Understanding the Molecular Mechanisms Underlying the Pathogenesis of Arthritis Pain Using Animal Models

Abstract
:1. Introduction
2. Osteoarthritis
2.1. Pain in Clinical OA
2.2. Animal Models for Studying the Pathogenesis of OA Pain
2.2.1. Animal Models
2.2.2. Behavioral Tests to Assess Pain in OA Animal Models
2.2.3. Understanding the Molecular Mechanism Underlying the Pathogenesis of OA Pain via Experiments in Animal Models
3. Rheumatoid Arthritis
3.1. Pain in Clinical RA
3.2. Animal Models for Studying the Pathogenesis of RA Pain
3.2.1. Animal Models
3.2.2. Behavioral Tests to Assess Pain in RA Animal Models
3.2.3. Understanding the Molecular Mechanism Underlying the Pathogenesis of RA Pain via Experiments in Animal Models
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [Green Version]
- Hadler, N. Knee pain is the malady—Not osteoarthritis. Ann. Intern. Med. 1992, 116, 598–599. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Kim, H.A.; Seo, Y.I.; Song, Y.W.; Jeong, J.Y.; Kim, D.H. The prevalence of knee osteoarthritis in elderly community residents in Korea. J. Korean Med. Sci. 2010, 25, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackerman, I.N.; Zomer, E.; Gilmartin-Thomas, J.F.; Liew, D. Forecasting the future burden of opioids for osteoarthritis. Osteoarthr. Cartil. 2018, 26, 350–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorlund, J.B.; Turkiewicz, A.; Prieto-Alhambra, D.; Englund, M. Opioid use in knee or hip osteoarthritis: A region-wide population-based cohort study. Osteoarthr. Cartil. 2019, 27, 871–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMik, D.E.; Bedard, N.A.; Dowdle, S.B.; Burnett, R.A.; McHugh, M.A.; Callaghan, J.J. Are We Still Prescribing Opioids for Osteoarthritis? J. Arthroplast. 2017, 32, 3578–3582. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Xie, F.; Smith, C.; Saag, K.G.; Chen, L.; Beukelman, T.; Mannion, M.; Yun, H.; Kertesz, S. Changing Trends in Opioid Use Among Patients with Rheumatoid Arthritis in the United States. Arthritis Rheumatol. 2017, 69, 1733–1740. [Google Scholar] [CrossRef]
- Rifbjerg-Madsen, S.; Christensen, A.W.; Christensen, R.; Hetland, M.L.; Bliddal, H.; Kristensen, L.E.; Danneskiold-Samsoe, B.; Amris, K. Pain and pain mechanisms in patients with inflammatory arthritis: A Danish nationwide cross-sectional DANBIO registry survey. PLoS ONE 2017, 12, e0180014. [Google Scholar] [CrossRef]
- Davis, M.A.; Ettinger, W.H.; Neuhaus, J.M.; Barclay, J.D.; Segal, M.R. Correlates of knee pain among US adults with and without radiographic knee osteoarthritis. J. Rheumatol. 1992, 19, 1943–1949. [Google Scholar]
- Creamer, P.; Lethbridge-Cejku, M.; Hochberg, M.C. Factors associated with functional impairment in symptomatic knee osteoarthritis. Rheumatology 2000, 39, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Bedson, J.; Croft, P.R. The discordance between clinical and radiographic knee osteoarthritis: A systematic search and summary of the literature. BMC Musculoskelet. Disord. 2008, 9, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neogi, T.; Felson, D.; Niu, J.; Nevitt, M.; Lewis, C.E.; Aliabadi, P.; Sack, B.; Torner, J.; Bradely, L.; Zhang, Y. Association between radiographic features of knee osteoarthritis and pain: Results from two cohort studies. BMJ 2009, 339, b2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Kim, H.A.; Felson, D.T.; Xu, L.; Kim, D.H.; Nevitt, M.C.; Yoshimura, N.; Kawaguchi, H.; Lin, J.; Kang, X.; et al. Radiographic Knee Osteoarthritis and Knee Pain: Cross-sectional study from Five Different Racial/Ethnic Populations. Sci. Rep. 2018, 8, 1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, K.M.; Lee, H.S.; Kim, D.H.; Paik, M.C.; Jang, D.G.; Lee, S.Y.; Kim, H.A. The Absence of Symptom in Subjects with Advanced Knee OA. Rheum. Dis. 2016, 75, 825–826. [Google Scholar] [CrossRef]
- Dye, S.F.; Vaupel, G.L.; Dye, C.C. Conscious neurosensory mapping of the internal structures of the human knee without intraarticular anesthesia. Am. J. Sports Med. 1998, 26, 773–777. [Google Scholar] [CrossRef]
- Zhang, Y.; Nevitt, M.; Niu, J.; Lewis, C.; Torner, J.; Guermazi, A.; Roemer, F.; McCulloch, C.; Felson, D.T. Fluctuation of knee pain and changes in bone marrow lesions, effusions, and synovitis on magnetic resonance imaging. Arthritis Rheum. 2011, 63, 692–699. [Google Scholar] [CrossRef] [Green Version]
- Englund, M.; Guermazi, A.; Gale, D.; Hunter, D.J.; Aliabadi, P.; Clancy, M.; Felson, D.T. Incidental meniscal findings on knee MRI in middle-aged and elderly persons. N. Engl. J. Med. 2008, 359, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.A.; Kim, I.; Song, Y.W.; Kim, D.H.; Niu, J.; Guermazi, A.; Crema, M.D.; Hunter, D.J.; Zhang, Y. The association between meniscal and cruciate ligament damage and knee pain in community residents. Osteoarthr. Cartil. 2011, 19, 1422–1428. [Google Scholar] [CrossRef] [Green Version]
- Felson, D.T.; Chaisson, C.E.; Hill, C.L.; Totterman, S.M.; Gale, M.E.; Skinner, K.M.; Kazis, L.; Gale, D.R. The association of bone marrow lesions with pain in knee osteoarthritis. Ann. Intern. Med. 2001, 134, 541–549. [Google Scholar] [CrossRef]
- Yusuf, E.; Kortekaas, M.C.; Watt, I.; Huizinga, T.W.; Kloppenburg, M. Do knee abnormalities visualised on MRI explain knee pain in knee osteoarthritis? A systematic review. Ann. Rheum. Dis. 2011, 70, 60–67. [Google Scholar] [CrossRef]
- Alliston, T.; Hernandez, C.J.; Findlay, D.M.; Felson, D.T.; Kennedy, O.D. Bone marrow lesions in osteoarthritis: What lies beneath. J. Orthop. Res. 2018, 36, 1818–1825. [Google Scholar] [CrossRef] [PubMed]
- Aso, K.; Shahtaheri, S.M.; Hill, R.; Wilson, D.; McWilliams, D.F.; Walsh, D.A. Associations of Symptomatic Knee Osteoarthritis with Histopathologic Features in Subchondral Bone. Arthritis Rheumatol. 2019, 71, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Arendt-Nielsen, L. Pain sensitisation in osteoarthritis. Clin. Exp. Rheumatol. 2017, 35 (Suppl. 107), 68–74. [Google Scholar] [PubMed]
- Neogi, T.; Frey-Law, L.; Scholz, J.; Niu, J.; Arendt-Nielsen, L.; Woolf, C.; Nevitt, M.; Bradley, L.; Felson, D.T.; Multicenter Osteoarthritis, S. Sensitivity and sensitisation in relation to pain severity in knee osteoarthritis: Trait or state? Ann. Rheum. Dis. 2015, 74, 682–688. [Google Scholar] [CrossRef] [Green Version]
- Steen Pettersen, P.; Neogi, T.; Magnusson, K.; Berner Hammer, H.; Uhlig, T.; Kvien, T.K.; Haugen, I.K. Peripheral and Central Sensitization of Pain in Individuals with Hand Osteoarthritis and Associations with Self-Reported Pain Severity. Arthritis Rheumatol. 2019, 71, 1070–1077. [Google Scholar] [CrossRef]
- Egsgaard, L.L.; Eskehave, T.N.; Bay-Jensen, A.C.; Hoeck, H.C.; Arendt-Nielsen, L. Identifying specific profiles in patients with different degrees of painful knee osteoarthritis based on serological biochemical and mechanistic pain biomarkers: A diagnostic approach based on cluster analysis. Pain 2015, 156, 96–107. [Google Scholar] [CrossRef]
- Eitner, A.; Hofmann, G.O.; Schaible, H.G. Mechanisms of Osteoarthritic Pain. Studies in Humans and Experimental Models. Front. Mol. Neurosci. 2017, 10, 349. [Google Scholar] [CrossRef] [Green Version]
- Felson, D.T. Developments in the clinical understanding of osteoarthritis. Arthritis Res. Ther. 2009, 11, 203. [Google Scholar] [CrossRef] [Green Version]
- Eitner, A.; Pester, J.; Nietzsche, S.; Hofmann, G.O.; Schaible, H.G. The innervation of synovium of human osteoarthritic joints in comparison with normal rat and sheep synovium. Osteoarthr. Cartil. 2013, 21, 1383–1391. [Google Scholar] [CrossRef] [Green Version]
- Suri, S.; Gill, S.E.; de Massena, C.S.; Wilson, D.; McWilliams, D.F.; Walsh, D.A. Neurovascular invasion at the osteochondral junction and in osteophytes in osteoarthritis. Ann. Rheum. Dis. 2007, 66, 1423–1428. [Google Scholar] [CrossRef] [Green Version]
- Pergolizzi, J.V., Jr.; Raffa, R.B.; Taylor, R., Jr.; Rodriguez, G.; Nalamachu, S.; Langley, P. A review of duloxetine 60 mg once-daily dosing for the management of diabetic peripheral neuropathic pain, fibromyalgia, and chronic musculoskeletal pain due to chronic osteoarthritis pain and low back pain. Pain Pract. 2013, 13, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Bannuru, R.R.; Osani, M.C.; Vaysbrot, E.E.; Arden, N.K.; Bennell, K.; Bierma-Zeinstra, S.M.A.; Kraus, V.B.; Lohmander, L.S.; Abbott, J.H.; Bhandari, M.; et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1578–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.E.; Malfait, A.M.; Block, J.A. Current status of nerve growth factor antibodies for the treatment of osteoarthritis pain. Clin. Exp. Rheumatol. 2017, 35 (Suppl. 107), 85–87. [Google Scholar] [PubMed]
- Dakin, P.; DiMartino, S.J.; Gao, H.; Maloney, J.; Kivitz, A.J.; Schnitzer, T.J.; Stahl, N.; Yancopoulos, G.D.; Geba, G.P. The Efficacy, Tolerability, and Joint Safety of Fasinumab in Osteoarthritis Pain: A Phase IIb/III Double-Blind, Placebo-Controlled, Randomized Clinical Trial. Arthritis Rheumatol. 2019, 71, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.E.; Schnitzer, T.J.; Birbara, C.A.; Mokhtarani, M.; Shelton, D.L.; Smith, M.D.; Brown, M.T. Tanezumab for the treatment of pain from osteoarthritis of the knee. N. Engl. J. Med. 2010, 363, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Little, C.B.; Zaki, S. What constitutes an “animal model of osteoarthritis”—The need for consensus? Osteoarthr. Cartil. 2012, 20, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Glasson, S.S.; Blanchet, T.J.; Morris, E.A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 2007, 15, 1061–1069. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Lu, W.; Chen, L.; Ge, Q.; Chen, D.; Xu, Z.; Shi, D.; Dai, J.; Li, J.; Ju, H.; et al. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci. Rep. 2017, 7, 43245. [Google Scholar] [CrossRef] [Green Version]
- Wondimu, E.B.; Culley, K.L.; Quinn, J.; Chang, J.; Dragomir, C.L.; Plumb, D.A.; Goldring, M.B.; Otero, M. Elf3 Contributes to Cartilage Degradation in vivo in a Surgical Model of Post-Traumatic Osteoarthritis. Sci. Rep. 2018, 8, 6438. [Google Scholar] [CrossRef] [Green Version]
- Maumus, M.; Roussignol, G.; Toupet, K.; Penarier, G.; Bentz, I.; Teixeira, S.; Oustric, D.; Jung, M.; Lepage, O.; Steinberg, R.; et al. Utility of a Mouse Model of Osteoarthritis to Demonstrate Cartilage Protection by IFNgamma-Primed Equine Mesenchymal Stem Cells. Front. Immunol. 2016, 7, 392. [Google Scholar] [CrossRef] [Green Version]
- Barve, R.A.; Minnerly, J.C.; Weiss, D.J.; Meyer, D.M.; Aguiar, D.J.; Sullivan, P.M.; Weinrich, S.L.; Head, R.D. Transcriptional profiling and pathway analysis of monosodium iodoacetate-induced experimental osteoarthritis in rats: Relevance to human disease. Osteoarthr. Cartil. 2007, 15, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, K.M.; Ball, A.D.; Jones, H.B.; Brinckmann, S.; Read, S.J.; Murray, F. Cellular and histopathological changes in the infrapatellar fat pad in the monoiodoacetate model of osteoarthritis pain. Osteoarthr. Cartil. 2009, 17, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Guzman, R.E.; Evans, M.G.; Bove, S.; Morenko, B.; Kilgore, K. Mono-iodoacetate-induced histologic changes in subchondral bone and articular cartilage of rat femorotibial joints: An animal model of osteoarthritis. Toxicol. Pathol. 2003, 31, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Ter, H.F.; Luiz, A.P.; Santana-Varela, S.; Chessell, I.P.; Welsh, F.; Wood, J.N.; Chenu, C. Noninvasive Mechanical Joint Loading as an Alternative Model for Osteoarthritic Pain. Arthritis Rheumatol. 2019, 71, 1078–1088. [Google Scholar] [CrossRef]
- Sokoloff, L.; Crittenden, L.B.; Yamamoto, R.S.; Jay, G.E., Jr. The genetics of degenerative joint disease in mice. Arthritis Rheum. 1962, 5, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Cheon, E.J. Animal Model of Osteoarthritis. J. Rheum. Dis. 2012, 19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.X.; Ren, K.; Dubner, R. Osteoarthritis pain mechanisms: Basic studies in animal models. Osteoarthr. Cartil. 2013, 21, 1308–1315. [Google Scholar] [CrossRef] [Green Version]
- Deuis, J.R.; Dvorakova, L.S.; Vetter, I. Methods Used to Evaluate Pain Behaviors in Rodents. Front. Mol. Neurosci. 2017, 10, 284. [Google Scholar] [CrossRef] [Green Version]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Pitcher, T.; Sousa-Valente, J.; Malcangio, M. The Monoiodoacetate Model of Osteoarthritis Pain in the Mouse. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, B.Y.; Allen, K.D. Factors affecting the reliability of behavioral assessments for rodent osteoarthritis models. Lab. Anim. 2019. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Kumar, P.; Jung, K.; Graf, I.; Menkhoff, H.; Schulz, X.; Bahr, M.; Hein, K. CatWalk gait analysis in a rat model of multiple sclerosis. BMC Neurosci. 2016, 17, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholami, M.; Saboory, E.; Mehraban, S.; Niakani, A.; Banihabib, N.; Azad, M.R.; Fereidoni, J. Time dependent antinociceptive effects of morphine and tramadol in the hot plate test: Using different methods of drug administration in female rats. Iran. J. Pharm. Res. 2015, 14, 303–311. [Google Scholar] [PubMed]
- O’Callaghan, J.P.; Holtzman, S.G. Quantification of the analgesic activity of narcotic antagonists by a modified hot-plate procedure. J. Pharmacol. Exp. Ther. 1975, 192, 497–505. [Google Scholar] [PubMed]
- Shiotsuki, H.; Yoshimi, K.; Shimo, Y.; Funayama, M.; Takamatsu, Y.; Ikeda, K.; Takahashi, R.; Kitazawa, S.; Hattori, N. A rotarod test for evaluation of motor skill learning. J. Neurosci. Methods 2010, 189, 180–185. [Google Scholar] [CrossRef]
- Lynch, J.J.; Castagne, V.; Moser, P.C.; Mittelstadt, S.W. Comparison of methods for the assessment of locomotor activity in rodent safety pharmacology studies. J. Pharmacol. Toxicol. Methods 2011, 64, 74–80. [Google Scholar] [CrossRef]
- Quinn, L.P.; Stean, T.O.; Trail, B.; Duxon, M.S.; Stratton, S.C.; Billinton, A.; Upton, N. LABORAS: Initial pharmacological validation of a system allowing continuous monitoring of laboratory rodent behaviour. J. Neurosci. Methods 2003, 130, 83–92. [Google Scholar] [CrossRef]
- Miller, R.E.; Tran, P.B.; Das, R.; Ghoreishi-Haack, N.; Ren, D.; Miller, R.J.; Malfait, A.M. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc. Natl. Acad. Sci. USA 2012, 109, 20602–20607. [Google Scholar] [CrossRef] [Green Version]
- Nwosu, L.N.; Mapp, P.I.; Chapman, V.; Walsh, D.A. Blocking the tropomyosin receptor kinase A (TrkA) receptor inhibits pain behaviour in two rat models of osteoarthritis. Ann. Rheum. Dis. 2016, 75, 1246–1254. [Google Scholar] [CrossRef] [Green Version]
- Harvey, V.L.; Dickenson, A.H. Behavioural and electrophysiological characterisation of experimentally induced osteoarthritis and neuropathy in C57Bl/6 mice. Mol. Pain 2009, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Wen, Z.H.; Chang, Y.C.; Huang, S.Y.; Tang, C.C.; Chen, W.F.; Hsieh, S.P.; Hsieh, C.S.; Jean, Y.H. Intra-articular magnesium sulfate (MgSO4) reduces experimental osteoarthritis and nociception: Association with attenuation of N-methyl-D-aspartate (NMDA) receptor subunit 1 phosphorylation and apoptosis in rat chondrocytes. Osteoarthr. Cartil. 2009, 17, 1485–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashraf, S.; Mapp, P.I.; Burston, J.; Bennett, A.J.; Chapman, V.; Walsh, D.A. Augmented pain behavioural responses to intra-articular injection of nerve growth factor in two animal models of osteoarthritis. Ann. Rheum. Dis. 2014, 73, 1710–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.C.; Zhu, B.; Jing, X.H.; Xiong, L.Z.; Wu, C.H.; Gao, F.; Li, H.P.; Xiang, H.C.; Zhu, H.; Zhou, B.; et al. Electroacupuncture Potentiates Cannabinoid Receptor-Mediated Descending Inhibitory Control in a Mouse Model of Knee Osteoarthritis. Front. Mol. Neurosci. 2018, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Comi, E.; Lanza, M.; Ferrari, F.; Mauri, V.; Caselli, G.; Rovati, L.C. Efficacy of CR4056, a first-in-class imidazoline-2 analgesic drug, in comparison with naproxen in two rat models of osteoarthritis. J. Pain Res. 2017, 10, 1033–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulet, B.; de Souza, R.; Knights, C.B.; Gentry, C.; Wilson, A.M.; Bevan, S.; Chang, Y.M.; Pitsillides, A.A. Modifications of gait as predictors of natural osteoarthritis progression in STR/Ort mice. Arthritis Rheumatol. 2014, 66, 1832–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Kraan, P.M.; Vitters, E.L.; van Beuningen, H.M.; van de Putte, L.B.; van den Berg, W.B. Degenerative knee joint lesions in mice after a single intra-articular collagenase injection. A new model of osteoarthritis. J. Exp. Pathol. 1990, 71, 19–31. [Google Scholar]
- Choi, W.S.; Lee, G.; Song, W.H.; Koh, J.T.; Yang, J.; Kwak, J.S.; Kim, H.E.; Kim, S.K.; Son, Y.O.; Nam, H.; et al. The CH25H-CYP7B1-RORalpha axis of cholesterol metabolism regulates osteoarthritis. Nature 2019, 566, 254–258. [Google Scholar] [CrossRef]
- Suter, E.; Herzog, W.; Leonard, T.R.; Nguyen, H. One-year changes in hind limb kinematics, ground reaction forces and knee stability in an experimental model of osteoarthritis. J. Biomech. 1998, 31, 511–517. [Google Scholar] [CrossRef]
- Janusz, M.J.; Hookfin, E.B.; Heitmeyer, S.A.; Woessner, J.F.; Freemont, A.J.; Hoyland, J.A.; Brown, K.K.; Hsieh, L.C.; Almstead, N.G.; De, B.; et al. Moderation of iodoacetate-induced experimental osteoarthritis in rats by matrix metalloproteinase inhibitors. Osteoarthr. Cartil. 2001, 9, 751–760. [Google Scholar] [CrossRef]
- Mason, R.M.; Chambers, M.G.; Flannelly, J.; Gaffen, J.D.; Dudhia, J.; Bayliss, M.T. The STR/ort mouse and its use as a model of osteoarthritis. Osteoarthr. Cartil. 2001, 9, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Huang, L.; Welch, I.; Norley, C.; Holdsworth, D.W.; Beier, F.; Cai, D. Early Changes of Articular Cartilage and Subchondral Bone in The DMM Mouse Model of Osteoarthritis. Sci. Rep. 2018, 8, 2855. [Google Scholar] [CrossRef] [PubMed]
- Knights, C.B.; Gentry, C.; Bevan, S. Partial medial meniscectomy produces osteoarthritis pain-related behaviour in female C57BL/6 mice. Pain 2012, 153, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.G.; Yu, B.; Mao, Y.Q.; Zhao, X.; Wang, X.Q.; Ding, H.F.; Cao, L.; Liu, G.W.; Nie, S.B.; Liu, S.; et al. Efficacy of zoledronic acid in treatment of teoarthritis is dependent on the disease progression stage in rat medial meniscal tear model. Acta Pharmacol. Sin. 2012, 33, 924–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cesare Mannelli, L.; Micheli, L.; Zanardelli, M.; Ghelardini, C. Low dose native type II collagen prevents pain in a rat osteoarthritis model. BMC Musculoskelet. Disord. 2013, 14, 228. [Google Scholar] [CrossRef] [Green Version]
- Ko, F.C.; Dragomir, C.; Plumb, D.A.; Goldring, S.R.; Wright, T.M.; Goldring, M.B.; van der Meulen, M.C. In vivo cyclic compression causes cartilage degeneration and subchondral bone changes in mouse tibiae. Arthritis Rheum. 2013, 65, 1569–1578. [Google Scholar] [CrossRef] [Green Version]
- Ruan, M.Z.; Patel, R.M.; Dawson, B.C.; Jiang, M.M.; Lee, B.H. Pain, motor and gait assessment of murine osteoarthritis in a cruciate ligament transection model. Osteoarthr. Cartil. 2013, 21, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Naik, S.R.; Wala, S.M. Arthritis, a complex connective and synovial joint destructive autoimmune disease: Animal models of arthritis with varied etiopathology and their significance. J. Postgrad. Med. 2014, 60, 309–317. [Google Scholar] [CrossRef]
- Huang, H.; Skelly, J.D.; Ayers, D.C.; Song, J. Age-dependent Changes in the Articular Cartilage and Subchondral Bone of C57BL/6 Mice after Surgical Destabilization of Medial Meniscus. Sci. Rep. 2017, 7, 42294. [Google Scholar] [CrossRef] [Green Version]
- Kung, L.H.W.; Ravi, V.; Rowley, L.; Bell, K.M.; Little, C.B.; Bateman, J.F. Comprehensive Expression Analysis of microRNAs and mRNAs in Synovial Tissue from a Mouse Model of Early Post-Traumatic Osteoarthritis. Sci. Rep. 2017, 7, 17701. [Google Scholar] [CrossRef] [Green Version]
- Van Dalen, S.C.; Blom, A.B.; Slöetjes, A.W.; Helsen, M.M.; Roth, J.; Vogl, T.; van de Loo, F.A.; Koenders, M.I.; van der Kraan, P.M.; van den Berg, W.B.; et al. Interleukin-1 is not involved in synovial inflammation and cartilage destruction in collagenase-induced osteoarthritis. Osteoarthr. Cartil. 2017, 25, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Song, D.H.; Kim, S.H.; Jung, Y.; Kim, S.J. Development and characterization of various osteoarthritis models for tissue engineering. PLoS ONE 2018, 13, e0194288. [Google Scholar] [CrossRef] [PubMed]
- Tawonsawatruk, T.; Sriwatananukulkit, O.; Himakhun, W.; Hemstapat, W. Comparison of pain behaviour and osteoarthritis progression between anterior cruciate ligament transection and osteochondral injury in rat models. Bone Joint Res. 2018, 7, 244–251. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, D.; Lin, Y.; Yuan, Q.; Zhou, X. Anterior Cruciate Ligament Transection-Induced Cellular and Extracellular Events in Menisci: Implications for Osteoarthritis. Am. J. Sports Med. 2018, 46, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhu, J.; Zhen, G.; Hu, Y.; An, S.; Li, Y.; Zheng, Q.; Chen, Z.; Yang, Y.; Wan, M.; et al. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J. Clin. Invest. 2019, 129, 1076–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, M.; Yang, M.; Yu, N.; Zhen, G.; Wan, M.; Liu, W.; Ji, B.; Ma, H.; Guo, Q.; Tong, P.; et al. Inhibition of cyclooxygenase-2 activity in subchondral bone modifies a subtype of osteoarthritis. Bone Res. 2019, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Carcole, M.; Zamanillo, D.; Merlos, M.; Fernandez-Pastor, B.; Cabanero, D.; Maldonado, R. Blockade of the Sigma-1 Receptor Relieves Cognitive and Emotional Impairments Associated to Chronic Osteoarthritis Pain. Front. Pharmacol. 2019, 10, 468. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, H.N.; Rathod, S.; Wolf, M.T.; Elisseeff, J.H. Intra-articular Injection of Urinary Bladder Matrix Reduces Osteoarthritis Development. AAPS J. 2017, 19, 141–149. [Google Scholar] [CrossRef]
- Fang, H.; Beier, F. Mouse models of osteoarthritis: Modelling risk factors and assessing outcomes. Nat. Rev. Rheumatol. 2014, 10, 413–421. [Google Scholar] [CrossRef]
- Bapat, S.; Hubbard, D.; Munjal, A.; Hunter, M.; Fulzele, S. Pros and cons of mouse models for studying osteoarthritis. Clin. Transl. Med. 2018, 7, 36. [Google Scholar] [CrossRef] [Green Version]
- Thysen, S.; Luyten, F.P.; Lories, R.J. Targets, models and challenges in osteoarthritis research. Dis. Model Mech. 2015, 8, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Fernihough, J.; Gentry, C.; Malcangio, M.; Fox, A.; Rediske, J.; Pellas, T.; Kidd, B.; Bevan, S.; Winter, J. Pain related behaviour in two models of osteoarthritis in the rat knee. Pain 2004, 112, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Huang, J.; Fan, Y.; Li, J.; You, T.; He, S.; Xiao, G.; Chen, D. Exploration of CRISPR/Cas9-based gene editing as therapy for osteoarthritis. Ann. Rheum. Dis. 2019, 78, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Malfait, A.M.; Miller, R.J. Emerging Targets for the Management of Osteoarthritis Pain. Curr. Osteoporos. Rep. 2016, 14, 260–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kc, R.; Li, X.; Kroin, J.S.; Liu, Z.; Chen, D.; Xiao, G.; Levine, B.; Li, J.; Hamilton, J.L.; van Wijnen, A.J.; et al. PKCdelta null mutations in a mouse model of osteoarthritis alter osteoarthritic pain independently of joint pathology by augmenting NGF/TrkA-induced axonal outgrowth. Ann. Rheum. Dis. 2016, 75, 2133–2141. [Google Scholar] [CrossRef] [Green Version]
- Malfait, A.M.; Ritchie, J.; Gil, A.S.; Austin, J.S.; Hartke, J.; Qin, W.; Tortorella, M.D.; Mogil, J.S. ADAMTS-5 deficient mice do not develop mechanical allodynia associated with osteoarthritis following medial meniscal destabilization. Osteoarthr. Cartil. 2010, 18, 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.E.; Tran, P.B.; Ishihara, S.; Larkin, J.; Malfait, A.M. Therapeutic effects of an anti-ADAMTS-5 antibody on joint damage and mechanical allodynia in a murine model of osteoarthritis. Osteoarthr. Cartil. 2016, 24, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.C.; Roberts, C.R.; Steeves, J.D.; Tetzlaff, W. Aggrecan components differentially modulate nerve growth factor-responsive and neurotrophin-3-responsive dorsal root ganglion neurite growth. J. Neurosci. Res. 2008, 86, 581–592. [Google Scholar] [CrossRef]
- Chen, D.; Shen, J.; Zhao, W.; Wang, T.; Han, L.; Hamilton, J.L.; Im, H.J. Osteoarthritis: Toward a comprehensive understanding of pathological mechanism. Bone Res. 2017, 5, 16044. [Google Scholar] [CrossRef]
- Miller, R.E.; Ishihara, S.; Tran, P.B.; Golub, S.B.; Last, K.; Miller, R.J.; Fosang, A.J.; Malfait, A.M. An aggrecan fragment drives osteoarthritis pain through Toll-like receptor 2. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Miller, R.E.; Belmadani, A.; Ishihara, S.; Tran, P.B.; Ren, D.; Miller, R.J.; Malfait, A.M. Damage-associated molecular patterns generated in osteoarthritis directly excite murine nociceptive neurons through Toll-like receptor 4. Arthritis Rheumatol. 2015, 67, 2933–2943. [Google Scholar] [CrossRef]
- Miller, R.E.; Scanzello, C.R.; Malfait, A.M. An emerging role for Toll-like receptors at the neuroimmune interface in osteoarthritis. Semin. Immunopathol. 2019, 41, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Ivanavicius, S.P.; Ball, A.D.; Heapy, C.G.; Westwood, F.R.; Murray, F.; Read, S.J. Structural pathology in a rodent model of osteoarthritis is associated with neuropathic pain: Increased expression of ATF-3 and pharmacological characterisation. Pain 2007, 128, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Tsuji, K.; Onuma, H.; Udo, M.; Ueki, H.; Akiyama, M.; Abula, K.; Katagiri, H.; Miyatake, K.; Watanabe, T.; et al. Persistent synovial inflammation plays important roles in persistent pain development in the rat knee before cartilage degradation reaches the subchondral bone. BMC Musculoskelet. Disord. 2018, 19, 291. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Saleh, R.; Achuthan, A.; Fleetwood, A.J.; Forster, I.; Hamilton, J.A.; Cook, A.D. CCL17 blockade as a therapy for osteoarthritis pain and disease. Arthritis Res. Ther. 2018, 20, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, G.J.; Welldon, K.J.; Halbout, P.; Findlay, D.M. Strontium ranelate treatment of human primary osteoblasts promotes an osteocyte-like phenotype while eliciting an osteoprotegerin response. Osteoporos. Int. 2009, 20, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Reginster, J.Y.; Badurski, J.; Bellamy, N.; Bensen, W.; Chapurlat, R.; Chevalier, X.; Christiansen, C.; Genant, H.; Navarro, F.; Nasonov, E.; et al. Efficacy and safety of strontium ranelate in the treatment of knee osteoarthritis: Results of a double-blind, randomised placebo-controlled trial. Ann. Rheum. Dis. 2013, 72, 179–186. [Google Scholar] [CrossRef]
- Pelletier, J.P.; Roubille, C.; Raynauld, J.P.; Abram, F.; Dorais, M.; Delorme, P.; Martel-Pelletier, J. Disease-modifying effect of strontium ranelate in a subset of patients from the Phase III knee osteoarthritis study SEKOIA using quantitative MRI: Reduction in bone marrow lesions protects against cartilage loss. Ann. Rheum. Dis. 2015, 74, 422–429. [Google Scholar] [CrossRef]
- Laslett, L.L.; Dore, D.A.; Quinn, S.J.; Boon, P.; Ryan, E.; Winzenberg, T.M.; Jones, G. Zoledronic acid reduces knee pain and bone marrow lesions over 1 year: A randomised controlled trial. Ann. Rheum. Dis. 2012, 71, 1322–1328. [Google Scholar] [CrossRef]
- O’Conor, C.J.; Griffin, T.M.; Liedtke, W.; Guilak, F. Increased susceptibility of Trpv4-deficient mice to obesity and obesity-induced osteoarthritis with very high-fat diet. Ann. Rheum. Dis. 2013, 72, 300–304. [Google Scholar] [CrossRef] [Green Version]
- Triantaphyllidou, I.E.; Kalyvioti, E.; Karavia, E.; Lilis, I.; Kypreos, K.E.; Papachristou, D.J. Perturbations in the HDL metabolic pathway predispose to the development of osteoarthritis in mice following long-term exposure to western-type diet. Osteoarthr. Cartil. 2013, 21, 322–330. [Google Scholar] [CrossRef] [Green Version]
- De Munter, W.; Blom, A.B.; Helsen, M.M.; Walgreen, B.; van der Kraan, P.M.; Joosten, L.A.; van den Berg, W.B.; van Lent, P.L. Cholesterol accumulation caused by low density lipoprotein receptor deficiency or a cholesterol-rich diet results in ectopic bone formation during experimental osteoarthritis. Arthritis Res. Ther. 2013, 15, R178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, T.M.; Huebner, J.L.; Kraus, V.B.; Guilak, F. Extreme obesity due to impaired leptin signaling in mice does not cause knee osteoarthritis. Arthritis Rheum. 2009, 60, 2935–2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louer, C.R.; Furman, B.D.; Huebner, J.L.; Kraus, V.B.; Olson, S.A.; Guilak, F. Diet-induced obesity significantly increases the severity of posttraumatic arthritis in mice. Arthritis Rheum. 2012, 64, 3220–3230. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Jain, D.; McNeill, J.N.; Little, D.; Anderson, J.A.; Huebner, J.L.; Kraus, V.B.; Rodriguiz, R.M.; Wetsel, W.C.; Guilak, F. Dietary fatty acid content regulates wound repair and the pathogenesis of osteoarthritis following joint injury. Ann. Rheum. Dis. 2015, 74, 2076–2083. [Google Scholar] [CrossRef]
- Jay, G.E., Jr.; Sokoloff, L. Natural history of degenerative joint disease in small laboratory animals. II. Epiphyseal maturation and osteoarthritis of the knee of mice of inbred strains. Ann. Arch. Pathol. 1956, 62, 129–135. [Google Scholar]
- Ochoa, J.; Mair, W.G. The normal sural nerve in man. II. Changes in the axons and Schwann cells due to ageing. Acta Neuropathol. 1969, 13, 217–239. [Google Scholar] [CrossRef]
- Ko, M.L.; King, M.A.; Gordon, T.L.; Crisp, T. The effects of aging on spinal neurochemistry in the rat. Brain Res. Bull. 1997, 42, 95–98. [Google Scholar] [CrossRef]
- Hamm, R.J.; Knisely, J.S. Environmentally induced analgesia: Age-related decline in a neurally mediated, nonopioid system. Psychol. Aging 1986, 1, 195–201. [Google Scholar] [CrossRef]
- Stannus, O.P.; Jones, G.; Blizzard, L.; Cicuttini, F.M.; Ding, C. Associations between serum levels of inflammatory markers and change in knee pain over 5 years in older adults: A prospective cohort study. Ann. Rheum. Dis. 2013, 72, 535–540. [Google Scholar] [CrossRef]
- Greene, M.A.; Loeser, R.F. Aging-related inflammation in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1966–1971. [Google Scholar] [CrossRef] [Green Version]
- Penninx, B.W.; Abbas, H.; Ambrosius, W.; Nicklas, B.J.; Davis, C.; Messier, S.P.; Pahor, M. Inflammatory markers and physical function among older adults with knee osteoarthritis. J. Rheumatol. 2004, 31, 2027–2031. [Google Scholar] [PubMed]
- De Hooge, A.S.; van de Loo, F.A.; Bennink, M.B.; Arntz, O.J.; de Hooge, P.; van den Berg, W.B. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthr. Cartil. 2005, 13, 66–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent cells and osteoarthritis: A painful connection. J. Clin. Investig. 2018, 128, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Ann. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.M.; Kim, S.J. Thymoquinone-induced reactive oxygen species causes apoptosis of chondrocytes via PI3K/Akt and p38kinase pathway. Exp. Biol. Med. 2013, 238, 811–820. [Google Scholar] [CrossRef]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Lorenz, J.; Grassel, S. Experimental osteoarthritis models in mice. Methods Mol. Biol. 2014, 1194, 401–419. [Google Scholar] [CrossRef]
- Ma, H.L.; Blanchet, T.J.; Peluso, D.; Hopkins, B.; Morris, E.A.; Glasson, S.S. Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthr. Cartil. 2007, 15, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Mahr, S.; Menard, J.; Krenn, V.; Muller, B. Sexual dimorphism in the osteoarthritis of STR/ort mice may be linked to articular cytokines. Ann. Rheum. Dis. 2003, 62, 1234–1237. [Google Scholar] [CrossRef] [Green Version]
- Van Osch, G.J.; van der Kraan, P.M.; Vitters, E.L.; Blankevoort, L.; van den Berg, W.B. Induction of osteoarthritis by intra-articular injection of collagenase in mice. Strain and sex related differences. Osteoarthr. Cartil. 1993, 1, 171–177. [Google Scholar] [CrossRef]
- Park, I.Y.; Hong, J.I.; Hwang, H.S.; Kim, H.A. Evaluation of cartilage degeneration and osteoarthritis pain on female and male mouse model of osteoarthritis. Osteoarthr. Cartil. 2018, 26, S360. [Google Scholar] [CrossRef] [Green Version]
- Sorge, R.E.; Totsch, S.K. Sex Differences in Pain. J. Neurosci. Res. 2017, 95, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.D.; Adeyemo, A.; O’Leary, M.E.; Bottaro, A. Animal models of rheumatoid pain: Experimental systems and insights. Arthritis Res. Ther. 2017, 19, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maini, R.; St Clair, E.W.; Breedveld, F.; Furst, D.; Kalden, J.; Weisman, M.; Smolen, J.; Emery, P.; Harriman, G.; Feldmann, M.; et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: A randomised phase III trial. ATTRACT Study Group. Lancet 1999, 354, 1932–1939. [Google Scholar] [CrossRef]
- Singh, J.A.; Christensen, R.; Wells, G.A.; Suarez-Almazor, M.E.; Buchbinder, R.; Lopez-Olivo, M.A.; Tanjong Ghogomu, E.; Tugwell, P. Biologics for rheumatoid arthritis: An overview of Cochrane reviews. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef] [Green Version]
- Hess, A.; Axmann, R.; Rech, J.; Finzel, S.; Heindl, C.; Kreitz, S.; Sergeeva, M.; Saake, M.; Garcia, M.; Kollias, G.; et al. Blockade of TNF-alpha rapidly inhibits pain responses in the central nervous system. Proc. Natl. Acad. Sci. USA 2011, 108, 3731–3736. [Google Scholar] [CrossRef] [Green Version]
- Smolen, J.S.; Strand, V.; Koenig, A.S.; Szumski, A.; Kotak, S.; Jones, T.V. Discordance between patient and physician assessments of global disease activity in rheumatoid arthritis and association with work productivity. Arthritis Res. Ther. 2016, 18, 114. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, D.F.; Walsh, D.A. Pain mechanisms in rheumatoid arthritis. Clin. Exp. Rheumatol. 2017, 35 (Suppl. 107), 94–101. [Google Scholar]
- Wolfe, F.; Hauser, W.; Hassett, A.L.; Katz, R.S.; Walitt, B.T. The development of fibromyalgia--I: Examination of rates and predictors in patients with rheumatoid arthritis (RA). Pain 2011, 152, 291–299. [Google Scholar] [CrossRef]
- Lee, Y.C.; Lu, B.; Edwards, R.R.; Wasan, A.D.; Nassikas, N.J.; Clauw, D.J.; Solomon, D.H.; Karlson, E.W. The role of sleep problems in central pain processing in rheumatoid arthritis. Arthritis Rheum. 2013, 65, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Joharatnam, N.; McWilliams, D.F.; Wilson, D.; Wheeler, M.; Pande, I.; Walsh, D.A. A cross-sectional study of pain sensitivity, disease-activity assessment, mental health, and fibromyalgia status in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krock, E.; Jurczak, A.; Svensson, C.I. Pain pathogenesis in rheumatoid arthritis-what have we learned from animal models? Pain 2018, 159 (Suppl. 1), S98–S109. [Google Scholar] [CrossRef] [PubMed]
- Bas, D.B.; Su, J.; Wigerblad, G.; Svensson, C.I. Pain in rheumatoid arthritis: Models and mechanisms. Pain Manag. 2016, 6, 265–284. [Google Scholar] [CrossRef]
- Buckley, B.J.; Ali, U.; Kelso, M.J.; Ranson, M. The Urokinase Plasminogen Activation System in Rheumatoid Arthritis: Pathophysiological Roles and Prospective Therapeutic Targets. Curr. Drug Targets 2019, 20, 970–981. [Google Scholar] [CrossRef]
- Asquith, D.L.; Miller, A.M.; McInnes, I.B.; Liew, F.Y. Animal models of rheumatoid arthritis. Eur. J. Immunol. 2009, 39, 2040–2044. [Google Scholar] [CrossRef]
- Caplazi, P.; Baca, M.; Barck, K.; Carano, R.A.; DeVoss, J.; Lee, W.P.; Bolon, B.; Diehl, L. Mouse Models of Rheumatoid Arthritis. Vet. Pathol. 2015, 52, 819–826. [Google Scholar] [CrossRef]
- Bevaart, L.; Vervoordeldonk, M.J.; Tak, P.P. Evaluation of therapeutic targets in animal models of arthritis: How does it relate to rheumatoid arthritis? Arthritis Rheum. 2010, 62, 2192–2205. [Google Scholar] [CrossRef]
- Moudgil, K.D.; Kim, P.; Brahn, E. Advances in rheumatoid arthritis animal models. Curr. Rheumatol. Rep. 2011, 13, 456–463. [Google Scholar] [CrossRef] [Green Version]
- Roy, T.; Ghosh, S. Animal models of rheumatoid arthritis: Correlation and usefulness with human rheumatoid arthritis. Indo. Am. J. Pharm. Res. 2013, 3, 6131–6142. [Google Scholar]
- Abramson, S.B.; Amin, A. Blocking the effects of IL-1 in rheumatoid arthritis protects bone and cartilage. Rheumatology 2002, 41, 972–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayer, S.; Bauer, G.; Willburger, M.; Sinn, K.; Alasti, F.; Plasenzotti, R.; Shvets, T.; Niederreiter, B.; Aschauer, C.; Steiner, G.; et al. Cartilage damage and bone erosion are more prominent determinants of functional impairment in longstanding experimental arthritis than synovial inflammation. Dis. Model. Mech. 2016, 9, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Impellizzeri, D.; Esposito, E.; Di Paola, R.; Ahmad, A.; Campolo, M.; Peli, A.; Morittu, V.M.; Britti, D.; Cuzzocrea, S. Palmitoylethanolamide and luteolin ameliorate development of arthritis caused by injection of collagen type II in mice. Arthritis Res. Ther. 2013, 15, R192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jochmann, E.; Boettger, M.K.; Anand, P.; Schaible, H.G. Antigen-induced arthritis in rats is associated with increased growth-associated protein 43-positive intraepidermal nerve fibres remote from the joint. Arthritis Res. Ther. 2015, 17, 299. [Google Scholar] [CrossRef] [Green Version]
- Christianson, C.A.; Corr, M.; Firestein, G.S.; Mobargha, A.; Yaksh, T.L.; Svensson, C.I. Characterization of the acute and persistent pain state present in K/BxN serum transfer arthritis. Pain 2010, 151, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Christianson, C.A.; Corr, M.; Yaksh, T.L.; Svensson, C.I. K/BxN serum transfer arthritis as a model of inflammatory joint pain. Methods Mol. Biol. 2012, 851, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Qu, L.; Caterina, M.J. Enhanced excitability and suppression of A-type K(+) currents in joint sensory neurons in a murine model of antigen-induced arthritis. Sci. Rep. 2016, 6, 28899. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.L.; Zhu, Y.M.; Qian, F.Y.; Yuan, C.C.; Yuan, D.P.; Zhou, X.P. MicroRNA-143-3p contributes to the regulation of pain responses in collagen-induced arthritis. Mol. Med. Rep. 2018, 18, 3219–3228. [Google Scholar] [CrossRef] [Green Version]
- Balkrishna, A.; Sakat, S.S.; Joshi, K.; Paudel, S.; Joshi, D.; Joshi, K.; Ranjan, R.; Gupta, A.; Bhattacharya, K.; Varshney, A. Anti-Inflammatory and Anti-Arthritic Efficacies of an Indian Traditional Herbo-Mineral Medicine “Divya Amvatari Ras” in Collagen Antibody-Induced Arthritis (CAIA) Mouse Model Through Modulation of IL-6/IL-1beta/TNF-alpha/NFkappaB Signaling. Front. Pharmacol. 2019, 10, 659. [Google Scholar] [CrossRef] [Green Version]
- Bersellini Farinotti, A.; Wigerblad, G.; Nascimento, D.; Bas, D.B.; Morado Urbina, C.; Nandakumar, K.S.; Sandor, K.; Xu, B.; Abdelmoaty, S.; Hunt, M.A.; et al. Cartilage-binding antibodies induce pain through immune complex-mediated activation of neurons. J. Exp. Med. 2019, 216, 1904–1924. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Zafra, T.; Gao, T.; Jurczak, A.; Sandor, K.; Hore, Z.; Agalave, N.M.; Su, J.; Estelius, J.; Lampa, J.; Hokfelt, T.; et al. Exploring the transcriptome of resident spinal microglia after collagen antibody-induced arthritis. Pain 2019, 160, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.S.; Seo, B.K.; Baek, Y.H. Analgesic effect of electroacupuncture on inflammatory pain in collagen-induced arthritis rats: Mediation by alpha2- and beta-adrenoceptors. Rheumatol. Int. 2013, 33, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Filippin, L.I.; Teixeira, V.N.; Viacava, P.R.; Lora, P.S.; Xavier, L.L.; Xavier, R.M. Temporal development of muscle atrophy in murine model of arthritis is related to disease severity. J. Cachexia Sarcopenia Muscle 2013, 4, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.S.; Williams, A.S.; Gilbert, S.J.; Harvey, A.K.; Evans, B.A.; Mason, D.J. AMPA/kainate glutamate receptors contribute to inflammation, degeneration and pain related behaviour in inflammatory stages of arthritis. Ann. Rheum. Dis. 2015, 74, 242–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujikado, N.; Saijo, S.; Iwakura, Y. Identification of arthritis-related gene clusters by microarray analysis of two independent mouse models for rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R100. [Google Scholar] [CrossRef] [Green Version]
- Akitsu, A.; Ishigame, H.; Kakuta, S.; Chung, S.H.; Ikeda, S.; Shimizu, K.; Kubo, S.; Liu, Y.; Umemura, M.; Matsuzaki, G.; et al. IL-1 receptor antagonist-deficient mice develop autoimmune arthritis due to intrinsic activation of IL-17-producing CCR2(+)Vgamma6(+)gammadelta T cells. Nat. Commun. 2015, 6, 7464. [Google Scholar] [CrossRef] [Green Version]
- Schinnerling, K.; Rosas, C.; Soto, L.; Thomas, R.; Aguillon, J.C. Humanized Mouse Models of Rheumatoid Arthritis for Studies on Immunopathogenesis and Preclinical Testing of Cell-Based Therapies. Front. Immunol. 2019, 10, 203. [Google Scholar] [CrossRef] [Green Version]
- Christianson, C.A.; Dumlao, D.S.; Stokes, J.A.; Dennis, E.A.; Svensson, C.I.; Corr, M.; Yaksh, T.L. Spinal TLR4 mediates the transition to a persistent mechanical hypersensitivity after the resolution of inflammation in serum-transferred arthritis. Pain 2011, 152, 2881–2891. [Google Scholar] [CrossRef] [Green Version]
- Bas, D.B.; Su, J.; Sandor, K.; Agalave, N.M.; Lundberg, J.; Codeluppi, S.; Baharpoor, A.; Nandakumar, K.S.; Holmdahl, R.; Svensson, C.I. Collagen antibody-induced arthritis evokes persistent pain with spinal glial involvement and transient prostaglandin dependency. Arthritis Rheum. 2012, 64, 3886–3896. [Google Scholar] [CrossRef]
- Inglis, J.J.; Notley, C.A.; Essex, D.; Wilson, A.W.; Feldmann, M.; Anand, P.; Williams, R. Collagen-induced arthritis as a model of hyperalgesia: Functional and cellular analysis of the analgesic actions of tumor necrosis factor blockade. Arthritis Rheum. 2007, 56, 4015–4023. [Google Scholar] [CrossRef]
- Clark, A.K.; Grist, J.; Al-Kashi, A.; Perretti, M.; Malcangio, M. Spinal cathepsin S and fractalkine contribute to chronic pain in the collagen-induced arthritis model. Arthritis Rheum. 2012, 64, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.G.; Cunha, T.M.; Vieira, S.M.; Lemos, H.P.; Verri, W.A., Jr.; Cunha, F.Q.; Ferreira, S.H. IL-17 mediates articular hypernociception in antigen-induced arthritis in mice. Pain 2010, 148, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Ebbinghaus, M.; Uhlig, B.; Richter, F.; von Banchet, G.S.; Gajda, M.; Brauer, R.; Schaible, H.G. The role of interleukin-1beta in arthritic pain: Main involvement in thermal, but not mechanical, hyperalgesia in rat antigen-induced arthritis. Arthritis Rheum. 2012, 64, 3897–3907. [Google Scholar] [CrossRef] [PubMed]
- Segond von Banchet, G.; Konig, C.; Patzer, J.; Eitner, A.; Leuchtweis, J.; Ebbinghaus, M.; Boettger, M.K.; Schaible, H.G. Long-Lasting Activation of the Transcription Factor CREB in Sensory Neurons by Interleukin-1beta During Antigen-Induced Arthritis in Rats: A Mechanism of Persistent Arthritis Pain? Arthritis Rheumatol. 2016, 68, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, K.; Faltus, R.; Robinson, G.; Sevilla, R.; Shin, J.; Zielstorff, M.; Byford, A.; Leccese, E.; Caniga, M.J.; Hseih, S.; et al. Etanercept ameliorates inflammation and pain in a novel mono-arthritic multi-flare model of streptococcal cell wall induced arthritis. BMC Musculoskelet. Disord. 2014, 15, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Jiang, X.; Zheng, Q.; Jeon, S.M.; Chen, T.; Liu, Y.; Kulaga, H.; Reed, R.; Dong, X.; Caterina, M.J.; et al. Neuronal FcgammaRI mediates acute and chronic joint pain. J. Clin. Invest. 2019, 130, 3754–3769. [Google Scholar] [CrossRef] [Green Version]
- Ebbinghaus, M.; Muller, S.; Segond von Banchet, G.; Eitner, A.; Wank, I.; Hess, A.; Hilger, I.; Kamradt, T.; Schaible, H.G. Contribution of Inflammation and Bone Destruction to Pain in Arthritis: A Study in Murine Glucose-6-Phosphate Isomerase-Induced Arthritis. Arthritis Rheumatol. 2019, 71, 2016–2026. [Google Scholar] [CrossRef]
- Frommholz, D.; Illges, H. Maximal locomotor depression follows maximal ankle swelling during the progression of arthritis in K/BxN mice. Rheumatol. Int. 2012, 32, 3999–4003. [Google Scholar] [CrossRef]



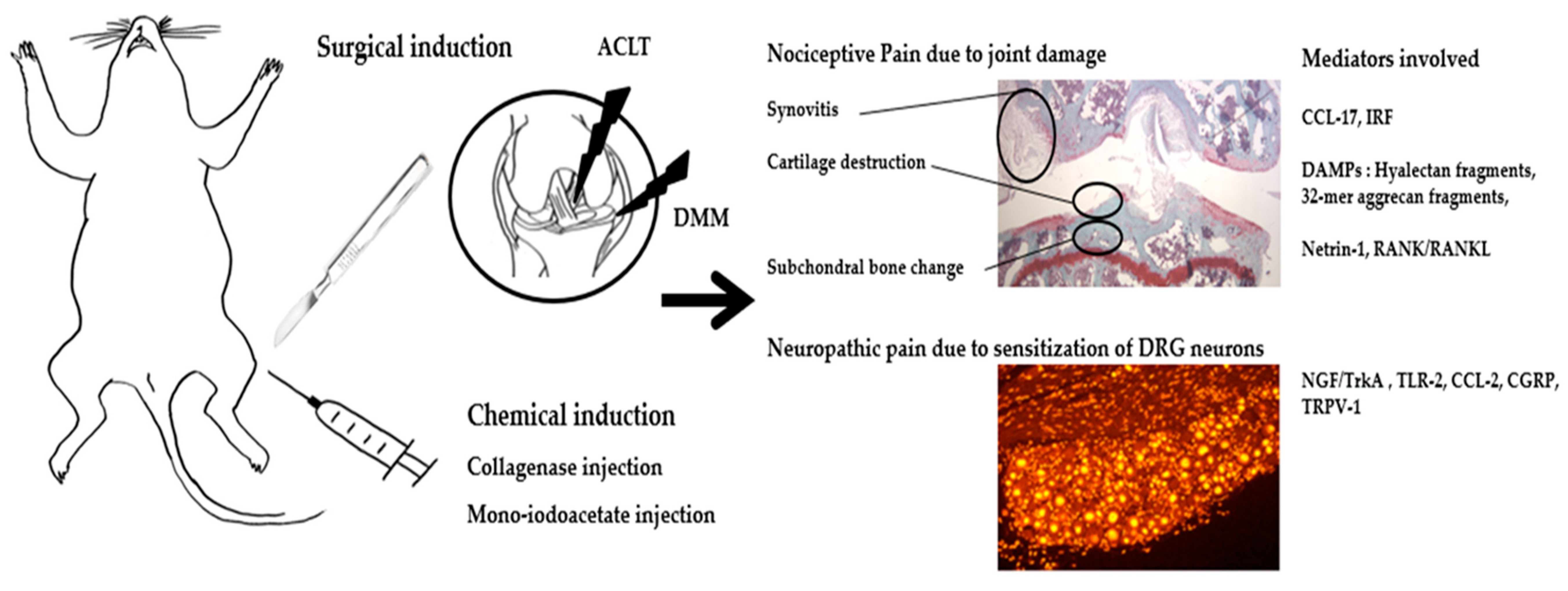{kind=link}
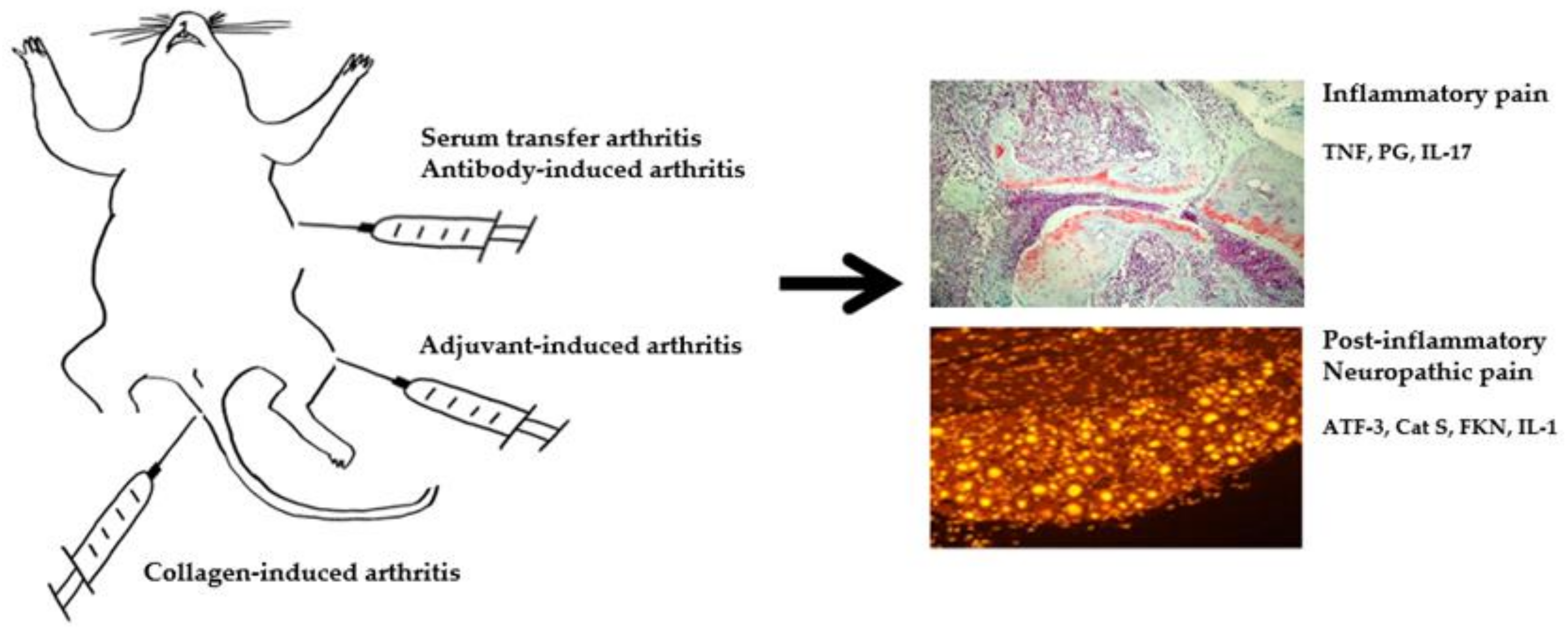{kind=link}
| Type | Model (Onset of Pathology) | Pros/Cons | Pathological Findings | Behavioral Assay (Pain Response Onset) | Findings | Molecular Pathogenesis Identified |
|---|---|---|---|---|---|---|
| Surgical | DMM (4 wk) | Mimics human post-traumatic OA, slow disease progression and mild cartilage damage, useful in assessing therapies. Need specially trained surgeon, risk of infection. | -Cartilage degradation -Synovial hyperplasia -Bone sclerosis -Osteophyte formation | von Frey (4 wk), incapacitance (4 wk), hot plate (8 wk), LABORAS (8 wk), gait abnormality (10 wk) | Pain response is progressively induced in early phase. Most representative method: von Frey | Loss of PKCδ exacerbates pain in DMM. ADAMTS-5 inhibition attenuates pain in DMM. Knockout of RANKL inhibits pain in ACLT mice. Knockout of Netrin-1 inhibits pain in ACLT mice. |
| MNX (2 wk) | More rapid disease onset and higher damage in the joint compared with human disease, useful in assessing therapies. Need specially trained surgeon, risk of infection. | von Frey (1 wk), incapacitance (3 d), cold plate (5 wk) | ||||
| ACLT (2 wk) | von Frey (1 wk), incapacitance (1 wk), hot plate (4 wk), rotarod (4 wk), LABORAS (1 wk), gait (8 w) | |||||
| Chemical | MIA (3–7 d) | Easy local injection method, rapid induction of severe joint degeneration, useful for studies in pain behavior. More difficult to translate to human setting. | -Inhibition of glycolysis and disruption of chondrocyte metabolism -Cartilage degradation -Synovial hyperplasia -Osteophyte formation | von Frey (1 wk), incapacitance (3 d), hot plate (6 d), rotarod (15 d), gait (6 d), LABORAS (14 d) | Pain response is induced rapidly within a week post-injection. Most representative methods: von Frey and incapacitance | Neutralization of CCL-17 ameliorates pain in CIOA. Increase of IRF-4, CCL-17 in CIOA. |
| CIOA (1–4 wk) | Most rapid progression, useful in assessing therapies. More difficult to translate to human setting. | -Cartilage degradation -Osteophyte formation | von Frey (1 wk), incapacitance (1 wk), hot plate (1 wk) | |||
| Spontaneous/noninvasive | STR/ort (8–16 wk) | Might mimic primary human OA, no specially trained personnel required. Long period for the development of OA, high variability of the disease phenotype and incidence. | -Cartilage degradation -Osteophyte formation | von Frey (no difference), gait (20 w), cold plate (no difference) | Pain response does not show significant variation with age. | STR/ort mice do not show any signs of pain even when treated with the opioid antagonist naloxone. |
| Mechanical joint loading (1 wk) | Noninvasive, suitable to study the effects of mechanical loading and intra-articular fracture, rapid induction of severe joint degeneration. Need specialized equipment. | -Cartilage degradation -Bone sclerosis -Osteophyte formation | von Frey (2 wk), incapacitance (4 wk), hot plate (no difference), rotarod (5 wk) | Pain response is progressively induced in early phase. | Anti-NGF alleviates pain in mechanical joint loading model. |
| Type | Model (Trigger, Onset of Pathology) | Pros/Cons | Pathological Findings | Behavioral Assay (Pain Response Onset) | Findings | Molecular Pathogenesis Identified |
|---|---|---|---|---|---|---|
| Induced (onset after first immunization) | CIA (CII/adjuvant) | Most common induced model, RA-like pathogenesis. Restricted strains in mice, severe progressive disease. | -Adaptive immune system activation against endogenous joint epitopes. -Inflammation -Immune cell infiltration -Joint destruction and synovial hyperplasia | von Frey (30 d), rotarod (5 d), locomotion (25 d), tail flick (1 wk), Hargreaves (24 d) | Pain response is induced rapidly after induction. Pain response increases in early phase and persists. Most representative methods: von Frey and Hargreaves | Anti-Cat S and FKN attenuate pain in CIA. Gabapentin and buprenorphine attenuate pain in CAIA. Knockout of TLR-4 inhibits pain in K/BxN. |
| CAIA (Anti-CII Ab) | Efficient and robust to study the effector phase of RA, diverse susceptible strains. Does not involve full spectrum of immune activation. | -Inflammation -Immune cell infiltration -Joint destruction | von Frey (6 d), hot plate (15 d), locomotion (5 d) | |||
| K/BxN (Serum/Anti-GPI Ab) | von Frey (2 d), Hargreaves (3 d), locomotion (3 d) | |||||
| AIA (mBSA/adjuvant) | Local RA-like pathogenesis, localized inflammation. Damage to cartilage less severe than in RA. | -Adaptive immune system activation against exogenous epitopes -Inflammation -Immune cell infiltration -Joint destruction and synovial hyperplasia | von Frey (22 d), Hargreaves (7 d), gait (15 d), incapacitance (17 d) | Pain response is induced rapidly after induction. Pain response increases in early phase and restores in late phase. Most representative methods: von Frey and incapacitance | Anti-IL-17 alleviates pain in AIA. Increase of IL-1RI and pCREB in AIA. | |
| Spontaneous | TNF-Tg (hTNF overexpression) | Useful for studies in effect of excess TNF in RA. Only been identified in mice, does not involve full spectrum of immune activation. | -Inflammation -Immune cell infiltration -Joint destruction and synovial hyperplasia -Pannus formation | von Frey (6 wk), tail flick (10 wk), Hargreaves (6 wk) | Pain response increases in early phase and persists. Most representative methods: von Frey and Hargreaves | Increase of the nociceptive brain activity in TNF-Tg mice. |
| IL-1RA−/− (Genetic deficiency of 1L-1Ra) | Useful for studies in effect of IL-1 signaling in RA. Only been identified in mice. | No behavioral data available to date. | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, J.-I.; Park, I.Y.; Kim, H.A. Understanding the Molecular Mechanisms Underlying the Pathogenesis of Arthritis Pain Using Animal Models. Int. J. Mol. Sci. 2020, 21, 533. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020533
Hong J-I, Park IY, Kim HA. Understanding the Molecular Mechanisms Underlying the Pathogenesis of Arthritis Pain Using Animal Models. International Journal of Molecular Sciences. 2020; 21(2):533. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020533
Chicago/Turabian StyleHong, Jeong-Im, In Young Park, and Hyun Ah Kim. 2020. "Understanding the Molecular Mechanisms Underlying the Pathogenesis of Arthritis Pain Using Animal Models" International Journal of Molecular Sciences 21, no. 2: 533. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020533