Sphingosine-1-Phosphate Receptor Modulators and Oligodendroglial Cells: Beyond Immunomodulation

 , ,
, ,  and
and
Abstract
:1. Introduction
1.1. Oligodendrocyte Differentiation
1.2. Oligodendrocytes and Remyelination
2. Sphingosine-1-Phosphate Receptors Expression
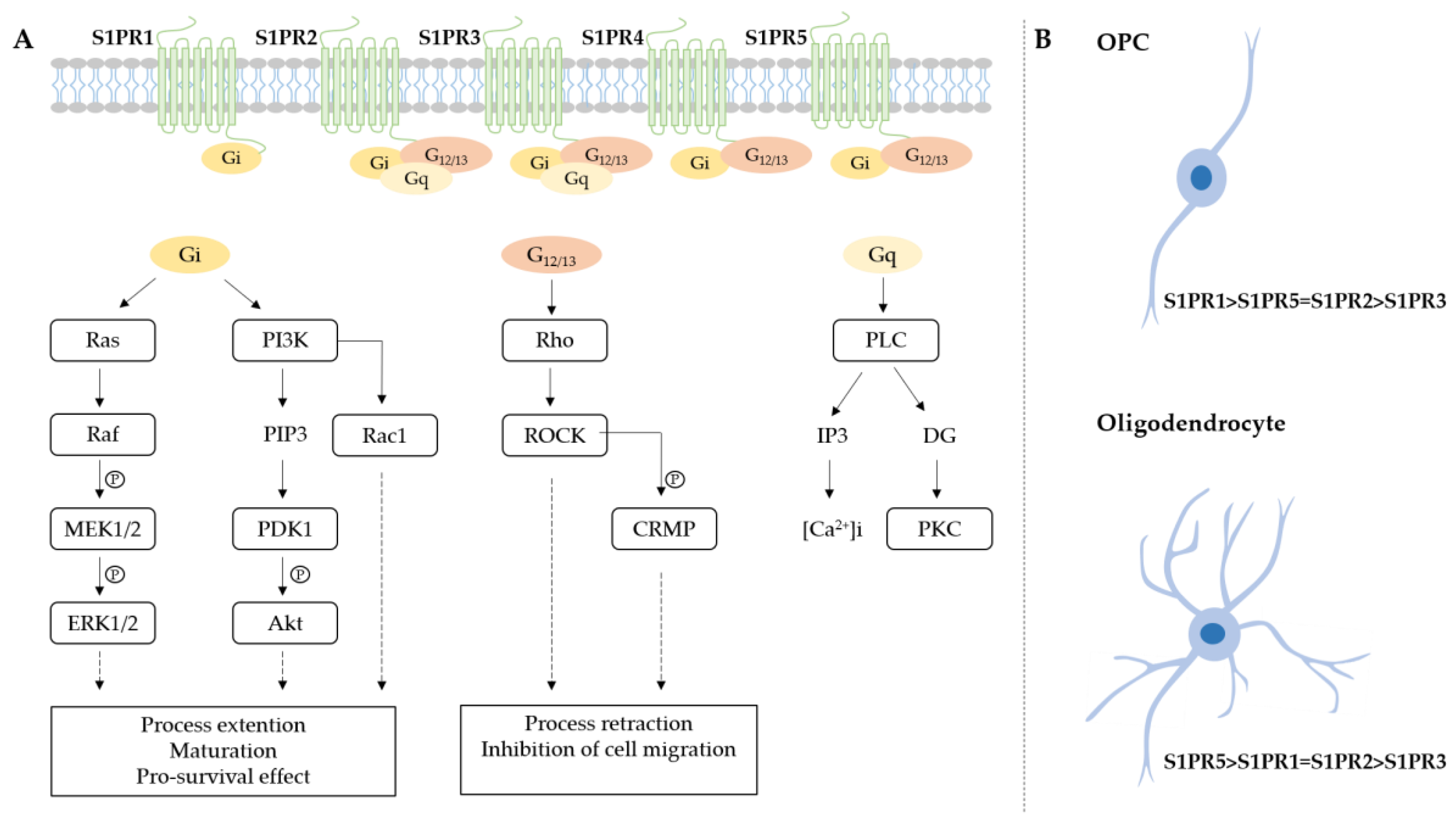
Sphingosine-1-Phosphate Receptor Signaling in Oligodendrolineage Cells
3. Sphingosine-1-Phosphate Receptors and Multiple Sclerosis Therapy
3.1. Sphingosine-1-Phosphate Receptor Modulators Approved for Multiple Sclerosis Treatment
3.1.1. Fingolimod
3.1.2. Siponimod
3.1.3. Ozanimod
3.2. New Modulators in Ongoing Trials
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MS | Multiple sclerosis |
| CNS | Central nervous system |
| S1PR | Sphingosine-1-phosphate receptor |
| OPC | Oligodendrocyte progenitor cell |
| Shh | Sonic hedgehog |
| Olig | Oligodendrocyte transcription factor |
| Nkx6.1 | Homeobox protein Nkx-6.1 |
| Sox | Sex determining region Y-box |
| PDGFα | Platelet-derived growth factor-alpha |
| PDGFRα | PDGFα receptor |
| PLP | Protein proteolipid protein |
| MBP | Myelin basic protein |
| MAG | Myelin-associated glycoprotein |
| GalC | Galactocerebroside |
| MOG | Myelin-oligodendrocyte glycoprotein |
| BBB | Blood-brain barrier |
| ROS | Reactive oxygen species |
| DMTs | Disease modifying therapies |
| SEMA4D | Semaphorin-4D |
| PMS | Progressive multiple sclerosis |
| SPMS | Secondary progressive multiple sclerosis |
| S1P | Sphingosine-1-phosphate |
| SphK | Sphingosine kinase |
| MAP | Mitogen-activated protein |
| PI3K | Phosphoinositide 3-kinase |
| PLC | Phospholipase C |
| NK | Natural killer |
| ERK | Extracellular signal-regulated kinase |
| Akt | Protein kinase B |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PDK1 | 3-phosphoinositide dependent protein kinase-1 |
| CREB | cAMP response element-binding protein |
| p38MAPK | p38 mitogen-activated protein kinase |
| MEK | Mitogen-activated protein kinase |
| ROCK | Rho-associated protein kinase 1 |
| CRMP | Collapsin response mediator protein |
| FDA | Food and drug administration |
| RRMS | Relapsing-remitting multiple sclerosis |
| EAE | Experimental autoimmune encephalomyelitis |
| CIS | Clinically isolated syndrome |
| BDNF | Brain-derived neurotrophic factor |
| LIF | Leukemia inhibitory factor |
| HBEGF | Heparin-binding EGF-like growth factor |
| LPC | Lysolecithin |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| HO-1 | Heme oxygenase |
| NQO1 | NAD(P)H: quinone acceptor oxidoreductases-1 |
| p.o. | post-onset |
| PPMS | Primary progressive form of MS |
| d.p.i | Day post immunization |
| GLAST | Glutamate-aspartate transporter |
| GLT-1 | Glutamate transporter-1 |
| IL | Interleukin |
| ARR | Annualized relapse rate |
| LPS | Lipopolysaccharides |
| TNFα | Tumor necrosis factor α |
References
- Azevedo, F.A.C.; Carvalho, L.R.B.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.L.; Leite, R.E.P.; Filho, W.J.; Lent, R.; Herculano-Houzel, S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Herculano-Houzel, S. The glia/neuron ratio: How it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 2014, 62, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valério-Gomes, B.; Guimarães, D.M.; Szczupak, D.; Lent, R. The Absolute Number of Oligodendrocytes in the Adult Mouse Brain. Front. Neuroanat. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cerdá, F.; Sánchez-Gómez, M.V.; Matute, C. Pío del Río Hortega and the discovery of the oligodendrocytes. Front. Neuroanat. 2015, 9. [Google Scholar] [CrossRef] [Green Version]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2016, 8, a020453. [Google Scholar] [CrossRef]
- Ravanelli, A.M.; Appel, B. Motor neurons and oligodendrocytes arise from distinct cell lineages by progenitor recruitment. Genes Dev. 2015, 29, 2504–2515. [Google Scholar] [CrossRef] [Green Version]
- Naruse, M.; Ishizaki, Y.; Ikenaka, K.; Tanaka, A.; Hitoshi, S. Origin of oligodendrocytes in mammalian forebrains: A revised perspective. J. Physiol. Sci. 2017, 67, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Nery, S.; Wichterle, H.; Fishell, G. Sonic hedgehog contributes to oligodendrocyte specification in the mammalian forebrain. Dev. Camb. Engl. 2001, 128, 527–540. [Google Scholar]
- Soula, C.; Danesin, C.; Kan, P.; Grob, M.; Poncet, C.; Cochard, P. Distinct sites of origin of oligodendrocytes and somatic motoneurons in the chick spinal cord: Oligodendrocytes arise from Nkx2.2-expressing progenitors by a Shh-dependent mechanism. Dev. Camb. Engl. 2001, 128, 1369–1379. [Google Scholar]
- Stolt, C.C.; Lommes, P.; Sock, E.; Chaboissier, M.-C.; Schedl, A.; Wegner, M. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev. 2003, 17, 1677–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farreny, M.-A.; Agius, E.; Bel-Vialar, S.; Escalas, N.; Khouri-Farah, N.; Soukkarieh, C.; Danesin, C.; Pituello, F.; Cochard, P.; Soula, C. FGF signaling controls Shh-dependent oligodendroglial fate specification in the ventral spinal cord. Neural Develop. 2018, 13, 3. [Google Scholar] [CrossRef]
- Frost, E.E.; Zhou, Z.; Krasnesky, K.; Armstrong, R.C. Initiation of oligodendrocyte progenitor cell migration by a PDGF-A activated extracellular regulated kinase (ERK) signaling pathway. Neurochem. Res. 2009, 34, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamashima, T.; Ishii, Y.; Nguyen, L.Q.; Okuno, N.; Sang, Y.; Matsushima, T.; Kurashige, Y.; Takebayashi, H.; Mori, H.; Fujimori, T.; et al. Oligodendrogenesis and Myelin Formation in the Forebrain Require Platelet-derived Growth Factor Receptor-alpha. Neuroscience 2020, 436, 11–26. [Google Scholar] [CrossRef]
- Tsai, H.-H.; Macklin, W.B.; Miller, R.H. Distinct modes of migration position oligodendrocyte precursors for localized cell division in the developing spinal cord. J. Neurosci. Res. 2009, 87, 3320–3330. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.-H.; Niu, J.; Munji, R.; Davalos, D.; Chang, J.; Zhang, H.; Tien, A.-C.; Kuo, C.J.; Chan, J.R.; Daneman, R.; et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science 2016, 351, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.R.L.; Polito, A.; Levine, J.M.; Reynolds, R. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 2003, 24, 476–488. [Google Scholar] [CrossRef]
- Wilson, H.C.; Scolding, N.J.; Raine, C.S. Co-expression of PDGF alpha receptor and NG2 by oligodendrocyte precursors in human CNS and multiple sclerosis lesions. J. Neuroimmunol. 2006, 176, 162–173. [Google Scholar] [CrossRef]
- Baracskay, K.L.; Kidd, G.J.; Miller, R.H.; Trapp, B.D. NG2-positive cells generate A2B5-positive oligodendrocyte precursor cells. Glia 2007, 55, 1001–1010. [Google Scholar] [CrossRef]
- Psachoulia, K.; Jamen, F.; Young, K.M.; Richardson, W.D. Cell cycle dynamics of NG2 cells in the postnatal and ageing brain. Neuron Glia Biol. 2009, 5, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Binamé, F.; Sakry, D.; Dimou, L.; Jolivel, V.; Trotter, J. NG2 regulates directional migration of oligodendrocyte precursor cells via Rho GTPases and polarity complex proteins. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 10858–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, I.; Schachner, M. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: An immunocytological study in the central nervous system. Dev. Biol. 1981, 83, 311–327. [Google Scholar] [CrossRef]
- Scolding, N.J.; Frith, S.; Linington, C.; Morgan, B.P.; Campbell, A.K.; Compston, D.A. Myelin-oligodendrocyte glycoprotein (MOG) is a surface marker of oligodendrocyte maturation. J. Neuroimmunol. 1989, 22, 169–176. [Google Scholar] [CrossRef]
- Elbaz, B.; Popko, B. Molecular Control of Oligodendrocyte Development. Trends Neurosci. 2019, 42, 263–277. [Google Scholar] [CrossRef]
- Zhou, L.; Shao, C.-Y.; Xie, Y.-J.; Wang, N.; Xu, S.-M.; Luo, B.-Y.; Wu, Z.-Y.; Ke, Y.H.; Qiu, M.; Shen, Y. Gab1 mediates PDGF signaling and is essential to oligodendrocyte differentiation and CNS myelination. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Tiane, A.; Schepers, M.; Rombaut, B.; Hupperts, R.; Prickaerts, J.; Hellings, N.; van den Hove, D.; Vanmierlo, T. From OPC to Oligodendrocyte: An Epigenetic Journey. Cells 2019, 8, 1236. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.A.; Li, A.M.; Grutzendler, J. Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci. 2018, 21, 683–695. [Google Scholar] [CrossRef]
- McKenzie, I.A.; Ohayon, D.; Li, H.; de Faria, J.P.; Emery, B.; Tohyama, K.; Richardson, W.D. Motor skill learning requires active central myelination. Science 2014, 346, 318–322. [Google Scholar] [CrossRef]
- Xiao, L.; Ohayon, D.; McKenzie, I.A.; Sinclair-Wilson, A.; Wright, J.L.; Fudge, A.D.; Emery, B.; Li, H.; Richardson, W.D. Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat. Neurosci. 2016, 19, 1210–1217. [Google Scholar] [CrossRef]
- Hasan, M.; Kanna, M.S.; Jun, W.; Ramkrishnan, A.S.; Iqbal, Z.; Lee, Y.; Li, Y. Schema-like learning and memory consolidation acting through myelination. FASEB J. 2019, 33, 11758–11775. [Google Scholar] [CrossRef] [Green Version]
- Kato, D.; Wake, H.; Lee, P.R.; Tachibana, Y.; Ono, R.; Sugio, S.; Tsuji, Y.; Tanaka, Y.H.; Tanaka, Y.R.; Masamizu, Y.; et al. Motor learning requires myelination to reduce asynchrony and spontaneity in neural activity. Glia 2020, 68, 193–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, E.G.; Kang, S.H.; Fukaya, M.; Bergles, D.E. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 2013, 16, 668–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, S. Demyelinating diseases. J. Clin. Pathol. 2006, 59, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Lutton, J.D.; Winston, R.; Rodman, T.C. Multiple sclerosis: Etiological mechanisms and future directions. Exp. Biol. Med. Maywood NJ 2004, 229, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Basivireddy, J.; Kollar, A.; Biron, K.E.; Reickmann, P.; Jefferies, W.A.; McQuaid, S. Blood-brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J. Neuroimmunol. 2010, 229, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.; Plumb, J.; Mirakhur, M.; McQuaid, S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood–brain barrier leakage and active demyelination. J. Pathol. 2003, 201, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, G.G.; Pacheco-Moisés, F.P.; Macías-Islas, M.Á.; Flores-Alvarado, L.J.; Mireles-Ramírez, M.A.; González-Renovato, E.D.; Hernández-Navarro, V.E.; Sánchez-López, A.L.; Alatorre-Jiménez, M.A. Role of the Blood–Brain Barrier in Multiple Sclerosis. Arch. Med. Res. 2014, 45, 687–697. [Google Scholar] [CrossRef]
- Cramer, S.P.; Simonsen, H.; Frederiksen, J.L.; Rostrup, E.; Larsson, H.B.W. Abnormal blood-brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. NeuroImage Clin. 2014, 4, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Duncan, I.D.; Radcliff, A.B.; Heidari, M.; Kidd, G.; August, B.K.; Wierenga, L.A. The adult oligodendrocyte can participate in remyelination. Proc. Natl. Acad. Sci. USA 2018, 115, E11807–E11816. [Google Scholar] [CrossRef] [Green Version]
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013, 125, 841–859. [Google Scholar] [CrossRef] [Green Version]
- Niu, J.; Tsai, H.-H.; Hoi, K.K.; Huang, N.; Yu, G.; Kim, K.; Baranzini, S.E.; Xiao, L.; Chan, J.R.; Fancy, S.P.J. Aberrant oligodendroglial-vascular interactions disrupt the blood-brain barrier, triggering CNS inflammation. Nat. Neurosci. 2019, 22, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Miron, V.; Cui, Q.; Cuo, Q.; Wegner, C.; Antel, J.; Brück, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain J. Neurol. 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yang, H.; Zang, C.; Dong, Y.; Shang, J.; Chen, J.; Wang, Y.; Liu, H.; Zhang, Z.; Xu, H.; et al. CXCR2 antagonism promotes oligodendrocyte precursor cell differentiation and enhances remyelination in a mouse model of multiple sclerosis. Neurobiol. Dis. 2020, 134, 104630. [Google Scholar] [CrossRef] [PubMed]
- Gruchot, J.; Weyers, V.; Göttle, P.; Förster, M.; Hartung, H.-P.; Küry, P.; Kremer, D. The Molecular Basis for Remyelination Failure in Multiple Sclerosis. Cells 2019, 8, 825. [Google Scholar] [CrossRef] [Green Version]
- Duncan, G.J.; Plemel, J.R.; Assinck, P.; Manesh, S.B.; Muir, F.G.W.; Hirata, R.; Berson, M.; Liu, J.; Wegner, M.; Emery, B.; et al. Myelin regulatory factor drives remyelination in multiple sclerosis. Acta Neuropathol. 2017, 134, 403–422. [Google Scholar] [CrossRef]
- Galloway, D.A.; Gowing, E.; Setayeshgar, S.; Kothary, R. Inhibitory milieu at the multiple sclerosis lesion site and the challenges for remyelination. Glia 2020, 68, 859–877. [Google Scholar] [CrossRef]
- French, H.M.; Reid, M.; Mamontov, P.; Simmons, R.A.; Grinspan, J.B. Oxidative Stress Disrupts Oligodendrocyte Maturation. J. Neurosci. Res. 2009, 87, 3076–3087. [Google Scholar] [CrossRef] [Green Version]
- Butts, B.D.; Houde, C.; Mehmet, H. Maturation-dependent sensitivity of oligodendrocyte lineage cells to apoptosis: Implications for normal development and disease. Cell Death Differ. 2008, 15, 1178–1186. [Google Scholar] [CrossRef]
- Back, S.A.; Gan, X.; Li, Y.; Rosenberg, P.A.; Volpe, J.J. Maturation-Dependent Vulnerability of Oligodendrocytes to Oxidative Stress-Induced Death Caused by Glutathione Depletion. J. Neurosci. 1998, 18, 6241–6253. [Google Scholar] [CrossRef]
- Juurlink, B.H.; Thorburne, S.K.; Hertz, L. Peroxide-scavenging deficit underlies oligodendrocyte susceptibility to oxidative stress. Glia 1998, 22, 371–378. [Google Scholar] [CrossRef]
- Giacci, M.K.; Bartlett, C.A.; Smith, N.M.; Iyer, K.S.; Toomey, L.M.; Jiang, H.; Guagliardo, P.; Kilburn, M.R.; Fitzgerald, M. Oligodendroglia Are Particularly Vulnerable to Oxidative Damage after Neurotrauma In Vivo. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 6491–6504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, Y.A.; Baer, A.; Hofer, M.P.; González, G.A.; Rundle, J.; Myrta, S.; Huang, J.K.; Zhao, C.; Rossner, M.J.; Trotter, M.W.B.; et al. Inhibition of phosphodiesterase-4 promotes oligodendrocyte precursor cell differentiation and enhances CNS remyelination. EMBO Mol. Med. 2013, 5, 1918–1934. [Google Scholar] [CrossRef]
- Ineichen, B.V.; Kapitza, S.; Bleul, C.; Good, N.; Plattner, P.S.; Seyedsadr, M.S.; Kaiser, J.; Schneider, M.P.; Zörner, B.; Martin, R.; et al. Nogo-A antibodies enhance axonal repair and remyelination in neuro-inflammatory and demyelinating pathology. Acta Neuropathol. 2017, 134, 423–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeb, S.; Azari, H.; Mostafavi-Pour, Z.; Ghanbari, A.; Ebrahimi, S.; Mokarram, P. 9-cis-Retinoic Acid and 1,25-dihydroxy Vitamin D3 Improve the Differentiation of Neural Stem Cells into Oligodendrocytes through the Inhibition of the Notch and Wnt Signaling Pathways. Iran. J. Med. Sci. 2018, 43, 523–532. [Google Scholar]
- Meffre, D.; Massaad, C.; Grenier, J. Lithium chloride stimulates PLP and MBP expression in oligodendrocytes via Wnt/β-catenin and Akt/CREB pathways. Neuroscience 2015, 284, 962–971. [Google Scholar] [CrossRef]
- Medina-Rodríguez, E.M.; Arenzana, F.J.; Pastor, J.; Redondo, M.; Palomo, V.; García de Sola, R.; Gil, C.; Martínez, A.; Bribián, A.; de Castro, F. Inhibition of endogenous phosphodiesterase 7 promotes oligodendrocyte precursor differentiation and survival. Cell. Mol. Life Sci. 2013, 70, 3449–3462. [Google Scholar] [CrossRef]
- LaGanke, C.; Samkoff, L.; Edwards, K.; Jung Henson, L.; Repovic, P.; Lynch, S.; Stone, L.; Mattson, D.; Galluzzi, A.; Fisher, T.L.; et al. Safety/tolerability of the anti-semaphorin 4D Antibody VX15/2503 in a randomized phase 1 trial. Neurol. Neuroimmunol. Neuroinflammation 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Wooliscroft, L.; Altowaijri, G.; Hildebrand, A.; Samuels, M.; Oken, B.; Bourdette, D.; Cameron, M. Phase I randomized trial of liothyronine for remyelination in multiple sclerosis: A dose-ranging study with assessment of reliability of visual outcomes. Mult. Scler. Relat. Disord. 2020, 41. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Greenberg, B.M.; Bowen, J.D.; Arnold, D.L.; Caggiano, A.O. A double-blind, placebo-controlled, single ascending-dose study of remyelinating antibody rHIgM22 in people with multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2017, 3, 2055217317743097. [Google Scholar] [CrossRef]
- Bove, R.M.; Green, A.J. Remyelinating Pharmacotherapies in Multiple Sclerosis. Neurother. J. Am. Soc. Exp. Neurother. 2017, 14, 894–904. [Google Scholar] [CrossRef] [Green Version]
- Wooliscroft, L.; Silbermann, E.; Cameron, M.; Bourdette, D. Approaches to Remyelination Therapies in Multiple Sclerosis. Curr. Treat. Options Neurol. 2019, 21, 34. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet Lond. Engl. 2017, 390, 2481–2489. [Google Scholar] [CrossRef] [Green Version]
- Schwartzbach, C.J.; Grove, R.A.; Brown, R.; Tompson, D.; Then Bergh, F.; Arnold, D.L. Lesion remyelinating activity of GSK239512 versus placebo in patients with relapsing-remitting multiple sclerosis: A randomised, single-blind, phase II study. J. Neurol. 2017, 264, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, M.; Aoki, H.; Ramanathan, R.; Hait, N.C.; Takabe, K. Sphingosine-1-Phosphate Signaling in Immune Cells and Inflammation: Roles and Therapeutic Potential. Mediators Inflamm. 2016, 2016, 8606878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, H.; Akiyama, T.; Irei, I.; Hamazaki, S.; Sadahira, Y. Cellular localization of sphingosine-1-phosphate receptor 1 expression in the human central nervous system. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2010, 58, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-phosphate signalling. Dev. Camb. Engl. 2014, 141, 5–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, S.E.; Milstien, S.; Spiegel, S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol. Metab. TEM 2007, 18, 300–307. [Google Scholar] [CrossRef]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and Intracellular Actions of Sphingosine-1-Phosphate. Adv. Exp. Med. Biol. 2010, 688, 141–155. [Google Scholar]
- Windh, R.T.; Lee, M.J.; Hla, T.; An, S.; Barr, A.J.; Manning, D.R. Differential coupling of the sphingosine 1-phosphate receptors Edg-1, Edg-3, and H218/Edg-5 to the G(i), G(q), and G(12) families of heterotrimeric G proteins. J. Biol. Chem. 1999, 274, 27351–27358. [Google Scholar] [CrossRef] [Green Version]
- Healy, L.M.; Antel, J.P. Sphingosine-1-Phosphate Receptors in the Central Nervous and Immune Systems. Curr. Drug Targets 2016, 17, 1841–1850. [Google Scholar] [CrossRef]
- Wang, L.; Dudek, S.M. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc. Res. 2009, 77, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, T.; Kajimoto, T.; Jahangeer, S.; Nakamura, S. Sphingosine kinase/sphingosine 1-phosphate signalling in central nervous system. Cell. Signal. 2009, 21, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.G.; Kim, H.J.; Miron, V.E.; Cook, S.; Kennedy, T.E.; Foster, C.A.; Antel, J.P.; Soliven, B. Functional consequences of S1P receptor modulation in rat oligodendroglial lineage cells. Glia 2007, 55, 1656–1667. [Google Scholar] [CrossRef]
- Coelho, R.P.; Payne, S.G.; Bittman, R.; Spiegel, S.; Sato-Bigbee, C. The Immunomodulator FTY720 Has a Direct Cytoprotective Effect in Oligodendrocyte Progenitors. J. Pharmacol. Exp. Ther. 2007, 323, 626–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novgorodov, A.S.; El-Alwani, M.; Bielawski, J.; Obeid, L.M.; Gudz, T.I. Activation of sphingosine-1-phosphate receptor S1P5 inhibits oligodendrocyte progenitor migration. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 1503–1514. [Google Scholar] [CrossRef]
- Yu, N.; Lariosa-Willingham, K.D.; Lin, F.-F.; Webb, M.; Rao, T.S. Characterization of lysophosphatidic acid and sphingosine-1-phosphate-mediated signal transduction in rat cortical oligodendrocytes. Glia 2004, 45, 17–27. [Google Scholar] [CrossRef]
- Miron, V.E.; Hall, J.A.; Kennedy, T.E.; Soliven, B.; Antel, J.P. Cyclical and Dose-Dependent Responses of Adult Human Mature Oligodendrocytes to Fingolimod. Am. J. Pathol. 2008, 173, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Jaillard, C.; Harrison, S.; Stankoff, B.; Aigrot, M.S.; Calver, A.R.; Duddy, G.; Walsh, F.S.; Pangalos, M.N.; Arimura, N.; Kaibuchi, K.; et al. Edg8/S1P5: An Oligodendroglial Receptor with Dual Function on Process Retraction and Cell Survival. J. Neurosci. 2005, 25, 1459–1469. [Google Scholar] [CrossRef]
- Miron, V.E.; Jung, C.G.; Kim, H.J.; Kennedy, T.E.; Soliven, B.; Antel, J.P. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann. Neurol. 2008, 63, 61–71. [Google Scholar] [CrossRef]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.H.M.; Okada, T.; Matloubian, M.; Lo, C.G.; Cyster, J.G. S1P1 receptor signaling overrides retention mediated by Gαi-coupled receptors to promote T cell egress. Immunity 2008, 28, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenne, C.N.; Enders, A.; Rivera, R.; Watson, S.R.; Bankovich, A.J.; Pereira, J.P.; Xu, Y.; Roots, C.M.; Beilke, J.N.; Banerjee, A.; et al. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J. Exp. Med. 2009, 206, 2469–2481. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366. [Google Scholar] [CrossRef]
- van Doorn, R.; Lopes Pinheiro, M.A.; Kooij, G.; Lakeman, K.; van het Hof, B.; van der Pol, S.M.A.; Geerts, D.; van Horssen, J.; van der Valk, P.; van der Kam, E.; et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J. Neuroinflammation 2012, 9, 133. [Google Scholar] [CrossRef] [Green Version]
- Miron, V.E.; Schubart, A.; Antel, J.P. Central nervous system-directed effects of FTY720 (fingolimod). J. Neurol. Sci. 2008, 274, 13–17. [Google Scholar] [CrossRef]
- Cui, Q.L.; Fang, J.; Kennedy, T.E.; Almazan, G.; Antel, J.P. Role of p38MAPK in S1P receptor-mediated differentiation of human oligodendrocyte progenitors. Glia 2014, 62, 1361–1375. [Google Scholar] [CrossRef]
- Kim, H.J.; Miron, V.E.; Dukala, D.; Proia, R.L.; Ludwin, S.K.; Traka, M.; Antel, J.P.; Soliven, B. Neurobiological effects of sphingosine 1-phosphate receptor modulation in the cuprizone model. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Dukala, D.E.; Soliven, B. S1P1 deletion in oligodendroglial lineage cells: Effect on differentiation and myelination. Glia 2016, 64, 570–582. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho family proteins: Coordinating cell responses. Trends Cell Biol. 2001, 11, 471–477. [Google Scholar] [CrossRef]
- Martínez-Morales, J.C.; Romero-Ávila, M.T.; Reyes-Cruz, G.; García-Sáinz, J.A. S1P1 receptor phosphorylation, internalization, and interaction with Rab proteins: Effects of sphingosine 1-phosphate, FTY720-P, phorbol esters, and paroxetine. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, K. FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol. Ther. 2005, 108, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Mullershausen, F.; Zecri, F.; Cetin, C.; Billich, A.; Guerini, D.; Seuwen, K. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat. Chem. Biol. 2009, 5, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Verzijl, D.; Peters, S.L.M.; Alewijnse, A.E. Sphingosine-1-phosphate receptors: Zooming in on ligand-induced intracellular trafficking and its functional implications. Mol. Cells 2010, 29, 99–104. [Google Scholar] [CrossRef]
- Albert, R.; Hinterding, K.; Brinkmann, V.; Guerini, D.; Müller-Hartwieg, C.; Knecht, H.; Simeon, C.; Streiff, M.; Wagner, T.; Welzenbach, K.; et al. Novel Immunomodulator FTY720 Is Phosphorylated in Rats and Humans To Form a Single Stereoisomer. Identification, Chemical Proof, and Biological Characterization of the Biologically Active Species and Its Enantiomer. J. Med. Chem. 2005, 48, 5373–5377. [Google Scholar] [CrossRef]
- Paugh, S.W.; Payne, S.G.; Barbour, S.E.; Milstien, S.; Spiegel, S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003, 554, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.-J.; Card, D.; Keohane, C.; et al. Alteration of Lymphocyte Trafficking by Sphingosine-1-Phosphate Receptor Agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef]
- Foster, C.A.; Howard, L.M.; Schweitzer, A.; Persohn, E.; Hiestand, P.C.; Balatoni, B.; Reuschel, R.; Beerli, C.; Schwartz, M.; Billich, A. Brain Penetration of the Oral Immunomodulatory Drug FTY720 and Its Phosphorylation in the Central Nervous System during Experimental Autoimmune Encephalomyelitis: Consequences for Mode of Action in Multiple Sclerosis. J. Pharmacol. Exp. Ther. 2007, 323, 469–475. [Google Scholar] [CrossRef] [Green Version]
- David, O.J.; Kovarik, J.M.; Schmouder, R.L. Clinical Pharmacokinetics of Fingolimod. Clin. Pharmacokinet. 2012, 51, 15–28. [Google Scholar] [CrossRef]
- David, O.J.; Behrje, R.; Pal, P.; Hara, H.; Lates, C.D.; Schmouder, R. Pharmacokinetic Interaction Between Fingolimod and Carbamazepine in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2018, 7, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Scott, F.L.; Clemons, B.; Brooks, J.; Brahmachary, E.; Powell, R.; Dedman, H.; Desale, H.G.; Timony, G.A.; Martinborough, E.; Rosen, H.; et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br. J. Pharmacol. 2016, 173, 1778–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Salama, Z.T. Siponimod: First Global Approval. Drugs 2019, 79, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, L.; Constantinescu, C.S.; Tanasescu, R. Siponimod for the treatment of secondary progressive multiple sclerosis. Expert Opin. Pharmacother. 2019, 20, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Shakeri-Nejad, K.; Gardin, A.; Gray, C.; Neelakantham, S.; Dumitras, S.; Legangneux, E. Safety, Tolerability, Pharmacodynamics and Pharmacokinetics of Intravenous Siponimod: A Randomized, Open-label Study in Healthy Subjects. Clin. Ther. 2020, 42, 175–195. [Google Scholar] [CrossRef] [Green Version]
- Glaenzel, U.; Jin, Y.; Nufer, R.; Li, W.; Schroer, K.; Adam-Stitah, S.; van Marle, S.P.; Legangneux, E.; Borell, H.; James, A.D.; et al. Metabolism and Disposition of Siponimod, a Novel Selective S1P1/S1P5 Agonist, in Healthy Volunteers and In Vitro Identification of Human Cytochrome P450 Enzymes Involved in Its Oxidative Metabolism. Drug Metab. Dispos. 2018, 46, 1001–1013. [Google Scholar] [CrossRef] [Green Version]
- Tran, J.Q.; Hartung, J.P.; Peach, R.J.; Boehm, M.F.; Rosen, H.; Smith, H.; Brooks, J.L.; Timony, G.A.; Olson, A.D.; Gujrathi, S.; et al. Results From the First-in-Human Study With Ozanimod, a Novel, Selective Sphingosine-1-Phosphate Receptor Modulator. J. Clin. Pharmacol. 2017, 57, 988–996. [Google Scholar] [CrossRef]
- Deogracias, R.; Yazdani, M.; Dekkers, M.P.J.; Guy, J.; Ionescu, M.C.S.; Vogt, K.E.; Barde, Y.-A. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 14230–14235. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, F.S.; Hofereiter, J.; Rübsamen, H.; Melms, J.; Schwarz, S.; Faber, H.; Weber, P.; Pütz, B.; Loleit, V.; Weber, F.; et al. Fingolimod induces neuroprotective factors in human astrocytes. J. Neuroinflammation 2015, 12, 184. [Google Scholar] [CrossRef] [Green Version]
- Van’t Veer, A.; Du, Y.; Fischer, T.Z.; Boetig, D.R.; Wood, M.R.; Dreyfus, C.F. Brain-Derived Neurotrophic Factor Effects on Oligodendrocyte Progenitors of the Basal Forebrain Are Mediated Through TrkB and the MAP Kinase Pathway. J. Neurosci. Res. 2009, 87, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; He, Q.; Wang, Y.; Cheng, X.; Howard, R.M.; Zhang, Y.; DeVries, W.H.; Shields, C.B.; Magnuson, D.S.K.; Xu, X.-M.; et al. Transplantation of Ciliary Neurotrophic Factor-Expressing Adult Oligodendrocyte Precursor Cells Promotes Remyelination and Functional Recovery after SpinalCord Injury. J. Neurosci. 2010, 30, 2989–3001. [Google Scholar] [CrossRef] [Green Version]
- VonDran, M.W.; Singh, H.; Honeywell, J.Z.; Dreyfus, C.F. Levels of BDNF Impact Oligodendrocyte Lineage Cells following a Cuprizone Lesion. J. Neurosci. 2011, 31, 14182–14190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, K.; Maki, T.; Saito, S.; Yamamoto, Y.; Kinoshita, H.; Choi, Y.K.; Arumugam, T.V.; Lim, Y.-A.; Chen, C.L.H.; Wong, P.T.-H.; et al. Effect of fingolimod on oligodendrocyte maturation under prolonged cerebral hypoperfusion. Brain Res. 2019, 1720, 146294. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, E. There is more to a lipid than just being a fat: Sphingolipid-guided differentiation of oligodendroglial lineage from embryonic stem cells. Neurochem. Res. 2011, 36, 1601–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.J.; Giovannoni, G.; Baker, D. Fingolimod modulates microglial activation to augment markers of remyelination. J. Neuroinflammation 2011, 8, 76. [Google Scholar] [CrossRef] [Green Version]
- Miron, V.E.; Ludwin, S.K.; Darlington, P.J.; Jarjour, A.A.; Soliven, B.; Kennedy, T.E.; Antel, J.P. Fingolimod (FTY720) enhances remyelination following demyelination of organotypic cerebellar slices. Am. J. Pathol. 2010, 176, 2682–2694. [Google Scholar] [CrossRef]
- Nystad, A.E.; Lereim, R.R.; Wergeland, S.; Oveland, E.; Myhr, K.-M.; Bø, L.; Torkildsen, Ø. Fingolimod downregulates brain sphingosine-1-phosphate receptor 1 levels but does not promote remyelination or neuroprotection in the cuprizone model. J. Neuroimmunol. 2020, 339, 577091. [Google Scholar] [CrossRef]
- Alme, M.N.; Nystad, A.E.; Bø, L.; Myhr, K.-M.; Vedeler, C.A.; Wergeland, S.; Torkildsen, Ø. Fingolimod does not enhance cerebellar remyelination in the cuprizone model. J. Neuroimmunol. 2015, 285, 180–186. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Lee, X.; Ji, B.; Guckian, K.; Apicco, D.; Pepinsky, R.B.; Miller, R.H.; Mi, S. Sphingosine 1-phosphate receptor modulator fingolimod (FTY720) does not promote remyelination in vivo. Mol. Cell. Neurosci. 2011, 48, 72–81. [Google Scholar] [CrossRef]
- Slowik, A.; Schmidt, T.; Beyer, C.; Amor, S.; Clarner, T.; Kipp, M. The sphingosine 1-phosphate receptor agonist FTY720 is neuroprotective after cuprizone-induced CNS demyelination. Br. J. Pharmacol. 2015, 172, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Bielawski, J.; Yang, H.; Kong, Y.; Zhou, B.; Li, J. Functional antagonism of sphingosine-1-phosphate receptor 1 prevents cuprizone-induced demyelination. Glia 2018, 66, 654–669. [Google Scholar] [CrossRef]
- Faizi, M.; Salimi, A.; Seydi, E.; Naserzadeh, P.; Kouhnavard, M.; Rahimi, A.; Pourahmad, J. Toxicity of cuprizone a Cu2+ chelating agent on isolated mouse brain mitochondria: A justification for demyelination and subsequent behavioral dysfunction. Toxicol. Mech. Methods 2016, 26, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, G.; Martínez-Bermúdez, A.K.; Liu, H.-N.; Khorchid, A.; Chemtob, S.; Mushynski, W.E.; Almazan, G. Developmental differences in HO-induced oligodendrocyte cell death: Role of glutathione, mitogen-activated protein kinases and caspase 3. J. Neurochem. 2004, 90, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.M.; Pinto, M.; Patel, S.; McCarthy, S.; Gao, H.; Taherian, M.; Karmally, S.; Pereira, C.V.; Dvoriantchikova, G.; Ivanov, D.; et al. Mitochondrial DNA Double-Strand Breaks in Oligodendrocytes Cause Demyelination, Axonal Injury, and CNS Inflammation. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 10185–10199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, K.-H.; Herrup, K. DNA damage in the oligodendrocyte lineage and its role in brain aging. Mech. Ageing Dev. 2017, 161, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Engedal, N.; Žerovnik, E.; Rudov, A.; Galli, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Monsurrò, V.; Betti, M.; Albertini, M.C. From Oxidative Stress Damage to Pathways, Networks, and Autophagy via MicroRNAs. Oxid. Med. Cell. Longev. 2018, 2018, 4968321. [Google Scholar] [CrossRef]
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Chen, Y.; Hoang, T.; Montgomery, R.L.; Zhao, X.; Bu, H.; Hu, T.; Taketo, M.M.; van Es, J.H.; Clevers, H.; et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the β-catenin–TCF interaction. Nat. Neurosci. 2009, 12, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Moyon, S.; Casaccia, P. DNA methylation in oligodendroglial cells during developmental myelination and in disease. Neurogenesis Austin Tex 2017, 4, e1270381. [Google Scholar] [CrossRef] [Green Version]
- van Horssen, J.; Witte, M.E.; Schreibelt, G.; de Vries, H.E. Radical changes in multiple sclerosis pathogenesis. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2011, 1812, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Rone, M.B.; Cui, Q.-L.; Fang, J.; Wang, L.-C.; Zhang, J.; Khan, D.; Bedard, M.; Almazan, G.; Ludwin, S.K.; Jones, R.; et al. Oligodendrogliopathy in Multiple Sclerosis: Low Glycolytic Metabolic Rate Promotes Oligodendrocyte Survival. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 4698–4707. [Google Scholar] [CrossRef] [Green Version]
- Martín-Montañez, E.; Pavia, J.; Valverde, N.; Boraldi, F.; Lara, E.; Oliver, B.; Hurtado-Guerrero, I.; Fernandez, O.; Garcia-Fernandez, M. The S1P mimetic fingolimod phosphate regulates mitochondrial oxidative stress in neuronal cells. Free Radic. Biol. Med. 2019, 137, 116–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Bassani, C.; De Angelis, A.; Ruffini, F.; Ottoboni, L.; Comi, G.; Martino, G.; Farina, C. Siponimod (BAF312) Activates Nrf2 While Hampering NFκB in Human Astrocytes, and Protects From Astrocyte-Induced Neurodegeneration. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Nellessen, A.; Nyamoya, S.; Zendedel, A.; Slowik, A.; Wruck, C.; Beyer, C.; Fragoulis, A.; Clarner, T. Nrf2 deficiency increases oligodendrocyte loss, demyelination, neuroinflammation and axonal damage in an MS animal model. Metab. Brain Dis. 2020, 35, 353–362. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Z.G.; Li, Y.; Ding, X.; Shang, X.; Lu, M.; Elias, S.B.; Chopp, M. Fingolimod treatment promotes proliferation and differentiation of oligodendrocyte progenitor cells in mice with experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2015, 76, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.; Baharvand, H.; Javan, M. Enhanced remyelination following lysolecithin-induced demyelination in mice under treatment with fingolimod (FTY720). Neuroscience 2015, 311, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.A.; Radue, E.-W.; Goodin, D.; Jeffery, D.; Rammohan, K.W.; Reder, A.T.; Vollmer, T.; Agius, M.A.; Kappos, L.; Stites, T.; et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014, 13, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Gajofatto, A.; Turatti, M.; Benedetti, M.D. Primary progressive multiple sclerosis: Current therapeutic strategies and future perspectives. Expert Rev. Neurother. 2017, 17, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Daniels, K.; van der Nat, P.B.; Frequin, S.T.F.M.; van der Wees, P.J.; Biesma, D.H.; Hoogervorst, E.L.J.; van de Garde, E.M.W. Real-World Results of Ocrelizumab Treatment for Primary Progressive Multiple Sclerosis. Mult. Scler. Int. 2020, 2020. [Google Scholar] [CrossRef]
- Lublin, F.; Miller, D.H.; Freedman, M.S.; Cree, B.A.C.; Wolinsky, J.S.; Weiner, H.; Lubetzki, C.; Hartung, H.-P.; Montalban, X.; Uitdehaag, B.M.J.; et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1075–1084. [Google Scholar] [CrossRef]
- Khatri, B.; Barkhof, F.; Comi, G.; Hartung, H.-P.; Kappos, L.; Montalban, X.; Pelletier, J.; Stites, T.; Wu, S.; Holdbrook, F.; et al. Comparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: A randomised extension of the TRANSFORMS study. Lancet Neurol. 2011, 10, 520–529. [Google Scholar] [CrossRef]
- Vargas, W.S.; Perumal, J.S. Fingolimod and cardiac risk: Latest findings and clinical implications. Ther. Adv. Drug Saf. 2013, 4, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Forrest, M.; Sun, S.-Y.; Hajdu, R.; Bergstrom, J.; Card, D.; Doherty, G.; Hale, J.; Keohane, C.; Meyers, C.; Milligan, J.; et al. Immune Cell Regulation and Cardiovascular Effects of Sphingosine 1-Phosphate Receptor Agonists in Rodents Are Mediated via Distinct Receptor Subtypes. J. Pharmacol. Exp. Ther. 2004, 309, 758–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gergely, P.; Nuesslein-Hildesheim, B.; Guerini, D.; Brinkmann, V.; Traebert, M.; Bruns, C.; Pan, S.; Gray, N.S.; Hinterding, K.; Cooke, N.G.; et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br. J. Pharmacol. 2012, 167, 1035–1047. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Vogelgesang Antje; Domanska Grazyna; Ruhnau Johanna; Dressel Alexander; Kirsch Michael; Schulze Juliane Siponimod (BAF312) Treatment Reduces Brain Infiltration but Not Lesion Volume in Middle-Aged Mice in Experimental Stroke. Stroke 2019, 50, 1224–1231. [CrossRef]
- Hundehege, P.; Cerina, M.; Eichler, S.; Thomas, C.; Herrmann, A.M.; Göbel, K.; Müntefering, T.; Fernandez-Orth, J.; Bock, S.; Narayanan, V.; et al. The next-generation sphingosine-1 receptor modulator BAF312 (siponimod) improves cortical network functionality in focal autoimmune encephalomyelitis. Neural Regen. Res. 2019, 14, 1950–1960. [Google Scholar] [CrossRef]
- Gentile, A.; Musella, A.; Bullitta, S.; Fresegna, D.; De Vito, F.; Fantozzi, R.; Piras, E.; Gargano, F.; Borsellino, G.; Battistini, L.; et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J. Neuroinflammation 2016, 13, 207. [Google Scholar] [CrossRef] [Green Version]
- Kipp, M. Does Siponimod Exert Direct Effects in the Central Nervous System? Cells 2020, 9, 1771. [Google Scholar] [CrossRef]
- O’Sullivan, C.; Schubart, A.; Mir, A.K.; Dev, K.K. The dual S1PR1/S1PR5 drug BAF312 (Siponimod) attenuates demyelination in organotypic slice cultures. J. Neuroinflammation 2016, 13. [Google Scholar] [CrossRef] [Green Version]
- Tiwari-Woodruff, S.; Yamate-Morgan, H.; Sekyi, M.; Lauderdale, K.; Hasselmann, J.; Schubart, A. The Sphingosine 1-phosphate (S1P) Receptor Modulator, Siponimod Decreases Oligodendrocyte Cell Death and Axon Demyelination in a Mouse Model of Multiple Sclerosis (I10.011). Neurology 2016, 86. [Google Scholar]
- Lamb, Y.N. Ozanimod: First Approval. Drugs 2020. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Arnold, D.L.; Comi, G.; Bar-Or, A.; Gujrathi, S.; Hartung, J.P.; Cravets, M.; Olson, A.; Frohna, P.A.; Selmaj, K.W. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 373–381. [Google Scholar] [CrossRef]
- Comi, G.; Kappos, L.; Selmaj, K.W.; Bar-Or, A.; Arnold, D.L.; Steinman, L.; Hartung, H.-P.; Montalban, X.; Kubala Havrdová, E.; Cree, B.A.C.; et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): A multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 2019, 18, 1009–1020. [Google Scholar] [CrossRef]
- Cohen, J.A.; Comi, G.; Selmaj, K.W.; Bar-Or, A.; Arnold, D.L.; Steinman, L.; Hartung, H.-P.; Montalban, X.; Kubala Havrdová, E.; Cree, B.A.C.; et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): A multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 2019, 18, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Inc, M.G. Ozanimod (RPC1063) Reduces the Plasma Biomarker Neurofilament Light… by Kristen R. Taylor Meadows. Available online: https://onlinelibrary.ectrims-congress.eu/ectrims/2017/ACTRIMS-ECTRIMS2017/199629/kristen.r.taylor.meadows.ozanimod.(rpc1063).reduces.the.plasma.biomarker.html (accessed on 11 October 2020).
- Inc, M.G. Ozanimod (RPC1063) is Potentially Neuroprotective through Direct… by Kristen R. Taylor Meadows. Available online: https://onlinelibrary.ectrims-congress.eu/ectrims/2017/ACTRIMS-ECTRIMS2017/200838/kristen.r.taylor.meadows.ozanimod.28rpc106329.is.potentially.neuroprotective.html (accessed on 13 May 2020).
- Dash, R.P.; Rais, R.; Srinivas, N.R. Ponesimod, a selective sphingosine 1-phosphate (S1P1) receptor modulator for autoimmune diseases: Review of clinical pharmacokinetics and drug disposition. Xenobiotica 2018, 48, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Pouzol, L.; Piali, L.; Bernard, C.C.; Martinic, M.M.; Steiner, B.; Clozel, M. Therapeutic Potential of Ponesimod Alone and in Combination with Dimethyl Fumarate in Experimental Models of Multiple Sclerosis. Innov. Clin. Neurosci. 2019, 16, 22–30. [Google Scholar] [PubMed]
- D’Ambrosio, D.; Freedman, M.S.; Prinz, J. Ponesimod, a selective S1P1 receptor modulator: A potential treatment for multiple sclerosis and other immune-mediated diseases. Ther. Adv. Chronic Dis. 2016, 7, 18–33. [Google Scholar] [CrossRef] [Green Version]
- Krösser, S.; Wolna, P.; Fischer, T.Z.; Boschert, U.; Stoltz, R.; Zhou, M.; Darpo, B. Effect of ceralifimod (ONO-4641) on lymphocytes and cardiac function: Randomized, double-blind, placebo-controlled trial with an open-label fingolimod arm. J. Clin. Pharmacol. 2015, 55, 1051–1060. [Google Scholar] [CrossRef]
- Komiya, T.; Sato, K.; Shioya, H.; Inagaki, Y.; Hagiya, H.; Kozaki, R.; Imai, M.; Takada, Y.; Maeda, T.; Kurata, H.; et al. Efficacy and immunomodulatory actions of ONO-4641, a novel selective agonist for sphingosine 1-phosphate receptors 1 and 5, in preclinical models of multiple sclerosis. Clin. Exp. Immunol. 2013, 171, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, K.; Maeda, Y.; Shimano, K.; Mogami, A.; Kataoka, H.; Ogawa, K.; Hikida, K.; Kumagai, H.; Asayama, M.; Yamamoto, T.; et al. Amiselimod, a novel sphingosine 1-phosphate receptor-1 modulator, has potent therapeutic efficacy for autoimmune diseases, with low bradycardia risk. Br. J. Pharmacol. 2017, 174, 15–27. [Google Scholar] [CrossRef]
- Kifuji, T.; Inoue, S.; Furukawa, M.; Perez Madera, B.; Goto, T.; Kumagai, H.; Mair, S.J.; Kawaguchi, A. Absorption, disposition and metabolic pathway of amiselimod (MT-1303) in healthy volunteers in a mass balance study. Xenobiotica 2019, 49, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gray, F.; Henderson, A.; Hicks, K.; Yang, J.; Thompson, P.; Oliver, J. Safety, pharmacokinetics, pharmacodynamics, and bioavailability of GSK2018682, a sphingosine-1-phosphate receptor modulator, in healthy volunteers. Clin. Pharmacol. Drug Dev. 2014, 3, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Nishi, T.; Miyazaki, S.; Takemoto, T.; Suzuki, K.; Iio, Y.; Nakajima, K.; Ohnuki, T.; Kawase, Y.; Nara, F.; Inaba, S.; et al. Discovery of CS-0777: A Potent, Selective, and Orally Active S1P1 Agonist. ACS Med. Chem. Lett. 2011, 2, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, D.K.; Yu, D.; Bernard, F.; Graham, D.E.; Boschert, U.; Dellovade, T. ONO-4641 (Ceralifimod) Prevents Evoked Potential Deficits in an Animal Model of Multiple Sclerosis (P1.218). Available online: https://www.semanticscholar.org/paper/ONO-4641-(Ceralifimod)-Prevents-Evoked-Potential-in-Crawford-Yu/878093219781feda7deef1ccc88355971b99aeec (accessed on 11 May 2020).
- Harada, T.; Wilbraham, D.; de La Borderie, G.; Inoue, S.; Bush, J.; Camm, A.J. Cardiac effects of amiselimod compared with fingolimod and placebo: Results of a randomised, parallel-group, phase I study in healthy subjects. Br. J. Clin. Pharmacol. 2017, 83, 1011–1027. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Compound | Classification | Mechanism of Action | Indication | Clinical Trial | References |
|---|---|---|---|---|---|
| Antisemaphorine 4D (VX15/2503) Catalent Pharma Solutions© | Humanizes monoclonal antibody | anti-SEMA4D. Blocks the interaction between SEMA4D and its receptors | PMS; RRMS | Phase I | [57] |
| Liothyronine (L-T3 liothyronine sodium) | Nuclear Hormone Agonists | Thyroid receptor agonist | MS | Phase I | [58] |
| rHIgM22 Acorda Therapeutics, Inc.© | Human IgM antibody | Not clarified | MS | Phase I | [59] |
| Domperidone | D2/D3 dopamine receptor antagonist | Increases prolactin serum levels | SPMS | Phase II | [60] |
| Clemastine (Clematine fumate meclastin) | Antihistamine | H1 antihistamine antagonist M1/M3 muscarinic receptors reverse antagonist | RRMS | Phase II | [61,62] |
| GSK239512 GlaxoSmithKline© | Antihistamine | H3 receptor antagonist | RRMS | Phase II | [63] |
| Compound | Receptor Affinity | T1/2 (Hours) | Tmax (Hours) | Pro-Drug (Phosphorylation) | Indication | References | |
|---|---|---|---|---|---|---|---|
| High | Low | ||||||
| Fingolimod (FTY720 Gilenya) Novartis© | S1PR1 (EC50 = 0.3 nM) S1PR3 (EC50 = 0.9 nM) S1PR5 (EC50 = 0.50 nM) | S1PR2 (EC50 > 10,000 nM) S1PR4 (EC50 = 345 nM) | 144–216 | 12–16 | + | RRMS | [99,100,101] |
| Siponimod (BAF312 Mayzent) Novartis© | S1PR1 (EC50 = 0.39 nM) S1PR5 (EC50 = 0.38 nM) | S1PR2 (EC50 > 10,000 nM) S1PR3 (EC50 > 1000 nM) S1PR4 (EC50 > 750 nM) | 26–33 | 6–8 | - | RRMS SPMS CIS | [102,103,104,105] |
| Ozanimod (RPC1063 Zeposia) Celgene© | S1PR1 (EC50 = 0.41 nM) S1PR5 (EC50 = 11 nM) | S1PR2 (EC50 > 10,000 nM) S1PR3 (EC50 > 10,000 nM) S1PR4 (EC50 > 7 nM) | 15–17 | 8–12 | - | RRMS SPMS CIS | [101,106] |
| Compound | Receptor Affinity | T1/2 (Hours) | Tmax (Hours) | Pro-Drug (Phosphorylation) | Indication | References | |
|---|---|---|---|---|---|---|---|
| High | Low | ||||||
| Ponesimod (ACT-128800) Actelion© | S1PR1 (EC50 = 5.7 nM) S1PR5 (EC50 = 11 nM) | S1PR2 (EC50 > 10,000 nM) S1PR3 (EC50 > 10,000 nM) S1PR4 (EC50 > 7000 nM) | 21–33 | 2–4 | - | RRMS (phase III) | [159] |
| Ceralifimod (ONO-4641) Ono Pharmaceutical© | S1PR1 (EC50 = 0.273 nM) S1PR5 (EC50 = 0.334 nM) | S1PR2 (EC50 > 30,000 nM) S1PR3 (EC50 > 30,000 nM) | 82–89 | 4–6 | - | RRMS (phase II) | [160,161] |
| Amiselimod (MT-1303) Mitsubishi tanabe pharma corporation© | S1PR1 (EC50 = 0.075 nM) S1PR5 (EC50 = 0.47 nM) S1PR4 (EC50 = 122 nM) | S1PR2 (EC50 > 10,000 nM) S1PR3 (EC50 > 10,000 nM) | 408 | 12–16 | + | RRMS (phase II) | [162,163] |
| GSK2018682 GlaxoSmithKline© | S1PR1 (EC50 = 0.07 nM) S1PR5 (EC50 = 0.072 nM) | S1PR3 (EC50 > 1000 nM) | 48–63 | 4–9 | - | RRMS (phase I) | [164] |
| CS-0777 Daiichi Sankyo© | S1PR1 (EC50 = 1.1 nM) S1PR5 (EC50 = 21 nM) | S1PR3 (EC50 = 350 nM) S1PR4 (no data) | 9–11 | 8–10 | + | MS (phase I) | [165] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roggeri, A.; Schepers, M.; Tiane, A.; Rombaut, B.; van Veggel, L.; Hellings, N.; Prickaerts, J.; Pittaluga, A.; Vanmierlo, T. Sphingosine-1-Phosphate Receptor Modulators and Oligodendroglial Cells: Beyond Immunomodulation. Int. J. Mol. Sci. 2020, 21, 7537. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207537
Roggeri A, Schepers M, Tiane A, Rombaut B, van Veggel L, Hellings N, Prickaerts J, Pittaluga A, Vanmierlo T. Sphingosine-1-Phosphate Receptor Modulators and Oligodendroglial Cells: Beyond Immunomodulation. International Journal of Molecular Sciences. 2020; 21(20):7537. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207537
Chicago/Turabian StyleRoggeri, Alessandra, Melissa Schepers, Assia Tiane, Ben Rombaut, Lieve van Veggel, Niels Hellings, Jos Prickaerts, Anna Pittaluga, and Tim Vanmierlo. 2020. "Sphingosine-1-Phosphate Receptor Modulators and Oligodendroglial Cells: Beyond Immunomodulation" International Journal of Molecular Sciences 21, no. 20: 7537. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207537