogenesis [14]. Mice raised in germ-free (GF) conditions do not develop DEN-induced HCC [24]. In agreement with this, mice deficient in TLR4 which sense LPS are less susceptible to HCC [23,24]. Depletion of gut microbiota with antibiotics also dramatically reduces tumor growth [24]. It appears that higher JNK activation in Nlrp12-/- tumors is due to increased activation of TLR pathways by LPS or other PAMPs. Consistently, Nlrp12-/- mice treated with antibiotics did not develop visible tumors [28]. In fact, microbial products are circulated to the liver through the portal vein after crossing the intestinal epithelial barrier. Therefore, our data further strengthens the growing body of evidence that gut-derived microbial products activate JNK and other inflammatory pathways in the liver, promoting HCC (Figure 1).
9. Concluding Remarks
Gut microbiota-derived PAMPs play a central role in HCC pathogenesis. Both parenchymal and non-parenchymal cells express TLRs, and therefore, may respond to PAMPs, leading to the activation of JNK and other inflammatory pathways. Therefore, suppression of this gut-liver axis of JNK pathway may constitute an effective means for inhibiting HCC pathogenesis. Our recent study points to NLRP12 as an important molecular brake of this signaling axis and HCC pathogenesis. It is interesting that NLRP12 regulates JNK but not NF-κB and ERK in hepatocytes. Whether NLRP12 negatively regulates the JNK pathway in a manner similar to its inhibition of NF-κB and ERK in myeloid cells remains unknown. Previous studies have documented that NLRP12 interacts with IRAK1 to suppress canonical NF-κB, and NIK and TRAF3 to inhibit non-canonical NF-κB in myeloid cells [69,91,92]. It would be interesting to see if NLRP12 interacts with these or other signaling adapters to attenuate JNK activation. Identification of kinases or interacting partners involved in NLRP12-mediated inhibition of the JNK pathway may facilitate finding new therapeutics for HCC.
Author Contributions
S.K. wrote the initial manuscript draft. H.Z. wrote, reviewed, and edited the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Cancer Prevention and Research Institute of Texas (CPRIT) Individual Investigator Award (RP160169), and UT Southwestern funding given to H.Z., S.K. and H.Z. are supported by Career In Immunology Fellowship, awarded by The American Association of Immunologists (AAI).
Acknowledgments
We would like to thank the UT Southwestern Animal Resource Center (ARC) and Simmons Comprehensive Cancer Center for supporting the original research discussed here.
Conflicts of Interest
Authors declared no conflict of interest to disclose.
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J. Natl. Cancer Inst. 2017, 109, djx030. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Andersen, J.B.; Thorgeirsson, S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 2015, 15, 653–667. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Karin, M. NF-kappaB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, W.N.; Chen, X.; Peng, X.D.; Jeon, S.M.; Birnbaum, M.J.; Guzman, G.; Hay, N. Spontaneous Hepatocellular Carcinoma after the Combined Deletion of Akt Isoforms. Cancer Cell 2016, 29, 523–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, L.; Zatloukal, K.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Investig. 2008, 118, 3943–3953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, G.; Kremer, M.; Hines, I.N. Contribution of gut bacteria to liver pathobiology. Gastroenterol. Res. Pract. 2010, 2010, 453563. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Brauner, B.; Bode, J.C.; Bode, C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: Reevaluation with an improved chromogenic assay. J. Hepatol. 1991, 12, 162–169. [Google Scholar] [CrossRef]
- Rutenburg, A.M.; Sonnenblick, E.; Koven, I.; Aprahamian, H.A.; Reiner, L.; Fine, J. The role of intestinal bacteria in the development of dietary cirrhosis in rats. J. Exp. Med. 1957, 106, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirera, I.; Bauer, T.M.; Navasa, M.; Vila, J.; Grande, L.; Taura, P.; Fuster, J.; Garcia-Valdecasas, J.C.; Lacy, A.; Suarez, M.J.; et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J. Hepatol. 2001, 34, 32–37. [Google Scholar] [CrossRef]
- Yoneyama, K.; Miyagishi, K.; Kiuchi, Y.; Shibata, M.; Mitamura, K. Risk factors for infections in cirrhotic patients with and without hepatocellular carcinoma. J. Gastroenterol. 2002, 37, 1028–1034. [Google Scholar] [CrossRef]
- Campillo, B.; Pernet, P.; Bories, P.N.; Richardet, J.P.; Devanlay, M.; Aussel, C. Intestinal permeability in liver cirrhosis: Relationship with severe septic complications. Eur. J. Gastroenterol. Hepatol. 1999, 11, 755–759. [Google Scholar] [CrossRef]
- Pascual, S.; Such, J.; Esteban, A.; Zapater, P.; Casellas, J.A.; Aparicio, J.R.; Girona, E.; Gutierrez, A.; Carnices, F.; Palazon, J.M.; et al. Intestinal permeability is increased in patients with advanced cirrhosis. Hepatogastroenterology 2003, 50, 1482–1486. [Google Scholar]
- Khan, S.; Godfrey, V.; Zaki, M.H. Cytosolic Nucleic Acid Sensors in Inflammatory and Autoimmune Disorders. Int. Rev. Cell Mol. Biol. 2019, 344, 215–253. [Google Scholar] [PubMed]
- Ramachandran, R.A.; Lupfer, C.; Zaki, H. The Inflammasome: Regulation of Nitric Oxide and Antimicrobial Host Defence. Adv. Microb. Physiol. 2018, 72, 65–115. [Google Scholar] [PubMed]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machida, K.; Tsukamoto, H.; Mkrtchyan, H.; Duan, L.; Dynnyk, A.; Liu, H.M.; Asahina, K.; Govindarajan, S.; Ray, R.; Ou, J.H.; et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc. Natl. Acad. Sci. USA 2009, 106, 1548–1553. [Google Scholar] [CrossRef] [Green Version]
- Paik, Y.H.; Schwabe, R.F.; Bataller, R.; Russo, M.P.; Jobin, C.; Brenner, D.A. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef]
- Hou, J.; Zhou, Y.; Zheng, Y.; Fan, J.; Zhou, W.; Ng, I.O.; Sun, H.; Qin, L.; Qiu, S.; Lee, J.M.; et al. Hepatic RIG-I predicts survival and interferon-alpha therapeutic response in hepatocellular carcinoma. Cancer Cell 2014, 25, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Udden, S.N.; Kwak, Y.T.; Godfrey, V.; Khan, M.A.W.; Khan, S.; Loof, N.; Peng, L.; Zhu, H.; Zaki, H. NLRP12 suppresses hepatocellular carcinoma via downregulation of cJun N-terminal kinase activation in the hepatocyte. Elife 2019, 8, e40396. [Google Scholar] [CrossRef]
- Nakagawa, H.; Maeda, S. Molecular mechanisms of liver injury and hepatocarcinogenesis: Focusing on the role of stress-activated MAPK. Pathol. Res. Int. 2012, 2012, 172894. [Google Scholar] [CrossRef] [Green Version]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef] [Green Version]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Derijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, S.; Bubici, C.; Zazzeroni, F.; Franzoso, G. Mechanisms of liver disease: Cross-talk between the NF-kappaB and JNK pathways. Biol. Chem. 2009, 390, 965–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, R.F. Cell death in the liver-all roads lead to JNK. Gastroenterology 2006, 131, 314–316. [Google Scholar] [CrossRef]
- Lin, C.P.; Liu, C.R.; Lee, C.N.; Chan, T.S.; Liu, H.E. Targeting c-Myc as a novel approach for hepatocellular carcinoma. World J. Hepatol. 2010, 2, 16–20. [Google Scholar] [CrossRef]
- Dang, C.V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Alarcon-Vargas, D.; Ronai, Z. c-Jun-NH2 kinase (JNK) contributes to the regulation of c-Myc protein stability. J. Biol. Chem. 2004, 279, 5008–5016. [Google Scholar] [CrossRef] [Green Version]
- Kawate, S.; Fukusato, T.; Ohwada, S.; Watanuki, A.; Morishita, Y. Amplification of c-myc in hepatocellular carcinoma: Correlation with clinicopathologic features, proliferative activity and p53 overexpression. Oncology 1999, 57, 157–163. [Google Scholar] [CrossRef]
- Stepniak, E.; Ricci, R.; Eferl, R.; Sumara, G.; Sumara, I.; Rath, M.; Hui, L.; Wagner, E.F. c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev. 2006, 20, 2306–2314. [Google Scholar] [CrossRef] [Green Version]
- Nagata, H.; Hatano, E.; Tada, M.; Murata, M.; Kitamura, K.; Asechi, H.; Narita, M.; Yanagida, A.; Tamaki, N.; Yagi, S.; et al. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor- suppression in rat hepatocellular carcinoma. Hepatology 2009, 49, 1944–1953. [Google Scholar] [CrossRef]
- Eferl, R.; Ricci, R.; Kenner, L.; Zenz, R.; David, J.P.; Rath, M.; Wagner, E.F. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell 2003, 112, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Sugioka, Y.; Watanabe, T.; Inagaki, Y.; Kushida, M.; Niioka, M.; Endo, H.; Higashiyama, R.; Okazaki, I. c-Jun NH2-terminal kinase pathway is involved in constitutive matrix metalloproteinase-1 expression in a hepatocellular carcinoma-derived cell line. Int. J. Cancer 2004, 109, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Westwick, J.K.; Weitzel, C.; Leffert, H.L.; Brenner, D.A. Activation of Jun kinase is an early event in hepatic regeneration. J. Clin. Investig. 1995, 95, 803–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, R.F.; Bradham, C.A.; Uehara, T.; Hatano, E.; Bennett, B.L.; Schoonhoven, R.; Brenner, D.A. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology 2003, 37, 824–832. [Google Scholar] [CrossRef]
- Kuntzen, C.; Sonuc, N.; De Toni, E.N.; Opelz, C.; Mucha, S.R.; Gerbes, A.L.; Eichhorst, S.T. Inhibition of c-Jun-N-terminal-kinase sensitizes tumor cells to CD95-induced apoptosis and induces G2/M cell cycle arrest. Cancer Res. 2005, 65, 6780–6788. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, M.; Kolbus, A.; Piu, F.; Szabowski, A.; Mohle-Steinlein, U.; Tian, J.; Karin, M.; Angel, P.; Wagner, E.F. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999, 13, 607–619. [Google Scholar] [CrossRef]
- Sakurai, T.; Maeda, S.; Chang, L.; Karin, M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA 2006, 103, 10544–10551. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Bakiri, L.; Mairhorfer, A.; Schweifer, N.; Haslinger, C.; Kenner, L.; Komnenovic, V.; Scheuch, H.; Beug, H.; Wagner, E.F. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat. Genet. 2007, 39, 741–749. [Google Scholar] [CrossRef]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Seki, E.; Brenner, D.A. Toll-like receptor signaling in the liver. Gastroenterology 2006, 130, 1886–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, M.; Garlick, D.S.; Greiner, D.L.; Davis, R.J. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011, 25, 634–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, G.M. A calculated response: Control of inflammation by the innate immune system. J. Clin. Investig. 2008, 118, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Wolf, N.; Lavelle, E.C. Innate Immune Receptors. Methods Mol. Biol. 2016, 1417, 1–43. [Google Scholar]
- Motta, V.; Soares, F.; Sun, T.; Philpott, D.J. NOD-like receptors: Versatile cytosolic sentinels. Physiol. Rev. 2015, 95, 149–178. [Google Scholar] [CrossRef] [Green Version]
- Kanneganti, T.D.; Lamkanfi, M.; Nunez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007, 27, 549–559. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes: Guardians of cytosolic sanctity. Immunol. Rev. 2009, 227, 95–105. [Google Scholar] [CrossRef]
- Allen, I.C. Non-Inflammasome Forming NLRs in Inflammation and Tumorigenesis. Front. Immunol. 2014, 5, 169. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Yeretssian, G. NOD-Like Receptors: Master Regulators of Inflammation and Cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Kinio, A.; Saleh, M. Functions of NOD-Like Receptors in Human Diseases. Front. Immunol. 2013, 4, 333. [Google Scholar] [CrossRef] [Green Version]
- Zaki, M.H.; Lamkanfi, M.; Kanneganti, T.D. Inflammasomes and Intestinal Tumorigenesis. Drug Discov. Today Dis. Mech. 2011, 8, e71–e78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, J.C.; Lich, J.D.; Ye, Z.; Allen, I.C.; Gris, D.; Wilson, J.E.; Schneider, M.; Roney, K.E.; O’Connor, B.P.; Moore, C.B.; et al. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J. Immunol. 2010, 185, 4515–4519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaki, M.H.; Vogel, P.; Malireddi, R.K.; Body-Malapel, M.; Anand, P.K.; Bertin, J.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 2011, 20, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Manji, G.A.; Grenier, J.M.; Al-Garawi, A.; Merriam, S.; Lora, J.M.; Geddes, B.J.; Briskin, M.; DiStefano, P.S.; Bertin, J. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J. Biol. Chem. 2002, 277, 29874–29880. [Google Scholar] [CrossRef] [Green Version]
- Vladimer, G.I.; Weng, D.; Paquette, S.W.; Vanaja, S.K.; Rathinam, V.A.; Aune, M.H.; Conlon, J.E.; Burbage, J.J.; Proulx, M.K.; Liu, Q.; et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 2012, 37, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.M.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef] [Green Version]
- Zaki, M.H.; Man, S.M.; Vogel, P.; Lamkanfi, M.; Kanneganti, T.D. Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc. Natl. Acad. Sci. USA 2014, 111, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Lukens, J.R.; Gurung, P.; Shaw, P.J.; Barr, M.J.; Zaki, M.H.; Brown, S.A.; Vogel, P.; Chi, H.; Kanneganti, T.D. The NLRP12 Sensor Negatively Regulates Autoinflammatory Disease by Modulating Interleukin-4 Production in T Cells. Immunity 2015, 42, 654–664. [Google Scholar] [CrossRef] [Green Version]
- Borghini, S.; Tassi, S.; Chiesa, S.; Caroli, F.; Carta, S.; Caorsi, R.; Fiore, M.; Delfino, L.; Lasiglie, D.; Ferraris, C.; et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum. 2011, 63, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Jeru, I.; Duquesnoy, P.; Fernandes-Alnemri, T.; Cochet, E.; Yu, J.W.; Lackmy-Port-Lis, M.; Grimprel, E.; Landman-Parker, J.; Hentgen, V.; Marlin, S.; et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc. Natl. Acad. Sci. USA 2008, 105, 1614–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeru, I.; Hentgen, V.; Normand, S.; Duquesnoy, P.; Cochet, E.; Delwail, A.; Grateau, G.; Marlin, S.; Amselem, S.; Lecron, J.C. Role of interleukin-1beta in NLRP12-associated autoinflammatory disorders and resistance to anti-interleukin-1 therapy. Arthritis Rheum. 2011, 63, 2142–2148. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.T.; Chen, L.; Lin, D.S.; Chen, S.Y.; Tsao, Y.P.; Guo, H.; Li, F.J.; Tseng, W.T.; Tam, J.W.; Chao, C.W.; et al. NLRP12 Regulates Anti-viral RIG-I Activation via Interaction with TRIM25. Cell Host Microbe 2019, 25, 602–616 e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauss, J.L.; Zeng, R.; Hickman-Brecks, C.L.; Wilson, J.E.; Ting, J.P.; Novack, D.V. NLRP12 provides a critical checkpoint for osteoclast differentiation. Proc. Natl. Acad. Sci. USA 2015, 112, 10455–10460. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Saxena, S.; Agrawal, I.; Singh, S.; Srinivasan, V.; Arvind, S.; Epari, S.; Paul, S.; Jha, S. Differential Expression Profile of NLRs and AIM2 in Glioma and Implications for NLRP12 in Glioblastoma. Sci. Rep. 2019, 9, 8480. [Google Scholar] [CrossRef]
- Chen, L.; Wilson, J.E.; Koenigsknecht, M.J.; Chou, W.C.; Montgomery, S.A.; Truax, A.D.; Brickey, W.J.; Packey, C.D.; Maharshak, N.; Matsushima, G.K.; et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat. Immunol. 2017, 18, 541–551. [Google Scholar] [CrossRef]
- Truax, A.D.; Chen, L.; Tam, J.W.; Cheng, N.; Guo, H.; Koblansky, A.A.; Chou, W.C.; Wilson, J.E.; Brickey, W.J.; Petrucelli, A.; et al. The Inhibitory Innate Immune Sensor NLRP12 Maintains a Threshold against Obesity by Regulating Gut Microbiota Homeostasis. Cell Host Microbe 2018, 24, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Maeda, S.; Yoshida, H.; Tateishi, R.; Masuzaki, R.; Ohki, T.; Hayakawa, Y.; Kinoshita, H.; Yamakado, M.; Kato, N.; et al. Serum IL-6 levels and the risk for hepatocarcinogenesis in chronic hepatitis C patients: An analysis based on gender differences. Int. J. Cancer 2009, 125, 2264–2269. [Google Scholar] [CrossRef]
- Liu, Z.C.; Ning, F.; Wang, H.F.; Chen, D.Y.; Cai, Y.N.; Sheng, H.Y.; Lash, G.E.; Liu, L.; Du, J. Epidermal growth factor and tumor necrosis factor alpha cooperatively promote the motility of hepatocellular carcinoma cell lines via synergistic induction of fibronectin by NF-kappaB/p65. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2568–2582. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.Y.; Lin, H.; Li, Y.S.; Lee, Y.H.; Chen, H.M.; Cheng, A.L.; Hsu, C.H. High plasma interleukin-6 levels associated with poor prognosis of patients with advanced hepatocellular carcinoma. Jpn. J. Clin. Oncol. 2017, 47, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Yamashita, T.; Terashima, T.; Suda, T.; Okada, H.; Asahina, Y.; Hayashi, T.; Hara, Y.; Nio, K.; Sunagozaka, H.; et al. Serum cytokine profiles predict survival benefits in patients with advanced hepatocellular carcinoma treated with sorafenib: A retrospective cohort study. BMC Cancer 2017, 17, 870. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; DeFrances, M. Liver regeneration. Adv. Biochem. Eng. Biotechnol. 2005, 93, 101–134. [Google Scholar]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [Green Version]
- Behrens, A.; Sibilia, M.; Wagner, E.F. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 1999, 21, 326–329. [Google Scholar] [CrossRef]
- Enomoto, A.; Suzuki, N.; Hirano, K.; Matsumoto, Y.; Morita, A.; Sakai, K.; Koyama, H. Involvement of SAPK/JNK pathway in X-ray-induced rapid cell death of human T-cell leukemia cell line MOLT-4. Cancer Lett. 2000, 155, 137–144. [Google Scholar] [CrossRef]
- Lich, J.D.; Williams, K.L.; Moore, C.B.; Arthur, J.C.; Davis, B.K.; Taxman, D.J.; Ting, J.P. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J. Immunol. 2007, 178, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Lich, J.D.; Moore, C.B.; Duncan, J.A.; Williams, K.L.; Ting, J.P. ATP binding by monarch-1/NLRP12 is critical for its inhibitory function. Mol. Cell Biol. 2008, 28, 1841–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
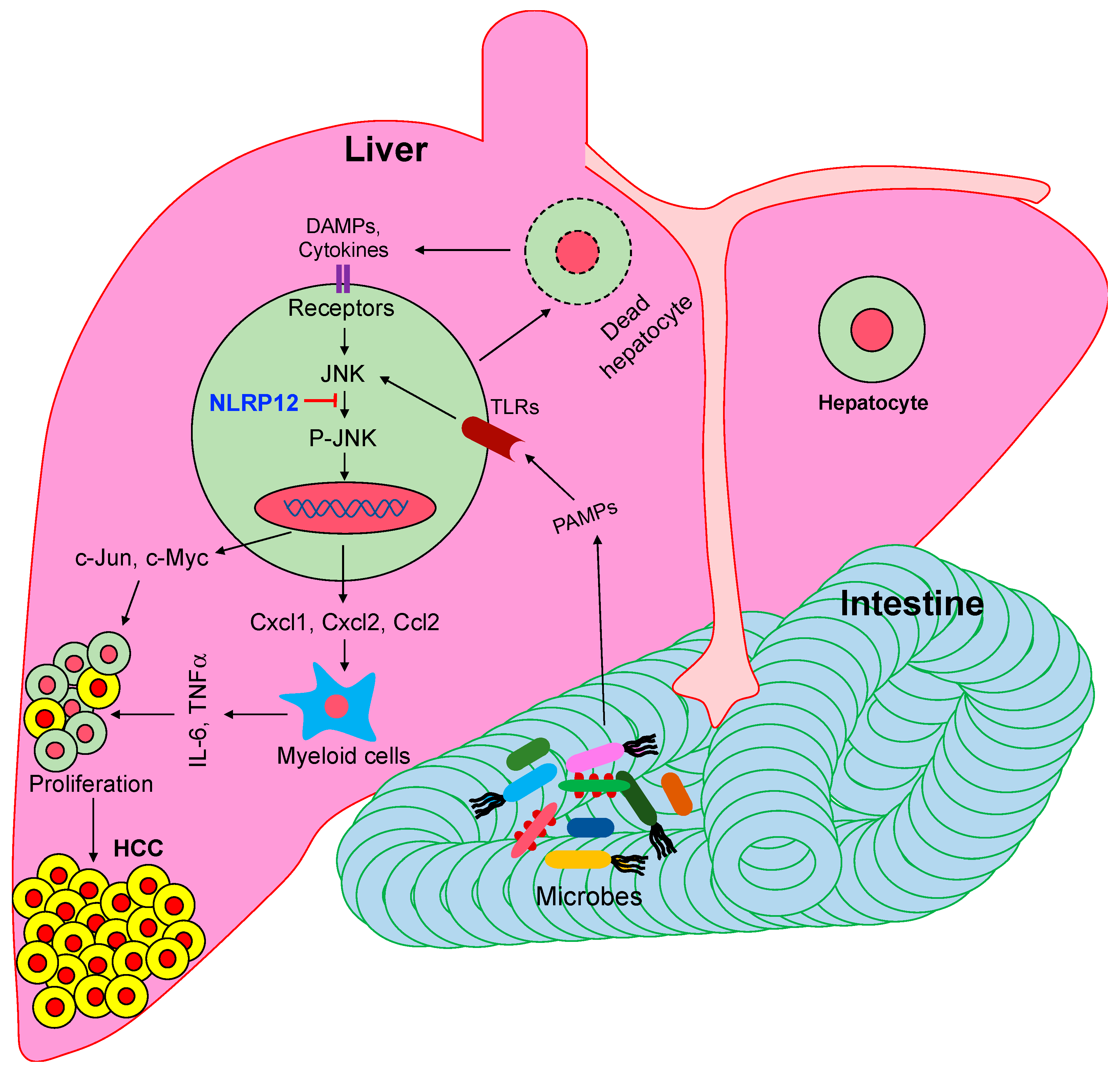
Figure 1.
NLRP12 attenuates hepatocellular carcinoma (HCC) pathogenesis via suppression of pathogen-associated molecular patterns (PAMPs)-mediated activation of c-Jun N-terminal kinase (JNK). Gut-derived microbial pattern molecules transport to the liver where they are sensed by toll-like receptors (TLRs), leading to the activation of the JNK pathway. Cytokines, chemokines, and protooncogenes induced by the activated JNK promote the proliferation of neoplastic cells in the liver. NLRP12 inhibits JNK activation, and thereby suppresses HCC pathogenesis.
Figure 1.
NLRP12 attenuates hepatocellular carcinoma (HCC) pathogenesis via suppression of pathogen-associated molecular patterns (PAMPs)-mediated activation of c-Jun N-terminal kinase (JNK). Gut-derived microbial pattern molecules transport to the liver where they are sensed by toll-like receptors (TLRs), leading to the activation of the JNK pathway. Cytokines, chemokines, and protooncogenes induced by the activated JNK promote the proliferation of neoplastic cells in the liver. NLRP12 inhibits JNK activation, and thereby suppresses HCC pathogenesis.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Khan, S.; Zaki, H. Crosstalk between NLRP12 and JNK during Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 496. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020496
AMA Style
Khan S, Zaki H. Crosstalk between NLRP12 and JNK during Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2020; 21(2):496. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020496
Chicago/Turabian StyleKhan, Shahanshah, and Hasan Zaki. 2020. "Crosstalk between NLRP12 and JNK during Hepatocellular Carcinoma" International Journal of Molecular Sciences 21, no. 2: 496. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020496
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.