Therapeutic Implications for PDE2 and cGMP/cAMP Mediated Crosstalk in Cardiovascular Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
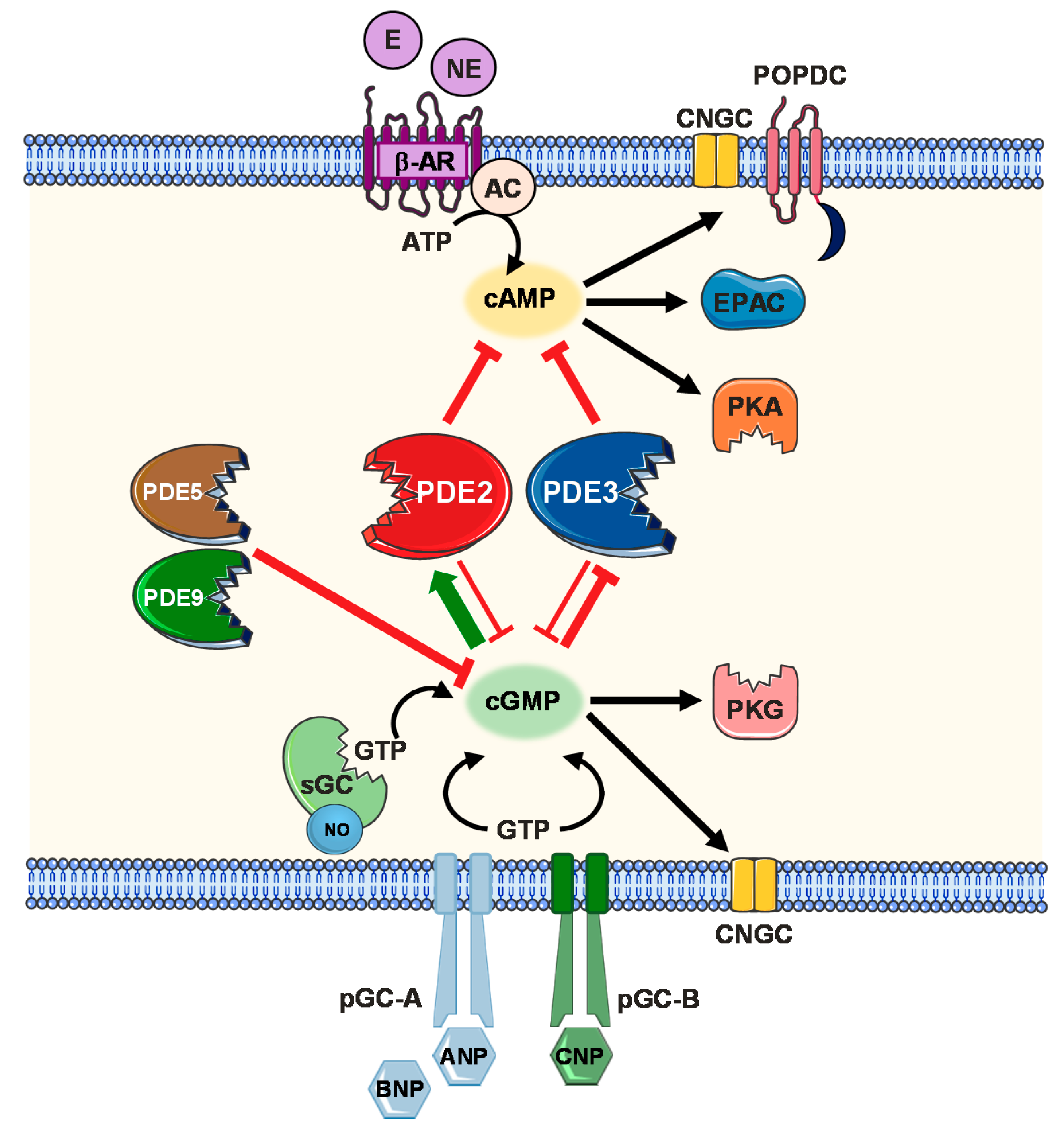
2. Cyclic Nucleotide Signalling and Compartmentalization
2.1. cAMP Signalling Pathway
2.2. cGMP Signalling Pathway (NO, Natriuretic Peptides)
2.3. Cellular Compartmentalization Mechanisms
- (i)
- Characteristic cardiomyocyte transverse tubules (T-tubules), caveolae and non-caveolae membrane microdomains contribute to ensure compartmentalized signal initiation [82]. Importantly, cholesterol and sphingolipid rich areas in the membrane, called lipid rafts, form gel-like, liquid-ordered domains that hinder membrane fluidity and prevent localized GPCRs to freely diffuse throughout the lipid bilayer [83]. Modern tools combining the use of scanning ion-conductance microscopy (SICM) and Förster resonance energy transfer (FRET) techniques aided to identify domain specific cAMP fluctuations upon generating a detailed topographical image of the cell surface. β1-AR specific cAMP responses were thereby detected at both T-tubule and non-T-tubule microdomains, whereas cAMP synthesis upon β2-AR specific activation was confined to the T-tubules only [8]. In a similar manner, cGMP responses after specific β3-AR stimulation were also observed to be restricted to the T-tubules [84]. Besides their influence on membrane fluidity, caveolae/lipid rafts localized within T-tubule and non-T-tubule regions also function to define AC isoform localization. In various cell types, AC1, AC3, AC5, AC6 and AC8 were reported to associate with lipid raft domains, while AC2, AC4, AC7 and AC9 were identified within non-raft microdomains [85]. Other studies also demonstrated that the β2-AR macromolecular complex including Gs, Gi, AC5, AC6 and PKA is specifically localized within caveolar membrane fragments. Contrastingly, β1-ARs are ubiquitously distributed in both caveolar and non-caveolar compartments in adult rat ventricular myocytes [86,87,88,89]. Furthermore, the β3-AR/eNOS/sGC macromachinery was also demonstrated to be localized within caveolae-enriched membrane fractions. On the other hand, heart failure contributes to β3-AR redistribution and altered co-localization of sGC and caveolin-3, disrupting compartmentalized cGMP synthesis [84]. Moreover, caveolae play a fundamental role to mediate appropriate signalling responses in VSMCs and the endothelium. In caveolin-1 deficient mice, impaired endothelium-dependent relaxation and reduced myogenic tone were observed [90,91]. Also, Sampson et al., demonstrated that KATP channels and AC co-localization in rat aortic smooth muscle caveolae is crucial for appropriate KATP channel modulation [92].
- (ii)
- Compartmentalized signalling also relies on the spatial organization of intracellular molecules coupling PKA activity to downstream effector targets as well as signal feedback regulatory elements. A-kinase anchoring proteins (AKAPs) are a superfamily of organizing scaffold proteins which are able to bind PKA and other signalling enzymes, directing their localization to specific cellular compartments [32,93,94]. 17 AKAPs have been detected and identified in cardiac tissues coordinating a plethora of signalling components besides PKA including PKC, PDEs, ACs, phosphatases and GTPases. Accordingly, their function has been highlighted to partake in various homeostatic as well as cardioprotective processes including calcium cycling, ECC coupling, heart rhythm and action potential regulation. Additionally, AKAPs coordinate signalling cascades involved in cardiac remodeling responses under pathophysiological settings [94,95,96,97]. Several AKAP-specific interactions with multiple AC and PDE isoforms have been identified, supporting precise compartment boundary construction [98,99,100]. Among others, AKAP79/150 was identified to associate with AC5 and AC6 [98] and mAKAP with AC5 [101]. Moreover, mAKAP was also demonstrated to associate with PDE4D3 [102,103]. Terrenoire et al., could also show that AKAP9 selectively formulates a complex between PDE4D3 and cardiac IKs channels, but not with PDE4D5 [104]. AKAP75 expression has also been demonstrated in VSMC promoting cAMP/PKA signalling [105]. AKAP-dependent PKA interactions with several ion channels (LTCC and KATP) have also been described in vascular tissue [106,107]. Such highly selective interactions mediated via AKAP scaffolds certainly fine tune the elicited intracellular responses. However, further investigations are still required to fully identify specific complex interactions and elucidate their role in cAMP/cGMP signal propagation.
- (iii)
- Localized cyclic nucleotide production alone remains insufficient to account for compartmentalized signalling responses if their diffusion throughout the cytosol is not restricted. Physical cytosolic barriers, cAMP buffering and export mechanisms via cardiac MRP4 efflux protein have been reported to limit cAMP diffusion in the cytosol [108,109]. More importantly, confined cyclic nucleotide distribution via PDE-mediated hydrolysis has been demonstrated in multiple studies [110,111]. 11 PDE subfamilies with over 100 different isoforms and splice variants have been identified [112]. Of the PDE superfamily, PDE1, 2, 3, 4, 5, 8 and 9 are fundamental constituents of the cardiac signalosome coordinating cardiac function under both physiological and pathophysiological conditions [113]. On the other hand PDE1, 2, 3, 4, 5 and 7 constitute the major PDE activity in the vasculature [114,115]. Only PDE2, PDE3 and to a lesser extent PDE1 have been shown to mediate a cGMP/cAMP crosstalk upon modulation of the PDE activities via cGMP. cGMP competitively inhibits the cAMP hydrolytic activity of both PDE1 and PDE3. Exceptionally, PDE2 is the only member of the PDE family that is activated upon allosteric cGMP binding, increasing its cAMP hydrolytic activity [110]. In this review, we will particularly focus to discuss PDE2 functional role in the cardiovascular system.
2.4. PDE2 Molecular Aspects and cGMP/cAMP-Mediated Crosstalk
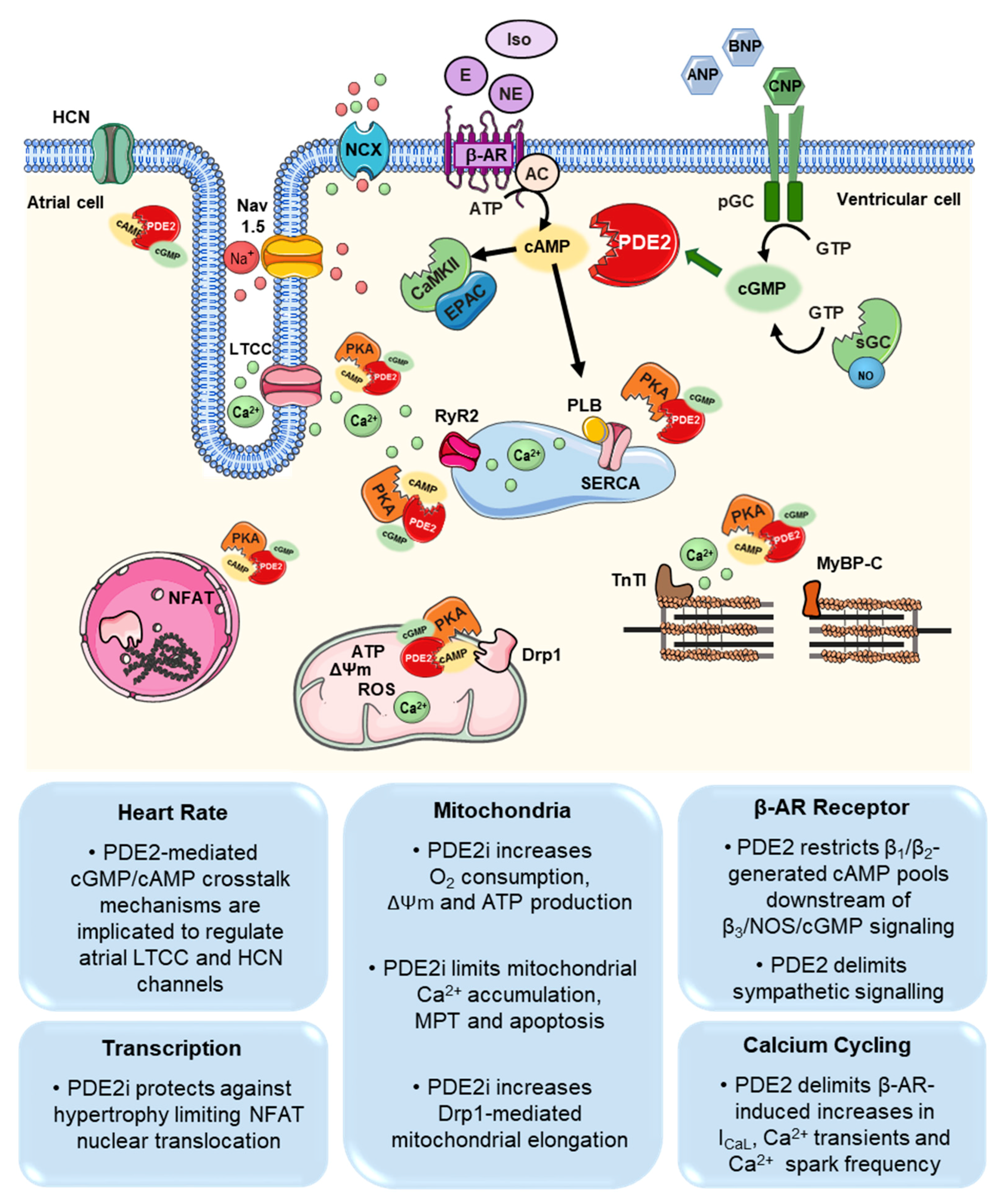
3. PDE2 Functions in the Cardiovascular System
3.1. Cardiomyocytes
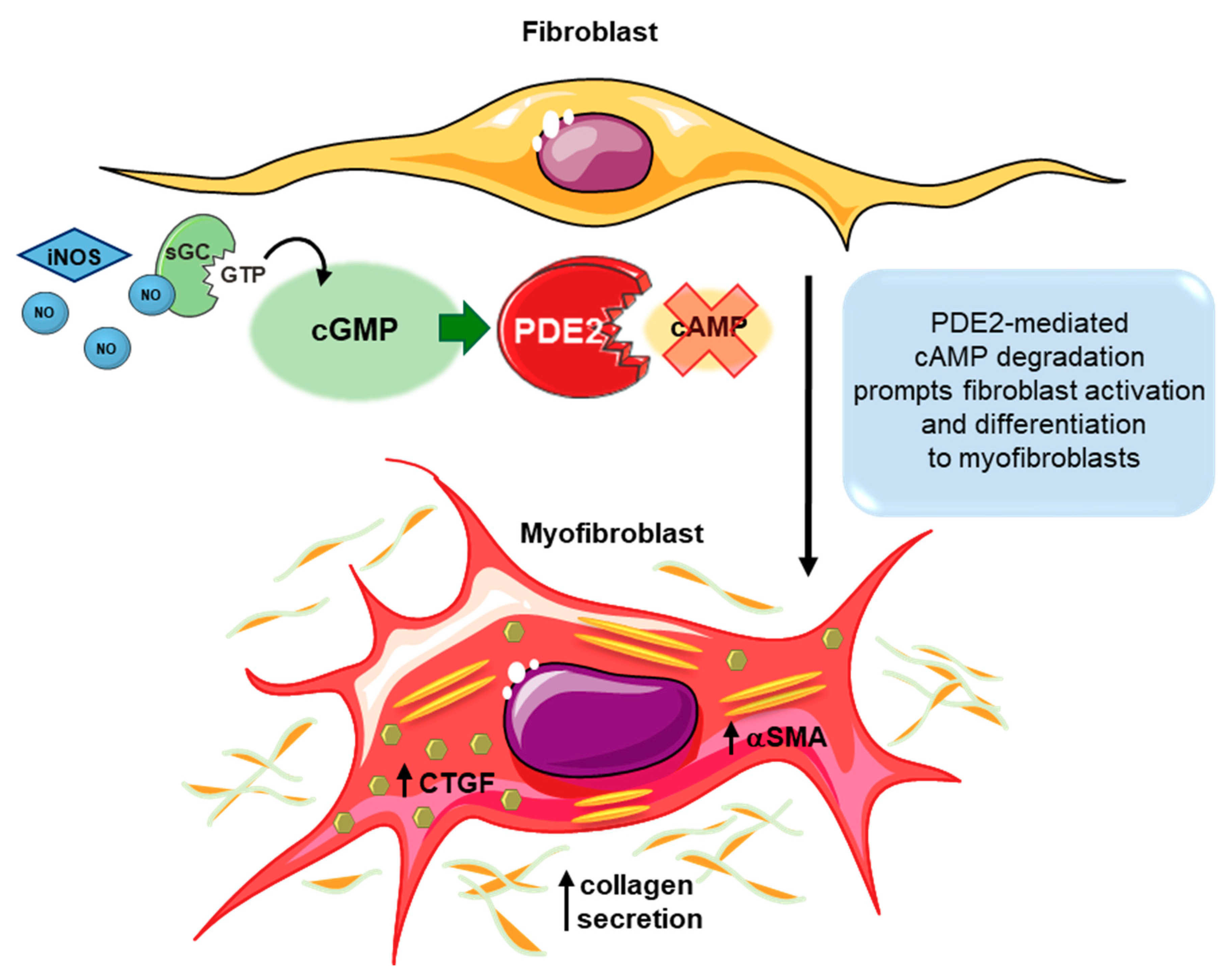
3.2. Fibroblasts
3.3. Sympathetic Neurons
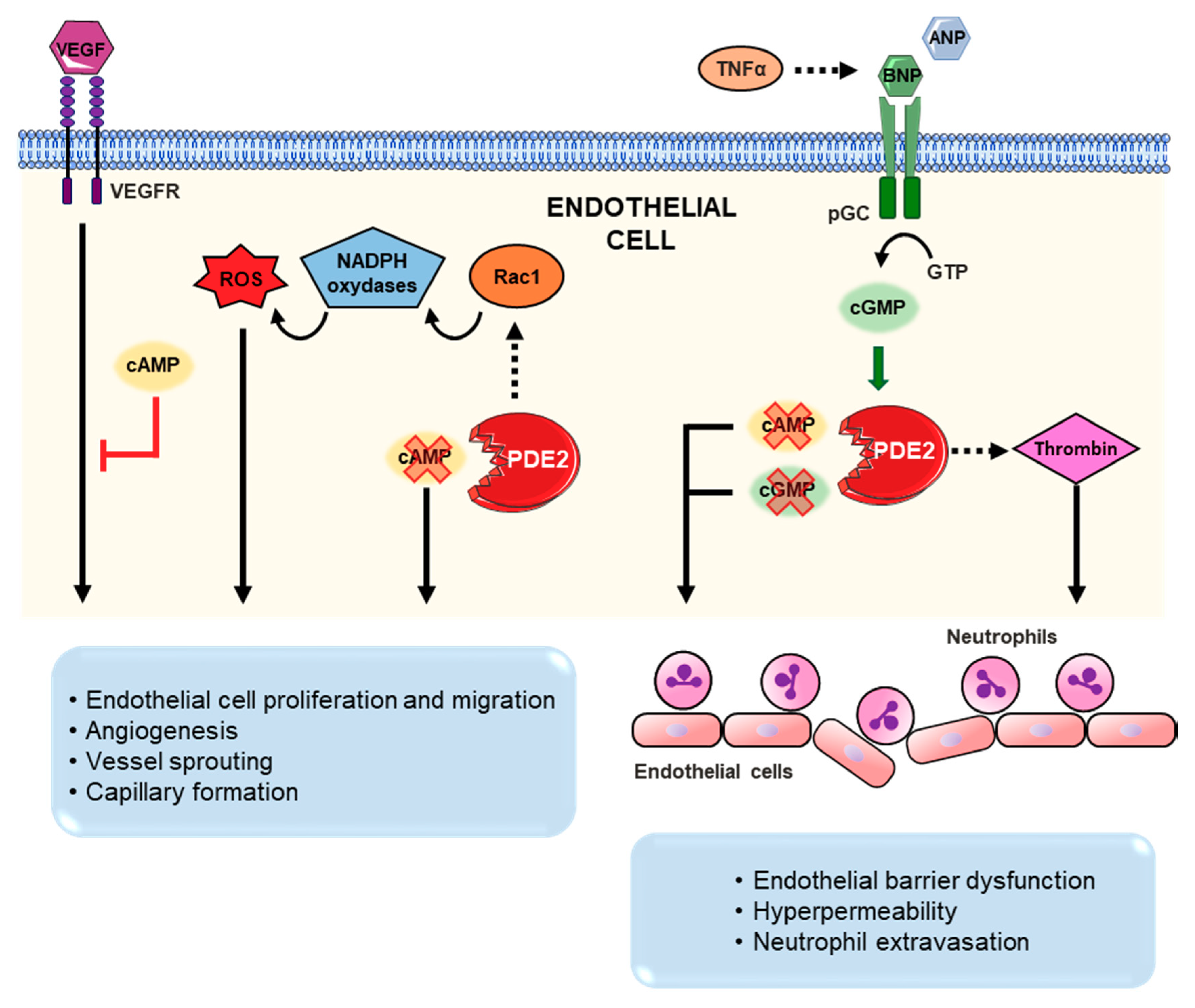
3.4. Vasculature and Circulating Blood Cells
4. Role of PDE2 in Cardiovascular Disease
4.1. Arrhythmia (Atria, Ventricular, Sinus Node)
4.2. Hypertrophy, Heart Failure
4.3. Myocardial Infarction (MI)/Reperfusion Injury
4.4. Angiogenesis
4.5. Inflammation/Sepsis
4.6. Pulmonary Hypertension
5. Clinical Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AC | Adenylyl cyclase |
| AhR | Aryl hydrocarbon receptor |
| ANP | Atrial natriuretic peptide |
| AKAP | A-kinase anchoring protein |
| β-AR | β-adrenoceptor |
| BNP | Brain natriuretic peptide |
| cAMP | 3′,5′-cyclic adenosine monophosphate |
| cGMP | 3′,5′-cyclic guanosine monophosphate |
| CNGCs | Cyclic nucleotide gated ion channels |
| CNP | C-type natriuretic peptide |
| CVD | Cardiovascular disease |
| DRP1 | Dynamin related protein 1 |
| ECC | Excitation-contraction coupling |
| EPAC | Exchange proteins directly activated by cAMP |
| GPCR | G protein coupled receptors |
| HUVEC | Human umbilical vein cells |
| LTCC | L-type Ca2+ channel |
| MI | Myocardial infarction |
| MPT | Mitochondrial permeability transition |
| MYBPC | Myosin-binding protein C |
| NP | Natriuretic peptide |
| NOS | Nitric oxide synthase |
| PDE | Phosphodiesterase |
| pGC | Particulate guanylyl cyclases |
| PH | Pulmonary hypertension |
| PLB | Phospholamban |
| POPDC | Popeye-domain-containing proteins |
| RyR | Ryanodine receptors |
| sAC | Soluble adenylyl cyclases |
| sGC | Soluble guanylyl cyclases |
| SNP | Sodium nitroprusside |
| SR | Sarcoplasmic reticulum |
| TNF-α | Tumor necrosis factor-α |
| VEGF | Vascular endothelial growth factor |
| VSMC | Vascular smooth muscle cell |
| VEC | Vascular endothelial cell |
References
- Kaptoge, S.; Pennells, L.; De Bacquer, D.; Cooney, M.T.; Kavousi, M.; Stevens, G.; Riley, L.M.; Savin, S.; Khan, T.; Altay, S.; et al. World Health Organization cardiovascular disease risk charts: Revised models to estimate risk in 21 global regions. Lancet Glob. Health 2019, 7, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.; Whitsel, L.; Khavjou, O.; Phelps, D.; Leib, A. Projections of Cardiovascular Disease Prevalence and Costs: 2015–2035; Technical Report for American Heart Association: Washington, DC, USA, 2016. [Google Scholar]
- Flora, G.D.; Nayak, M.K. A Brief Review of Cardiovascular Diseases, Associated Risk Factors and Current Treatment Regimes. Curr. Pharm. Des. 2019, 25, 4063–4084. [Google Scholar] [CrossRef] [PubMed]
- Tzoulaki, I.; Elliott, P.; Kontis, V.; Ezzati, M. Worldwide Exposures to Cardiovascular Risk Factors and Associated Health Effects: Current Knowledge and Data Gaps. Circulation 2016, 133, 2314–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viacheslav, N.; Manuela, Z. (Eds.) Microdomains in the Cardiovascular System; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Perera, R.K.; Sprenger, J.U.; Steinbrecher, J.H.; Hübscher, D.; Lehnart, S.E.; Abesser, M.; Schuh, K.; El-Armouche, A.; Nikolaev, V.O. Microdomain switch of cGMP-regulated phosphodiesterases leads to ANP-induced augmentation of β-adrenoceptor-stimulated contractility in early cardiac hypertrophy. Circ. Res. 2015, 116, 1304–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef]
- Zakhary, D.R.; Moravec, C.S.; Bond, M. Regulation of PKA binding to AKAPs in the heart: Alterations in human heart failure—Alterations in Human Heart Failure. Circulation 2000, 101, 1459–1464. [Google Scholar] [CrossRef] [Green Version]
- Mongillo, M.; McSorley, T.; Evellin, S.; Sood, A.; Lissandron, V.; Terrin, A.; Huston, E.; Hannawacker, A.; Lohse, M.J.; Pozzan, T.; et al. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ. Res. 2004, 95, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Surdo, N.C.; Berrera, M.; Koschinski, A.; Brescia, M.; MacHado, M.R.; Carr, C.; Wright, P.; Gorelik, J.; Morotti, S.; Grandi, E.; et al. FRET biosensor uncovers cAMP nano-domains at b-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Berisha, F.; Nikolaev, V.O. Cyclic nucleotide imaging and cardiovascular disease. Pharmacol. Ther. 2017, 175, 107–115. [Google Scholar] [CrossRef]
- Bedioune, I.; Bobin, P.; Leroy, J.; Fischmeister, R.; Vandecasteele, G. Cyclic Nucleotide Phosphodiesterases and Compartmentation in Normal and Diseased Heart. In Microdomains in the Cardiovascular System; Viacheslav, N., Manuela, Z., Eds.; Springer: Cham, Switzerland, 2017; pp. 97–116. [Google Scholar]
- Kim, G.E.; Kass, D.A. Cardiac phosphodiesterases and their modulation for treating heart disease. In Heart Failure; Springer: Cham, Switzerland, 2016; Volume 243, pp. 249–269. [Google Scholar]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef] [PubMed]
- Mehel, H.; Emons, J.; Vettel, C.; Wittköpper, K.; Seppelt, D.; Dewenter, M.; Lutz, S.; Sossalla, S.; Maier, L.S.; Lechêne, P.; et al. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts β-adrenergic responses in cardiomyocytes. J. Am. Coll. Cardiol. 2013, 62, 1596–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vettel, C.; Lindner, M.; Dewenter, M.; Lorenz, K.; Schanbacher, C.; Riedel, M.; Lämmle, S.; Meinecke, S.; Mason, F.E.; Sossalla, S.; et al. Phosphodiesterase 2 Protects Against Catecholamine-Induced Arrhythmia and Preserves Contractile Function after Myocardial Infarction. Circ. Res. 2017, 120, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Zoccarato, A.; Surdo, N.C.; Aronsen, J.M.; Fields, L.A.; Mancuso, L.; Dodoni, G.; Stangherlin, A.; Livie, C.; Jiang, H.; Sin, Y.Y.; et al. Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2. Circ. Res. 2015, 117, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Baliga, R.S.; Preedy, M.E.J.; Dukinfield, M.S.; Chu, S.M.; Aubdool, A.A.; Bubb, K.J.; Moyes, A.J.; Tones, M.A.; Hobbs, A.J. Phosphodiesterase 2 inhibition preferentially promotes NO/guanylyl cyclase/cGMP signaling to reverse the development of heart failure. Proc. Natl. Acad. Sci. USA 2018, 115, 7428–7437. [Google Scholar] [CrossRef] [Green Version]
- Judina, A.; Gorelik, J.; Wright, P.T. Studying signal compartmentation in adult cardiomyocytes. Biochem. Soc. Trans. 2020, 48, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Madamanchi, A. β-adrenergic receptor signaling in cardiac function and heart failure. McGill J. Med. 2007, 10, 99–104. [Google Scholar]
- Keely, S.L. Prostaglandin E1 Activation of Heart cAMP-dependent Protein Kinase: Apparent Dissociation of Protein Kinase Activation from Increases in Phosphorylase Activity and Contractile Force. Mol. Pharmacol. 1979, 15, 235–245. [Google Scholar]
- Honda, A.; Sekiguchi, Y.; Mori, Y. Prostaglandin E2 stimulates cyclic AMP-mediated hyaluronan synthesis in rabbit pericardial mesothelial cells. Biochem. J. 1993, 292, 497–502. [Google Scholar] [CrossRef]
- Méry, P.F.; Brechler, V.; Pavoine, C.; Pecker, F.; Fischmeister, R. Glucagon stimulates the cardiac Ca2+ current by activation of adenylyl cyclase and inhibition of phosphodiesterase. Nature 1990, 345, 158–161. [Google Scholar] [CrossRef]
- Vila Petroff, M.G.; Egan, J.M.; Wang, X.; Sollott, S.J. Glucagon-Like Peptide-1 Increases cAMP but Fails to Augment Contraction in Adult Rat Cardiac Myocytes. Circ. Res. 2001, 89, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessauer, C.W.; Watts, V.J.; Ostrom, R.S.; Conti, M.; Dove, S.; Seifert, R. International union of basic and clinical pharmacology. CI. structures and small molecule modulators of mammalian adenylyl cyclases. Pharmacol. Rev. 2017, 69, 96–139. [Google Scholar] [CrossRef] [PubMed]
- Birnbaumer, L.; Abramowitz, J.; Brown, A.M. Receptor-effector coupling by G proteins. Biochim. Biophys. Acta 1990, 1031, 163–224. [Google Scholar] [CrossRef]
- Tilley, D.G. G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ. Res. 2011, 109, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, I.; Bualeong, T.; Budde, H.; Cimiotti, D.; Appukuttan, A.; Klein, N.; Steinwascher, P.; Reusch, P.; Mügge, A.; Meyer, R.; et al. Soluble adenylyl cyclase: A novel player in cardiac hypertrophy induced by isoprenaline or pressure overload. PLoS ONE 2018, 13, e0192322. [Google Scholar] [CrossRef] [Green Version]
- Boularan, C.; Gales, C. Cardiac cAMP: Production, hydrolysis, modulation and detection. Front. Pharmacol. 2015, 6, 203. [Google Scholar] [CrossRef] [Green Version]
- Nakano, S.J.; Sucharov, J.; van Dusen, R.; Cecil, M.; Nunley, K.; Wickers, S.; Karimpur-Fard, A.; Stauffer, B.L.; Miyamoto, S.D.; Sucharov, C.C. Cardiac Adenylyl Cyclase and Phosphodiesterase Expression Profiles Vary by Age, Disease, and Chronic Phosphodiesterase Inhibitor Treatment. J. Card. Fail. 2017, 23, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, T.A.; Dessauer, C.W. Function of Adenylyl Cyclase in Heart: The AKAP Connection. J. Cardiovasc. Dev. Dis. 2018, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium Cycling and Signaling in Cardiac Myocytes. Annu. Rev. Physiol. 2007, 70, 23–49. [Google Scholar] [CrossRef] [Green Version]
- Indolfi, C.; Avvedimento, E.V.; Di Lorenzo, E.; Esposito, G.; Rapacciuolo, A.; Giuliano, P.; Grieco, D.; Cavuto, L.; Stingone, A.M.; Ciullo, I.; et al. Activation of cAMP-PKA signaling in vivo inhibits smooth muscle cell proliferation induced by vascular injury. Nat. Med. 1997, 3, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Morgado, M.; Cairrão, E.; Santos-Silva, A.J.; Verde, I. Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Cell. Mol. Life Sci. 2012, 69, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.E.; Lum, H.; Schaphorst, K.L.; Verin, A.D.; Garcia, J.G.N. Regulation of endothelial barrier function by the cAMP-dependent protein kinase. Endothelium 2000, 7, 287–308. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.L.; Fitzsimons, D.P.; Ralphe, J.C. Cardiac MyBP-C Regulates the Rate and Force of Contraction in Mammalian Myocardium. Circ. Res. 2015, 116, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Desantiago, J.; Chu, G.; Kranias, E.G.; Bers, D.M. Phosphorylation of phospholamban and troponin I in β-adrenergic-induced acceleration of cardiac relaxation. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, 69–79. [Google Scholar] [CrossRef]
- El-Armouche, A.; Eschenhagen, T. β-Adrenergic stimulation and myocardial function in the failing heart. Heart Fail. Rev. 2009, 14, 225–241. [Google Scholar] [CrossRef]
- Saucerman, J.J.; McCulloch, A.D. Cardiac beta-Adrenergic Signaling: From Subcellular Microdomains to Heart Failure. Ann. N. Y. Acad. Sci. 2006, 1080, 348–361. [Google Scholar] [CrossRef]
- Yang, J.H.; Polanowska-Grabowska, R.K.; Smith, J.S.; Shields, C.W.; Saucerman, J.J. PKA catalytic subunit compartmentation regulates contractile and hypertrophic responses to β-adrenergic signaling. J. Mol. Cell. Cardiol. 2014, 66, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Tomita, H.; Nazmy, M.; Kajimoto, K.; Yehia, G.; Molina, C.A.; Sadoshima, J. Inducible cAMP early repressor (ICER) is a negative-feedback regulator of cardiac hypertrophy and an important mediator of cardiac myocyte apoptosis in response to β-adrenergic receptor stimulation. Circ. Res. 2003, 93, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Roberts, O.L.; Dart, C. CAMP signalling in the vasculature: The role of Epac (exchange protein directly activated by cAMP). Biochem. Soc. Trans. 2014, 42, 89–97. [Google Scholar] [CrossRef]
- Fujita, T.; Umemura, M.; Yokoyama, U.; Okumura, S.; Ishikawa, Y. The role of Epac in the heart. Cell. Mol. Life Sci. 2017, 74, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Bucchi, A.; Piantoni, C.; Barbuti, A.; DiFrancesco, D.; Baruscotti, M. HCN Channels and Cardiac Pacemaking. In Channelopathies in Heart Disease; Thomas, D., Remme, C.A., Eds.; Springer: Cham, Switzerland, 2018; pp. 97–126. [Google Scholar]
- Shen, B.; Cheng, K.T.; Leung, Y.K.; Kwok, Y.C.; Kwan, H.Y.; Wong, C.O.; Chen, Z.Y.; Huang, Y.; Yao, X. Epinephrine-induced Ca2+ influx in vascular endothelial cells is mediated by CNGA2 channels. J. Mol. Cell. Cardiol. 2008, 45, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.K.; Du, J.; Huang, Y.; Yao, X. Cyclic Nucleotide-Gated Channels Contribute to Thromboxane A2-Induced Contraction of Rat Small Mesenteric Arteries. PLoS ONE 2010, 5, e11098. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.; Breher, S.S.; Waldeyer, C.; Schindler, R.F.R.; Nikolaev, V.O.; Rinné, S.; Wischmeyer, E.; Schlueter, J.; Becher, J.; Simrick, S.; et al. Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J. Clin. Investig. 2012, 122, 1119–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, T. POPDC proteins and cardiac function. Biochem. Soc. Trans. 2019, 47, 1393–1404. [Google Scholar] [CrossRef]
- Schindler, R.; Scotton, C.; French, V.; Ferlini, A.; Brand, T. The Popeye Domain Containing Genes and Their Function in Striated Muscle. J. Cardiovasc. Dev. Dis. 2016, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Sandner, P.; Krieg, T. cGMP at the centre of attention: Emerging strategies for activating the cardioprotective PKG pathway. Basic Res. Cardiol. 2018, 113, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Potter, L.R. Guanylyl cyclase structure, function and regulation. Cell. Signal. 2011, 23, 1921–1926. [Google Scholar] [CrossRef] [Green Version]
- Potter, L.R. Regulation and therapeutic targeting of peptide-activated receptor guanylyl cyclases. Pharmacol. Ther. 2011, 130, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Denninger, J.W.; Marletta, M.A. Guanylate cyclase and the .NO/cGMP signaling pathway. Biochim. Biophys. Acta 1999, 1411, 334–350. [Google Scholar] [CrossRef] [Green Version]
- Russwurm, M.; Behrends, S.; Harteneck, C.; Koesling, D. Functional properties of a naturally occurring isoform of soluble guanylyl cyclase. Biochem. J. 1998, 335, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mergia, E.; Stegbauer, J. Role of Phosphodiesterase 5 and Cyclic GMP in Hypertension. Curr. Hypertens. Rep. 2016, 18, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M. Molecular physiology of membrane guanylyl cyclase receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef]
- Matsuo, A.; Nagai-Okatani, C.; Nishigori, M.; Kangawa, K.; Minamino, N. Natriuretic peptides in human heart: Novel insight into their molecular forms, functions, and diagnostic use. Peptides 2019, 111, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Minamino, N.; Horio, T.; Nishikimi, T. Natriuretic Peptides in the Cardiovascular System. In Handbook of Biologically Active Peptides; Kastin, A.J., Ed.; Academic Press: Burlington, MA, USA, 2006; pp. 1199–1207. [Google Scholar]
- Stangherlin, A.; Gesellchen, F.; Zoccarato, A.; Terrin, A.; Fields, L.A.; Berrera, M.; Surdo, N.C.; Craig, M.A.; Smith, G.; Hamilton, G.; et al. CGMP signals modulate camp levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ. Res. 2011, 108, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Cuello, F.; Nikolaev, V.O. Cardiac cGMP Signaling in Health and Disease. J. Cardiovasc. Pharmacol. 2020, 75, 399–409. [Google Scholar] [CrossRef]
- Blanton, R.M. cGMP Signaling and Modulation in Heart Failure. J. Cardiovasc. Pharmacol. 2020, 75, 385–398. [Google Scholar] [CrossRef]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef] [Green Version]
- Krüger, M.; Kötter, S.; Grützner, A.; Lang, P.; Andresen, C.; Redfield, M.M.; Butt, E.; dos Remedios, C.G.; Linke, W.A. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 2009, 104, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, B.; Lohmann, S.M.; Smolenski, A.; Linnemüller, S.; Pieske, B.; Schröder, F.; Molkentin, J.D.; Drexler, H.; Wollert, K.C. Inhibition of calcineurin-NFAT hypertrophy signaling by cGMP-dependent protein kinase type I in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 11363–11368. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, H.; Kuwahara, K.; Nishida, M.; Jian, Z.; Rong, X.; Kiyonaka, S.; Kuwabara, Y.; Kurose, H.; Inoue, R.; Mori, Y.; et al. Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-a signaling in the heart. Circ. Res. 2010, 106, 1849–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaiber, M.; Kruse, M.; Völker, K.; Schröter, J.; Feil, R.; Freichel, M.; Gerling, A.; Feil, S.; Dietrich, A.; Londoño, J.E.C.; et al. Novel insights into the mechanisms mediating the local antihypertrophic effects of cardiac atrial natriuretic peptide: Role of cGMP-dependent protein kinase and RGS2. Basic Res. Cardiol. 2010, 105, 583–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokudome, T.; Kishimoto, I.; Horio, T.; Arai, Y.; Schwenke, D.O.; Hino, J.; Okano, I.; Kawano, Y.; Kohno, M.; Miyazato, M.; et al. Regulator of G-protein signaling subtype 4 mediates antihypertrophic effect of locally secreted natriuretic peptides in the heart. Circulation 2008, 117, 2329–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukreja, R.C.; Salloum, F.N.; Das, A.; Koka, S.; Ockaili, R.A.; Xi, L. Emerging new uses of phosphodiesterase-5 inhibitors in cardiovascular diseases. Exp. Clin. Cardiol. 2011, 16, e30. [Google Scholar] [PubMed]
- Kitakaze, M.; Asakura, M.; Kim, J.; Shintani, Y.; Asanuma, H.; Hamasaki, T.; Seguchi, O.; Myoishi, M.; Minamino, T.; Ohara, T.; et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): Two randomised trials. Lancet 2007, 370, 1483–1493. [Google Scholar] [CrossRef]
- Madhani, M.; Hall, A.R.; Cuello, F.; Charles, R.L.; Burgoyne, J.R.; Fuller, W.; Hobbs, A.J.; Shattock, M.J.; Eaton, P. Phospholemman Ser69 phosphorylation contributes to sildenafil-induced cardioprotection against reperfusion injury. Am. J. Physiol. Circ. Physiol. 2010, 299, H827–H836. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.D.T.; Garlid, K.D.; West, I.C.; Lincoln, T.M.; Downey, J.M.; Cohen, M.V.; Critz, S.D. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ. Res. 2005, 97, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Xi, L.; Kukreja, R.C. Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3β. J. Biol. Chem. 2008, 283, 29572–29585. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.D.T.; Pierre, S.V.; Cohen, M.V.; Downey, J.M.; Garlid, K.D. cGMP signalling in pre-and post-conditioning: The role of mitochondria. Cardiovasc. Res. 2008, 77, 344–352. [Google Scholar] [CrossRef]
- Kass, D.A.; Takimoto, E.; Nagayama, T.; Champion, H.C. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc. Res. 2007, 75, 303–314. [Google Scholar] [CrossRef]
- VanSchouwen, B.; Melacini, G. Regulation of HCN ion channels by non-canonical cyclic nucleotides. Handb. Exp. Pharmacol. 2017, 238, 123–133. [Google Scholar] [PubMed]
- Zhao, C.Y.; Greenstein, J.L.; Winslow, R.L. Roles of phosphodiesterases in the regulation of the cardiac cyclic nucleotide cross-talk signaling network. J. Mol. Cell. Cardiol. 2016, 91, 215–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.S.; Brunton, L.L.; Brown, J.H.; Reese, J.B.; Mayer, S.E. Hormonally specific expression of cardiac protein kinase activity. Proc. Natl. Acad. Sci. USA 1979, 76, 1570–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoccarato, A.; Zaccolo, M. cAMP Compartmentalisation and Hypertrophy of the Heart: ‘Good’ Pools of cAMP and ‘Bad’ Pools of cAMP Coexist in the Same Cardiac Myocyte. In Microdomains in the Cardiovascular System; Nikolaev, V., Zaccolo, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 117–141. [Google Scholar]
- Perera, R.K.; Nikolaev, V.O. Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol. 2013, 207, 650–662. [Google Scholar] [CrossRef]
- Bhogal, N.; Hasan, A.; Gorelik, J. The Development of Compartmentation of cAMP Signaling in Cardiomyocytes: The Role of T-Tubules and Caveolae Microdomains. J. Cardiovasc. Dev. Dis. 2018, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.R.; Gratwohl, J.; Cozad, M.; Yang, P.-C.; Clancy, C.E.; Harvey, R.D. Compartmentalized cAMP Signaling Associated With Lipid Raft and Non-raft Membrane Domains in Adult Ventricular Myocytes. Front. Pharmacol. 2018, 9, 332. [Google Scholar] [CrossRef]
- Schobesberger, S.; Wright, P.T.; Poulet, C.; Sanchez-Alonso, J.L.; Mansfield, C.; Friebe, A.; Harding, S.E.; Balligand, J.L.; Nikolaev, V.O.; Gorelik, J. β3-Adrenoceptor redistribution impairs NO/cGMP/PDE2 signalling in failing cardiomyocytes. Elife 2020, 9, e52221. [Google Scholar] [CrossRef]
- Johnstone, T.B.; Agarwal, S.R.; Harvey, R.D.; Ostrom, R.S. cAMP Signaling Compartmentation: Adenylyl Cyclases as Anchors of Dynamic Signaling Complexes. Mol. Pharmacol. 2018, 93, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Rybin, V.O.; Pak, E.; Alcott, S.; Steinberg, S.F. Developmental changes in β2-adrenergic receptor signaling in ventricular myocytes: The role of Gi proteins and caveolae microdomains. Mol. Pharmacol. 2003, 63, 1338–1348. [Google Scholar] [CrossRef]
- Head, B.P.; Pateli, H.H.; Roth, D.M.; Lai, N.C.; Niesman, I.R.; Farquhar, M.G.; Insel, P.A. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J. Biol. Chem. 2005, 280, 31036–31044. [Google Scholar] [CrossRef] [Green Version]
- Calaghan, S.; White, E. Caveolae modulate excitation-contraction coupling and β2- adrenergic signalling in adult rat ventricular myocytes. Cardiovasc. Res. 2006, 69, 816–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.R.; MacDougall, D.A.; Tyser, R.; Pugh, S.D.; Calaghan, S.C.; Harvey, R.D. Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 50, 500–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293, 2449–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razani, B.; Engelman, J.A.; Wang, X.B.; Schubert, W.; Zhang, X.L.; Marks, C.B.; Macalusol, F.; Russell, R.G.; Li, M.; Pestell, R.G.; et al. Caveolin-1 Null Mice Are Viable but Show Evidence of Hyperproliferative and Vascular Abnormalities. J. Biol. Chem. 2001, 276, 38121–38138. [Google Scholar] [CrossRef]
- Sampson, L.J.; Hayabuchi, Y.; Standen, N.B.; Dart, C. Caveolae localize protein kinase A signaling to arterial ATP-sensitive potassium channels. Circ. Res. 2004, 95, 1012–1018. [Google Scholar] [CrossRef] [Green Version]
- Redden, J.M.; Dodge-Kafka, K.L.; Kapiloff, M.S. Function to Failure: Compartmentalization of Cardiomyocyte Signaling by A-Kinase-Anchoring Proteins. In Microdomains in the Cardiovascular System; Nikolaev, V., Zaccolo, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 37–57. [Google Scholar]
- Zhu, Y.; Jiang, X.; Zheng, Y.; Xiong, J.; Wei, D.; Zhang, D. Cardiac function modulation depends on the A-kinase anchoring protein complex. J. Cell. Mol. Med. 2019, 23, 7170–7179. [Google Scholar] [CrossRef]
- Diviani, D.; Osman, H.; Reggi, E. A-Kinase Anchoring Protein-Lbc: A Molecular Scaffold Involved in Cardiac Protection. J. Cardiovasc. Dev. Dis. 2018, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Diviani, D.; Reggi, E.; Arambasic, M.; Caso, S.; Maric, D. Emerging roles of A-kinase anchoring proteins in cardiovascular pathophysiology. Biochim. Biophys. Acta 2016, 1863, 1926–1936. [Google Scholar] [CrossRef]
- Kritzer, M.D.; Li, J.; Dodge-Kafka, K.; Kapiloff, M.S. AKAPs: The architectural underpinnings of local cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Efendiev, R.; Samelson, B.K.; Nguyen, B.T.; Phatarpekar, P.V.; Baameur, F.; Scott, J.D.; Dessauer, C.W. AKAP79 interacts with multiple Adenylyl Cyclase (AC) isoforms and scaffolds AC5 and -6 to α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J. Biol. Chem. 2010, 285, 14450–14458. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.X.; Cooper, D.M.F. AKAP79, PKC, PKA and PDE4 participate in a Gq-linked muscarinic receptor and adenylate cyclase 2 cAMP signalling complex. Biochem. J. 2013, 455, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delint-Ramirez, I.; Willoughby, D.; Hammond, G.V.R.; Ayling, L.J.; Cooper, D.M.F. Palmitoylation targets AKAP79 protein to lipid rafts and promotes its regulation of calcium-sensitive adenylyl cyclase type 8. J. Biol. Chem. 2011, 286, 32962–32975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapiloff, M.S.; Piggott, L.A.; Sadana, R.; Li, J.; Heredia, L.A.; Henson, E.; Efendiev, R.; Dessauer, C.W. An adenylyl cyclase-mAKAP β signaling complex regulates cAMP levels in cardiac myocytes. J. Biol. Chem. 2009, 284, 23540–23546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnart, S.E.; Wehrens, X.H.T.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.L.C.; Conti, M.; Marks, A.R. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodge, K.L.; Khouangsathiene, S.; Kapiloff, M.S.; Mouton, R.; Hill, E.V.; Houslay, M.D.; Langeberg, L.K.; Scott, J.D. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001, 20, 1921–1930. [Google Scholar] [CrossRef] [Green Version]
- Terrenoire, C.; Houslay, M.D.; Baillie, G.S.; Kass, R.S. The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J. Biol. Chem. 2009, 284, 9140–9146. [Google Scholar] [CrossRef] [Green Version]
- Indolfi, C.; Stabile, E.; Coppola, C.; Gallo, A.; Perrino, C.; Allevato, G.; Cavuto, L.; Torella, D.; Di Lorenzo, E.; Troncone, G.; et al. Membrane-Bound Protein Kinase A Inhibits Smooth Muscle Cell Proliferation In Vitro and In Vivo by Amplifying cAMP–Protein Kinase A Signals. Circ. Res. 2001, 88, 319–324. [Google Scholar] [CrossRef]
- Zhong, J.; Hume, J.R.; Keef, K.D. Anchoring protein is required for cAMP-dependent stimulation of L-type Ca2+ channels in rabbit portal vein. Am. J. Physiol. 1999, 277, C840–C844. [Google Scholar] [CrossRef]
- Hayabuchi, Y.; Dart, C.; Standen, N.B. Evidence for involvement of A-kinase anchoring protein in activation of rat arterial KATP channels by protein kinase A. J. Physiol. 2001, 536, 421–427. [Google Scholar] [CrossRef]
- Richards, M.; Lomas, O.; Jalink, K.; Ford, K.L.; Vaughan-Jones, R.D.; Lefkimmiatis, K.; Swietach, P. Intracellular tortuosity underlies slow cAMP diffusion in adult ventricular myocytes. Cardiovasc. Res. 2016, 110, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Sassi, Y.; Abi-Gerges, A.; Fauconnier, J.; Mougenot, N.; Reiken, S.; Haghighi, K.; Kranias, E.G.; Marks, A.R.; Lacampagne, A.; Engelhardt, S.; et al. Regulation of cAMP homeostasis by the efflux protein MRP4 in cardiac myocytes. FASEB J. 2012, 26, 1009–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaccolo, M.; Movsesian, M.A. cAMP and cGMP signaling cross-talk: Role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res. 2007, 100, 1569–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, M.; Mika, D.; Richter, W. Cyclic AMP compartments and signaling specificity: Role of cyclic nucleotide phosphodiesterases. J. Gen. Physiol. 2014, 143, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Omori, K.; Kotera, J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, K.; Kass, D.A. Nanodomain Regulation of Cardiac Cyclic Nucleotide Signaling by Phosphodiesterases. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 455–479. [Google Scholar] [CrossRef] [PubMed]
- Rybalkin, S.D.; Yan, C.; Bornfeldt, K.E.; Beavo, J.A. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ. Res. 2003, 93, 280–291. [Google Scholar] [CrossRef]
- Surapisitchat, J.; Beavo, J.A. Regulation of endothelial barrier function by cyclic nucleotides: The role of phosphodiesterases. Handb. Exp. Pharmacol. 2011, 204, 193–210. [Google Scholar] [CrossRef] [Green Version]
- Martins, T.J.; Mumby, M.C.; Beavog, J.A. Purification and Characterization of a Cyclic GMP-stimulated Cyclic Nucleotide Phosphodiesterase from Bovine Tissues. J. Biol. Chem. 1982, 257, 1973–1979. [Google Scholar]
- Pandit, J.; Forman, M.D.; Fennell, K.F.; Dillman, K.S.; Menniti, F.S. Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc. Natl. Acad. Sci. USA 2009, 106, 18225–18230. [Google Scholar] [CrossRef] [Green Version]
- Rascón, A.; Soderling, S.H.; Schaefer, J.B.; Beavo, J.A. Cloning and characterization of cAMP-specific phosphodiesterase (TbPDE2B) from Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 2002, 99, 4714–4719. [Google Scholar] [CrossRef] [Green Version]
- Rosman, G.J.; Martins, T.J.; Sonnenburg, W.K.; Beavo, J.A.; Ferguson, K.; Loughney, K. Isolation and characterization of human cDNAs encoding a cGMP-stimulated 3′,5′-cyclic nucleotide phosphodiesterase. Gene 1997, 191, 89–95. [Google Scholar] [CrossRef]
- Russwurm, C.; Zoidl, G.; Koesling, D.; Russwurm, M. Dual acylation of PDE2A splice variant 3. Targeting to synaptic membranes. J. Biol. Chem. 2009, 284, 25782–25790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavo, J.A.; Francis, S.H.; Houslay, M.D. (Eds.) Cyclic Nucleotide Phosphodiesterases in Health and Disease, 1st ed.; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Monterisi, S.; Lobo, M.J.; Livie, C.; Castle, J.C.; Weinberger, M.; Baillie, G.; Surdo, N.C.; Musheshe, N.; Stangherlin, A.; Gottlieb, E.; et al. PDE2A2 regulates mitochondria morphology and apoptotic cell death via local modulation of cAMP/PKA signalling. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, V.; Fouque, F.; Nivet, V.; Clot, J.-P.; Lugnier, C.; Desbuquois, B.; Benelli, C. Activation of a cGMP-stimulated cAMP phosphodiesterase by protein kinase C in a liver Golgi-endosomal fraction. Eur. J. Biochem. 2001, 259, 892–900. [Google Scholar] [CrossRef]
- Mongillo, M.; Tocchetti, C.G.; Terrin, A.; Lissandron, V.; Cheung, Y.F.; Dostmann, W.R.; Pozzan, T.; Kass, D.A.; Paolocci, N.; Houslay, M.D.; et al. Compartmentalized phosphodiesterase-2 activity blunts β-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ. Res. 2006, 98, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Lugnier, C.; Keravis, T.; Le Bec, A.; Pauvert, O.; Proteau, S.; Rousseau, E. Characterization of cyclic nucleotide phosphodiesterase isoforms associated to isolated cardiac nuclei. Biochim. Biophys. Acta 1999, 1472, 431–446. [Google Scholar] [CrossRef]
- Martinez, S.E.; Wu, A.Y.; Glavas, N.A.; Tang, X.B.; Turley, S.; Hol, W.G.J.; Beavo, J.A. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc. Natl. Acad. Sci. USA 2002, 99, 13260–13265. [Google Scholar] [CrossRef] [Green Version]
- Martinez, S.E.; Beavo, J.A.; Hol, W.G.J. GAF domains: Two-billion-year-old molecular switches that bind cyclic nucleotides. Mol. Interv. 2002, 2, 317–323. [Google Scholar] [CrossRef]
- Wu, A.Y.; Tang, X.B.; Martinez, S.E.; Ikeda, K.; Beavo, J.A. Molecular determinants for cyclic nucleotide binding to the regulatory domains of phosphodiesterase 2A. J. Biol. Chem. 2004, 279, 37928–37938. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yang, Q.; Dai, D.; Huang, Q. X-ray crystal structure of phosphodiesterase 2 in complex with a highly selective, nanomolar inhibitor reveals a binding-induced pocket important for selectivity. J. Am. Chem. Soc. 2013, 135, 11708–11711. [Google Scholar] [CrossRef]
- Card, G.L.; England, B.P.; Suzuki, Y.; Fong, D.; Powell, B.; Lee, B.; Luu, C.; Tabrizizad, M.; Gillette, S.; Ibrahim, P.N.; et al. Structural basis for the activity of drugs that inhibit phosphodiesterases. Structure 2004, 12, 2233–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osadchii, O.E. Myocardial Phosphodiesterases and Regulation of Cardiac Contractility in Health and Cardiac Disease. Cardiovasc. Drugs Ther. 2007, 21, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, W.L.; Appleman, M.M. The role of cyclic GMP in the regulation of cyclic AMP hydrolysis. Metabolism 1975, 24, 311–319. [Google Scholar] [CrossRef]
- Weber, S.; Zeller, M.; Guan, K.; Wunder, F.; Wagner, M.; El-Armouche, A. PDE2 at the crossway between cAMP and cGMP signalling in the heart. Cell. Signal. 2017, 38, 76–84. [Google Scholar] [CrossRef]
- De Oliveira, S.K.; Hoffmeister, M.; Gambaryan, S.; Müller-Esterl, W.; Guimaraes, J.A.; Smolenski, A.P. Phosphodiesterase 2A forms a complex with the co-chaperone XAP2 and regulates nuclear translocation of the aryl hydrocarbon receptor. J. Biol. Chem. 2007, 282, 13656–13663. [Google Scholar] [CrossRef] [Green Version]
- Walker, M.K.; Thackaberry, E.A.; Gabaldon, D.M.; Smith, S.M. Aryl Hydrocarbon Receptor Null Mice Develop Cardiac Hypertrophy and Increased Hypoxia-Inducible Factor-1a in the Absence of Cardiac Hypoxia. Cardiovasc. Toxicol. 2002, 2, 263–273. [Google Scholar]
- Mery, P.F.; Pavoine, C.; Belhassen, L.; Pecker, F.; Fischmeister, R. Nitric oxide regulates cardiac Ca2+ current. Involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. J. Biol. Chem. 1993, 268, 26286–26295. [Google Scholar] [PubMed]
- Kim, K.H.; Kim, H.K.; Hwang, I.C.; Cho, H.J.; Je, N.; Kwon, O.M.; Choi, S.J.; Lee, S.P.; Kim, Y.J.; Sohn, D.W. PDE 5 inhibition with udenafil improves left ventricular systolic/diastolic functions and exercise capacity in patients with chronic heart failure with reduced ejection fraction; A 12-week, randomized, double-blind, placebo-controlled trial. Am. Heart J. 2015, 169, 813–822.e3. [Google Scholar] [CrossRef]
- Redfield, M.M.; Chen, H.H.; Borlaug, B.A.; Semigran, M.J.; Lee, K.L.; Lewis, G.; LeWinter, M.M.; Rouleau, J.L.; Bull, D.A.; Mann, D.L.; et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: A randomized clinical trial. JAMA 2013, 309, 1268–1277. [Google Scholar] [CrossRef]
- Herring, N.; Rigg, L.; Terrar, D.A.; Paterson, D.J. NO-cGMP pathway increases the hyperpolarisation-activated current, If, and heart rate during adrenergic stimulation. Cardiovasc. Res. 2001, 52, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, Å.B.; Brunton, L.L. Attenuation of cAMP accumulation in adult rat cardiac fibroblasts by IL-1β and NO: Role of cGMP-stimulated PDE2. Am. J. Physiol. Physiol. 2002, 283, C463–C471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozmaritsa, N.; Christ, T.; Van Wagoner, D.R.; Haase, H.; Stasch, J.-P.; Matschke, K.; Ravens, U. Attenuated response of L-type calcium current to nitric oxide in atrial fibrillation. Cardiovasc. Res. 2014, 101, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Román Moltzau, L.; Meier, S.; Magnus Aronsen, J.; Afzal, F.; Sjaastad, I.; Skomedal, T.; Osnes, J.-B.; Olav Levy, F.; Qvigstad, E. Differential regulation of C-type natriuretic peptide-induced cGMP and functional responses by PDE2 and PDE3 in failing myocardium. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Coskran, T.M.; Wilhelms, M.B.; Adamowicz, W.O.; O’Donnell, M.M.; Muravnick, K.B.; Menniti, F.S.; Kleiman, R.J.; Morton, D. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J. Histochem. Cytochem. 2009, 57, 933–949. [Google Scholar] [CrossRef] [Green Version]
- Vettel, C.; Lämmle, S.; Ewens, S.; Cervirgen, C.; Emons, J.; Ongherth, A.; Dewenter, M.; Lindner, D.; Westermann, D.; Nikolaev, V.O.; et al. PDE2-mediated cAMP hydrolysis accelerates cardiac fibroblast to myofibroblast conversion and is antagonized by exogenous activation of cGMP signaling pathways. Am. J. Physiol. Circ. Physiol. 2014, 306, H1246–H1252. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Henrich, M.; Buckler, K.J.; McMenamin, M.; Mee, C.J.; Sattelle, D.B.; Paterson, D.J. Neuronal nitric oxide synthase gene transfer decreases [Ca2+]i in cardiac sympathetic neurons. J. Mol. Cell. Cardiol. 2007, 43, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Pan, J.; Sun, J.; Ding, L.; Ruan, L.; Reed, M.; Yu, X.; klabnik, J.; Lin, D.; Li, J.; et al. Inhibition of phosphodiesterase 2 reverses impaired cognition and neuronal remodeling caused by chronic stress. Neurobiol. Aging 2015, 36, 955–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Li, D.; Hao, G.; McCaffary, D.; Neely, O.; Woodward, L.; Ioannides, D.; Lu, C.J.; Brescia, M.; Zaccolo, M.; et al. Phosphodiesterase 2A as a therapeutic target to restore cardiac neurotransmission during sympathetic hyperactivity. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Li, D.; Lu, C.J.; Hao, G.; Wright, H.; Woodward, L.; Liu, K.; Vergari, E.; Surdo, N.C.; Herring, N.; Zaccolo, M.; et al. Efficacy of B-type natriuretic peptide is coupled to phosphodiesterase 2A in cardiac sympathetic neurons. Hypertension 2015, 66, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.T.; Beavo, J.A. Specific localized expression of cGMP PDEs in Purkinje neurons and macrophages. Neurochem. Int. 2004, 45, 853–857. [Google Scholar] [CrossRef]
- Bender, A.T.; Ostenson, C.L.; Giordano, D.; Beavo, J.A. Differentiation of human monocytes in vitro with granulocyte-macrophage colony-stimulating factor and macrophage colony-stimulating factor produces distinct changes in cGMP phosphodiesterase expression. Cell. Signal. 2004, 16, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Michie, A.M.; Lobban, M.; Müller, T.; Harnett, M.M.; Houslay, M.D. Rapid regulation of PDE-2 and PDE-4 cyclic AMP phosphodiesterase activity following ligation of the T cell antigen receptor on thymocytes: Analysis using the selective inhibitors erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA) and rolipram. Cell. Signal. 1996, 8, 97–110. [Google Scholar] [CrossRef]
- Witwicka, H.; Kobiałka, M.; Siednienko, J.; Mitkiewicz, M.; Gorczyca, W.A. Expression and activity of cGMP-dependent phosphodiesterases is up-regulated by lipopolysaccharide (LPS) in rat peritoneal macrophages. Biochim. Biophys. Acta 2007, 1773, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seybold, J.; Thomas, D.; Witzenrath, M.; Boral, Ş.; Hocke, A.C.; Bürger, A.; Hatzelmann, A.; Tenor, H.; Schudt, C.; Krüll, M.; et al. Tumor necrosis factor-α-dependent expression of phosphodiesterase 2: Role in endothelial hyperpermeability. Blood 2005, 105, 3569–3576. [Google Scholar] [CrossRef] [Green Version]
- Preedy, M.E.J. Cardiac Cyclic Nucleotide Phosphodiesterases: Roles and Therapeutic Potential in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 401–417. [Google Scholar] [CrossRef] [Green Version]
- Mika, D.; Bobin, P.; Pomé Rance, M.; Lechê Ne, P.; Westenbroek, R.E.; Catterall, W.A.; Goire Vandecasteele, G.; Rô Me Leroy, J.; Fischmeister, R. Differential regulation of cardiac excitation-contraction coupling by cAMP phosphodiesterase subtypes. Cardiovasc. Res. 2013, 100, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Vandecasteele, G.; Verde, I.; Rücker-Martin, C.; Donzeau-Gouge, P.; Fischmeister, R. Cyclic GMP regulation of the L-type Ca2+ channel current in human atrial myocytes. J. Physiol. 2001, 533, 329–340. [Google Scholar] [CrossRef]
- Rivet-Bastide, M.; Vandecasteele, G.; Hatem, S.; Verde, I.; Bénardeau, A.; Mercadier, J.-J.; Fischmeister, R. cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes. J. Clin. Investig. 1997, 99, 2710–2718. [Google Scholar] [CrossRef] [Green Version]
- Dittrich, M.; Jurevicius, J.; Georget, M.; Rochais, F.; Fleischmann, B.K.; Hescheler, J.; Fischmeister, R. Local response of L-type Ca2+ current to nitric oxide in frog ventricular myocytes. J. Physiol. 2001, 534, 109–121. [Google Scholar] [CrossRef]
- Han, X.; Shimoni, Y.; Giles, W.R. A cellular mechanism for nitric oxide-mediated cholinergic control of mammalian heart rate. J. Gen. Physiol. 1995, 106, 45–65. [Google Scholar] [CrossRef] [Green Version]
- Bastug-Özel, Z.; Wright, P.T.; Kraft, A.E.; Pavlovic, D.; Howie, J.; Froese, A.; Fuller, W.; Gorelik, J.; Shattock, M.J.; Nikolaev, V.O. Heart failure leads to altered β2-adrenoceptor/cyclic adenosine monophosphate dynamics in the sarcolemmal phospholemman/Na,K ATPase microdomain. Cardiovasc. Res. 2019, 115, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Dawei, L.; Zhenyu, W.; Valérie, N.; Marta, L.; Delphine, M.; Grégoire, V.; Rodolphe, F.; Catherine, B. PDE2 regulates membrane potential, respiration and permeability transition of rodent subsarcolemmal cardiac mitochondria. Mitochondrion 2019, 47, 64–75. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Russwurm, M.; Günnewig, K.; Gertz, M.; Zoidl, G.; Ramos, L.; Buck, J.; Levin, L.R.; Rassow, J.; Manfredi, G.; et al. A Phosphodiesterase 2A Isoform Localized to Mitochondria Regulates Respiration. J. Biol. Chem. 2011, 286, 30423–30432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, D.; Varin, A.; Nicolas, V.; Courilleau, D.; Mateo, P.; Caubere, C.; Rouet, P.; Gomez, A.M.; Vandecasteele, G.; et al. A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death. Cell Death Dis. 2016, 7, e2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozdniakova, S.; Guitart-Mampel, M.; Garrabou, G.; Di Benedetto, G.; Ladilov, Y.; Regitz-Zagrosek, V. 17β-Estradiol reduces mitochondrial cAMP content and cytochrome oxidase activity in a phosphodiesterase 2-dependent manner. Br. J. Pharmacol. 2018, 175, 3876–3890. [Google Scholar] [CrossRef]
- Gustafsson, Å.B.; Brunton, L.L. Interactions of the Cyclic AMP and Nitric Oxide Pathways in Cardiac Fibroblasts. In Pathophysiology of Cardiovascular Disease: Progress in Experimental Cardiology; Dhalla, N.S., Rupp, H., Angel, A., Pierce, G., Eds.; Springer: Boston, MA, USA, 2004; pp. 109–123. [Google Scholar]
- Gustafsson, Å.B.; Brunton, L.L. β-Adrenergic stimulation of rat cardiac fibroblasts enhances induction of nitric-oxide synthase by interleukin-1β via message stabilization. Mol. Pharmacol. 2000, 58, 1470–1478. [Google Scholar] [CrossRef] [Green Version]
- Delaunay, M.; Osman, H.; Kaiser, S.; Diviani, D. The Role of Cyclic AMP Signaling in Cardiac Fibrosis. Cells 2019, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Rowley, J.W.; Oler, A.J.; Tolley, N.D.; Hunter, B.N.; Low, E.N.; Nix, D.A.; Yost, C.C.; Zimmerman, G.A.; Weyrich, A.S. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 2011, 118, e101. [Google Scholar] [CrossRef] [Green Version]
- Rondina, M.T.; Weyrich, A.S. Targeting Phosphodiesterases in Anti-platelet Therapy. Handb. Exp. Pharmacol. 2012, 225–238. [Google Scholar]
- Xiang, Q.; Pang, X.; Liu, Z.; Yang, G.; Tao, W.; Pei, Q.; Cui, Y. Progress in the development of antiplatelet agents: Focus on the targeted molecular pathway from bench to clinic. Pharmacol. Ther. 2019, 203, 107393. [Google Scholar] [CrossRef]
- Dickinson, N.T.; Jang, E.K.; Haslam, R.J. Activation of cGMP-stimulated phosphodiesterase by nitroprusside limits cAMP accumulation in human platelets: Effects on platelet aggregation. Biochem. J. 1997, 323, 371–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favot, L.; Keravis, T.M.; Lugnier, C. VEGF-induced HUVEC migration and proliferation are decreased by PDE2 and PDE4 inhibitors. Thromb. Haemost. 2003, 90, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Trevor, A.; Katzung, B.; Masters, S.; Kruidering-Hall, M. Pharmacology Examination & Board Review, 11th ed.; McGraw-Hill Medical: New York, NY, USA, 2010; pp. 121–132. [Google Scholar]
- Cuffe, M.S.; Califf, R.M.; Adams, K.F.; Benza, R.; Bourge, R.; Colucci, W.S.; Massie, B.M.; O’Connor, C.M.; Pina, I.; Quigg, R.; et al. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: A randomized controlled trial. J. Am. Med. Assoc. 2002, 287, 1541–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rita Assenza, M.; Barbagallo, F.; Barrios, F.; Cornacchione, M.; Campolo, F.; Vivarelli, E.; Gianfrilli, D.; Auletta, L.; Soricelli, A.; Isidori, A.M.; et al. Critical role of phosphodiesterase 2A in mouse congenital heart defects. Cardiovasc. Res. 2018, 114, 830–845. [Google Scholar] [CrossRef]
- Hua, R.; Adamczyk, A.; Robbins, C.; Ray, G.; Rose, R.A. Distinct Patterns of Constitutive Phosphodiesterase Activity in Mouse Sinoatrial Node and Atrial Myocardium. PLoS ONE 2012, 7, e47652. [Google Scholar] [CrossRef] [Green Version]
- Isidori, A.M.; Cornacchione, M.; Barbagallo, F.; Di Grazia, A.; Barrios, F.; Fassina, L.; Monaco, L.; Giannetta, E.; Gianfrilli, D.; Garofalo, S.; et al. Inhibition of type 5 phosphodiesterase counteracts β2-adrenergic signalling in beating cardiomyocytes. Cardiovasc. Res. 2015, 106, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Yanaka, N.; Kurosawa, Y.; Minami, K.; Kawai, E.; Omori, K. Cgmp-Phosphodiesterase Activity Is Up-regulated in Response to Pressure Overload of Rat Ventricles. Biosci. Biotechnol. Biochem. 2003, 67, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Nash, C.A.; Brown, L.M.; Malik, S.; Cheng, X.; Smrcka, A.V. Compartmentalized cyclic nucleotides have opposing effects on regulation of hypertrophic phospholipase Cε signaling in cardiac myocytes. J. Mol. Cell. Cardiol. 2018, 121, 51–59. [Google Scholar] [CrossRef]
- Wagner, M.; Mehel, H.; Fischmeister, R.; El-Armouche, A. Phosphodiesterase 2: Anti-adrenergic friend or hypertrophic foe in heart disease? Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 1139–1141. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Sinovas, A.; Yaser, A.E.; Ae, A.; Michael, H.; Ae, P.; Garcia-Dorado, D. Reperfusion injury as a therapeutic challenge in patients with acute myocardial infarction. Heart Fail. Rev. 2007, 12, 207–216. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Inserte, J.; Rodriguez-Sinovas, A.; Piper, H.M. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 2012, 94, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, L.; Pozdniakova, S.; Jayarajan, V.; Troidl, C.; Abdallah, Y.; Aslam, M.; Ladilov, Y. Protective role of soluble adenylyl cyclase against reperfusion-induced injury of cardiac cells. Biochim. Biophys. Acta 2019, 1865, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP Produced inside Mitochondria Regulates Oxidative Phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Spitzl, A.; Mathes, D.; Nikolaev, V.O.; Werner, F.; Weirather, J.; Špiranec, K.; Röck, K.; Fischer, J.W.; Kämmerer, U.; et al. Endothelial Actions of ANP Enhance Myocardial Inflammatory Infiltration in the Early Phase after Acute Infarction. Circ. Res. 2016, 119, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Oberwinkler, H.; Werner, F.; Ganer, B.; Nakagawa, H.; Feil, R.; Hofmann, F.; Schlossmann, J.; Dietrich, A.; Gudermann, T.; et al. Atrial natriuretic peptide-mediated inhibition of microcirculatory endothelial Ca2+ and permeability response to histamine involves cGMP-dependent protein kinase i and TRPC6 channels. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2121–2129. [Google Scholar] [CrossRef]
- Oka, T.; Akazawa, H.; Naito, A.T.; Komuro, I. Angiogenesis and Cardiac Hypertrophy. Circ. Res. 2014, 114, 565–571. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Izumiya, Y.; Shiojima, I.; Sato, K.; Sawyer, D.B.; Colucci, W.S.; Walsh, K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension 2006, 47, 887–893. [Google Scholar] [CrossRef]
- Friehs, I.; Margossian, R.E.; Moran, A.M.; Cao-Danh, H.; Moses, M.A.; Del Nido, P.J.; Friehs, I.; Cao-Danh, H.; Del Nido, P.J.; Margossian, R.E.; et al. Vascular endothelial growth factor delays onset of failure in pressure-overload hyper-trophy through matrix metalloproteinase activation and angiogenesis. Basic Res. Cardiol. 2006, 101, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Sahara, M.; Sata, M.; Morita, T.; Nakajima, T.; Hirata, Y.; Nagai, R. A phosphodiesterase-5 inhibitor vardenafil enhances angiogenesis through a protein kinase g-dependent hypoxia-inducible factor-1/vascular endothelial growth factor pathway. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1315–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diebold, I.; Djordjevic, T.; Petry, A.; Hatzelmann, A.; Tenor, H.; Hess, J.; Görlach, A. Phosphodiesterase 2 mediates redox-sensitive endothelial cell proliferation and angiogenesis by thrombin via rac1 and NADPH oxidase 2. Circ. Res. 2009, 104, 1169–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neviere, R.; Delguste, F.; Durand, A.; Inamo, J.; Boulanger, E.; Preau, S. Abnormal Mitochondrial cAMP/PKA Signaling Is Involved in Sepsis-Induced Mitochondrial and Myocardial Dysfunction. Int. J. Mol. Sci. 2016, 17, 2075. [Google Scholar] [CrossRef] [PubMed]
- Farber, H.W.; Loscalzo, J. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2004, 351, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Bubb, K.J.; Trinder, S.L.; Baliga, R.S.; Patel, J.; Clapp, L.H.; MacAllister, R.J.; Hobbs, A.J. Inhibition of phosphodiesterase 2 augments cGMP and cAMP signaling to ameliorate pulmonary hypertension. Circulation 2014, 130, 496–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, V.V.; McGoon, M.D. Pulmonary arterial hypertension. Circulation 2006, 114, 1417–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishak Gabra, N.B.; Mahmoud, O.; Ishikawa, O.; Shah, V.; Altshul, E.; Oron, M.; Mina, B. Pulmonary Arterial Hypertension and Therapeutic Interventions. Int. J. Angiol. 2019, 28, 80–82. [Google Scholar] [CrossRef]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360. [Google Scholar] [CrossRef]
- Jing, Z.C.; Yu, Z.X.; Shen, J.Y.; Wu, B.X.; Xu, K.F.; Zhu, X.Y.; Pan, L.; Zhang, Z.L.; Liu, X.Q.; Zhang, Y.S.; et al. Vardenafil in pulmonary arterial hypertension: A randomized, double-blind, placebo-controlled study. Am. J. Respir. Crit. Care Med. 2011, 183, 1723–1729. [Google Scholar] [CrossRef]
- Rubin, L.J.; Badesch, D.B.; Fleming, T.R.; Galiè, N.; Simonneau, G.; Ghofrani, H.A.; Oakes, M.; Layton, G.; Serdarevic-Pehar, M.; McLaughlin, V.V.; et al. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: The SUPER-2 study. Chest 2011, 140, 1274–1283. [Google Scholar] [CrossRef]
- Zhang, C.; Lueptow, L.M.; Zhang, H.T.; O’Donnell, J.M.; Xu, Y. The role of phosphodiesterase-2 in psychiatric and neurodegenerative disorders. Adv. Neurobiol. 2017, 17, 307–347. [Google Scholar] [PubMed]
- Domek-Łopacińska, K.; Strosznajder, J.B. The effect of selective inhibition of cyclic GMP hydrolyzing phosphodiesterases 2 and 5 on learning and memory processes and nitric oxide synthase activity in brain during aging. Brain Res. 2008, 1216, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.S.R.; Rutten, K.; Sydlik, S.; Rostamian, S.; Steinbusch, H.W.M.; Van Den Hove, D.L.A.; Prickaerts, J. Chronic phosphodiesterase type 2 inhibition improves memory in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Neuropharmacology 2013, 64, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Helal, C.J.; Arnold, E.; Boyden, T.; Chang, C.; Chappie, T.A.; Fisher, E.; Hajos, M.; Harms, J.F.; Hoffman, W.E.; Humphrey, J.M.; et al. Identification of a Potent, Highly Selective, and Brain Penetrant Phosphodiesterase 2A Inhibitor Clinical Candidate. J. Med. Chem. 2018, 61, 1001–1018. [Google Scholar] [CrossRef]
- Mikami, S.; Nakamura, S.; Ashizawa, T.; Nomura, I.; Kawasaki, M.; Sasaki, S.; Oki, H.; Kokubo, H.; Hoffman, I.D.; Zou, H.; et al. Discovery of Clinical Candidate N-((1S)-1-(3-Fluoro-4-(trifluoromethoxy)phenyl)-2-methoxyethyl)-7-methoxy-2-oxo-2,3-dihydropyrido[2,3-b]pyrazine-4(1H)-carboxamide (TAK-915): A Highly Potent, Selective, and Brain-Penetrating Phosphodiesterase 2A Inhibitor for the Treatment of Cognitive Disorders. J. Med. Chem. 2017, 60, 7677–7702. [Google Scholar] [CrossRef]
- Mikami, S.; Sasaki, S.; Asano, Y.; Ujikawa, O.; Fukumoto, S.; Nakashima, K.; Oki, H.; Kamiguchi, N.; Imada, H.; Iwashita, H.; et al. Discovery of an Orally Bioavailable, Brain-Penetrating, in Vivo Active Phosphodiesterase 2A Inhibitor Lead Series for the Treatment of Cognitive Disorders. J. Med. Chem. 2017, 60, 7658–7676. [Google Scholar] [CrossRef]
- Griffiths, G.J. Exisulind Cell Pathways. Curr. Opin. Investig. Drugs 2000, 1, 386–391. [Google Scholar]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadek, M.S.; Cachorro, E.; El-Armouche, A.; Kämmerer, S. Therapeutic Implications for PDE2 and cGMP/cAMP Mediated Crosstalk in Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 7462. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207462
Sadek MS, Cachorro E, El-Armouche A, Kämmerer S. Therapeutic Implications for PDE2 and cGMP/cAMP Mediated Crosstalk in Cardiovascular Diseases. International Journal of Molecular Sciences. 2020; 21(20):7462. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207462
Chicago/Turabian StyleSadek, Mirna S., Eleder Cachorro, Ali El-Armouche, and Susanne Kämmerer. 2020. "Therapeutic Implications for PDE2 and cGMP/cAMP Mediated Crosstalk in Cardiovascular Diseases" International Journal of Molecular Sciences 21, no. 20: 7462. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207462