Glucose-6-Phosphate Dehydrogenase Deficiency Activates Endothelial Cell and Leukocyte Adhesion Mediated via the TGFβ/NADPH Oxidases/ROS Signaling Pathway


Abstract
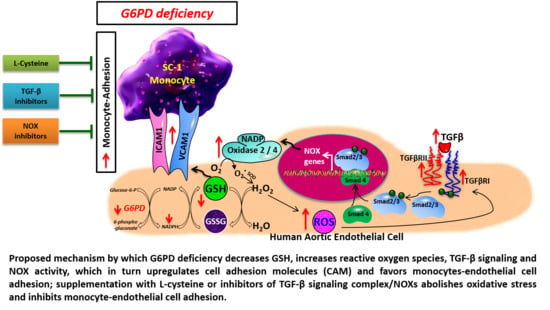
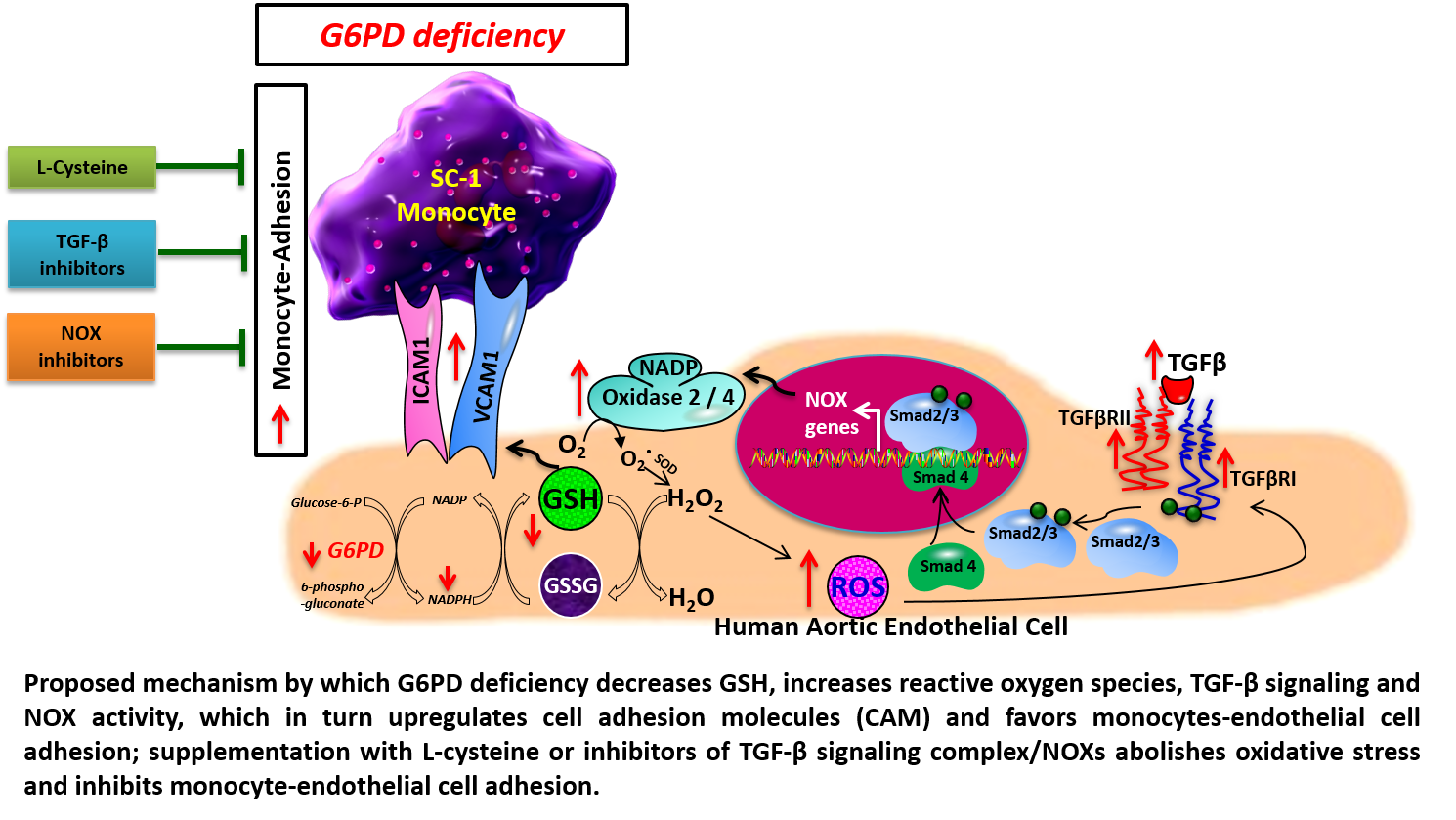
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
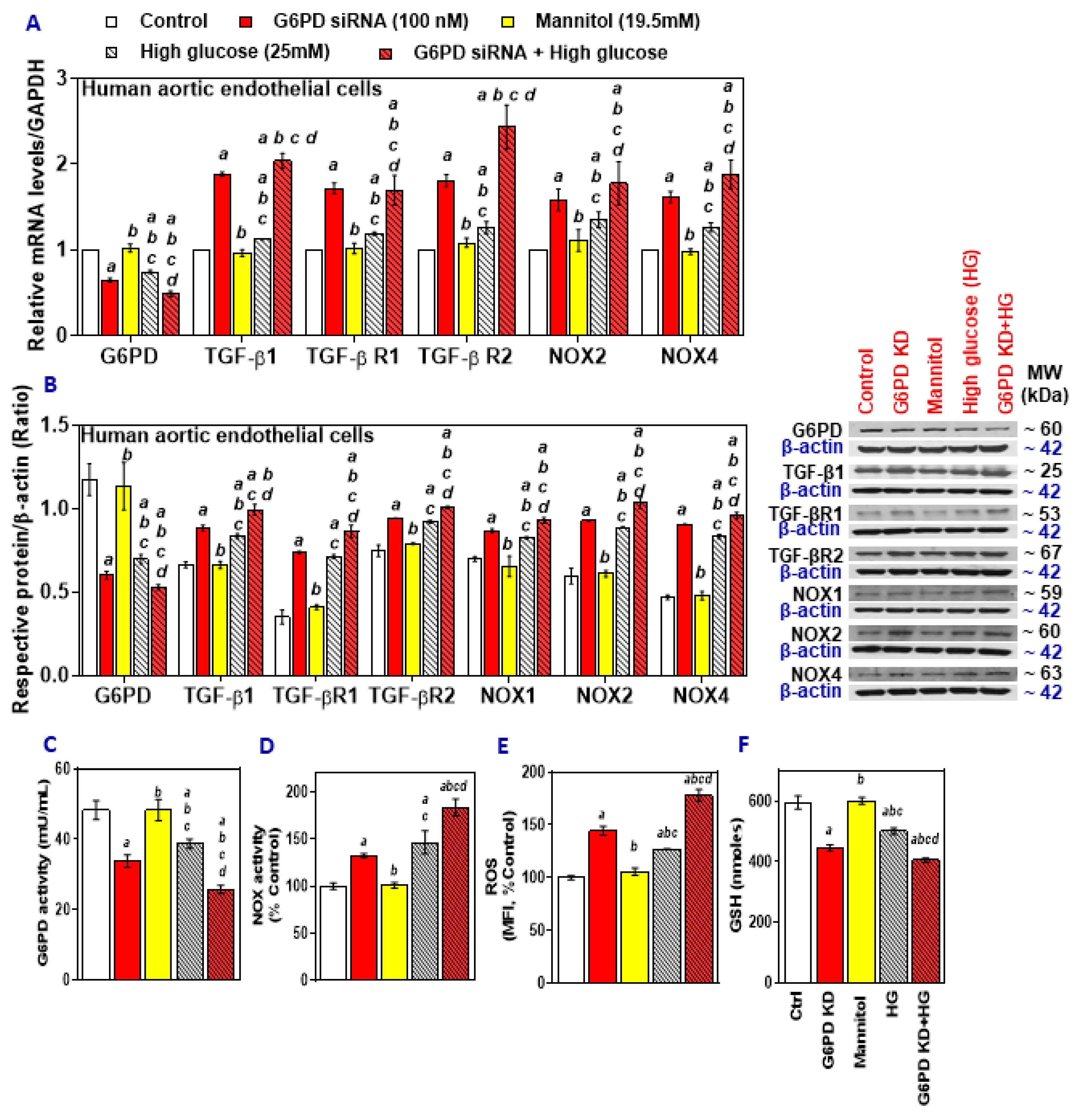
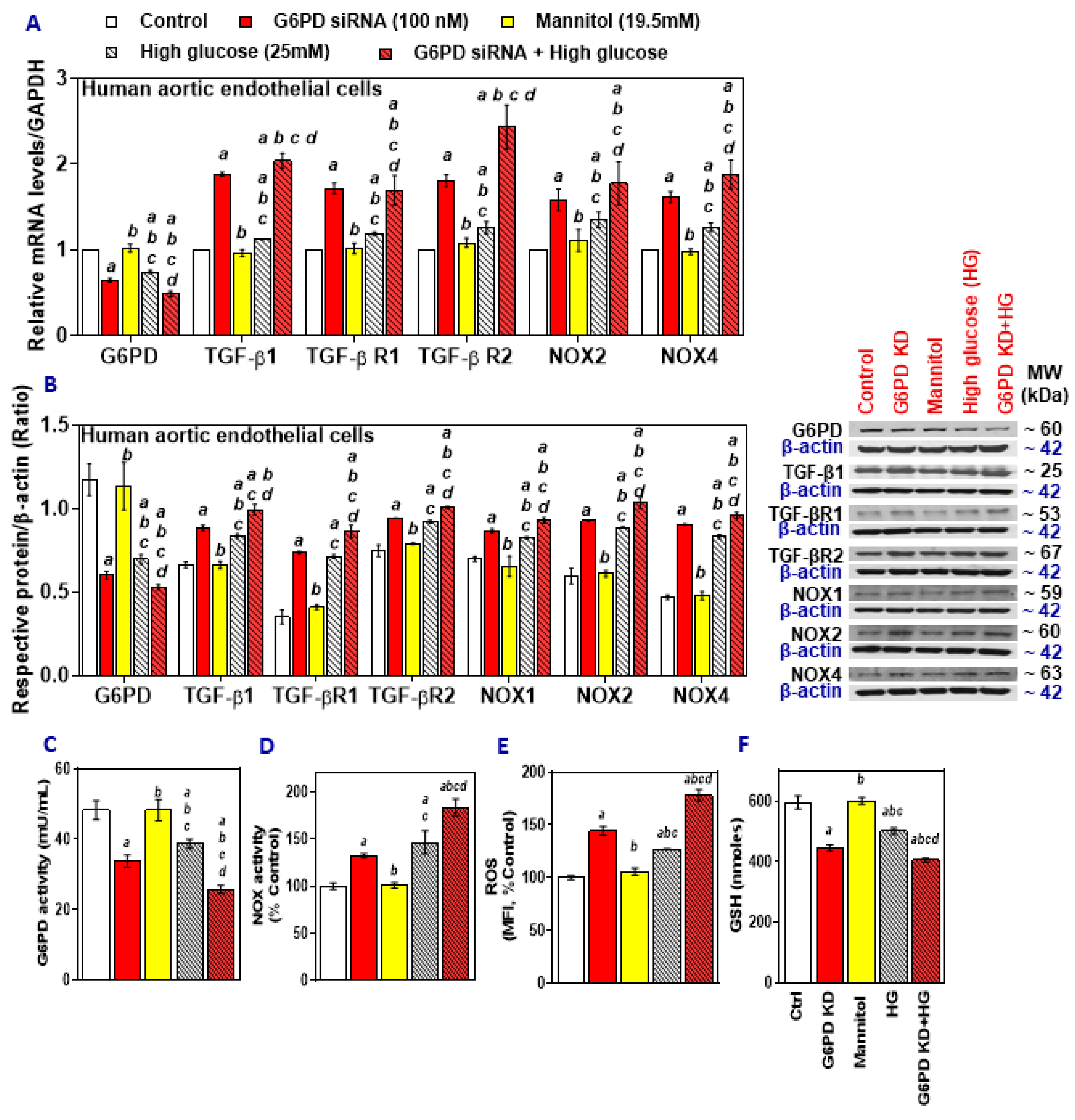
2.1. High Glucose and Palmitate Mitigate G6PD and Increases Oxidative Stress
2.2. G6PD Deficiency Exacerbates the Effects of High Glucose
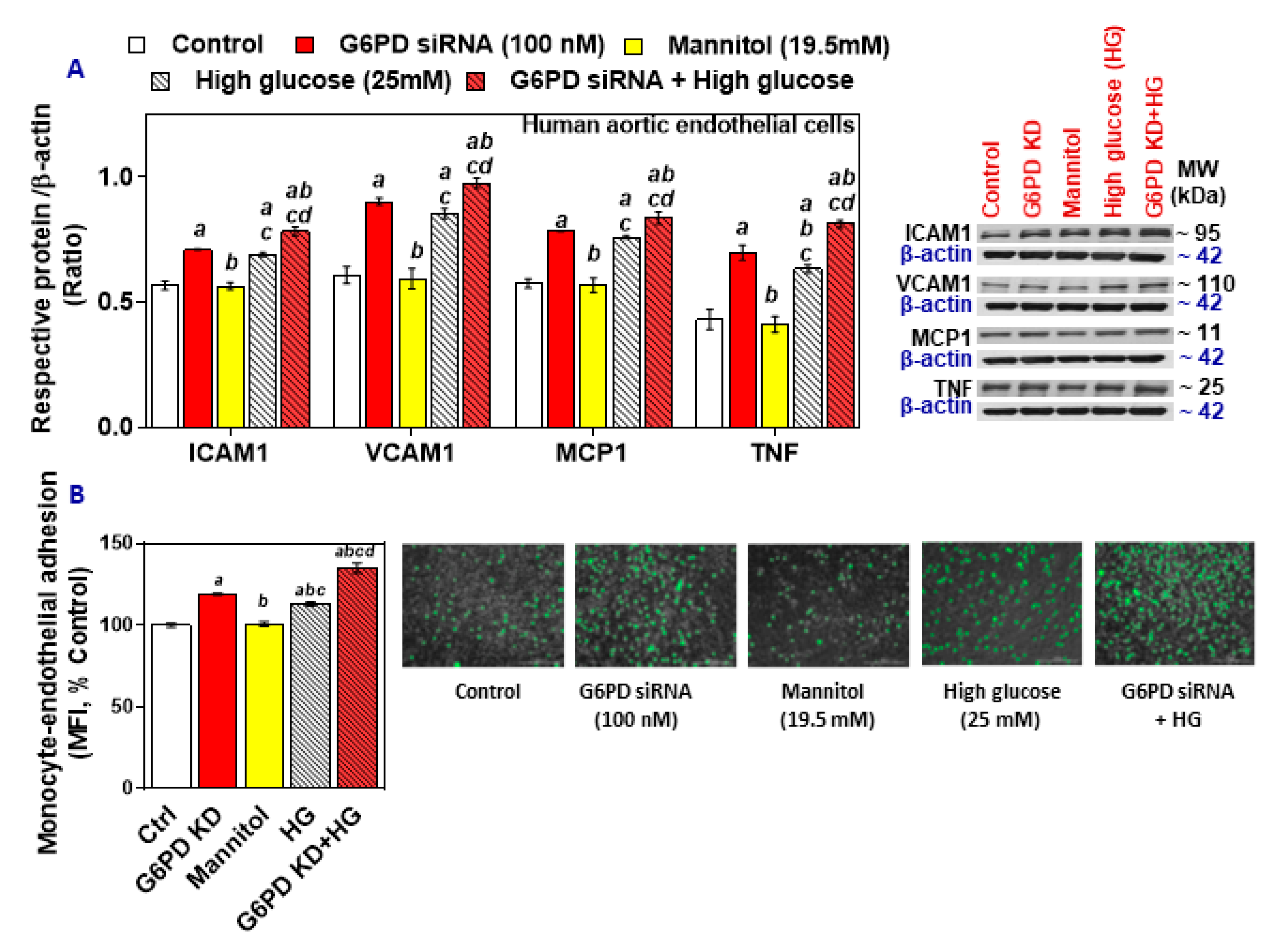
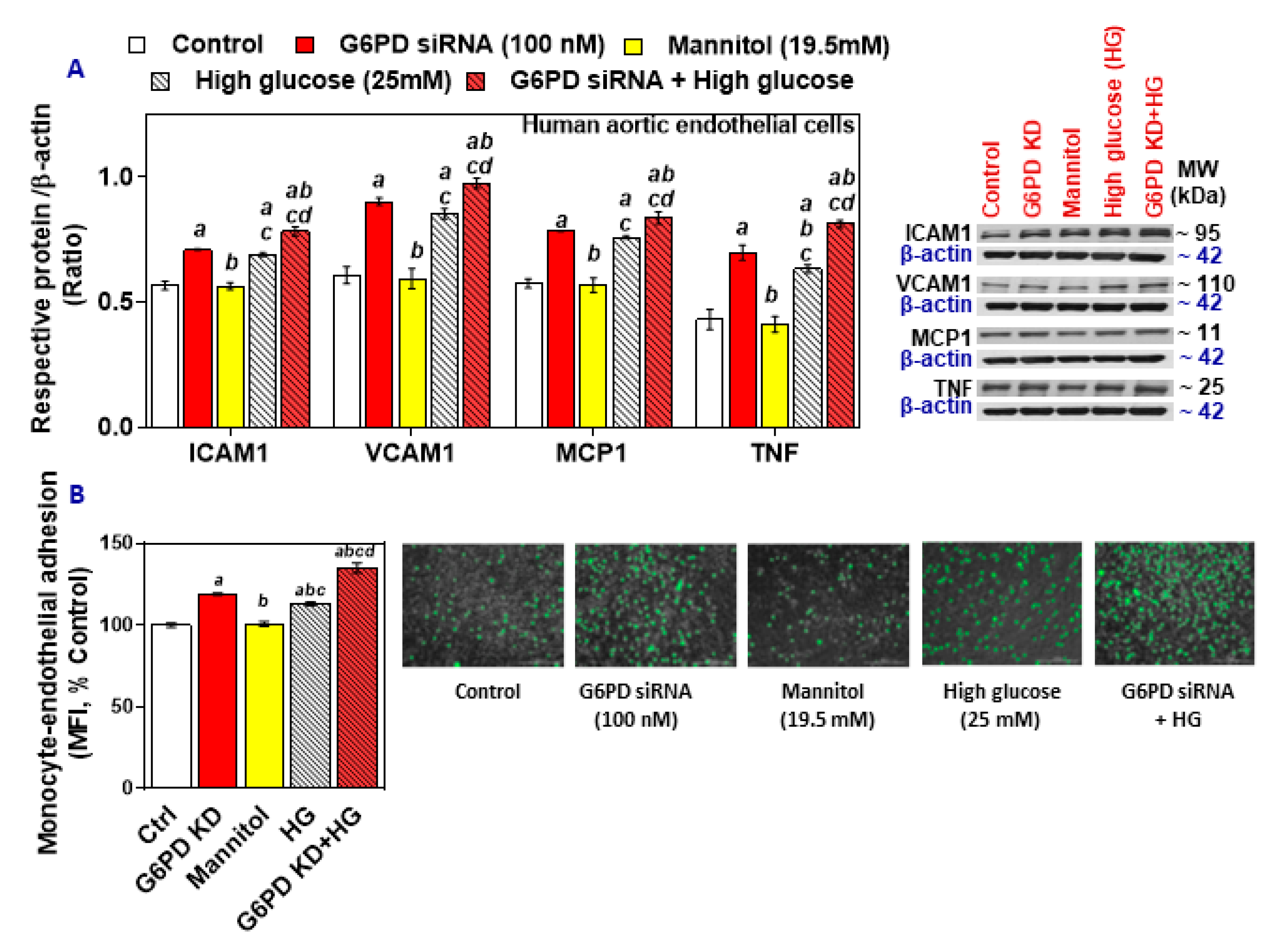
2.3. G6PD Deficiency and Treatment with High Glucose Increases Cell Adhesion Molecules and Inflammatory Cytokines in HAEC In Vitro
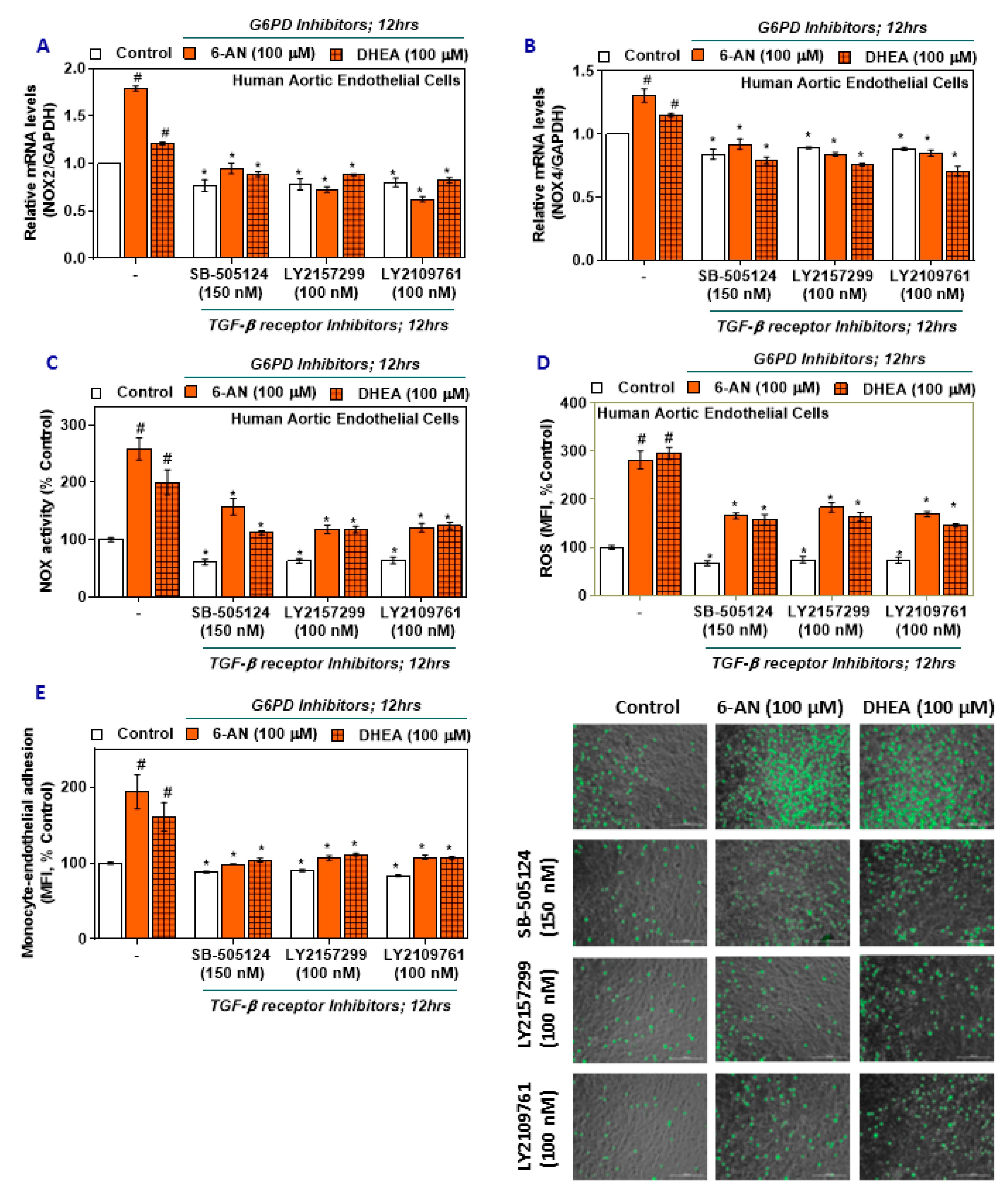
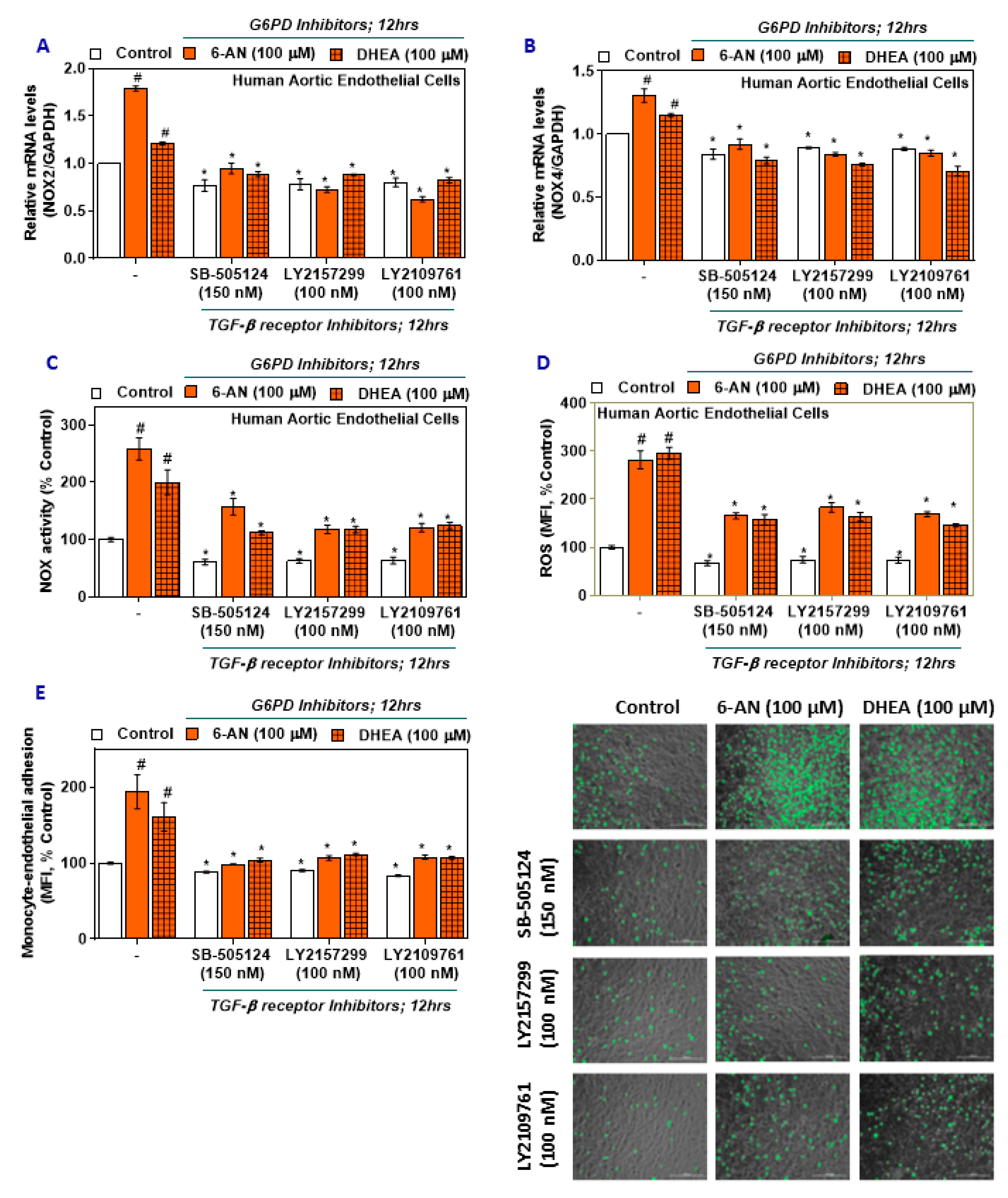
2.4. TGF-β Inhibitors Lower NOX2/4 Expression and Activity and Oxidative Stress in G6PD-Deficient HAEC
2.5. TGF-β Inhibitors Reduce Monocyte-Endothelial Adhesion in G6PD-Deficient HAEC
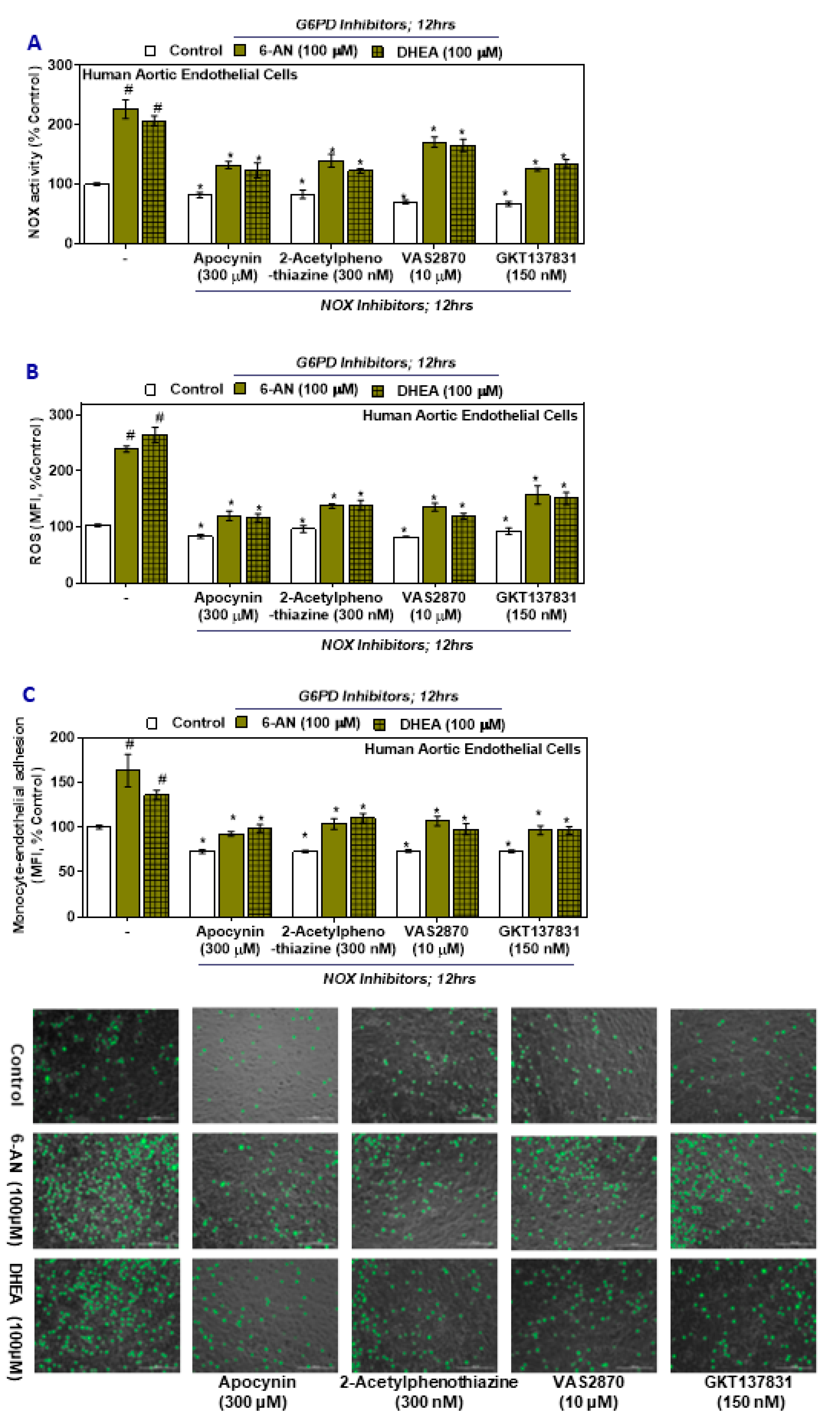
2.6. NOX Inhibitors Impede Oxidative Stress and Monocyte-Endothelial Cell Adhesion in G6PD Deficient Cells
2.7. Effects of L-Cysteine Ethyl Ester (LC ee) on mRNA Expression of TGF-β/NOX, NOX Activity, ROS Levels, and Monocyte-Endothelial Adhesion
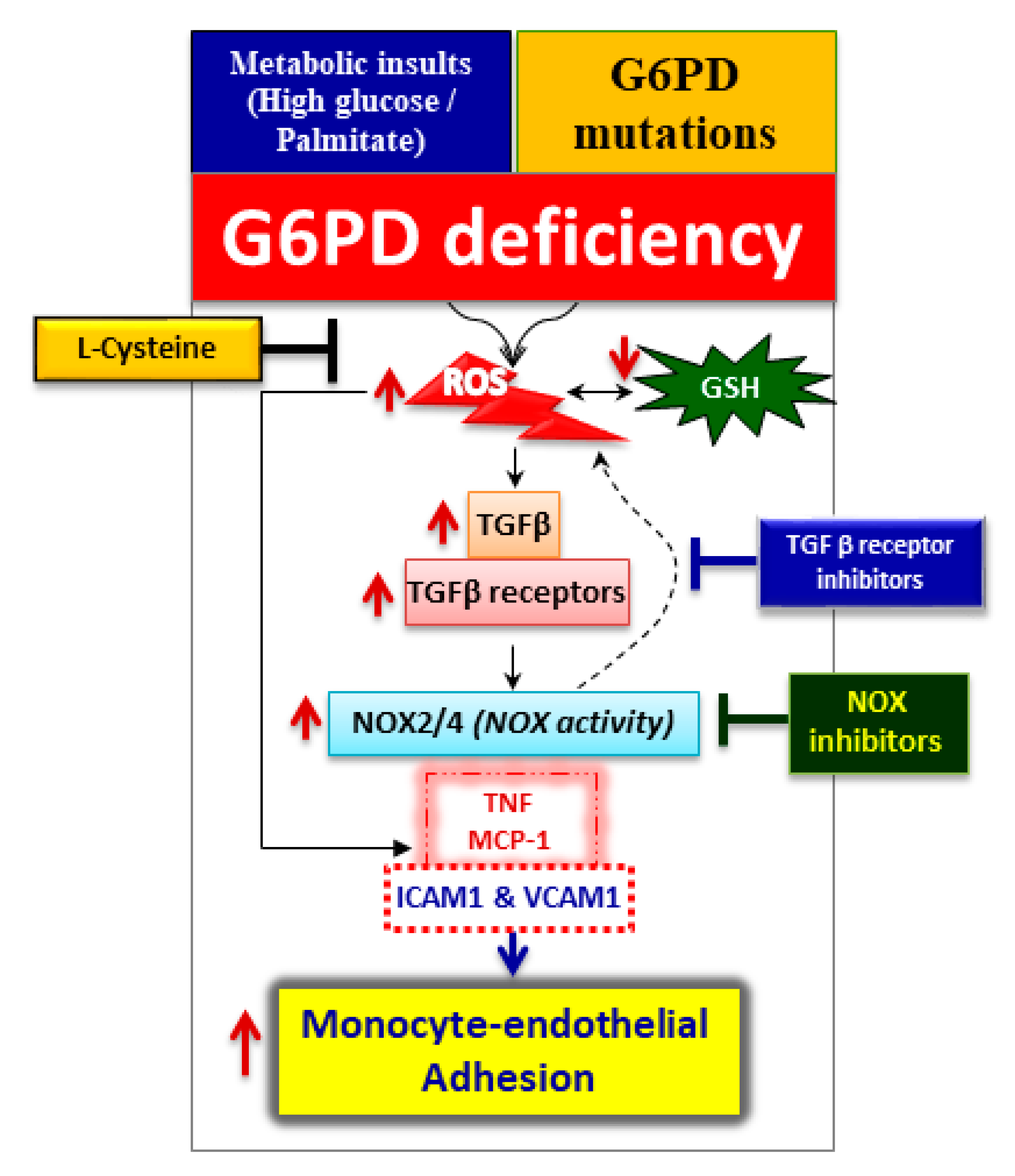
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Treatments
4.3. G6PD siRNA Knockdown Assay
4.4. Cell Viability Assay
4.5. Monocyte-Endothelial Adhesion Assay
4.6. Cellular Reactive Oxygen Species (ROS) Measurement
4.7. Analysis of mRNA Expression Using Quantitative PCR
4.8. Preparation of Whole-Cell Extracts
4.9. NADPH Oxidase Activity
4.10. G6PD Activity
4.11. GSH Assay
4.12. Western Blot Analysis
4.13. Statistical Analysis and Software
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-AN | 6-aminonicotinamide |
| AA | African American |
| CAM | Cell adhesion molecule |
| CVD | Cardiovascular disease |
| DHEA | Dehydroepiandrosterone |
| ED | Endothelial dysfunction |
| G6PD | Glucose-6-phosphate dehydrogenase |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GSH | Glutathione |
| HAEC | Human aortic endothelial cells |
| HG | High glucose |
| ICAM-1 | Intercellular adhesion molecule 1 |
| LC ee | L-cysteine ethyl ester |
| MCP-1 | monocyte chemoattractant protein 1 |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| OS | Oxidative stress |
| RBC | Red blood cell |
| ROS | Reactive oxygen species |
| TGF-β | Transforming growth factor-beta |
| TGF-βR1 | TGF-β receptor 1 |
| TGF-βR2 | TGF-β receptor 2 |
| TNF | tumor necrosis factor |
| VCAM-1 | Vascular adhesion molecule 1 |
References
- Layton, M.; Ramachandran, M.; O’Shaughnessy, D.; Luzzatto, L. Glucose-6-phosphate dehydrogenase deficiency. Curr. Paediatr. 1995, 5, 190–194. [Google Scholar] [CrossRef]
- Parsanathan, R.; Jain, S.K. Glucose-6-phosphate dehydrogenase (G6PD) deficiency is linked with cardiovascular disease. Hypertens. Res. 2020, 43, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.E.; Kang, S.; Wyatt, C.J.; Kim, F.S.; Mangelsdorff, A.D.; Weigel, F.K. Glucose-6-phosphate dehydrogenase deficiency is associated with cardiovascular disease in U.S. military centers. Tex. Hear. Inst. J. 2018, 45, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Chinevere, T.D.; Murray, C.K.; Grant, E.; Johnson, G.A.; Duelm, F.; Hospenthal, D.R. Prevalence of glucose-6-phosphate dehydrogenase deficiency in U.S. army personnel. Mil. Med. 2006, 171, 905–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pes, G.M.; Parodi, G.; Dore, M.P. Glucose-6-phosphate dehydrogenase deficiency and risk of cardiovascular disease: A propensity score-matched study. Atherosclerosis 2019, 282, 148–153. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, X.; Guan, T.; Wang, X.; Zhang, H.; Zeng, X.; Dai, Q.; Wang, Y.; Zhou, L.; Ma, X. The association between glucose-6-phosphate dehydrogenase deficiency and abnormal blood pressure among prepregnant reproductive-age Chinese females. Hypertens. Res. 2018, 42, 75–84. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart disease and stroke statistics-2017 update: A report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- Gaskin, R.S.; Estwick, D.; Peddi, R. G6PD deficiency: Its role in the high prevalence of hypertension and diabetes mellitus. Ethn. Dis. 2001, 11, 749–754. [Google Scholar]
- Jain, S.K.; Palmer, M. Effect of glucose-6-phosphate dehydrogenase deficiency on reduced and oxidized glutathione and lipid peroxide levels in the blood of African-Americans. Clin. Chim. Acta 1996, 253, 181–183. [Google Scholar] [CrossRef]
- Ho, H.-Y.; Cheng, M.-L.; Lu, F.-J.; Chou, Y.-H.; Stern, A.; Liang, C.-M.; Chiu, D.T.-Y. Enhanced oxidative stress and accelerated cellular senescence in glucose-6-phosphate dehydrogenase (G6PD)-deficient human fibroblasts. Free Radic. Biol. Med. 2000, 29, 156–169. [Google Scholar] [CrossRef]
- Leopold, J.A.; Cap, A.; Scribner, A.W.; Stanton, R.C.; Loscalzo, J. Glucose-6-phosphate dehydrogenase deficiency promotes endothelial oxidant stress and decreases endothelial nitric oxide bioavailability. FASEB J. 2001, 15, 1771–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsanathan, R.; Jain, S.K. Novel invasive and noninvasive cardiac-specific biomarkers in obesity and cardiovascular diseases. Metab. Syndr. Relat. Disord. 2020, 18, 10–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goumans, M.J.; Ten Dijke, P. TGF-beta signaling in control of cardiovascular function. Cold Spring Harb. Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ortega, M.; Rodriguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-beta signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nóbrega-Pereira, S.; Fernandez-Marcos, P.J.; Brioche, T.; Gómez-Cabrera, M.C.; Salvador-Pascual, A.; Flores, J.M.; Viña, J.; Serrano, M. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016, 7, 10894. [Google Scholar] [CrossRef] [Green Version]
- Nkhoma, E.T.; Poole, C.; Vannappagari, V.; Hall, S.A.; Beutler, E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: A systematic review and meta-analysis. Blood Cells Mol. Dis. 2009, 42, 267–278. [Google Scholar] [CrossRef]
- Liu, R.-M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Jaffer, O.A.; Carter, A.B.; Sanders, P.N.; Dibbern, M.E.; Winters, C.J.; Murthy, S.; Ryan, A.J.; Rokita, A.G.; Prasad, A.M.; Zabner, J.; et al. Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-beta-mediated collagen production in a murine asthma model. Am. J. Respir. Cell Mol. Biol. 2015, 52, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Ishizaka, N.; Aizawa, T.; Sata, M.; Iso-o, N.; Noiri, E.; Mori, I.; Ohno, M.; Nagai, R. Iron chelation and a free radical scavenger suppress angiotensin II-induced upregulation of TGF-beta1 in the heart. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1836–H1843. [Google Scholar] [CrossRef]
- Richter, K.; Konzack, A.; Pihlajaniemi, T.; Heljasvaara, R.; Kietzmann, T. Redox-fibrosis: Impact of TGFbeta1 on ROS generators, mediators and functional consequences. Redox Biol. 2015, 6, 344–352. [Google Scholar] [CrossRef]
- Montorfano, I.; Becerra, A.; Cerro, R.; Echeverria, C.; Saez, E.; Morales, M.G.; Fernandez, R.; Cabello-Verrugio, C.; Simon, F. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-beta1 and TGF-beta2-dependent pathway. Lab. Investig. 2014, 94, 1068–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanna, F.; Bonatesta, R.R.; Frongia, B.; Uda, S.; Banni, S.; Melis, M.P.; Collu, M.; Madeddu, C.; Serpe, R.; Puddu, S.; et al. Production of inflammatory molecules in peripheral blood mononuclear cells from severely glucose-6-phosphate dehydrogenase-deficient subjects. J. Vasc. Res. 2007, 44, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Parsanathan, R.; Jain, S.K. G6PD deficiency shifts polarization of monocytes/macrophages towards a proinflammatory and profibrotic phenotype. Cell. Mol. Immunol. 2020, 1–3. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to mesenchymal transition: Role in physiology and in the pathogenesis of human diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Congdon, P.J.; Aggarwal, R.K.; Littlewood, J.M.; Shapiro, H. Glucose 6-phosphate dehydrogenase deficiency and cystic fibrosis. Postgrad. Med. J. 1981, 57, 453–454. [Google Scholar] [CrossRef] [Green Version]
- Elko, E.A.; Mahoney, J.M.; Vacek, P.; Van Der Vliet, A.; Anathy, V.; Van Der Velden, J.L.; Janssen-Heininger, Y.M.; Seward, D.J.; Van Der Vilet, A. Age-dependent dysregulation of redox genes may contribute to fibrotic pulmonary disease susceptibility. Free Radic. Biol. Med. 2019, 141, 438–446. [Google Scholar] [CrossRef]
- Kurdyukov, S.; Eccles, C.A.; Desai, A.A.; Gonzalez-Garay, M.L.; Yuan, J.X.-J.; Garcia, J.G.N.; Rafikova, O.; Rafikov, R. New cases of glucose-6-phosphate dehydrogenase deficiency in pulmonary arterial hypertension. PLoS ONE 2018, 13, e0203493. [Google Scholar] [CrossRef]
- Touyz, R.M. Apocynin, NADPH oxidase, and vascular cells: A complex matter. Hypertension 2008, 51, 172–174. [Google Scholar] [CrossRef] [Green Version]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential roles of the NADPH-Oxidase 1 and 2 in platelet activation and thrombosis. Arter. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Park, J.; Lee, W.T.; Yenari, M.A. NOX inhibitors—A promising avenue for ischemic stroke. Exp. Neurobiol. 2017, 26, 195–205. [Google Scholar] [CrossRef]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.M.; Wingler, K.; Schmidt, H.H.H.W. Evolution of NADPH oxidase inhibitors: Selectivity and mechanisms for target engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Atkuri, K.R.; Mantovani, J.J.; Herzenberg, L.A. N-Acetylcysteine—A safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol. 2007, 7, 355–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badaloo, A.; Reid, M.; Forrester, T.; Heird, W.C.; Jahoor, F. Cysteine supplementation improves the erythrocyte glutathione synthesis rate in children with severe edematous malnutrition. Am. J. Clin. Nutr. 2002, 76, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Parsanathan, R.; Jain, S.K. l-Cysteine In Vitro can restore cellular glutathione and inhibits the expression of cell adhesion molecules in G6PD-deficient monocytes. Amino Acids 2018, 50, 909–921. [Google Scholar] [CrossRef]
- Parsanathan, R.; Jain, S.K. Glucose-6-phosphate dehydrogenase deficiency increases cell adhesion molecules and activates human monocyte-endothelial cell adhesion: Protective role of l-cysteine. Arch. Biochem. Biophys. 2019, 663, 11–21. [Google Scholar] [CrossRef]
- Zhang, Z.; Apse, K.; Pang, J.; Stanton, R.C. High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J. Biol. Chem. 2000, 275, 40042–40047. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Osborne, B.W.; Stanton, R.C. Diabetes causes inhibition of glucose-6-phosphate dehydrogenase via activation of PKA, which contributes to oxidative stress in rat kidney cortex. Am. J. Physiol. Physiol. 2005, 289, F1040–F1047. [Google Scholar] [CrossRef] [Green Version]
- Parsanathan, R.; Jain, S.K. Hydrogen sulfide regulates circadian-clock genes in C2C12 myotubes and the muscle of high-fat-diet-fed mice. Arch. Biochem. Biophys. 2019, 672, 108054. [Google Scholar] [CrossRef]
- Parsanathan, R.; Jain, S.K. Glutathione deficiency induces epigenetic alterations of vitamin D metabolism genes in the livers of high-fat diet-fed obese mice. Sci. Rep. 2019, 9, 14784. [Google Scholar] [CrossRef] [Green Version]
- Parsanathan, R.; Jain, S.K. Hydrogen sulfide increases glutathione biosynthesis, and glucose uptake and utilisation in C2C12 mouse myotubes. Free Radic. Res. 2018, 52, 288–303. [Google Scholar] [CrossRef]
- Jain, S.K.; Parsanathan, R.; Achari, A.E.; Kanikarla-Marie, P.; Bocchini, J.A. Glutathione stimulates vitamin d regulatory and glucose-metabolism genes, lowers oxidative stress and inflammation, and increases 25-hydroxy-vitamin D levels in blood: A novel approach to treat 25-hydroxyvitamin D deficiency. Antioxid. Redox Signal. 2018, 29, 1792–1807. [Google Scholar] [CrossRef] [PubMed]
- Abid, R.; Spokes, K.C.; Shih, S.-C.; Aird, W.C. NADPH oxidase activity selectively modulates vascular endothelial growth factor signaling pathways. J. Biol. Chem. 2007, 282, 35373–35385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsanathan, R.; Jain, S.K. Glutathione deficiency alters the vitamin D-metabolizing enzymes CYP27B1 and CYP24A1 in human renal proximal tubule epithelial cells and kidney of HFD-fed mice. Free Radic. Biol. Med. 2019, 131, 376–381. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parsanathan, R.; Jain, S.K. Glucose-6-Phosphate Dehydrogenase Deficiency Activates Endothelial Cell and Leukocyte Adhesion Mediated via the TGFβ/NADPH Oxidases/ROS Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 7474. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207474
Parsanathan R, Jain SK. Glucose-6-Phosphate Dehydrogenase Deficiency Activates Endothelial Cell and Leukocyte Adhesion Mediated via the TGFβ/NADPH Oxidases/ROS Signaling Pathway. International Journal of Molecular Sciences. 2020; 21(20):7474. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207474
Chicago/Turabian StyleParsanathan, Rajesh, and Sushil K. Jain. 2020. "Glucose-6-Phosphate Dehydrogenase Deficiency Activates Endothelial Cell and Leukocyte Adhesion Mediated via the TGFβ/NADPH Oxidases/ROS Signaling Pathway" International Journal of Molecular Sciences 21, no. 20: 7474. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207474