DNA Methylation Is Correlated with Gene Expression during Diapause Termination of Early Embryonic Development in the Silkworm (Bombyx mori)

 ,
,
Abstract
:1. Introduction
2. Results
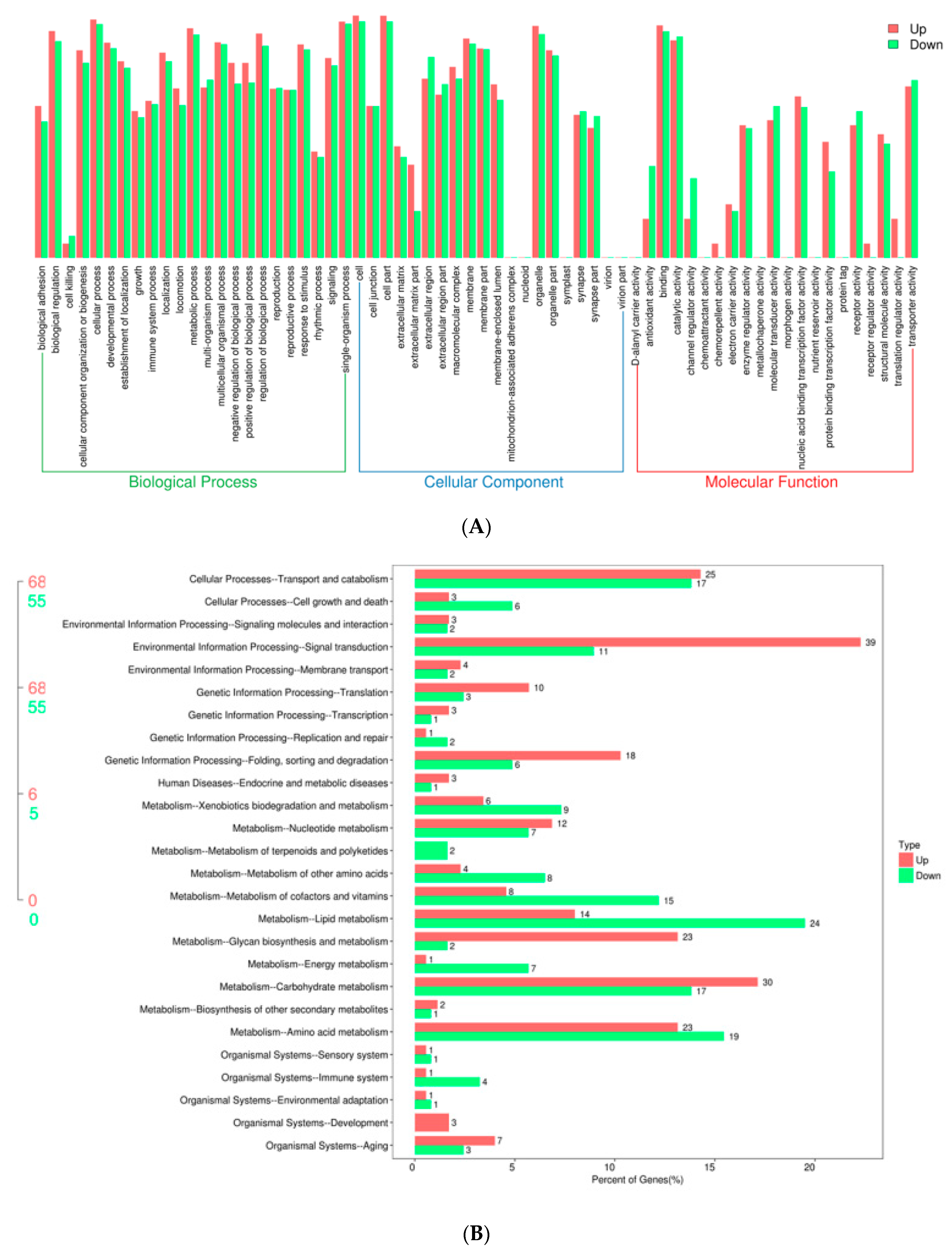
2.1. Transcriptional Profiles of Silkworm Diapause-Destined Eggs and Diapause-Terminated Eggs
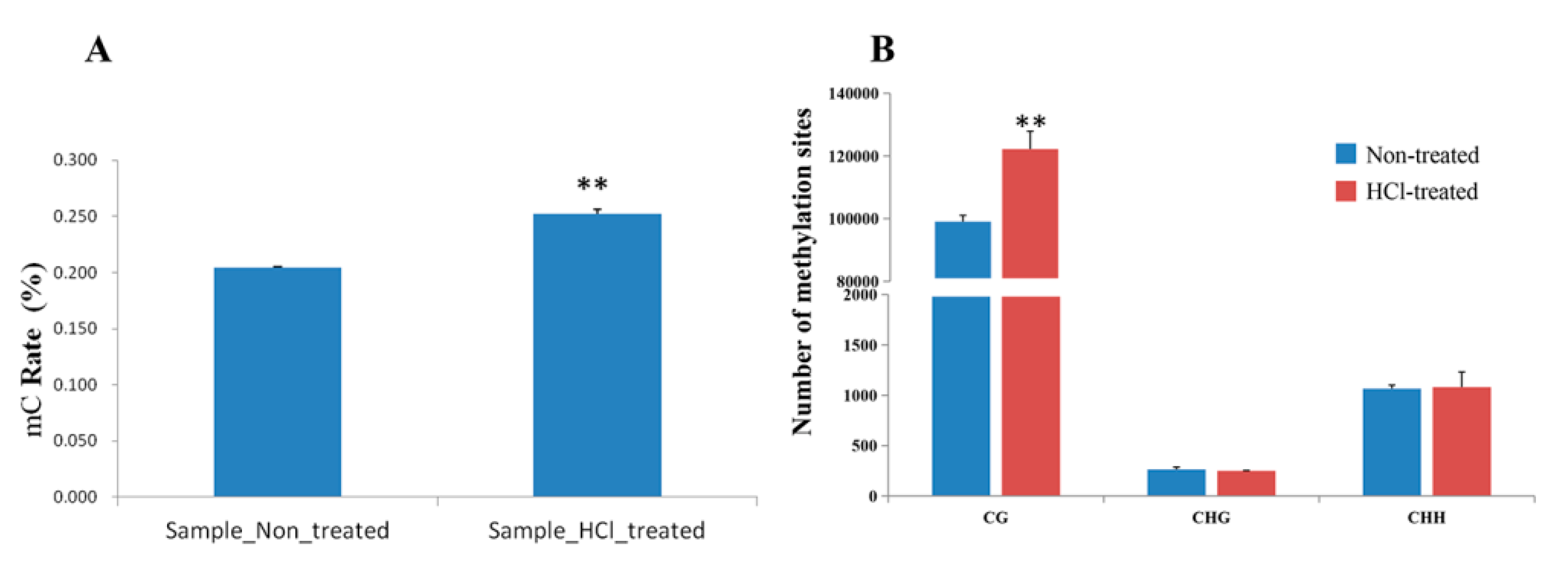
2.2. Overview of Methylation Landscapes of Silkworm Diapause-Destined Eggs and Diapause-Terminated Eggs
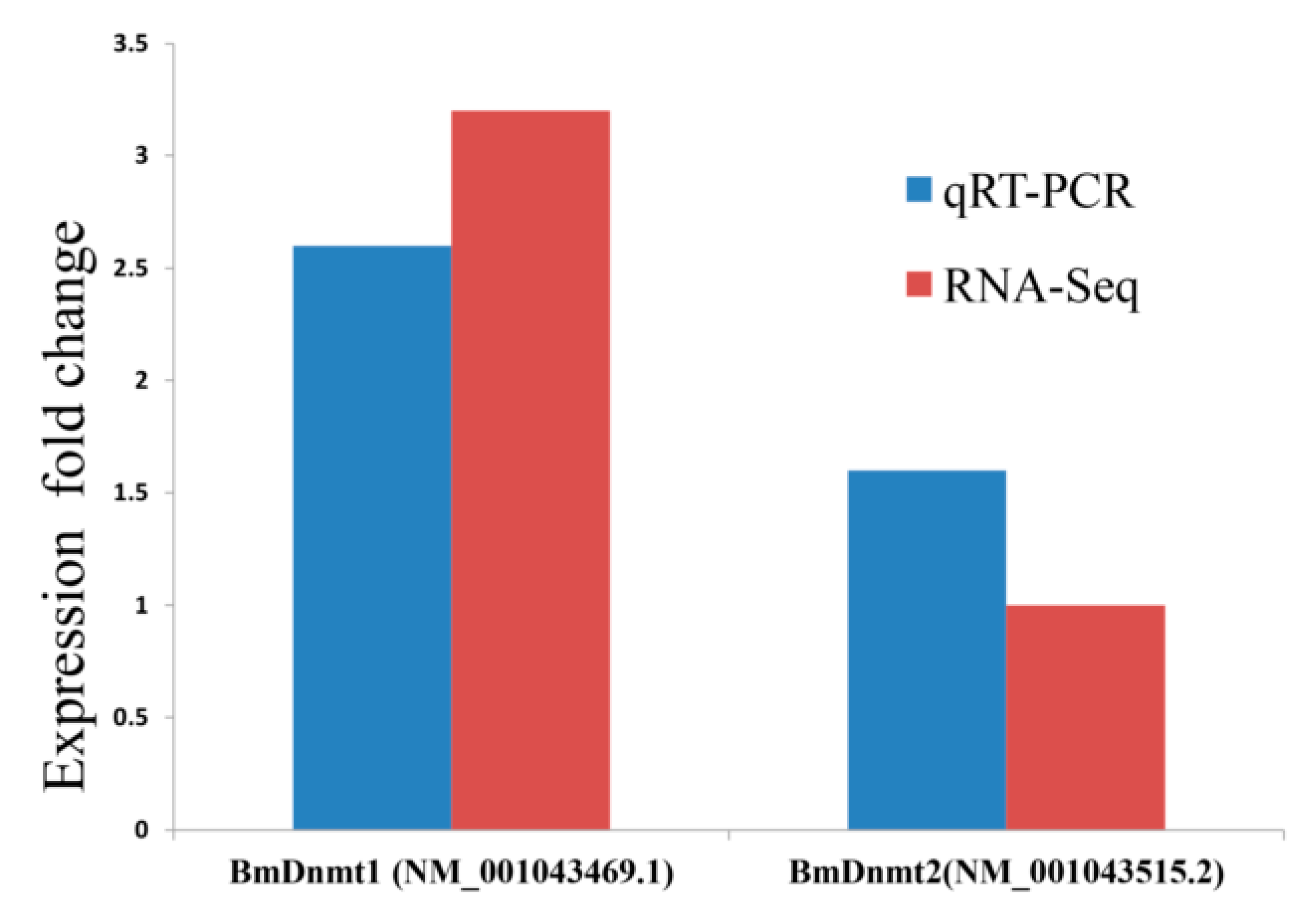
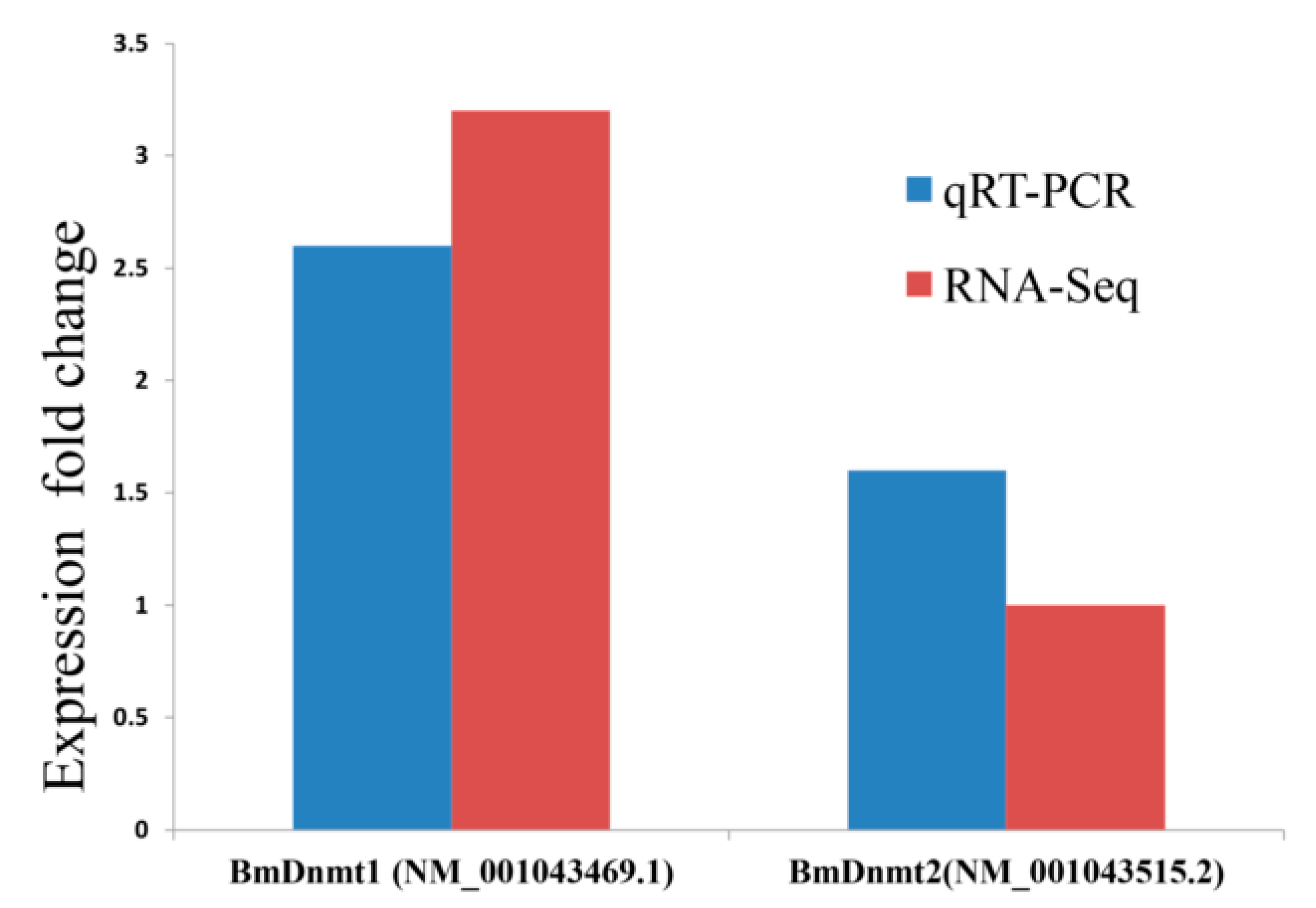
2.3. Validation of RNA-seq and WGBS
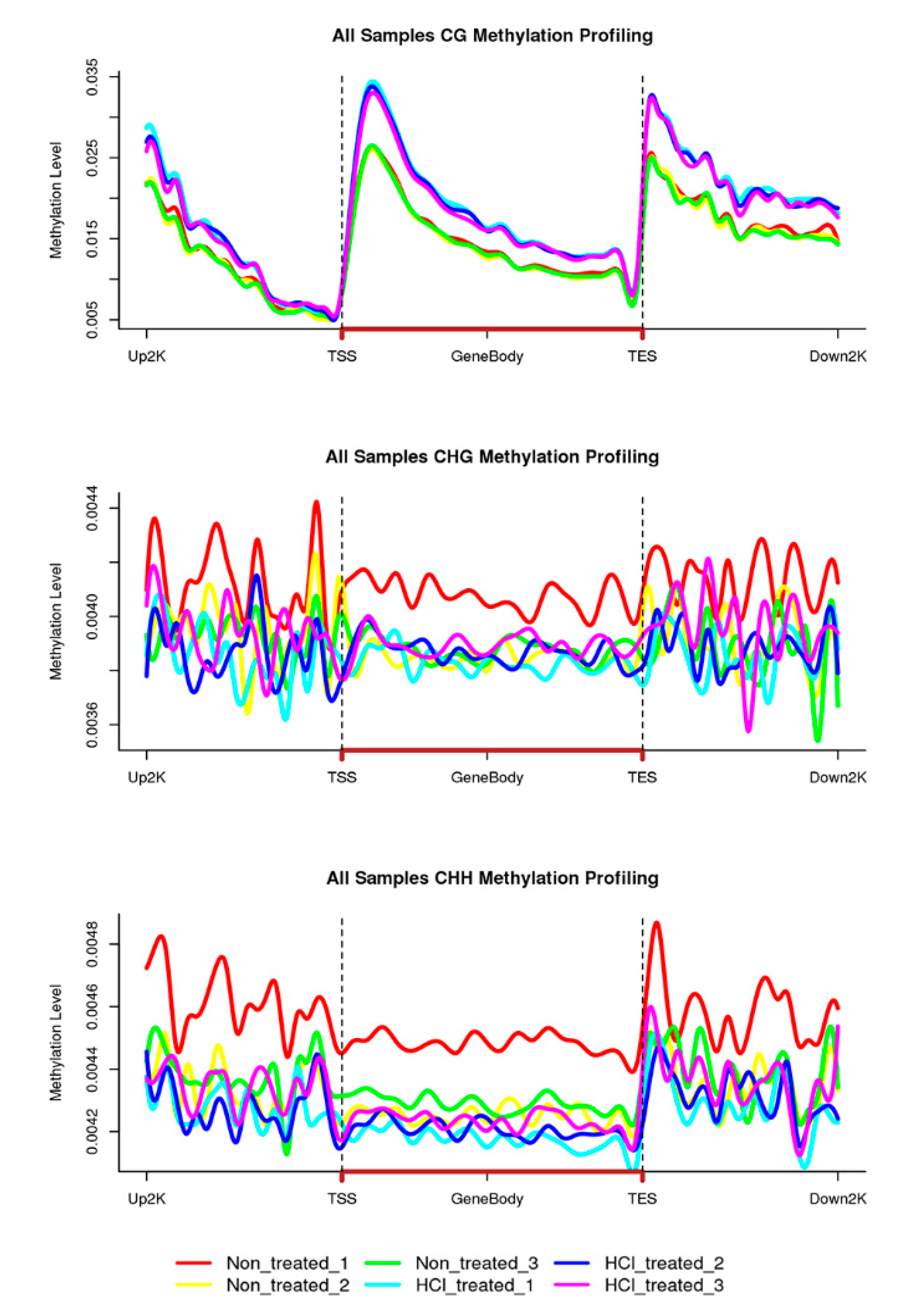
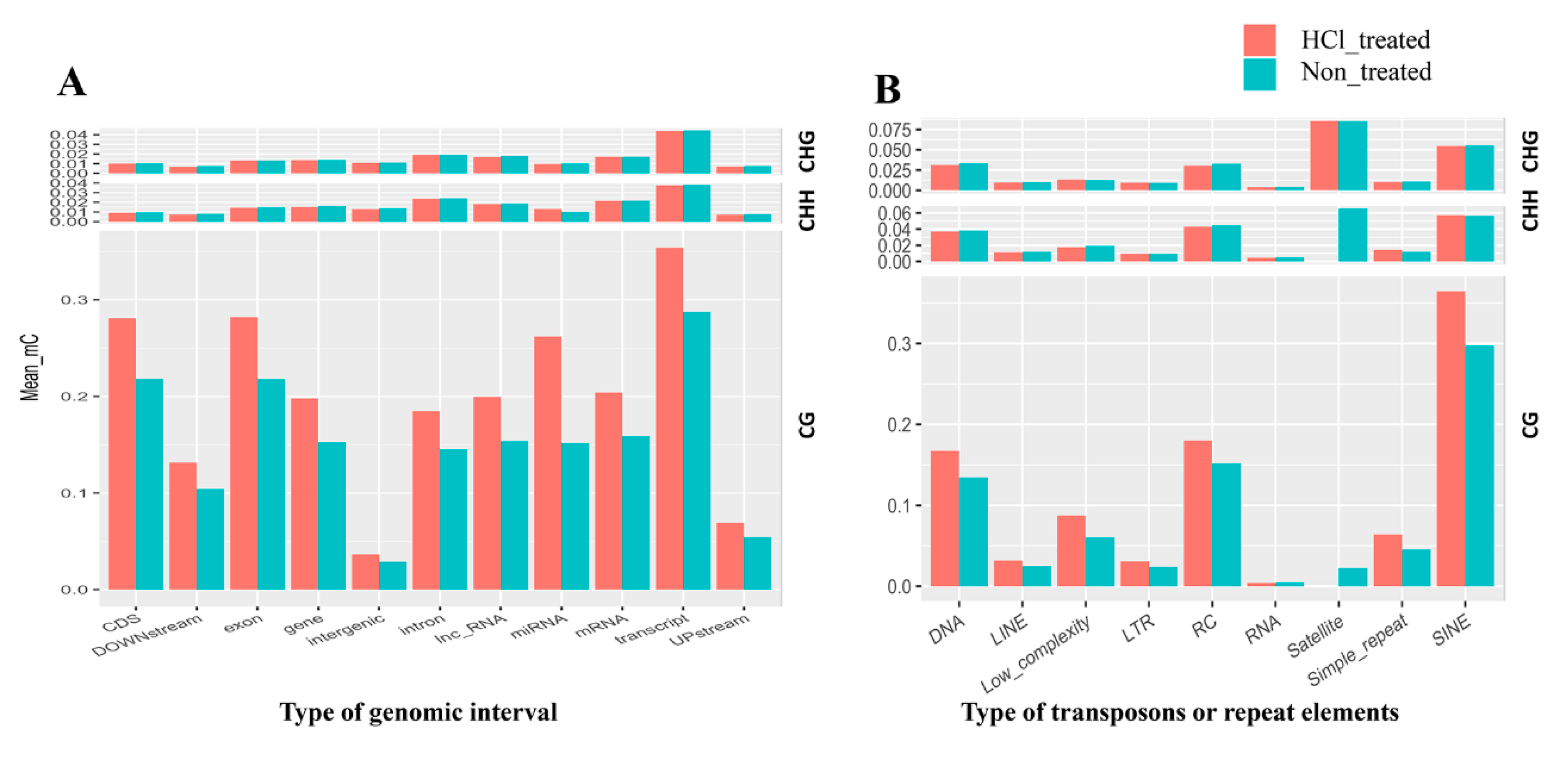
2.4. DNA Methylation Patterns in Genomic Regions of Diapause-Destined Eggs and Diapause-Terminated Eggs
2.5. Differentially Methylated Regions (DMRs) and Differentially Methylated Promoters (DMPs) Responding to Embryonic Development
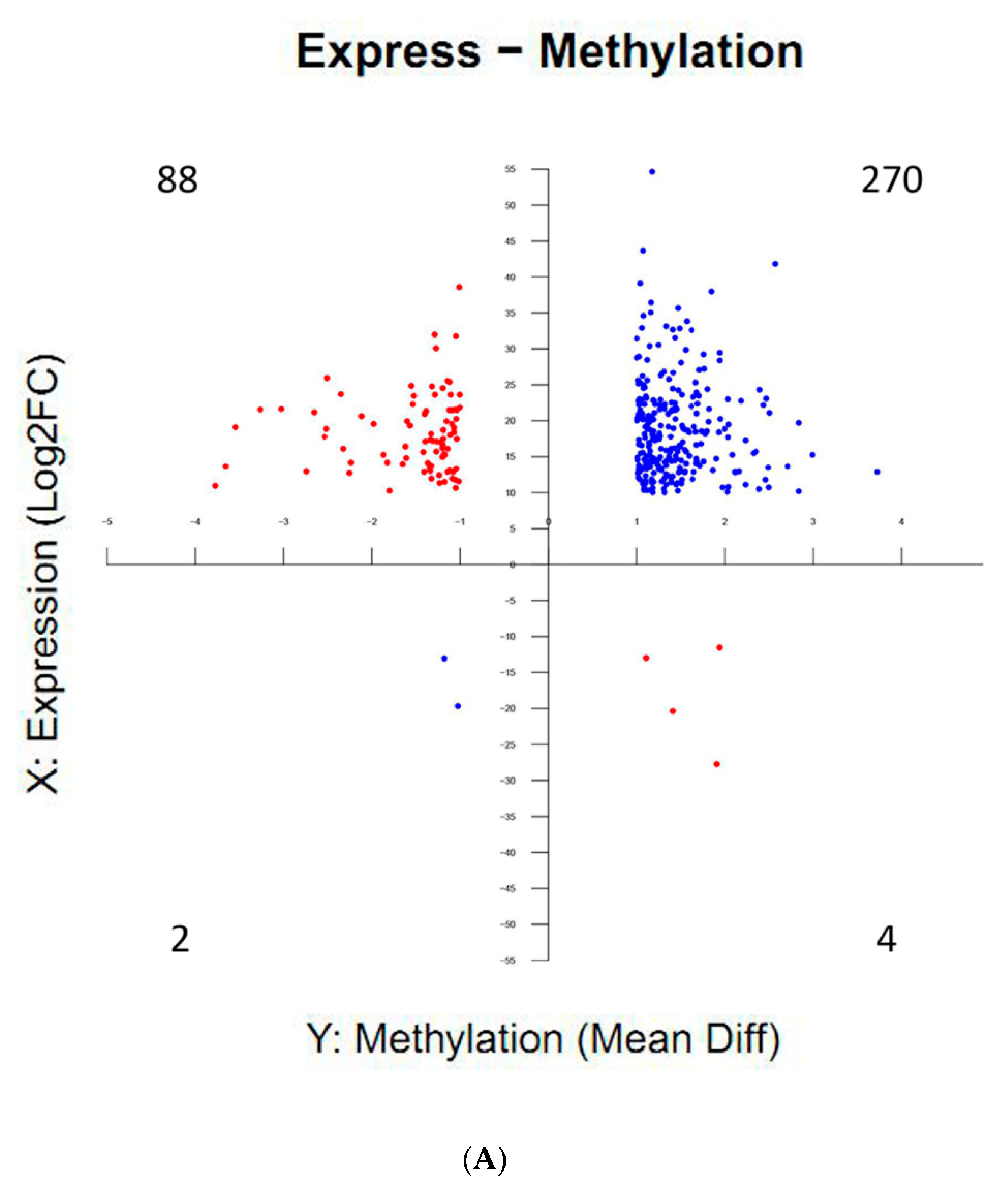
2.6. Correlations Between DNA Methylation and Gene Expression during Silkworm Early Embryonic Development
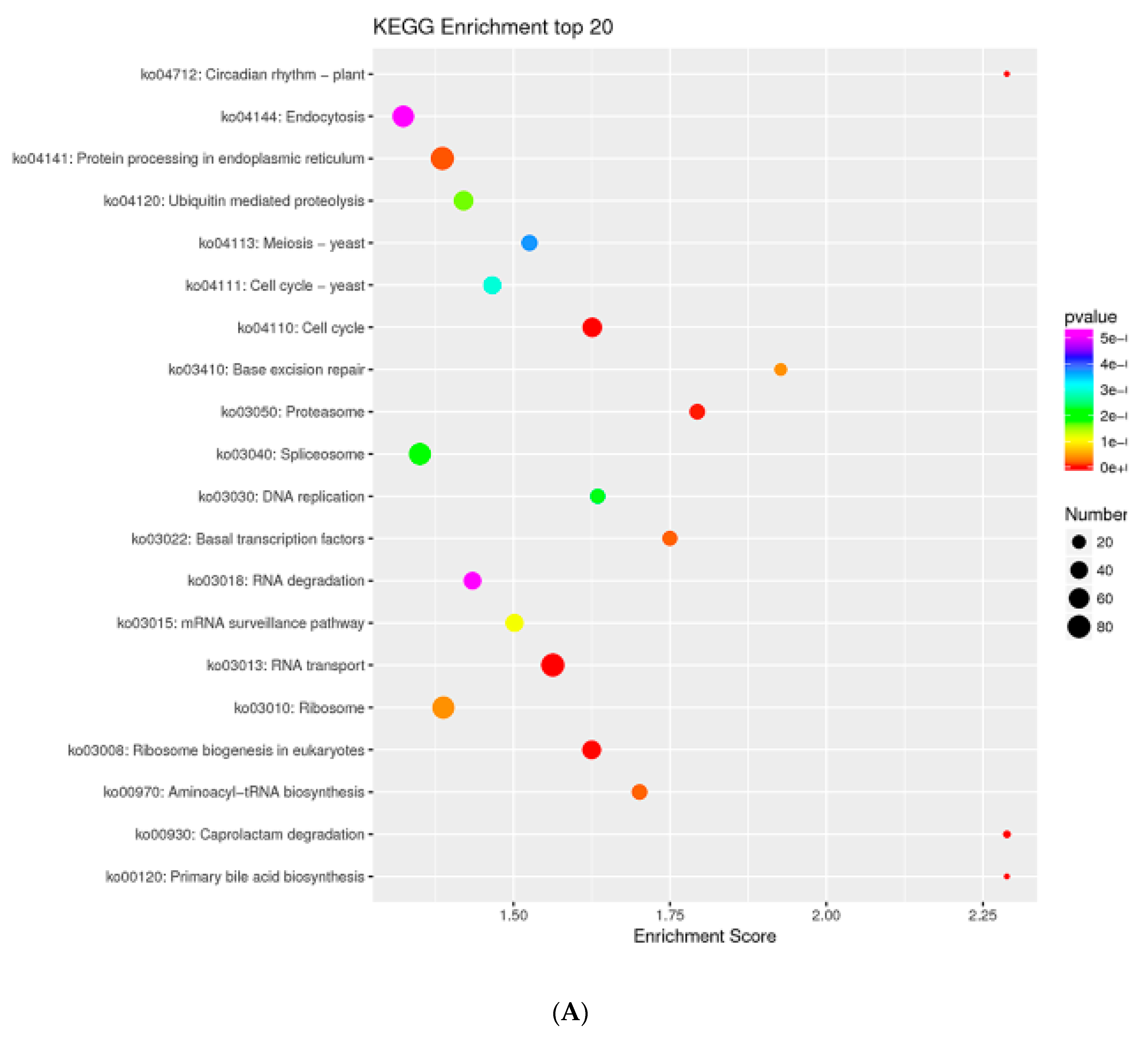
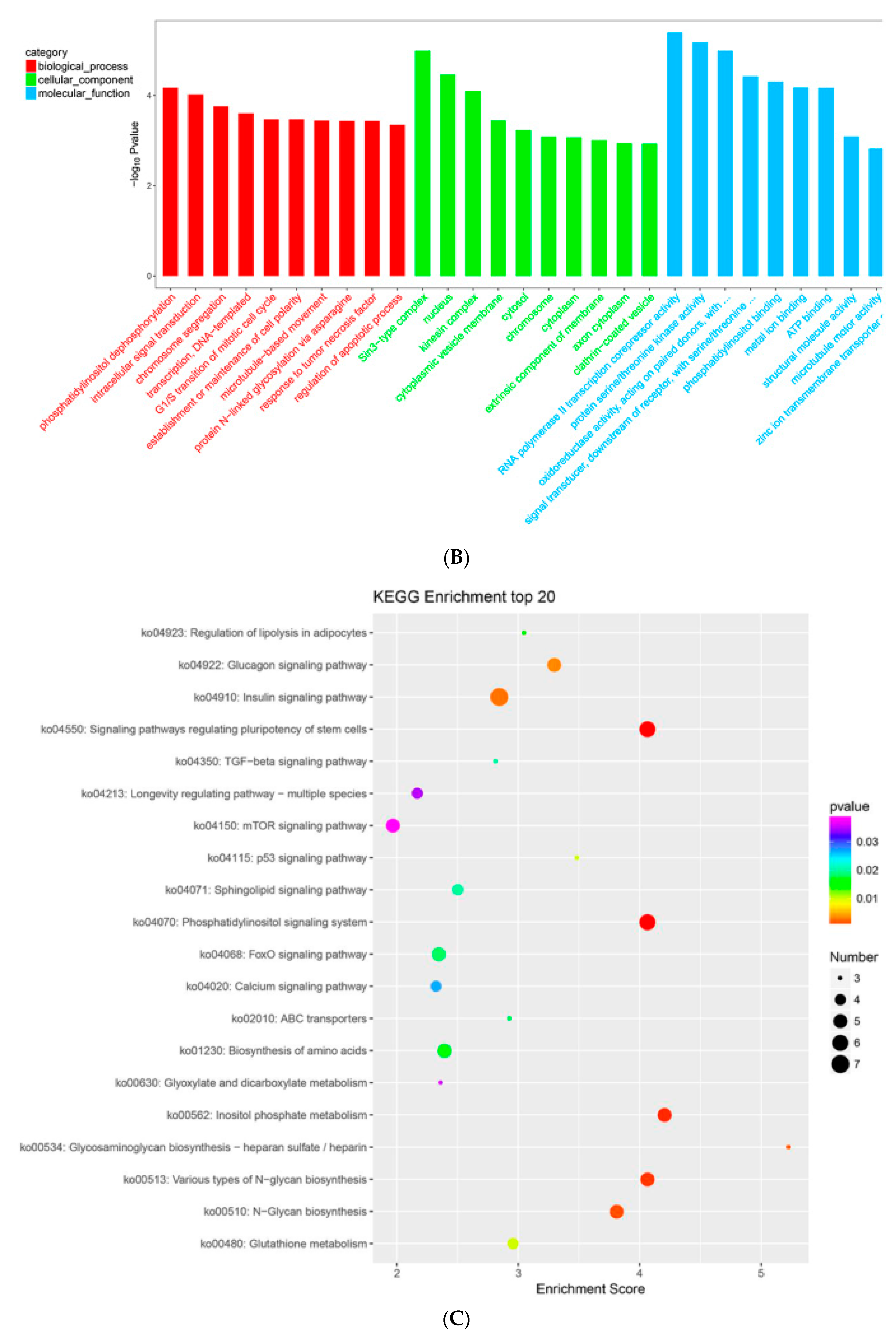
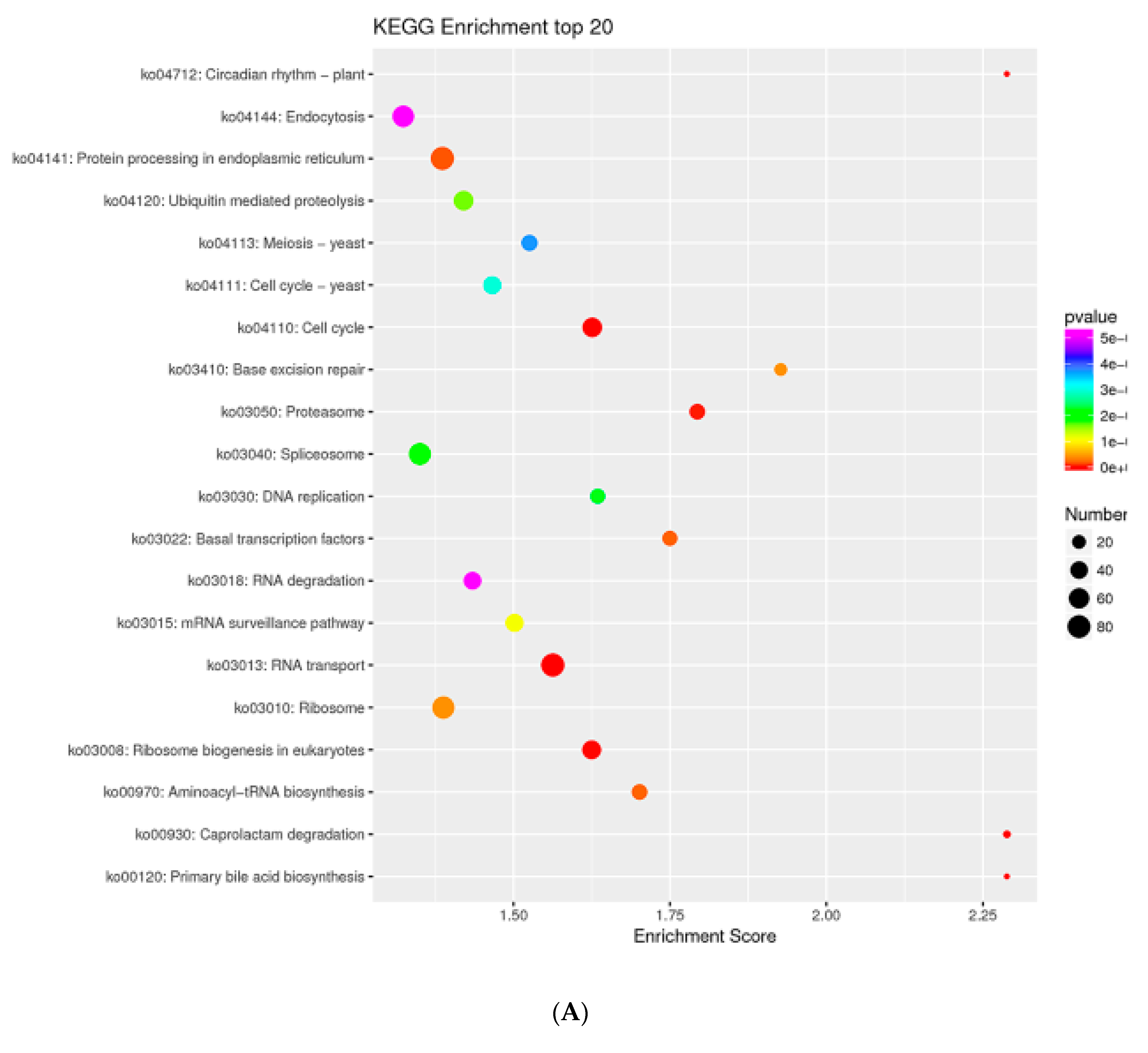
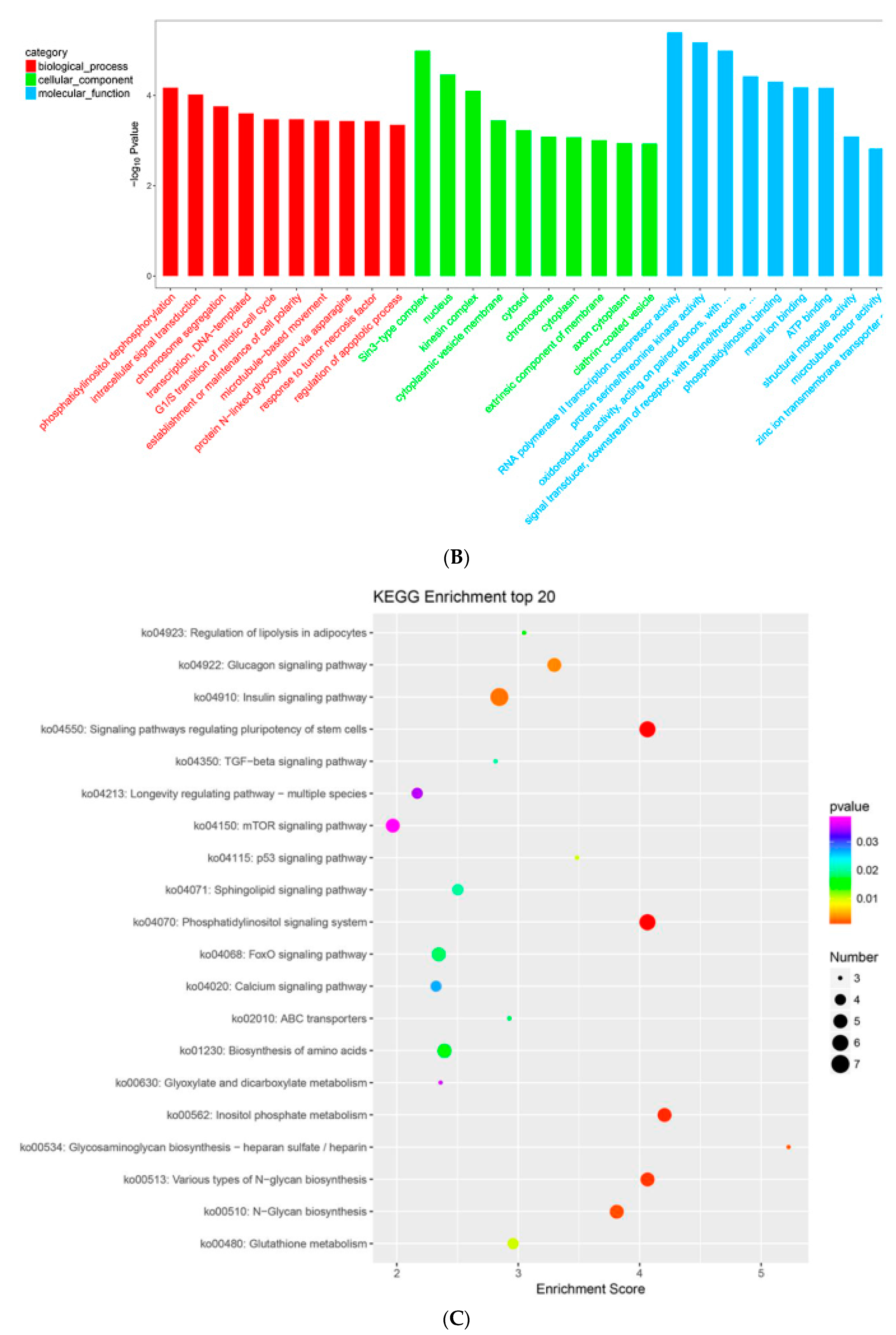
2.7. MTGs Are Involved in Metabolic Biosynthesis
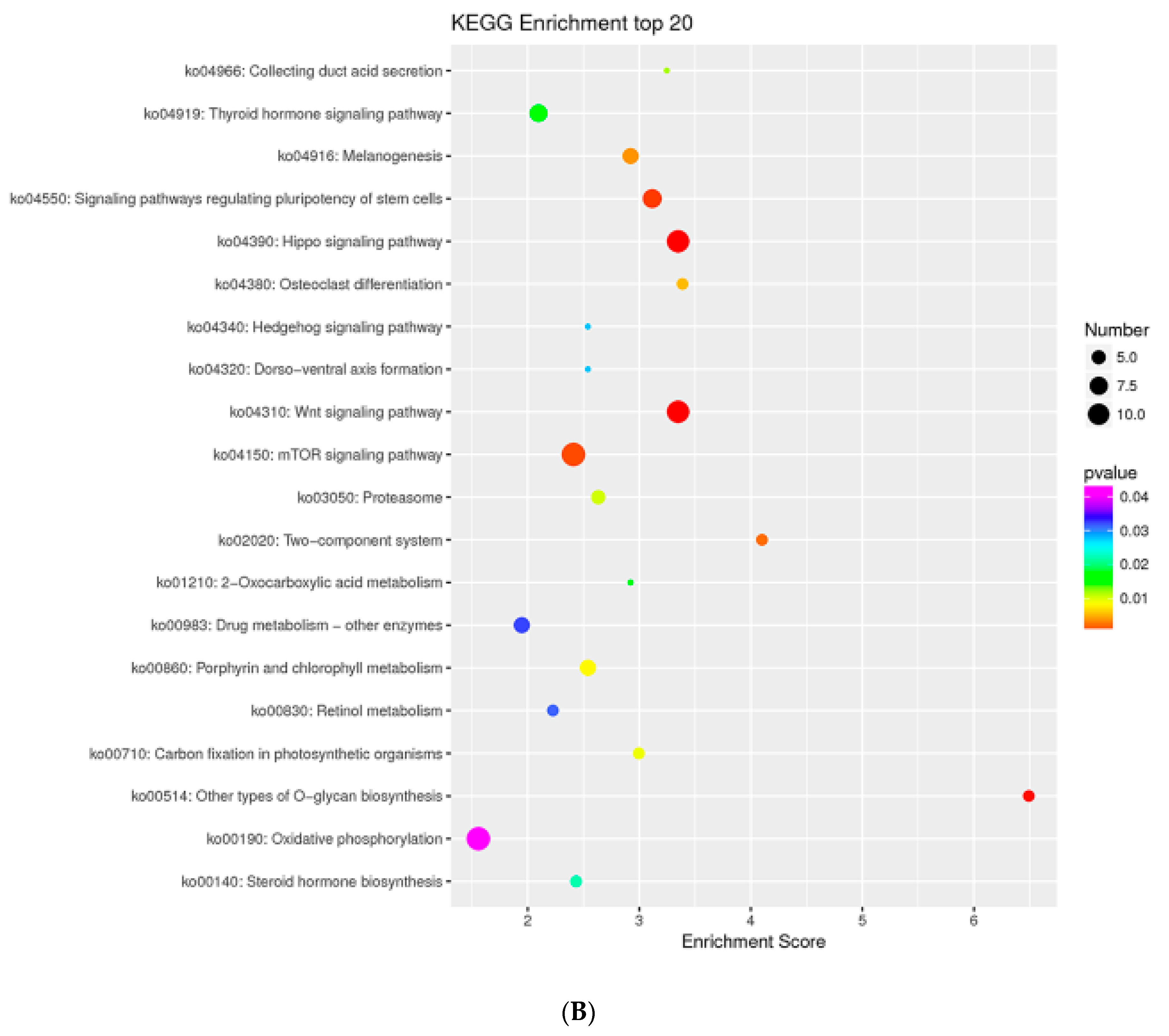
2.8. MTGs Are Involved in Phosphorylation
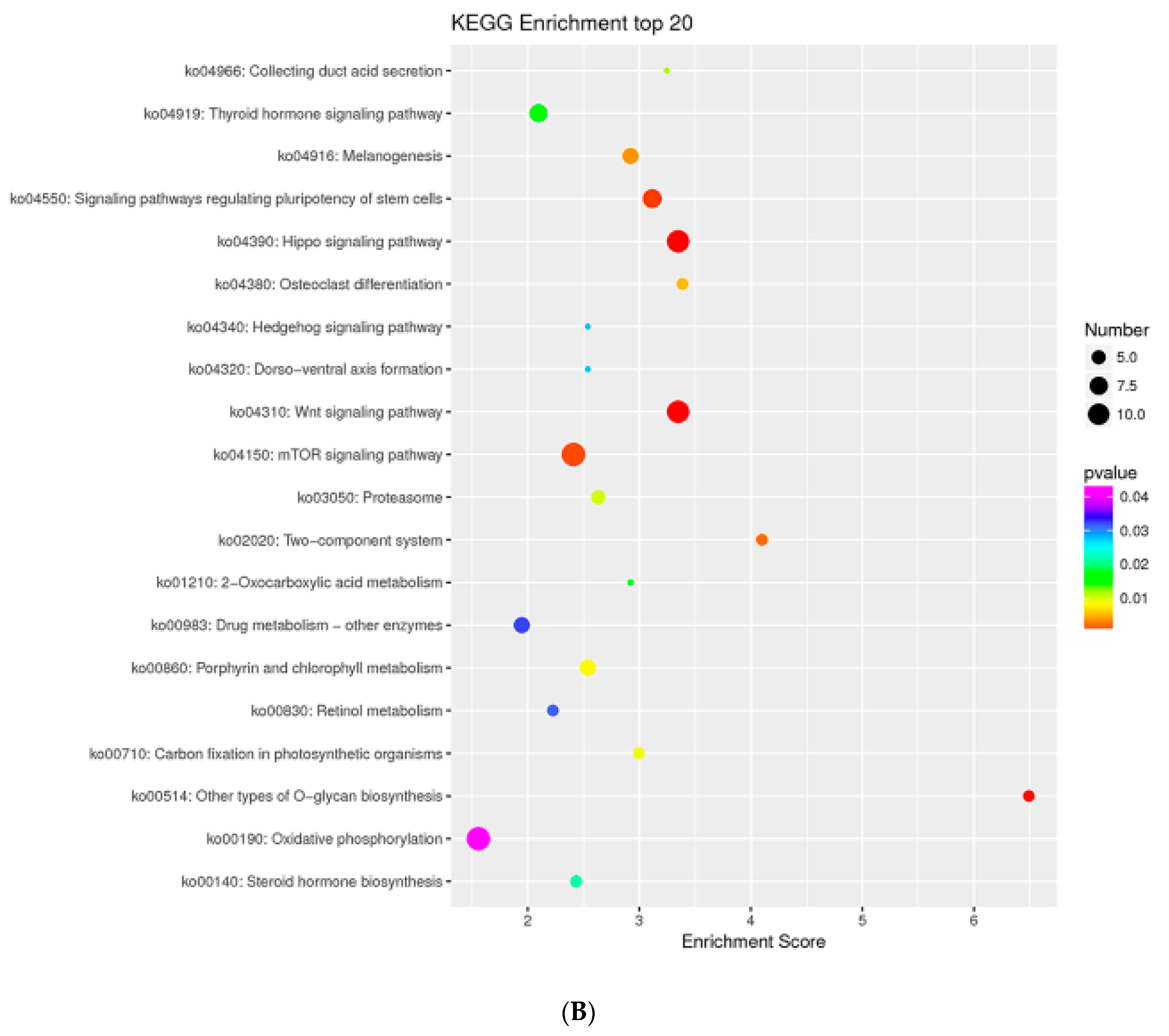
2.9. MTGs are Involved in the Cell Cycle, Apoptosis and Stem Cell Pluripotency
3. Discussion
4. Materials and Methods
4.1. Silkworm Feeding and Artificial Diapause Termination
4.2. Sample Preparation for RNA-seq and WGBS
4.3. WGBS
4.4. Transcriptome Analysis
4.5. GO and KEGG Enrichment Analysis
4.6. RT-qPCR and McrBC-qPCR Analysis
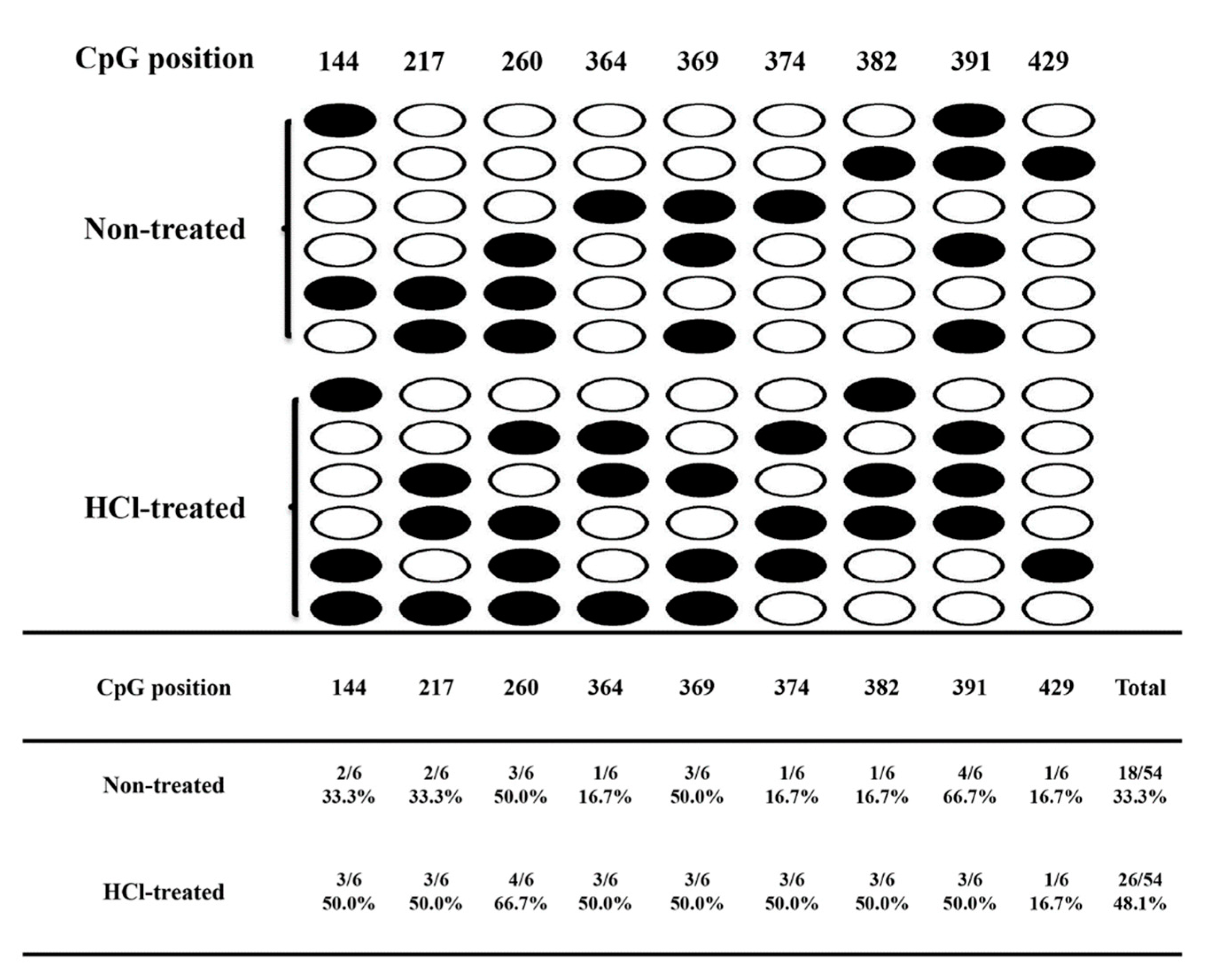
4.7. Bisulfite Sequencing Validation of Different Methylation Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fu, Q.; Li, S.; He, N. Silkworm egg proteins at the germ-band formation stage and a functional analysis of BmEP80 protein. Insect Biochem. Mol. Biol. 2011, 41, 572–581. [Google Scholar] [CrossRef]
- Paulsen, M.; Ferguson-Smith, A.C. DNA methylation in genomic imprinting, development, and disease. J. Pathol. 2001, 195, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.L.; Koch, C.M.; Clelland, G.K.; Dunham, I.; Antoniou, M. DNA methylation-histone modification relationships across the desmin locus in human primary cells. BMC Mol. Biol. 2009, 10, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellman, A.; Chess, A. Gene body-specific methylation on the active X chromosome. Science 2007, 315, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D. Global Epigenomic Reconfiguration during Mammalian Brain Development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef]
- Lyko, F.; Foret, S.; Kucharski, R.; Wolf, S.; Falckenhayn, C.; Maleszka, R. The honey bee epigenomes: Differential methylation of brain DNA in queens and workers. PLoS Biol. 2011, 9, e1000506. [Google Scholar] [CrossRef]
- Rivière, G.; Wu, G.-C.; Fellous, A.; Goux, D.; Sourdaine, P.; Favrel, P. DNA Methylation Is Crucial for the Early Development in the Oyster, C. gigas. Mar. Biotechnol. 2013, 15, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Ceccaldi, A.; Zhang, Y.; Arimondo, P.B.; Jeltsch, A. DNA methyltransferase assays. Methods Mol. Biol. 2011, 791, 157–177. [Google Scholar] [PubMed]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, D.L.; Ding, R.R.; Hu, X.Y.; Yang, X.R.; Xu, S.Q.; Gu, W.; Zhang, M. Cadmium-induced genome-wide DNA methylation changes in growth and oxidative metabolism in Drosophila melanogaster. BMC Genom. 2019, 20, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glastad, K.M.; Hunt, B.G.; Goodisman, M.A. Evolutionary insights into DNA methylation in insects. Curr. Opin. Insect Sci. 2014, 1, 25–30. [Google Scholar] [CrossRef]
- Glastad, K.M.; Hunt, B.; Yi, S.; Goodisman, M. DNA methylation in insects: On the brink of the epigenomic era. Insect Mol. Biol. 2011, 20, 553–565. [Google Scholar] [CrossRef]
- Sarda, S.; Zeng, J.; Hunt, B.; Yi, S. The Evolution of Invertebrate Gene Body Methylation. Mol. Biol. Evol. 2012, 29, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Hunt, B.G.; Glastad, K.M.; Yi, S.V.; Goodisman, M.A. The Function of Intragenic DNA Methylation: Insights from Insect Epigenomes. Integr. Comp. Biol. 2013, 53, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Bewick, A.; Vogel, K.; Moore, A.; Schmitz, R. Evolution of DNA Methylation across Insects. Mol. Biol. Evol. 2016, 34, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jorda, M.; Jones, P.L.; Maleszka, R.; Ling, X.; Robertson, H.M.; Mizzen, C.A.; Peinado, M.A.; Robinson, G.E. Functional CpG Methylation System in a Social Insect. Science 2006, 314, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Patalano, S.; Hore, T.A.; Reik, W.; Sumner, S. Shifting behaviour: Epigenetic reprogramming in eusocial insects. Curr. Opin. Cell Biol. 2012, 24, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Li-Byarlay, H.; Li, Y.; Stroud, H.; Feng, S.; Newman, T.C.; Kaneda, M.; Hou, K.K.; Worley, K.C.; Elsik, C.G.; Wickline, S.A.; et al. RNA interference knockdown of DNA methyl-transferase 3 affects gene alternative splicing in the honey bee. Proc. Natl. Acad. Sci. USA 2013, 110, 12750–12755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Dong, Y.; Lin, L.; Saqib, H.S.A.; Ma, X.; Xu, X.; Zhang, L.; Jing, X.; Peng, L.; Wang, Y.; et al. Implication for DNA methylation involved in the host transfer of diamondback moth, Plutellaxylostella (L.). Arch. Insect Biochem. Physiol. 2019, 102, e21600. [Google Scholar]
- Wang, X.; Wheeler, D.; Avery, A.; Rago, A.; Werren, J.H. Function and Evolution of DNA Methylation in Nasonia vitripennis. PLoS Genet. 2013, 9, e1003872. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Li, X.; Dai, F.; Xu, X.; Tan, A.; Chen, L.; Zhang, G.; Ding, Y.; Li, Q.; Lian, J. Comparative methylomics between domesticated and wild silkworms implies possible epigenetic influences on silkworm domestication. BMC Genom. 2013, 14, 646. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.M.; Lim, K.S.; Chapman, J.W.; Bass, C. Genome-Wide Characterisation of DNA Methylation in an Invasive Lepidopteran Pest, the Cotton Bollworm Helicoverpa armigera. G3 2018, 8, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Zhu, J.D.; Chen, Q.; Dai, F.Y.; Li, X.; Li, M.W.; Zhang, H.Y.; Zhang, G.J.; Li, D.; Yang, D. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010, 28, 516–520. [Google Scholar] [CrossRef]
- Wu, P.; Jie, W.; Shang, Q.; Annan, E.; Jiang, X.; Hou, C.; Chen, T.; Guo, X. DNA methylation in silkworm genome may provide insights into epigenetic regulation of response to Bombyx moricypovirus infection. Sci. Rep. 2017, 7, 16013. [Google Scholar] [CrossRef] [Green Version]
- Uno, T.; Nomura, Y.; Nakamura, M.; Nakao, A.; Tajima, S.; Kanamaru, K.; Yamagata, H.; Iwanaga, Y. Expression, purification, and characterization of methyl DNA binding protein from Bombyx mori. J. Insect Sci. 2005, 5, 8. [Google Scholar]
- Mitsudome, T.; Mon, H.; Xu, J.; Li, Z.; Lee, J.M.; Patil, A.A.; Masuda, A.; Iiyama, K.; Morokuma, D.; Kusakabe, T. Biochemical characterization of maintenance DNA methyltransferase DNMT-1 from silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2015, 58, 55–65. [Google Scholar] [CrossRef]
- Patalano, S.; Vlasova, A.; Wyatt, C.; Ewels, P.; Camara, F.; Ferreira, P.; Asher, C.; Jurkowski, T.; Segonds-Pichon, A.; Bachman, M.; et al. Molecular signatures of plastic phenotypes in two eusocial insect species with simple societies. Proc. Natl. Acad. Sci. USA 2015, 112, 13970–13975. [Google Scholar] [CrossRef] [Green Version]
- Kay, S.; Skowronski, D.; Hunt, B.G. Developmental DNA methyltransferase expression in the fire ant Solenopsis invicta. Insect Sci. 2018, 25, 57–65. [Google Scholar] [CrossRef]
- Tauber, M.J.; Tauber, C.A. Insect Seasonality: Diapause Maintenance, Termination, and Postdiapause Development. J. Ann. Rev. Ent. 1976, 21, 81–107. [Google Scholar] [CrossRef]
- Kostál, V. Eco-physiological phases of insect diapause. J. Insect Physiol. 2006, 52, 113–127. [Google Scholar] [CrossRef]
- Ohtsuki, Y. Silkworm Eggs. In Japanese Society of Sericultural Science Ageneral Text Book of Sericulture Nihon SansiShinbun-Sha; Toyo Co. LTD: Tokyo, Japan, 1979; pp. 156–173. (In Japanese) [Google Scholar]
- Yamashita, O.; Yaginuma, T. Silkworm Eggs at Low Temperatures: Implications for Sericulture. In Insects at Low Temperature; Jieisha Corporation: Tokyo, Japan, 1991; pp. 424–445. [Google Scholar]
- Nakagaki, M.; Takei, R.; Nagashima, E.; Yaginuma, T. Cell cycles in embryos of the silkworm, Bombyx mori: G2-arrest at diapause stage. Dev. Genes Evol. 1991, 200, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Sonobe, H.; Matsumoto, A.; Fukuzaki, Y.; Fujiwara, S. Carbohydrate metabolism and restricted oxygen supply in the eggs of the silkworm, Bombyx mori. J. Insect Physiol. 1979, 25, 381–388. [Google Scholar] [CrossRef]
- Li, B.; Hu, P.; Zhang, S.Z.; Toufeeq, S.; Wang, J.; Zhao, K.; Xu, X.; Xu, J.; Huang, S. DNA methyltransferase BmDnmt1 and BmDnmt2 in silkworm (Bombyx mori) and the regulation of silkworm embryonic development. Arch. Insect Biochem. Physiol. 2018, 100, e201529. [Google Scholar]
- Egger, G.; Wielscher, M.; Pulverer, W.; Kriegner, A.; Weinhäusel, A. DNA methylation testing and marker validation using PCR: Diagnostic applications. Expert Rev. Mol. Diagn. 2012, 12, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J.-K.; Lang, Z. Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. USA 2019, 116, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, O.; Yaginuma, T.; Hasegawa, K. Hormonal and metabolic control of egg diapause of the silkworm, Bombyx mori (Lepidoptera: Bombycidae). Entomol. Gen. 1981, 7, 195–211. [Google Scholar]
- Ponnuvel, K.M.; Murthy, G.N.; Awasthi, A.K.; Rao, G.; Vijayaprakash, N.B. Differential gene expression during early embryonic development in diapause and non-diapause eggs of multivoltine silkworm Bombyx mori. Indian J. Exp. Biol. 2010, 48, 1143–1151. [Google Scholar]
- Conduit, S.; Dyson, J.; Mitchell, C. Inositol polyphosphate 5-phosphatases; New players in the regulation of cilia and ciliopathies. FEBS Lett. 2012, 586, 2846–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, L.; Riddiford, L.; Kiguchi, K. Morphogenesis in the Early Embryo of the Lepidopteran Bombyx mori. Dev. Biol. 1994, 165, 137–151. [Google Scholar] [CrossRef]
- Bewick, A.; Sanchez, Z.; Mckinney, E.; Moore, A.; Moore, P.; Schmitz, R. Gene-regulatory independent functions for insect DNA methylation. bioRxiv 2018. [Google Scholar] [CrossRef]
- Bonasio, R.; Li, Q.; Lian, J.; Mutti, N.S.; Jin, L.; Zhao, H.; Zhang, P.; Wen, P.; Xiang, H.; Ding, Y.; et al. Genome-wide and Caste-Specific DNA Methylomes of the Ants Camponotus floridanus and Harpegnathos saltator. Curr. Biol. 2012, 22, 1755–1764. [Google Scholar] [CrossRef] [Green Version]
- Mandrioli, M.; Volpi, N. The Genome of the Lepidopteran Mamestra brassicae has a Vertebrate-like Content of Methyl-cytosine. Genetica 2003, 119, 187–191. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; Van Nocker, S.; Ma, F.; Guan, Q. Single-Base Methylome Analysis Reveals Dynamic Epigenomic Differences Associated with Water Deficit in Apple. Plant Biotechnol. J. 2017, 16, 672–687. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef]
- Harris, C.J.; Scheibe, M.; Wongpalee, S.P.; Liu, W.; Cornett, E.M.; Vaughan, R.M.; Li, X.; Chen, W.; Xue, Y.; Zhong, Z.; et al. A DNA methylation reader complex that enhances gene transcription. Science 2018, 362, 1182. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, Z.; Zhang, Q.; Li, B.; Lu, C.; Li, W.; Cheng, T.; Xia, Q.; Zhao, P. DNA methylation on N6-adenine in lepidopteran Bombyx mori. Biochim. Biophys. Acta 2018, 1861, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Shawn, C.; Zhang, X.; Chen, P.-Y.; Bostick, M.; Mary, G.; Hetzel, J.; Jain, J.; Steven, S.; Marnie, H. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaud, P.; Goubely, C.; Pélissier, T.; Deragon, J.M. SINE retroposons can be used in vivo as nucleation centers for de novo methylation. Mol. Cell. Biol. 2000, 20, 3434–3441. [Google Scholar] [CrossRef] [PubMed]
- Hasse, A.; Schulz, W.A. Enhancement of reporter gene de novo methylation by DNA fragments from the alpha-fetoprotein control region. J. Biol. Chem. 1994, 269, 1821–1826. [Google Scholar] [PubMed]
- Yates, P.A.; Burman, R.W.; Mummaneni, P.; Krussel, S.; Turker, M.S. Tandem B1 elements located in a mouse methylation center provide a target for de novo DNA methylation. J. Biol. Chem. 1999, 274, 36357–36361. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.A.; Ge, L.; Eichler, E.E. An Alu transposition model for the origin and expansion of human segmental duplications. J. Am. J. Hum. Genet. 2003, 73, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.X.; Lee, M.P.; Chen, A.D.; Brown, S.D.; Hsieh, T.S. Isolation and characterization of a Drosophila gene essential for early embryonic development and formation of cortical cleavage furrows. J. Cell Biol. 1996, 134, 923–934. [Google Scholar] [CrossRef] [Green Version]
- Loncar, D.; Singer, S. Tyrosine phosphorylation accompanying the cellularization of the syncytial blastoderm of Drosophila. Proc. Natl. Acad. Sci. USA 1995, 92, 8154–8157. [Google Scholar] [CrossRef] [Green Version]
- Denisenko, O.N.; Bomsztyk, K. The product of the murine homolog of the Drosophila extra sex combs gene displays transcriptional repressor activity. Mol. Cell. Biol. 1997, 17, 4707–4717. [Google Scholar] [CrossRef] [Green Version]
- Schlitt, T.; Brazma, A. Current approaches to gene regulatory network modeling. BMC Bioinform. 2007, 8, S9. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Wang, F.; Dong, Z.; Hua, X.; Xia, Q. Label-free quantitative phosphoproteomic profiling of cellular response induced by an insect cytokine paralytic peptide. J. Proteom. 2016, 154, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.; Helton, E.; Banerjee, S.; Venable, M.; Johnson, L.; Schoeb, T.R.; Kesterson, R.F.; Crawford, D. G2E3 Is a Dual Function Ubiquitin Ligase Required for Early Embryonic Development. J. Biol. Chem. 2008, 283, 22304–22315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAP ping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Marc, L.; Bjoern, U. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Yu, S.; Liu, H.; Luo, L. Analysis of relative gene expression using different real-time quantitative PCR. Acta Agron. Sin. 2007, 12, 1214–1218. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SampleName | Raw Reads | Clean Reads | Clean Bases | Total Mapped | ≥Q30 | GC Content |
|---|---|---|---|---|---|---|
| HCl-treated_1 | 48,961,256 | 47,914,231 | 7,012,354,124 | 95.49% | 95.17% | 43.33% |
| HCl-treated_2 | 49,151,346 | 48,032,314 | 7,032,354,785 | 95.33% | 95.02% | 43.62% |
| HCl-treated_3 | 49,564,521 | 48,462,345 | 7,094,523,651 | 95.41% | 95.00% | 43.46% |
| Non-treated_1 | 49,254,256 | 48,108,659 | 7,045,898,541 | 95.33% | 94.88% | 43.63% |
| Non-treated_2 | 49,422,345 | 48,188,754 | 7,034,562,547 | 94.78% | 94.84% | 43.77% |
| Non-treated_3 | 49,802,145 | 48,657,854 | 7,117,854,216 | 95.11% | 95.03% | 43.66% |
| HCl-treated vs. Non-treated | DEGs | Upregulated | Downregulated | |||
| Quantity | 1732 | 856 | 876 | |||
| Samples Name | Clean Reads | Clean Base | Clean Reads Percentage | Clean Base Percentage | GC Content | >Q30 |
|---|---|---|---|---|---|---|
| Non-treated_1 | 93,793,430 | 13,090,963,533 | 98.79% | 98.49% | 19.29% | 94.81% |
| Non-treated_2 | 96,273,278 | 13,450,975,096 | 98.08% | 97.88% | 19.27% | 92.81% |
| Non-treated_3 | 92,904,772 | 12,983,974,042 | 98.08% | 97.91% | 19.22% | 92.82% |
| HCl-treated_1 | 98,947,490 | 13,832,834,274 | 98.03% | 97.89% | 19.18% | 92.85% |
| HCl-treated_2 | 95,316,260 | 13,313,691,168 | 98.38% | 98.15% | 19.26% | 93.48% |
| HCl-treated_3 | 89,068,596 | 12,445,567,794 | 98.12% | 97.93% | 19.26% | 92.95% |
| GeneID | Synonym | Methylation Difference Level (%) | Methylation p-Value | Expression Foldchange | Expression p-Value | Description |
|---|---|---|---|---|---|---|
| N-glycan biosynthesis | ||||||
| LOC101745965 | FucT6 | 15.19663 | 2.85 × 10−25 | 4.233024 | 8.12 × 10−23 | alpha-(1,6)-fucosyltransferase-like [Bombyx mori (domestic silkworm)] |
| LOC101739008 | ALG9 | 17.21088 | 1.25 × 10−25 | 2.306166 | 6.43 × 10−28 | alpha-1,2-mannosyltransferase ALG9 [Bombyx mori (domestic silkworm)] |
| LOC101742247 | Stt3b | 22.27647 | 2.30 × 10−10 | 2.125494 | 3.94 × 10−28 | dolichyl-diphosphooligosaccharide--protein glycosyltransferase subunit STT3B [Bombyx mori (domestic silkworm)] |
| LOC101744335 | alpha-Man-Ia | 12.0725 | 3.80 × 10−7 | 2.024585 | 1.34 × 10−17 | mannosyl-oligosaccharide alpha-1,2-mannosidase IA [Bombyx mori (domestic silkworm)] |
| LOC101742043 | GmII | 10.32376 | 5.80 × 10−16 | 2.1135 | 2.50 × 10−65 | alpha-mannosidase 2 [Bombyx mori (domestic silkworm)] |
| Glycosaminoglycan biosynthesis—heparan sulfate/heparin | ||||||
| LOC101741346 | EXT2 | 22.1426 | 1.38 × 10−16 | 2.396993 | 7.06 × 10−6 | exostosin-2 [Bombyx mori (domestic silkworm)] |
| LOC101736578 | Hs2st | 21.04841 | 3.40 × 10−15 | 2.298892 | 2.05 × 10−14 | heparin sulfate O-sulfotransferase [Bombyx mori (domestic silkworm)] |
| LOC101737390 | HS6ST2 | 21.02652 | 1.21 × 10−8 | 3.199666 | 4.66 × 10−7 | heparan-sulfate 6-O-sulfotransferase 2 [Bombyx mori (domestic silkworm)] |
| Glutathione metabolism | ||||||
| LOC101745413 | Oplah | 14.92435 | 2.49 × 10−6 | 2.405576 | 3.76 × 10−14 | 5-oxoprolinase [Bombyx mori (domestic silkworm)] |
| LOC101742094 | Srm | 10.35505 | 1.84 × 10−5 | 2.181386 | 3.31 × 10−11 | spermidine synthase [Bombyx mori (domestic silkworm)] |
| LOC692521 | GSTd2 | 11.33547 | 5.47 × 10−5 | 0.426264 | 1.12 × 10−13 | glutathione S-transferase delta 2[Bombyx mori (domestic silkworm)] |
| LOC100862776 | Ggt1 | 12.9659 | 2.17 × 10−7 | 0.149641 | 9.93 × 10−42 | gamma-glutamyl transpeptidase [Bombyx mori (domestic silkworm)] |
| Regulation of lipolysis in adipocytes | ||||||
| LOC101737606 | Ac76E | 10.18663 | 5.91 × 10−5 | 7.139607 | 1.27 × 10−6 | adenylate cyclase type 2 [Bombyx mori (domestic silkworm)] |
| LOC101745072 | irs1-b | 23.61161 | 1.20 × 10−12 | 2.851851 | 2.82 × 10−20 | insulin receptor substrate 1-B [Bombyx mori (domestic silkworm)] |
| LOC100158253 | Pi3k60 | 20.1611 | 8.42 × 10−13 | 2.09716 | 7.71 × 10−44 | phosphatidylinositol 3-kinase 60 [Bombyx mori (domestic silkworm)] |
| Biosynthesis of amino acids | ||||||
| LOC101740956 | ACO1 | 25.61842 | 7.63 × 10−8 | 2.171117 | 6.59 × 10−24 | cytoplasmic aconitate hydratase [Bombyx mori (domestic silkworm)] |
| LOC101740405 | Gs2 | 17.49432 | 3.22 × 10−12 | 0.438415 | 4.33 × 10−29 | glutamine synthetase 2 cytoplasmic [Bombyx mori (domestic silkworm)] |
| LOC101743550 | Ald | 24.49332 | 1.70 × 10−14 | 0.436375 | 5.81 × 10−37 | fructose-bisphosphate aldolase [Bombyx mori (domestic silkworm)] |
| LOC101738188 | argF | 20.62089 | 5.46 × 10−9 | 0.230464 | 1.88 × 10−79 | ornithine carbamoyltransferase-like [Bombyx mori (domestic silkworm)] |
| LOC101738186 | ACO2 | 17.0617 | 6.27 × 10−14 | 0.415694 | 0 | probable aconitate hydratase, mitochondrial [Bombyx mori (domestic silkworm)] |
| Glyoxylate and dicarboxylate metabolism | ||||||
| LOC101740956 | ACO1 | 25.61842 | 7.63 × 10−8 | 2.171117 | 6.59 × 10−24 | cytoplasmic aconitate hydratase [Bombyx mori (domestic silkworm)] |
| LOC101740405 | Gs2 | 17.49432 | 3.22 × 10−12 | 0.438415 | 4.33 × 10−29 | glutamine synthetase 2 cytoplasmic [Bombyx mori (domestic silkworm)] |
| LOC101738186 | ACO2 | 17.0617 | 6.27 × 10−14 | 0.415694 | 0 | probable aconitate hydratase, mitochondrial [Bombyx mori (domestic silkworm)] |
| GeneID | Synonym | Methylation Difference Level (%) | Methylation p-Value | Expression Foldchange | Expression p-Value | Description |
|---|---|---|---|---|---|---|
| Phosphatidylinositol-signaling system | ||||||
| LOC101740091 | Inpp5e | 29.18555 | 0.000496 | 3.378116 | 6.58 × 10−6 | 72 kDa inositol polyphosphate 5-phosphatase [Bombyx mori (domestic silkworm)] |
| LOC101740803 | Inpp5j | 22.42952 | 1.11 × 10−6 | 2.701747 | 6.94 × 10−9 | inositol polyphosphate 5-phosphatase K [Bombyx mori (domestic silkworm)] |
| LOC101745952 | PTEN | 14.48683 | 7.92 × 10−6 | 2.540607 | 7.55 × 10−75 | phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase PTEN [Bombyx mori (domestic silkworm)] |
| LOC101735646 | Plce1 | 16.95742 | 7.17 × 10−28 | 2.1977 | 7.13 × 10−10 | 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase epsilon-1 [Bombyx mori (domestic silkworm)] |
| LOC100158253 | Pi3k60 | 20.1611 | 8.42 × 10−13 | 2.09716 | 7.71 × 10−44 | phosphatidylinositol 3-kinase 60 [Bombyx mori (domestic silkworm)] |
| LOC101743221 | Inpp5a | 22.18242 | 3.06 × 10−19 | 2.035092 | 7.69 × 10−8 | type I inositol 1,4,5-trisphosphate 5-phosphatase [Bombyx mori (domestic silkworm)] |
| GeneID | Synonym | Methylation Difference Level (%) | Methylation p-Value | Expression Foldchange | Expression p-Value | Description |
|---|---|---|---|---|---|---|
| G1/S transition of mitotic cell cycle | ||||||
| LOC101746803 | Phf8 | 22.76216 | 1.34 × 10−9 | 4.539773 | 9.05 × 10−12 | histone lysine demethylase PHF8 [Bombyx mori (domestic silkworm)] |
| LOC 100216493 | Cyce | 16.97983 | 6.34 × 10−6 | 2.868805 | 1.35 × 10−97 | cyclin E [Bombyx mori (domestic silkworm)] |
| LOC101741790 | SKP2 | 26.22085 | 0.001772 | 2.090556 | 9.75 × 10−24 | S-phase kinase-associated protein 2 [Bombyx mori (domestic silkworm)] |
| LOC101738704 | ACVR1 | 13.23511 | 1.51 × 10−5 | 2.028722 | 5.10 × 10−59 | activin receptor type-1 [Bombyx mori (domestic silkworm)] |
| Regulation of apoptotic process | ||||||
| LOC101742044 | CG14226 | 10.69776 | 1.84 × 10−11 | 5.6281 | 4.06 × 10−93 | cytokine receptor [Bombyx mori (domestic silkworm)] |
| LOC101739208 | G2e3 | 16.07484 | 2.73 × 10−14 | 2.619197 | 4.72 × 10−51 | G2/M phase-specific E3 ubiquitin-protein ligase [Bombyx mori (domestic silkworm)] |
| LOC101745221 | DLG5 | 11.60066 | 1.46 × 10−06 | 2.141483 | 6.91 × 10−51 | disks large homolog 5 [Bombyx mori (domestic silkworm)] |
| LOC101741790 | SKP2 | 26.22085 | 0.001772 | 2.090556 | 9.75 × 10−24 | S-phase kinase-associated protein 2 [Bombyx mori (domestic silkworm)] |
| LOC101740336 | Sh3kbp1 | 22.78633 | 0.001078 | 2.027284 | 2.31 × 10−45 | CD2-associated protein [Bombyx mori (domestic silkworm)] |
| LOC101738660 | Ppid | 21.54647 | 3.27 × 10−19 | 0.480788 | 6.92 × 10−53 | peptidyl-prolyl cis-trans isomerase D [Bombyx mori (domestic silkworm)] |
| Signaling pathways regulating pluripotency of stem cells | ||||||
| LOC101742798 | Mad | 12.83938 | 0.001686 | 4.322594 | 2.95 × 10−21 | protein mothers against dpp [Bombyx mori (domestic silkworm)] |
| LOC101744534 | BMPR2 | 23.0032 | 1.24 × 10−6 | 4.090361 | 2.39 × 10−30 | probable serine/threonine-protein kinase DDB_G0278901 [Bombyx mori (domestic silkworm)] |
| LOC101740154 | RIF1 | 12.9223 | 2.47 × 10−9 | 2.924023 | 4.05 × 10−19 | telomere-associated protein RIF1 [Bombyx mori (domestic silkworm)] |
| LOC100158253 | Pi3k60 | 20.1611 | 8.42 × 10−13 | 2.09716 | 7.71 × 10−44 | phosphatidylinositol 3-kinase 60 [Bombyx mori (domestic silkworm)] |
| LOC101735864 | KIAA0445 | 15.4856 | 8.89 × 10−7 | 2.064435 | 2.90 × 10−47 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A containing DEAD/H box 1 homolog [Bombyx mori (domestic silkworm)] |
| LOC101738704 | ACVR1 | 13.23511 | 1.51 × 10−5 | 2.028722 | 5.10 × 10−59 | activin receptor type-1 [Bombyx mori (domestic silkworm)] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Hu, P.; Zhu, L.-B.; You, L.-L.; Cao, H.-H.; Wang, J.; Zhang, S.-Z.; Liu, M.-H.; Toufeeq, S.; Huang, S.-J.; et al. DNA Methylation Is Correlated with Gene Expression during Diapause Termination of Early Embryonic Development in the Silkworm (Bombyx mori). Int. J. Mol. Sci. 2020, 21, 671. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020671
Li B, Hu P, Zhu L-B, You L-L, Cao H-H, Wang J, Zhang S-Z, Liu M-H, Toufeeq S, Huang S-J, et al. DNA Methylation Is Correlated with Gene Expression during Diapause Termination of Early Embryonic Development in the Silkworm (Bombyx mori). International Journal of Molecular Sciences. 2020; 21(2):671. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020671
Chicago/Turabian StyleLi, Bing, Pei Hu, Lin-Bao Zhu, Ling-Ling You, Hui-Hua Cao, Jie Wang, Shang-Zhi Zhang, Ming-Hui Liu, Shahzad Toufeeq, Shou-Jun Huang, and et al. 2020. "DNA Methylation Is Correlated with Gene Expression during Diapause Termination of Early Embryonic Development in the Silkworm (Bombyx mori)" International Journal of Molecular Sciences 21, no. 2: 671. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020671