Genetic Analyses in Dent Disease and Characterization of CLCN5 Mutations in Kidney Biopsies

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
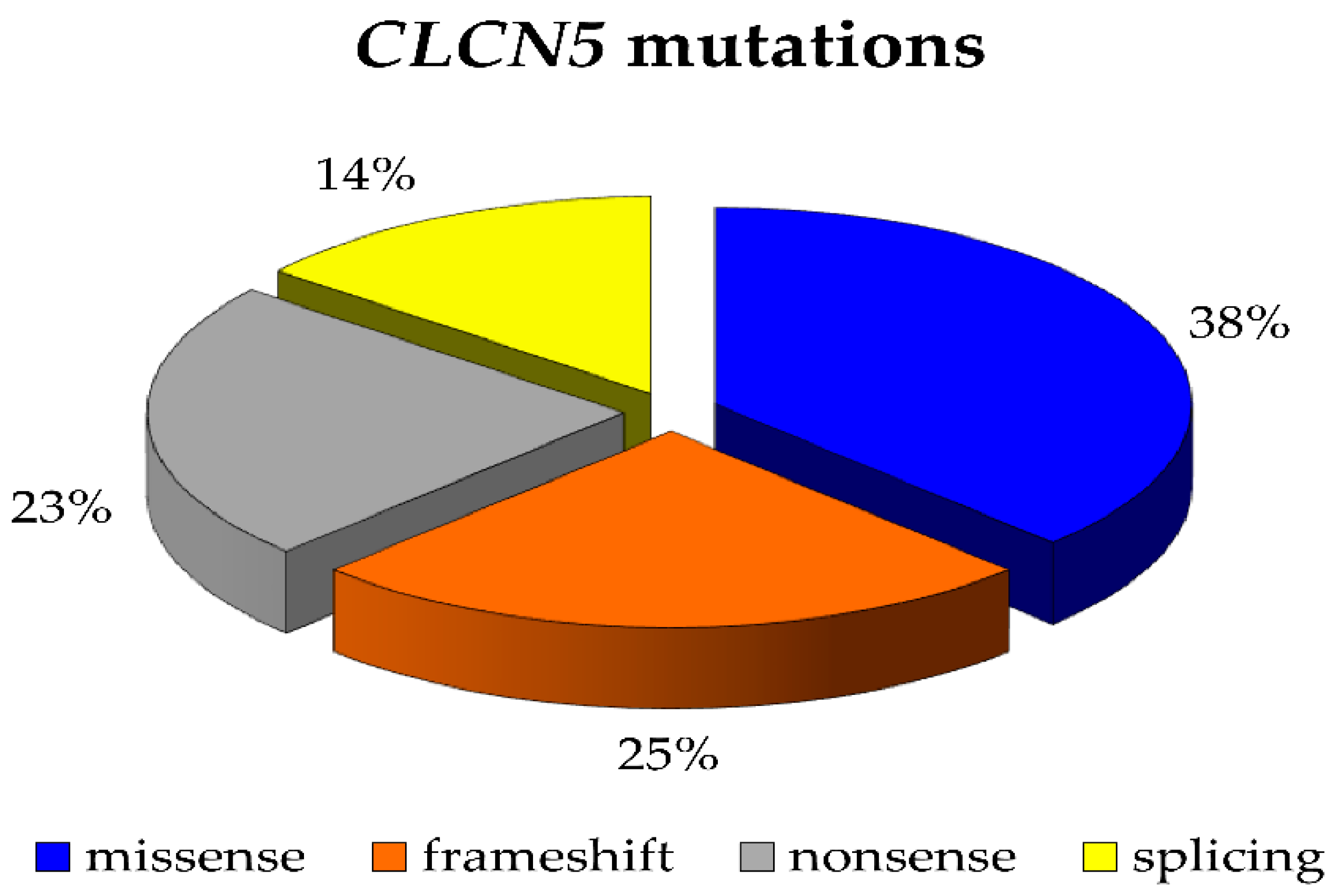
2.1. CLCN5 Gene Mutation Analysis
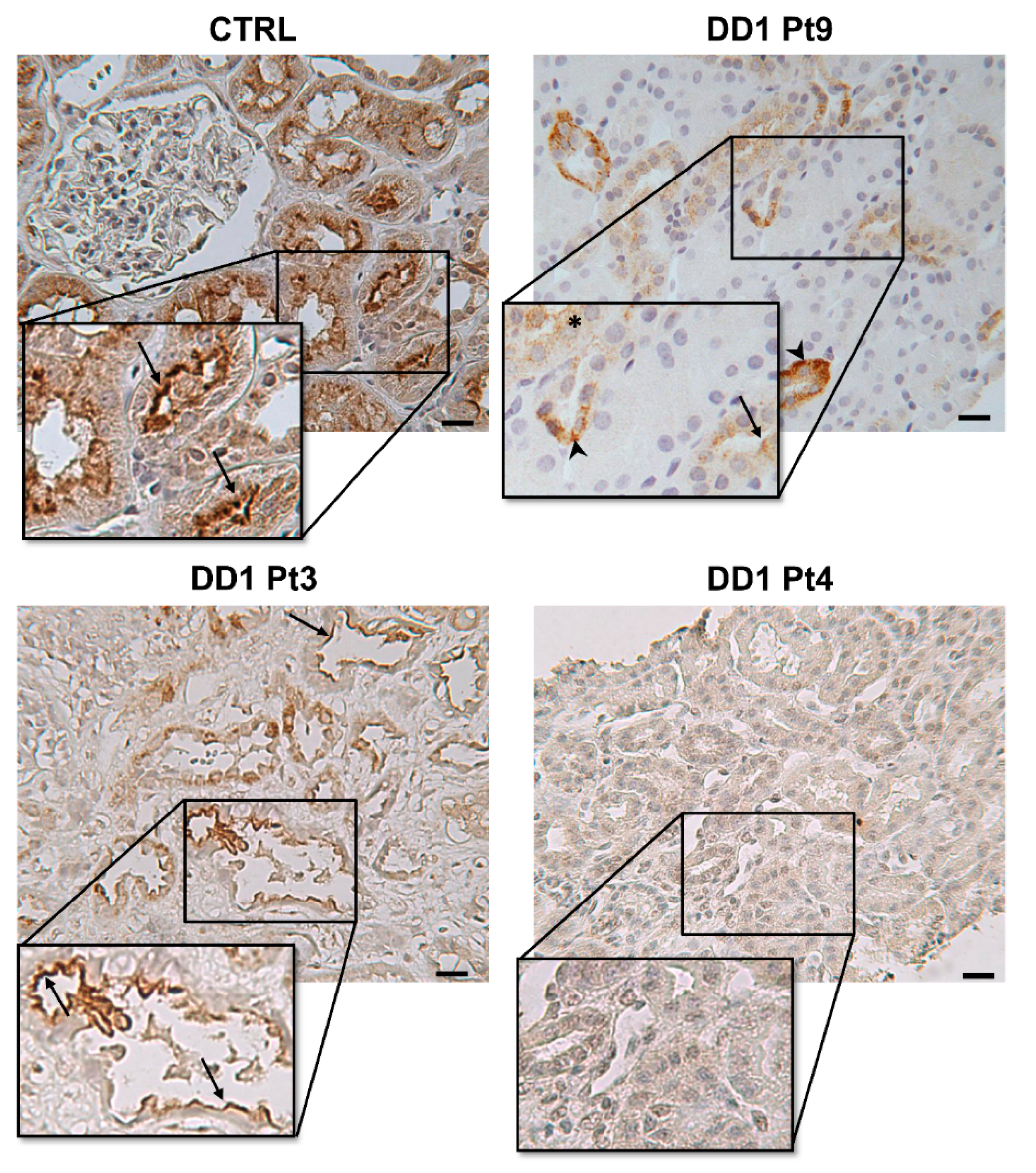
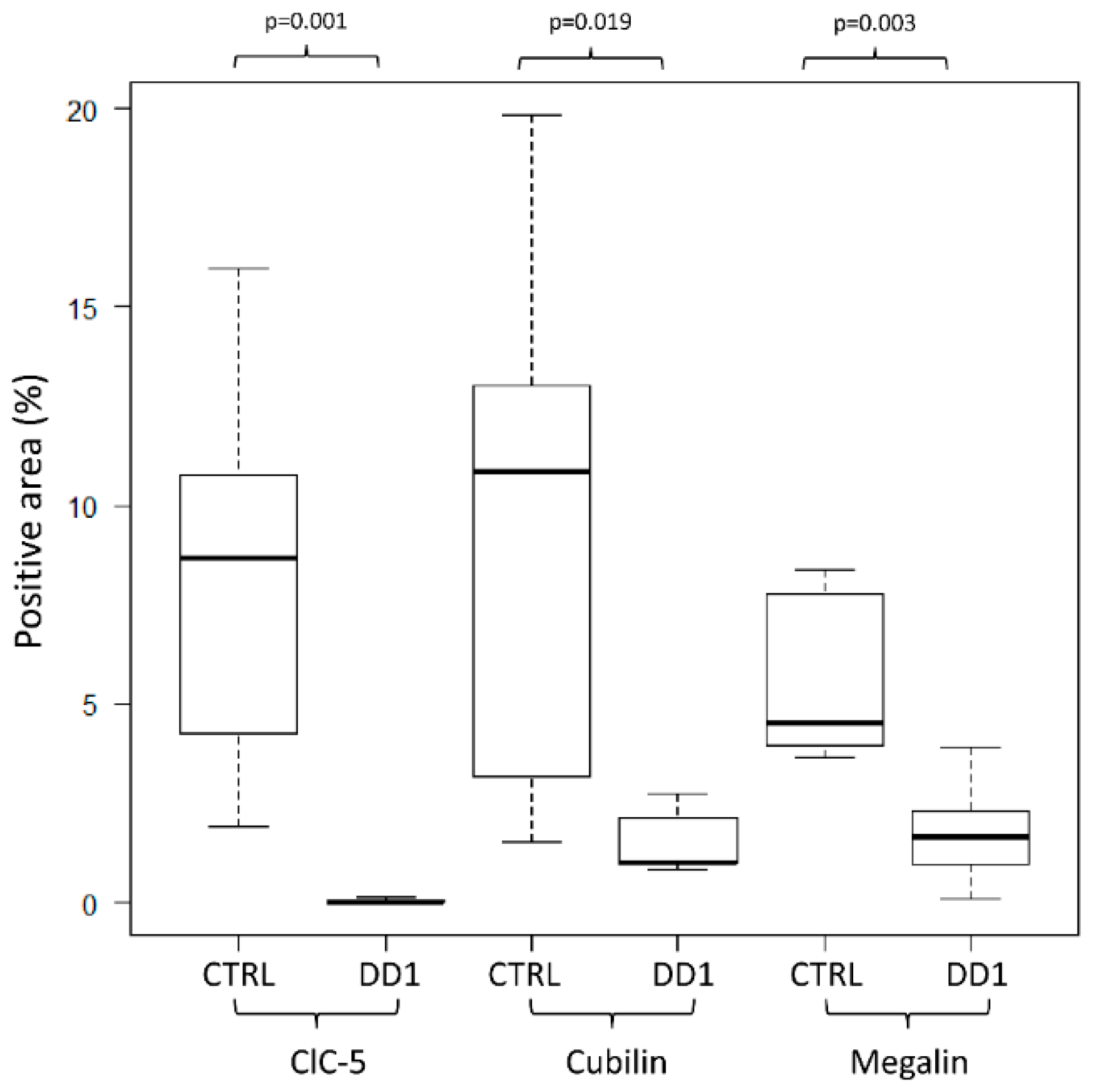
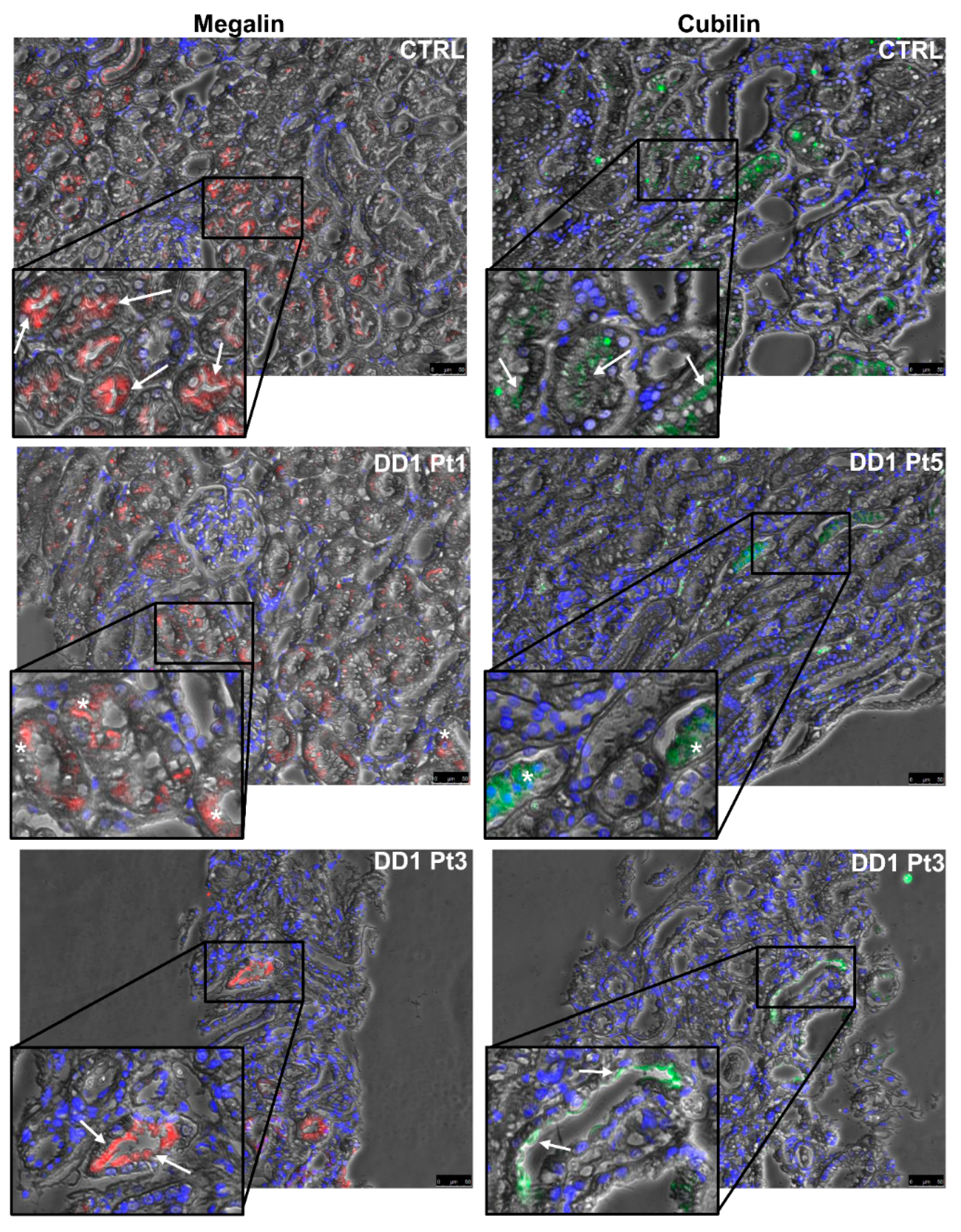
2.2. ClC-5, Megalin, and Cubilin Immunolabeling in DD1 Kidney Biopsies
2.3. Whole Exome Sequencing (WES) Study
3. Discussion
4. Material and Methods
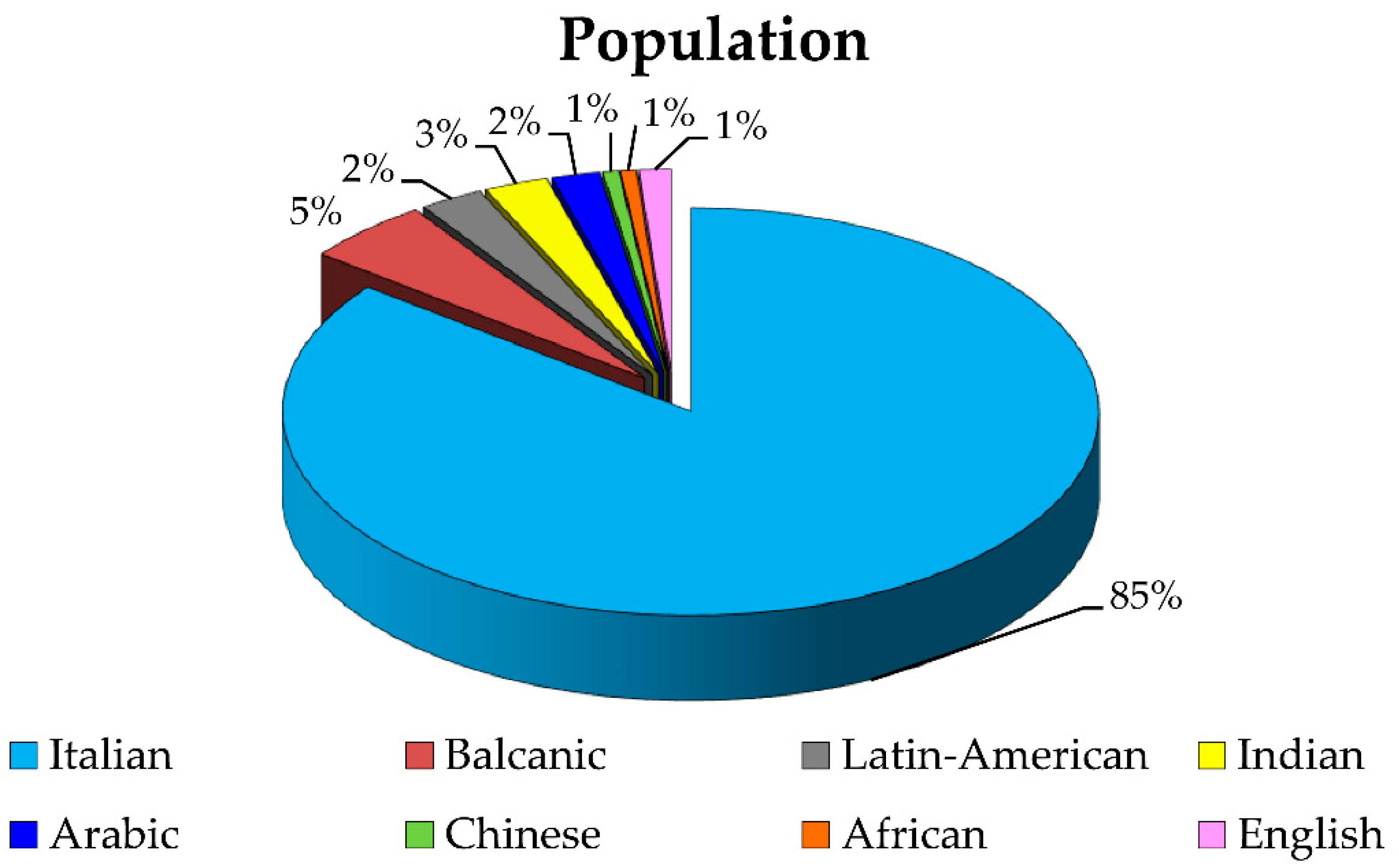
4.1. Patients
4.1.1. DNA Samples
4.1.2. Biopsies
4.2. Sanger Sequencing
4.3. Whole-Exome Sequencing (WES)
4.4. Immunohistochemistry (IHC)
4.5. Immunofluorescence (IF)
4.6. Morphometric Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lloyd, S.E.; Pearce, S.H.; Fisher, S.E.; Steinmeyer, K.; Schwappach, B.; Scheinman, S.J.; Harding, B.; Bolino, A.; Devoto, M.; Goodyer, P.; et al. A common molecular basis for three inherited kidney stone diseases. Nature 1996, 379, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Thakker, R.V. Pathogenesis of Dent’s disease and related syndromes of X-linked nephrolithiasis. Kidney Int. 2000, 57, 787–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrong, O.M.; Norden, A.G.; Feest, T.G. Dent’s disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. Q. J. Med. 1994, 87, 473–493. [Google Scholar]
- Reinhart, S.C.; Norden, A.G.; Lapsley, M.; Thakker, R.V.; Pang, J.; Moses, A.M.; Frymoyer, P.A.; Favus, M.J.; Hoepner, J.A.; Scheinman, S.J. Characterization of carrier females and affected males with X-linked recessive nephrolithiasis. J. Am. Soc. Nephrol. 1995, 5, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, S.E.; Gunther, W.; Pearce, S.H.; Thomson, A.; Bianchi, M.L.; Bosio, M.; Craig, I.W.; Fisher, S.E.; Scheinman, S.J.; Wrong, O.; et al. Characterization of renal chloride channel, CLCN5, mutations in hypercalciuric nephrolithiasis (kidney stones) disorders. Hum. Mol. Genet. 1997, 6, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J.; Günther, W.; Pusch, M.; Schwappach, B. Properties of voltage-gated chloride channels of the ClC gene family. J. Physiol. 1995, 482, 19S–25S. [Google Scholar] [CrossRef] [Green Version]
- Thakker, R.V. Chloride channels cough up. Nat. Genet. 1997, 17, 125. [Google Scholar] [CrossRef]
- Picollo, A.; Pusch, M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 2005, 436, 420–423. [Google Scholar] [CrossRef]
- Mansour-Hendili, L.; Blanchard, A.; Le Pottier, N.; Roncelin, I.; Lourdel, S.; Treard, C.; González, W.; Vergara-Jaque, A.; Morin, G.; Colin, E.; et al. Mutation update of the CLCN5 gene responsible for Dent disease 1. Hum. Mutat. 2015, 36, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Jouret, F.; Igarashi, T.; Gofflot, F.; Wilson, P.D.; Karet, F.E.; Thakker, R.V.; Devuyst, O. Comparative ontogeny; processing; and segmental distribution of the renal chloride channel; ClC-5. Kidney Int. 2004, 65, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Christensen, E.I.; Devuyst, O.; Dom, G.; Nielsen, R.; Van Der Smissen, P.; Verroust, P.; Leruth, M.; Guggino, W.B.; Courtoy, P.J. Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc. Natl. Acad. Sci. USA 2003, 100, 8472–8477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunther, W.; Luchow, A.; Cluzeaud, F.; Vandewalle, A.; Jentsch, T.J. ClC-5, the chloride channel mutated in Dent’s disease; colocalizes with the proton pump in endocytically active kidney cells. Proc. Natl. Acad. Sci. USA 1998, 95, 8075–8080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Antonio, C.; Molinski, S.; Ahmadi, S.; Huan, L.J.; Wellhauser, L.; Bear, C.E. Conformational defects underlie proteasomal degradation of Dent’s disease-causing mutants of ClC-5. Biochem. J. 2013, 452, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourdel, S.; Grand, T.; Burgos, J.; González, W.; Sepúlveda, F.V.; Teulon, J. ClC-5 mutations associated with Dent’s disease: A major role of the dimer interface. Pflug. Arch. 2012, 463, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Grand, T.; L’Hoste, S.; Mordasini, D.; Defontaine, N.; Keck, M.; Pennaforte, T.; Genete, M.; Laghmani, K.; Teulon, J.; Lourdel, S. Heterogeneity in the processing of CLCN5 mutants related to Dent disease. Hum. Mutat. 2011, 32, 476–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.J.; Reed, A.A.; Loh, N.Y.; Thakker, R.V.; Lippiat, J.D. Characterization of Dent’s disease mutations of CLC-5 reveals a correlation between functional and cell biological consequences and protein structure. Am. J. Physiol. Ren. Physiol. 2009, 296, F390–F397. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, M.; Doroszewicz, J.; Seyberth, H.W.; Bökenkamp, A.; Balluch, B.; Nuutinen, M.; Utsch, B.; Waldegger, S. Functional evaluation of Dent’s disease-causing mutations: Implications for ClC-5 channel trafficking and internalization. Hum. Genet. 2005, 117, 228–237. [Google Scholar] [CrossRef]
- Hoopes, R.R., Jr.; Raja, K.M.; Koich, A.; Hueber, P.; Reid, R.; Knohl, S.J.; Scheinman, S.J. Evidence for genetic heterogeneity in Dent’s disease. Kidney Int. 2004, 65, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Hoopes, R.R., Jr.; Shrimpton, A.E.; Knohl, S.J.; Hueber, P.; Reed, A.A.; Christie, P.T.; Igarashi, T.; Lee, P.; Lehman, A.; White, C.; et al. Dent disease with mutations in OCRL1. Am. J. Hum. Genet. 2005, 76, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Hichri, H.; Rendu, J.; Monnier, N.; Coutton, C.; Dorseuil, O.; Poussou, R.V.; Baujat, G.; Blanchard, A.; Nobili, F.; Ranchin, B.; et al. From Lowe syndrome to Dent disease: Correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum. Mutat. 2011, 32, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Shrimpton, A.E.; Hoopes, R.R., Jr.; Knohl, S.J.; Hueber, P.; Reed, A.A.; Christie, P.T.; Igarashi, T.; Lee, P.; Lehman, A.; White, C.; et al. OCRL1 mutations in Dent 2 patients suggest a mechanism for phenotypic variability. Nephron Physiol. 2009, 112, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J. Chloride channels are different. Nature 2002, 415, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, G.; Tran, D.V.; Donini, M.; Navarin, N.; Aiolli, F.; Sperduti, A.; Valle, G. Scuba: Scalable kernel-based gene prioritization. BMC Bioinform. 2018, 19, 23. [Google Scholar] [CrossRef] [Green Version]
- Merriman, T.R.; Dalbeth, N. The genetic basis of hyperuricaemia and gout. Jt. Bone Spine 2011, 78, 35–40. [Google Scholar] [CrossRef]
- Iharada, M.; Miyaji, T.; Fujimoto, T.; Hiasa, M.; Anzai, N.; Omote, H.; Moriyama, Y. Type 1 sodium-dependent phosphate transporter (SLC17A1 Protein) is a Cl(-)-dependent urate exporter. J. Biol. Chem. 2010, 285, 26107–26113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, T.; Matsuo, H.; Kawamura, Y.; Nagamori, S.; Nishiyama, T.; Wei, L.; Nakayama, A.; Nakamura, T.; Sakiyama, M.; Takada, T.; et al. NPT1/SLC17A1 is a renal urate exporter in humans and its common gain-of-function variant decreases the risk of renal under-excretion gout. Arthritis Rheumatol. 2015, 67, 281–287. [Google Scholar] [CrossRef]
- Higashino, T.; Matsuo, H.; Sakiyama, M.; Nakayama, A.; Nakamura, T.; Takada, T.; Ogata, H.; Kawamura, Y.; Kawaguchi, M.; Naito, M.; et al. Common variant of PDZ domain containing 1 (PDZK1) gene is associated with gout susceptibility: A replication study and meta-analysis in Japanese population. Drug Metab. Pharm. 2016, 31, 464–466. [Google Scholar] [CrossRef] [Green Version]
- Anzai, N.; Kanai, Y.; Endou, H. New insights into renal transport of urate. Curr. Opin. Rheumatol. 2007, 19, 151–157. [Google Scholar] [CrossRef]
- De Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [Green Version]
- Alexander, R.T.; Dimke, H.; Cordat, E. Proximal tubular NHEs: Sodium, protons and calcium? Am. J. Physiol. Ren. Physiol. 2013, 305, F229–F236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biemesderfer, D.; Nagy, T.; DeGray, B.; Aronson, P. Specific association of megalin and the Na+/H+ exchanger isoform NHE3 in the proximal tubule. J. Biol. Chem. 1999, 274, 17518–17524. [Google Scholar] [CrossRef] [Green Version]
- Gekle, M.; Völker, K.; Mildenberger, S.; Freudinger, R.; Shull, G.E.; Wiemann, M. NHE3 Na+/H+ exchanger supports proximal tubular protein reabsorption in vivo. Am. J. Physiol. Ren. Physiol. 2004, 287, F469–F473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanska, M.; Zaniew, M.; Recker, F.; Mizerska-Wasiak, M.; Zaluska-Lesniewska, I.; Kilis-Pstrusinska, K.; Adamczyk, P.; Zawadzki, J.; Pawlaczyk, K.; Ludwig, M.; et al. Dent disease in children: Diagnostic and therapeutic considerations. Clin. Nephrol. 2015, 84, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Brown, M.R.; Cogal, A.G.; Gauvin, D.; Harris, P.C.; Lieske, J.C.; Romero, M.F.; Chang, M.H. Functional and transport analyses of CLCN5 genetic changes identified in Dent disease patients. Physiol. Rep. 2016, 4, e12776. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yue, Z.; Xu, T.; Chen, M.; Zhong, L.; Liu, T.; Jing, X.; Deng, J.; Hu, B.; Liu, Y.; et al. Dent Disease in Chinese Children and Findings from Heterozygous Mothers: Phenotypic Heterogeneity, Fetal Growth, and 10 Novel Mutations. J. Pediatr. 2016, 174, 204–210.e1. [Google Scholar] [CrossRef]
- Kubo, K.; Aizawa, T.; Watanabe, S.; Tsugawa, K.; Tsuruga, K.; Ito, E.; Joh, K.; Tanaka, H. Does Dent disease remain an underrecognized cause for young boys with focal glomerulosclerosis? Pediatr. Int. 2016, 58, 747–749. [Google Scholar] [CrossRef]
- Wong, W.; Poke, G.; Stack, M.; Kara, T.; Prestidge, C.; Flintoff, K. Phenotypic variability of Dent disease in a large New Zealand kindred. Pediatr. Nephrol. 2017, 32, 365–369. [Google Scholar] [CrossRef]
- Guven, A.; Al-Rijjal, R.A.; BinEssa, H.A.; Dogan, D.; Kor, Y.; Zou, M.; Kaya, N.; Alenezi, A.F.; Hancili, S.; Tarım, Ö.; et al. Mutational analysis of PHEX, FGF23 and CLCN5 in patients with hypophosphataemic rickets. Clin. Endocrinol. 2017, 87, 103–112. [Google Scholar] [CrossRef]
- Günthner, R.; Wagner, M.; Thurm, T.; Ponsel, S.; Höfele, J.; Lange-Sperandio, B. Identification of co-occurrence in a patient with Dent’s disease and ADA2-deficiency by exome sequencing. Gene 2018, 649, 23–26. [Google Scholar] [CrossRef]
- Sancakli, O.; Kulu, B.; Sakallioglu, O. A novel mutation of Dent’s disease in an 11-year-old male with nephrolithiasis and nephrocalcinosis. Arch. Argent. Pediatr. 2018, 116, e442–e444. [Google Scholar]
- Bignon, Y.; Alekov, A.; Frachon, N.; Lahuna, O.; Jean-Baptiste Doh-Egueli, C.; Deschênes, G.; Vargas-Poussou, R.; Lourdel, S. A novel CLCN5 pathogenic mutation supports Dent disease with normal endosomal acidification. Hum. Mutat. 2018, 39, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, M.; Shen, T.; Wang, Y.; Li, Y.; Shi, X.; Dang, X. Next-Generation Sequencing in Early Diagnosis of Dent Disease 1: Two Case Reports. Front. Med. 2018, 5, 347. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Matsui, I.; Mori, T.; Sakaguchi, Y.; Mizui, M.; Ueda, Y.; Takahashi, A.; Doi, Y.; Shimada, K.; Yamaguchi, S.; et al. Severe Osteomalacia with Dent Disease Caused by a Novel Intronic Mutation of the CLCN5 gene. Intern. Med. 2018, 57, 3603–3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Shen, Q.; Rao, J.; Zhang, A.; Zheng, B.; Liu, X.; Shen, Y.; Chen, Z.; Wu, Y.; Hou, L.; et al. Multicenter study of the clinical features and mutation gene spectrum of Chinese children with Dent disease. Clin. Genet. 2019. [Google Scholar] [CrossRef]
- Wu, F.; Roche, P.; Christie, P.T.; Loh, N.Y.; Reed, A.A.; Esnouf, R.M.; Thakker, R.V. Modeling study of human renal chloride channel (hCLC-5) mutations suggests a structural-functional relationship. Kidney Int. 2003, 63, 1426–1432. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.; Savaresi, S.; Forster, I.C.; Dutzler, R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat. Struct. Mol. Biol. 2007, 14, 60–67. [Google Scholar] [CrossRef]
- Wellhauser, L.; Luna-Chavez, C.; D’Antonio, C.; Tainer, J.; Bear, C.E. ATP induces conformational changes in the carboxylterminal region of ClC-5. J. Biol. Chem. 2011, 286, 6733–6741. [Google Scholar] [CrossRef] [Green Version]
- Zifarelli, G.; Pusch, M. Intracellular regulation of human ClC-5 by adenine nucleotides. EMBO Rep. 2009, 10, 1111–1116. [Google Scholar] [CrossRef] [Green Version]
- Grand, T.; Mordasini, D.; L’Hoste, S.; Pennaforte, T.; Genete, M.; Biyeyeme, M.J.; Vargas-Poussou, R.; Blanchard, A.; Teulon, J.; Lourdel, S. Novel CLCN5 mutations in patients with Dent’s disease result in altered ion currents or impaired exchanger processing. Kidney Int. 2009, 76, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Pusch, M.; Ludewig, U.; Jentsch, T.J. Temperature dependence of fast and slow gating relaxations of CLC-0 chloride channels. J. Gen. Physiol. 1997, 109, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, T.; Günther, W.; Sekine, T.; Inatomi, J.; Shiraga, H.; Takahashi, S.; Suzuki, J.; Tsuru, N.; Yanagihara, T.; Shimazu, M.; et al. Functional characterization of renal chloride channel, CLCN5, mutations associated with Dent’s Japan disease. Kidney Int. 1998, 54, 1850–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devuyst, O.; Christie, P.T.; Courtoy, P.J.; Beauwens, R.; Thakker, R.V. Intra-renal and subcellular distribution of the human chloride channel, CLC-5, reveals a pathophysiological basis for Dent’s disease. Hum. Mol. Genet. 1999, 8, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cai, H.; Cebotaru, L.; Hryciw, D.H.; Weinman, E.J.; Donowitz, M.; Guggino, S.E.; Guggino, W.B. ClC-5: Role in endocytosis in the proximal tubule. Am. J. Physiol. Ren. Physiol. 2005, 289, F850–F862. [Google Scholar] [CrossRef] [Green Version]
- Hryciw, D.H.; Wang, Y.; Devuyst, O.; Pollock, C.A.; Poronnik, P.; Guggino, W.B. Cofilin interacts with ClC-5 and regulates albumin uptake in proximal tubule cell lines. J. Biol. Chem. 2003, 278, 40169–40176. [Google Scholar] [CrossRef] [Green Version]
- Kellermayer, R. Translational readthrough induction of pathogenic nonsense mutations. Eur. J. Med. Genet. 2006, 49, 445–450. [Google Scholar] [CrossRef]
- Oren, Y.S.; Pranke, I.M.; Kerem, B.; Sermet-Gaudelus, I. The suppression of premature termination codons and the repair of splicing mutations in CFTR. Curr. Opin. Pharmacol. 2017, 34, 125–131. [Google Scholar] [CrossRef]
- Piwon, N.; Günther, W.; Schwake, M.; Bösl, M.R.; Jentsch, T.J. ClC-5 Cl−-channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature 2000, 408, 369–373. [Google Scholar] [CrossRef]
- Norden, A.G.; Lapsley, M.; Igarashi, T.; Kelleher, C.L.; Lee, P.J.; Matsuyama, T.; Scheinman, S.J.; Shiraga, H.; Sundin, D.P.; Thakker, R.V.; et al. Urinary megalin deficiency implicates abnormal tubular endocytic function in Fanconi syndrome. J. Am. Soc. Nephrol. 2002, 13, 125–133. [Google Scholar]
- Tanuma, A.; Sato, H.; Takeda, T.; Hosojima, M.; Obayashi, H.; Hama, H.; Iino, N.; Hosaka, K.; Kaseda, R.; Imai, N.; et al. Functional characterization of a novel missense CLCN5 mutation causing alterations in proximal tubular endocytic machinery in Dent’s disease. Nephron Physiol. 2007, 107, p87–p97. [Google Scholar] [CrossRef]
- Santo, Y.; Hirai, H.; Shima, M.; Yamagata, M.; Michigami, T.; Nakajima, S.; Ozono, K. Examination of megalin in renal tubular epithelium from patients with Dent disease. Pediatr. Nephrol. 2004, 19, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Gorvin, C.M.; Wilmer, M.J.; Piret, S.E.; Harding, B.; van den Heuvel, L.P.; Wrong, O.; Jat, P.S.; Lippiat, J.D.; Levtchenko, E.N.; Thakker, R.V. Receptor-mediated endocytosis and endosomal acidification is impaired in proximal tubule epithelial cells of Dent disease patients. Proc. Natl. Acad. Sci. USA 2013, 110, 7014–7019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anglani, F.; D’Angelo, A.; Bertizzolo, L.M.; Tosetto, E.; Ceol, M.; Cremasco, D.; Bonfante, L.; Addis, M.A.; Del Prete, D. Dent Disease Italian Network. Nephrolithiasis; kidney failure and bone disorders in Dent disease patients with and without CLCN5 mutations. SpringerPlus 2015, 4, 492. [Google Scholar] [CrossRef] [Green Version]
- Anglani, F.; Terrin, L.; Brugnara, M.; Battista, M.; Cantaluppi, V.; Ceol, M.; Bertoldi, L.; Valle, G.; Joy, M.P.; Pober, B.R.; et al. Hypercalciuria and nephrolithiasis: Expanding the renal phenotype of Donnai-Barrow syndrome. Clin. Genet. 2018, 94, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Köttgen, A.; Albrecht, E.; Teumer, A.; Vitart, V.; Krumsiek, J.; Hundertmark, C.; Pistis, G.; Ruggiero, D.; O’Seaghdha, C.M.; Haller, T.; et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 2013, 45, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phipps-Green, A.J.; Merriman, M.E.; Topless, R.; Altaf, S.; Montgomery, G.W.; Franklin, C.; Jones, G.T.; van Rij, A.M.; White, D.; Stamp, L.K.; et al. Twenty-eight loci that influence serum urate levels: Analysis of association with gout. Ann. Rheum. Dis. 2016, 75, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Ketharnathan, S.; Leask, M.; Boocock, J.; Phipps-Green, A.J.; Antony, J.; O’Sullivan, J.M.; Merriman, T.R.; Horsfield, J.A. A non-coding genetic variant maximally associated with serum urate levels is functionally linked to HNF4A-dependent PDZK1 expression. Hum. Mol. Genet. 2018, 27, 3964–3973. [Google Scholar] [CrossRef]
- Gisler, S.M.; Pribanic, S.; Bacic, D.; Forrer, P.; Gantenbein, A.; Sabourin, L.A.; Tsuji, A.; Zhao, Z.S.; Manser, E.; Biber, J.; et al. PDZK1: I. a major scaffolder in brush borders of proximal tubular cells. Kidney Int. 2003, 64, 1733–1745. [Google Scholar] [CrossRef] [Green Version]
- Capuano, P.; Bacic, D.; Stange, G.; Hernando, N.; Kaissling, B.; Pal, R.; Kocher, O.; Biber, J.; Wagner, C.A.; Murer, H. Expression and regulation of the renal Na/phosphate cotransporter NaPi-IIa in a mouse model deficient for the PDZ protein PDZK1. Pflug. Arch. 2005, 449, 392–402. [Google Scholar] [CrossRef]
- Slattery, C.; Jenkin, K.A.; Lee, A.; Simcocks, A.C.; McAinch, A.J.; Poronnik, P.; Hryciw, D.H. Na+-H+ exchanger regulatory factor 1 (NHERF1) PDZ scaffold binds an internal binding site in the scavenger receptor megalin. Cell. Physiol. Biochem. 2011, 27, 171–178. [Google Scholar] [CrossRef]
- Sakiyama, M.; Matsuo, H.; Nagamori, S.; Ling, W.; Kawamura, Y.; Nakayama, A.; Higashino, T.; Chiba, T.; Ichida, K.; Kanai, Y.; et al. Expression of a human NPT1/SLC17A1 missense variant which increases urate export. Nucleosides Nucleotides Nucleic Acids 2016, 35, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Böger, C.A.; Chen, M.H.; Tin, A.; Olden, M.; Köttgen, A.; de Boer, I.H.; Fuchsberger, C.; O’Seaghdha, C.M.; Pattaro, C.; Teumer, A.; et al. CUBN is a gene locus for albuminuria. J. Am. Soc. Nephrol. 2011, 22, 555–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovunc, B.; Otto, E.A.; Vega-Warner, V.; Saisawat, P.; Ashraf, S.; Ramaswami, G.; Fathy, H.M.; Schoeb, D.; Chernin, G.; Lyons, R.H.; et al. Exome sequencing reveals cubilin mutation as a single-gene cause of proteinuria. J. Am. Soc. Nephrol. 2011, 22, 1815–1820. [Google Scholar] [CrossRef] [PubMed]
- Tanner, S.M.; Sturm, A.C.; Baack, E.C.; Liyanarachchi, S.; de la Chapelle, A. Inherited cobalamin malabsorption. Mutations in three genes reveal functional and ethnic patterns. Orphanet J. Rare Dis. 2012, 7, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäffer, A.A. Digenic inheritance in medical genetics. J. Med. Genet. 2013, 50, 641–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deltas, C. Digenic inheritance and genetic modifiers. Clin. Genet. 2018, 93, 429–438. [Google Scholar] [CrossRef]
- Janecke, A.R.; Heinz-Erian, P.; Yin, J.; Petersen, B.S.; Franke, A.; Lechner, S.; Fuchs, I.; Melancon, S.; Uhlig, H.H.; Travis, S.; et al. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum. Mol. Genet. 2015, 24, 6614–6623. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Jin, S.; Duan, X.; Wang, T.; Martini, S.; Hulamm, P.; Cha, B.; Hubbard, A.; Donowitz, M.; Guggino, S.E. Chloride channel (Clc)-5 is necessary for exocytic trafficking of Na+/H+ exchanger 3 (NHE3). J. Biol. Chem. 2011, 286, 22833–22845. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Borovac, J.; Spicer, Z.; Hoenderop, J.G.; Bindels, R.J.; Shull, G.E.; Doschak, M.R.; Cordat, E.; Alexander, R.T. The epithelial sodium/proton exchanger; NHE3; is necessary for renal and intestinal calcium (re)absorption. Am. J. Physiol. Ren. Physiol. 2012, 302, F943–F956. [Google Scholar] [CrossRef] [Green Version]
- Katsanis, N. The continuum of causality in human genetic disorders. Genome Biol. 2016, 17, 233. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.N.; Krawczak, M.; Polychronakos, C.; Tyler-Smith, C.; Kehrer-Sawatzki, H. Where genotype is not predictive of phenotype: Towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum. Genet. 2013, 132, 1077–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosetto, E.; Ghiggeri, G.M.; Emma, F.; Barbano, G.; Carrea, A.; Vezzoli, G.; Torregrossa, R.; Cara, M.; Ripanti, G.; Ammenti, A.; et al. Phenotypic and genetic heterogeneity in Dent’s disease: The results of an Italian collaborative study. Nephrol. Dial. Transplant. 2006, 21, 2452–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Berkel, Y.; Ludwig, M.; van Wijk, J.A.E.; Bökenkamp, A. Proteinuria in Dent disease: A review of the literature. Pediatr. Nephrol. 2017, 32, 1851–1859. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Bertoldi, L.; Forcato, C.; Vitulo, N.; Birolo, G.; De Pascale, F.; Feltrin, E.; Schiavon, R.; Anglani, F.; Negrisolo, S.; Zanetti, A.; et al. QueryOR: A comprehensive web platform for genetic variant analysis and prioritization. BMC Bioinform. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [Green Version]
- Yongwook, C.; Agnes, P.C. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Ceol, M.; Tiralongo, E.; Baelde, H.J.; Vianello, D.; Betto, G.; Marangelli, A.; Bonfante, L.; Valente, M.; Della Barbera, M.; D’Angelo, A.; et al. Involvement of the tubular ClC-type exchanger ClC-5 in glomeruli of human proteinuric nephropathies. PLoS ONE 2012, 7, e45605. [Google Scholar] [CrossRef] [PubMed]
- Gianesello, L.; Priante, G.; Ceol, M.; Radu, C.M.; Saleem, M.A.; Simioni, P.; Terrin, L.; Anglani, F.; Del Prete, D. Albumin uptake in human podocytes: A possible role for the cubilin-amnionless (CUBAM) complex. Sci. Rep. 2017, 7, 13705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008; ISBN 3-900051-07-0. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Mutation | Nucleotide | Exon-Intron | Protein | Pathogenicity Assessment | Protein Domain |
|---|---|---|---|---|---|
| Frameshift | c.100_101insG | Exon 2 | p.(Glu35fs) | Pathogenic (Ib) | A helix, stop in Loop A-B |
| Frameshift | c.125delA | Exon 3 | p.(Glu42fs) | Pathogenic (Ib) | Loop A-B |
| Frameshift | c.266_267insT | Exon 4 | p.(Ile89fs) | Pathogenic (Ib) | B helix, stop in Loop B-C |
| Frameshift | c.518delT | Exon 6 | p.(Ile173fs) | Pathogenic (Ib) | D helix, stop in Loop D-E |
| Frameshift § | c.691delA | Exon 6 | p.(Lys231fs) | Pathogenic (Ia) | Loop F-G, stop at the end of helix G |
| Frameshift | c.1164_1165insAG | Exon 8 | p.(Lys388fs) | Pathogenic (Ib) | L helix, stop in helix M |
| Frameshift | c.1635_1638delCAAG | Exon 10 | p.(Ser545fs) | Pathogenic (Ib) | Q helix, stop in cytoplasmic |
| Frameshift | c.1657delG | Exon 10 | p.(Arg554fs) | Pathogenic (Ib) | Cytoplasmic, stop at the beginning of CBS1 cytoplasmic domain |
| Frameshift | c.1920delC | Exon 10 | p.(Ile641fs) | Pathogenic (Ib) | CBS1 cytoplasmic domain, stop in cytoplasmic |
| Nonsense | c.1287G>A | Exon 8 | p.(Trp429*) | Pathogenic (Ib) | M helix |
| Nonsense | c.2016C>G | Exon 11 | p.(Tyr672*) | Pathogenic (Ib) | Cytoplasmic |
| Nonsense | c.2128C>T | Exon 11 | p.(Gln710*) | Pathogenic (Ib) | Cytoplasmic-beta strand in CBS2 domain |
| Missense | c.262G>A | Exon 4 | p.(Gly88Ser) | Likely pathogenic (IV) | B helix |
| Missense | c.305G>T | Exon 4 | p.(Cys102Phe) | Likely pathogenic (V) | Loop B-C |
| Missense | c.518T>A | Exon 6 | p.(Ile173Lys) | Likely pathogenic (V) | D helix |
| Missense § | c.608C>G | Exon 6 | p.(Ser203Trp) | Pathogenic (II) | E helix |
| Missense | c.809G>A | Exon 8 | p.(Ser270Asn) | Likely pathogenic (IV) | Loop H-I |
| Missense § | c.922G>A | Exon 8 | p.(Val308Met) | Pathogenic (IIIb) | Loop I-J |
| Missense | c.1565T>A | Exon 10 | p.(Val522Asp) | Likely pathogenic (V) | P helix |
| Missense | c.1619C>T | Exon 10 | p.(Ala540Val) | Likely pathogenic (IV) | Q helix |
| Missense | c.2192A>C | Exon 12 | p.(His731Pro) | Likely pathogenic (V) | Cytoplasmic-CBS2 domain |
| Splicing | c.105+5G>C | Intron 2-splice site | p.? | Likely pathogenic (II) | |
| Splicing | c.1348-1G>A | Intron 8-splice site | p.? | Pathogenic (Ic) |
| Patient | CLCN5 Mutation | Age at Biopsy (Years) | Indication for Biopsy | Histopathological Findings | ClC-5 Immunolabeling Morphometric Evaluation (% Positive Area) |
|---|---|---|---|---|---|
| 1 | p.(Thr44fs) | 2 | Proteinuria | Minimal changes | 0.01 |
| 2 | p.(Lys231fs) (novel) | 14 | Proteinuria | Normal | 0.00 |
| 3 | p.(Arg34*) | 11 | Nephrotic syndrome | Chronic interstitial nephritis with global glomerulosclerosis | 0.12 |
| 4 | p.(Arg34*) | 6 | Proteinuria | Global glomerulosclerosis and IgM nephropathy | 0.00 |
| 5 | p.(Gln600*) | 6 | Proteinuria | Tubulointerstitial injury with focal glomerulosclerosis | 0.05 |
| 6 | p.(Ser203Trp) (novel) | 3 | Proteinuria | Normal | 0.01 |
| 7 | p.(Ser261Arg) | 4 | Heavy proteinuria | Proliferative mesangial glomerulonephritis | 0.07 |
| 8 | p.(Tyr272Cys) | NA | Proteinuria | Normal | 0.01 |
| 9 | p.(Val308Met) (novel) | 9 | Proteinuria and hematuria | Normal | 0.08 |
| 10 | p.(Trp547Arg) | 1 | Proteinuria | Normal | 0.00 |
| Pt ID | Transcript Level Variation | Codon Substitution | Frequency ExAC (European) | Mutation Taster | PROVEAN | DANN | ClinVar | ACMG/AMP Variant Interpretation |
|---|---|---|---|---|---|---|---|---|
| SLC17A1 (NM_005074.3) | ||||||||
| AMS | c.1309G>A | p.(Ala437Thr) | rs1189357572 0.000003 (GnomAD) (0.000008) | Polymorphism (1.000) | Neutral (−1.97) | 0.967 | NA | VUS |
| PDZK1 (NM_002614.4) | ||||||||
| AMS | c.22C>T | p.(Arg8*) homozygous | rs191362962 0.0001157 (0.0001799) | Disease causing automatic | NA | 0.998 | NA | VUS |
| LRP2 (NM_004525.2) | ||||||||
| BDA | c.6727C>T | p.(Arg2243*) | novel | Disease causing automatic (1.000) | NA | 0.996 | NA | Pathogenic (Ib) |
| BDA | c.242T>A | p.(Ile81Asn) | novel | Disease causing (1.000) | Damaging (−4.62) | 0.993 | NA | Likely pathogenic (V) |
| AMV | c.6160G>A | p.(Asp2054Asn) | rs138269726 0.0011 (0.0017) | Disease causing (1.000) | Neutral (−1.85) | 0.999 | Pathogenic allele | Likely pathogenic (II) |
| AMT | c.2006G>A | p.(Gly669Asp) | rs34291900 0.0285 (0.0434) | Disease causing (1.000) | Damaging (−5.44) | 0.998 | Likely benign allele | Likely benign |
| AMT | c.7894A>G | p.(Asn2632Asp) | rs17848169 0.02951 (0.0426) | Disease causing (1.000) | Neutral (−1.73) | 0.452 | Likely benign allele | Likely benign |
| SLC3A1 (NM_000341.4) | ||||||||
| AMS | c.680G>A | p.(Arg227Gln) | rs142469446 0.0002 (0.0002) | Disease causing (1.000) | Neutral (−1.63) | 1 | NA | Likely pathogenic (IV) |
| AMS | c.797T>C | p.(Phe266Ser) | rs141587158 0.003 (0.004) | Disease causing (1.000) | Damaging (−4.7) | 0.998 | NA | Likely pathogenic (IIIb) |
| CUBN (NM_001081.3) | ||||||||
| AMT | c.10265C>T | p.(Thr3422Ile) | rs1801230 0.01832 (0.02829) | Disease causing (1.000) | Damaging (−2.94) | 0.985 | Likely benign allele | Benign |
| AMV | c.7040_7042del | p.(Val2347del) | rs1279549461 (TOPmed) 0.0000001 | Disease causing (0.998) | Deleterious (−9.81) | NA | NA | VUS |
| SLC9A3 (NM_004174.3) | ||||||||
| AMT | c.848G>A | p.(Arg283His) | rs146899318 0.00033 (0.00036) | Disease causing (0.853) | Damaging (−3.85) | 0.999 | NA | VUS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gianesello, L.; Ceol, M.; Bertoldi, L.; Terrin, L.; Priante, G.; Murer, L.; Peruzzi, L.; Giordano, M.; Paglialonga, F.; Cantaluppi, V.; et al. Genetic Analyses in Dent Disease and Characterization of CLCN5 Mutations in Kidney Biopsies. Int. J. Mol. Sci. 2020, 21, 516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020516
Gianesello L, Ceol M, Bertoldi L, Terrin L, Priante G, Murer L, Peruzzi L, Giordano M, Paglialonga F, Cantaluppi V, et al. Genetic Analyses in Dent Disease and Characterization of CLCN5 Mutations in Kidney Biopsies. International Journal of Molecular Sciences. 2020; 21(2):516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020516
Chicago/Turabian StyleGianesello, Lisa, Monica Ceol, Loris Bertoldi, Liliana Terrin, Giovanna Priante, Luisa Murer, Licia Peruzzi, Mario Giordano, Fabio Paglialonga, Vincenzo Cantaluppi, and et al. 2020. "Genetic Analyses in Dent Disease and Characterization of CLCN5 Mutations in Kidney Biopsies" International Journal of Molecular Sciences 21, no. 2: 516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020516