Hypoxic Melanoma Cells Deliver microRNAs to Dendritic Cells and Cytotoxic T Lymphocytes through Connexin-43 Channels

 ,
, 
Abstract
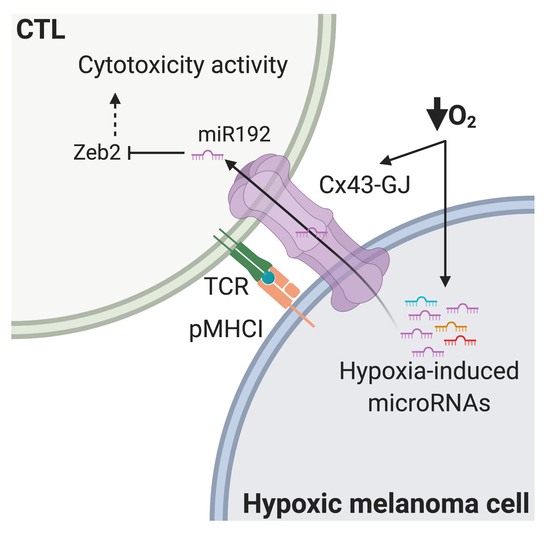
:
1. Introduction
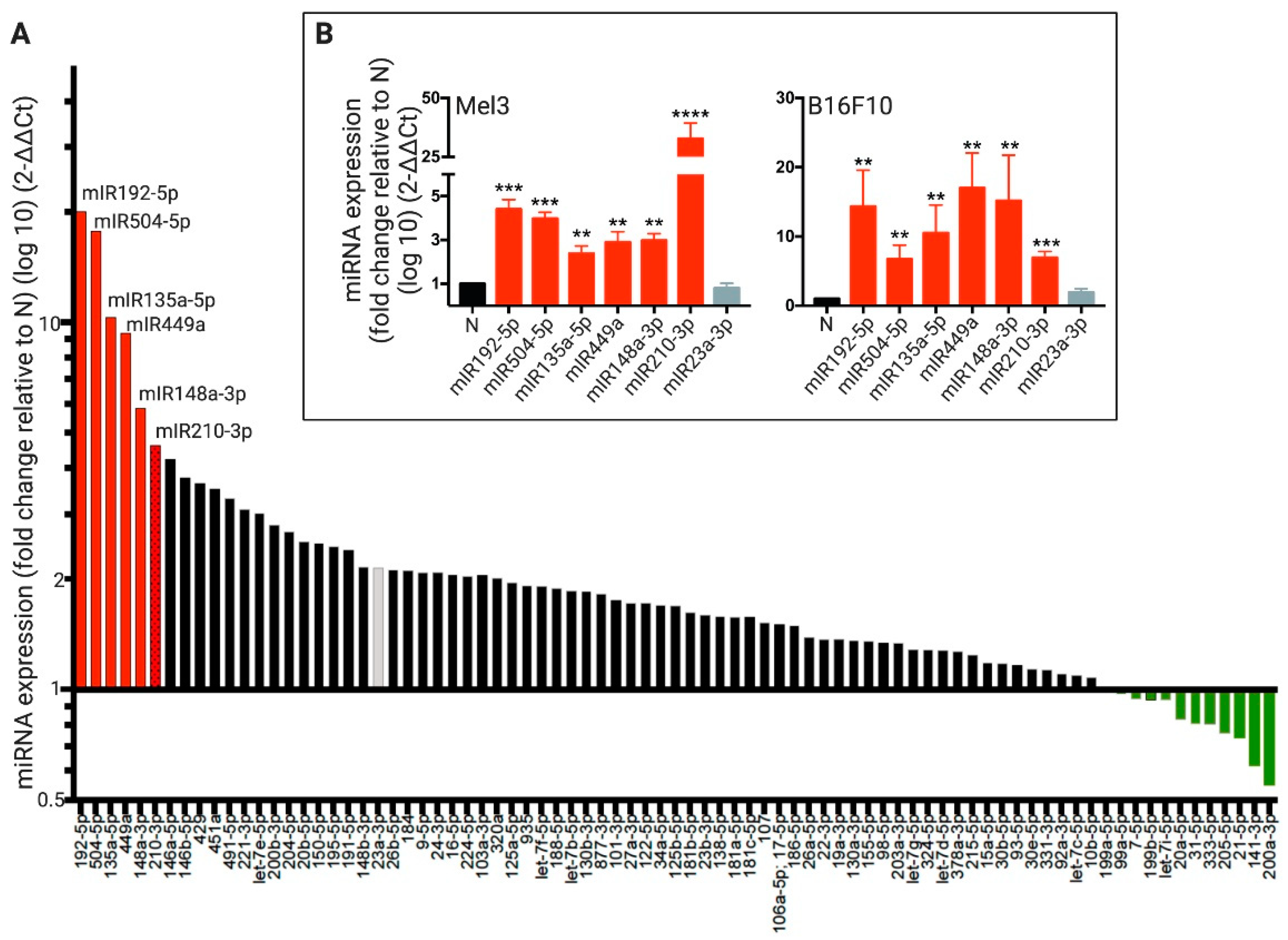
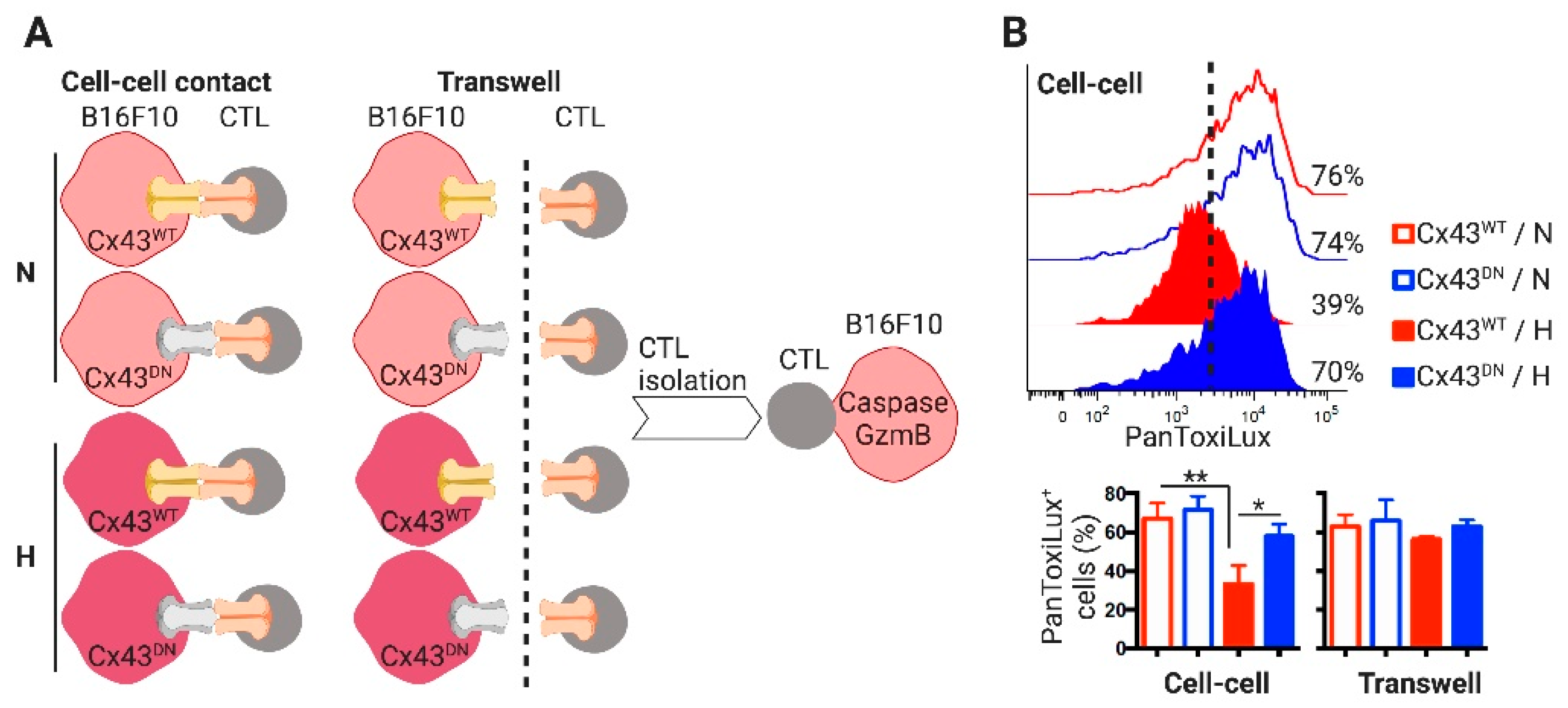
2. Results
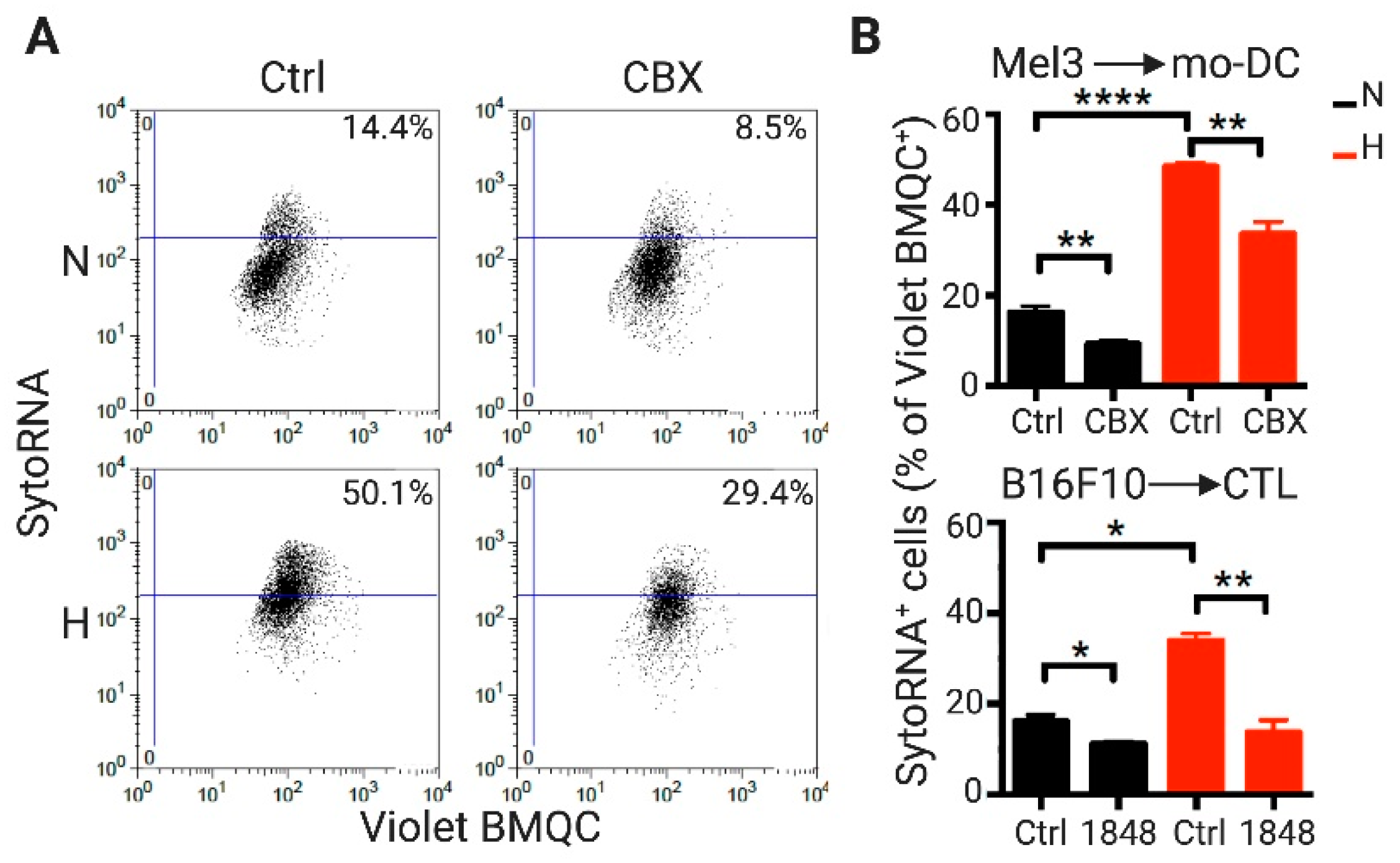
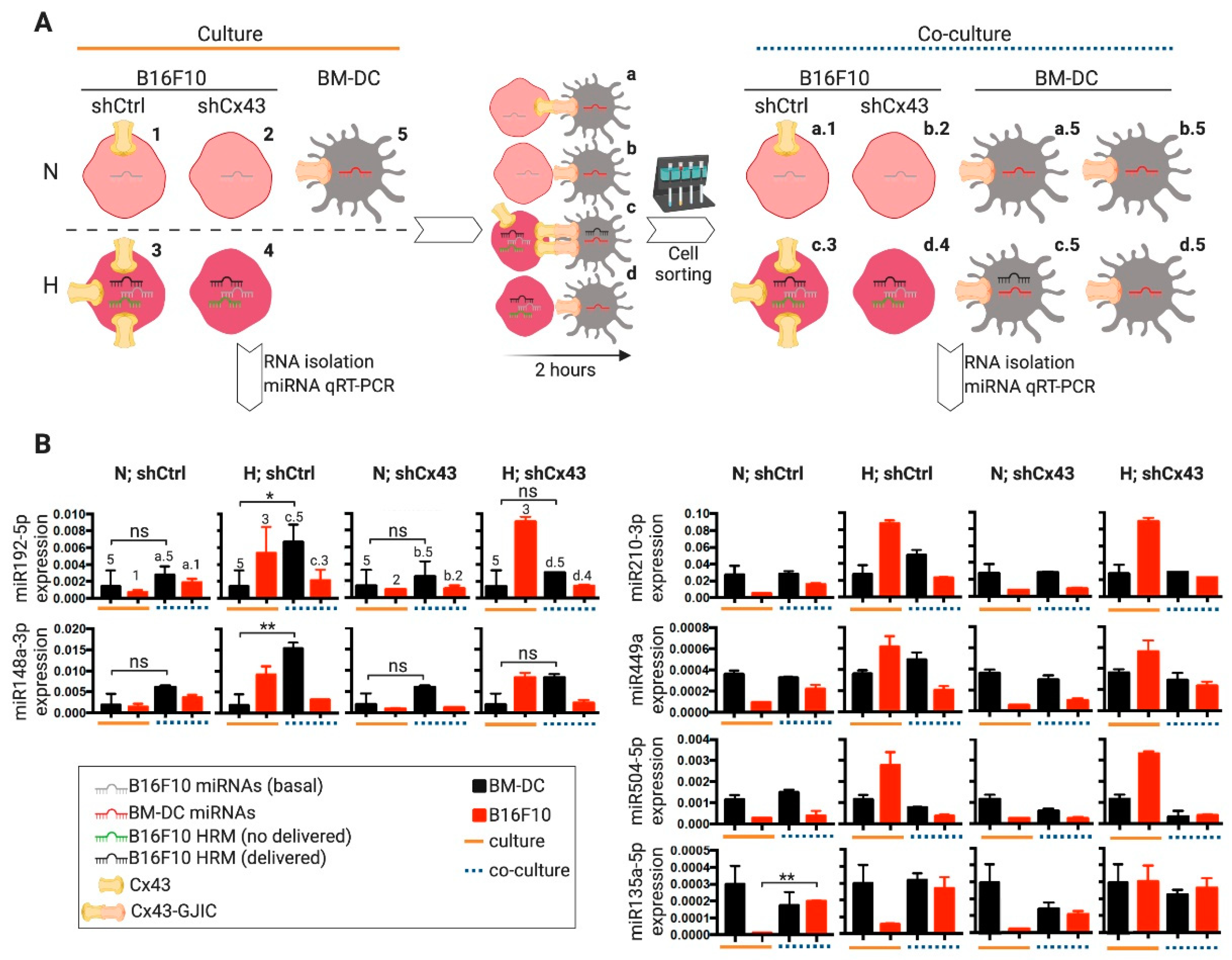
2.1. DCs and CTLs Acquire RNA Molecules from Hypoxic Melanoma Cells through a Cx43-GJ-Dependent Mechanism
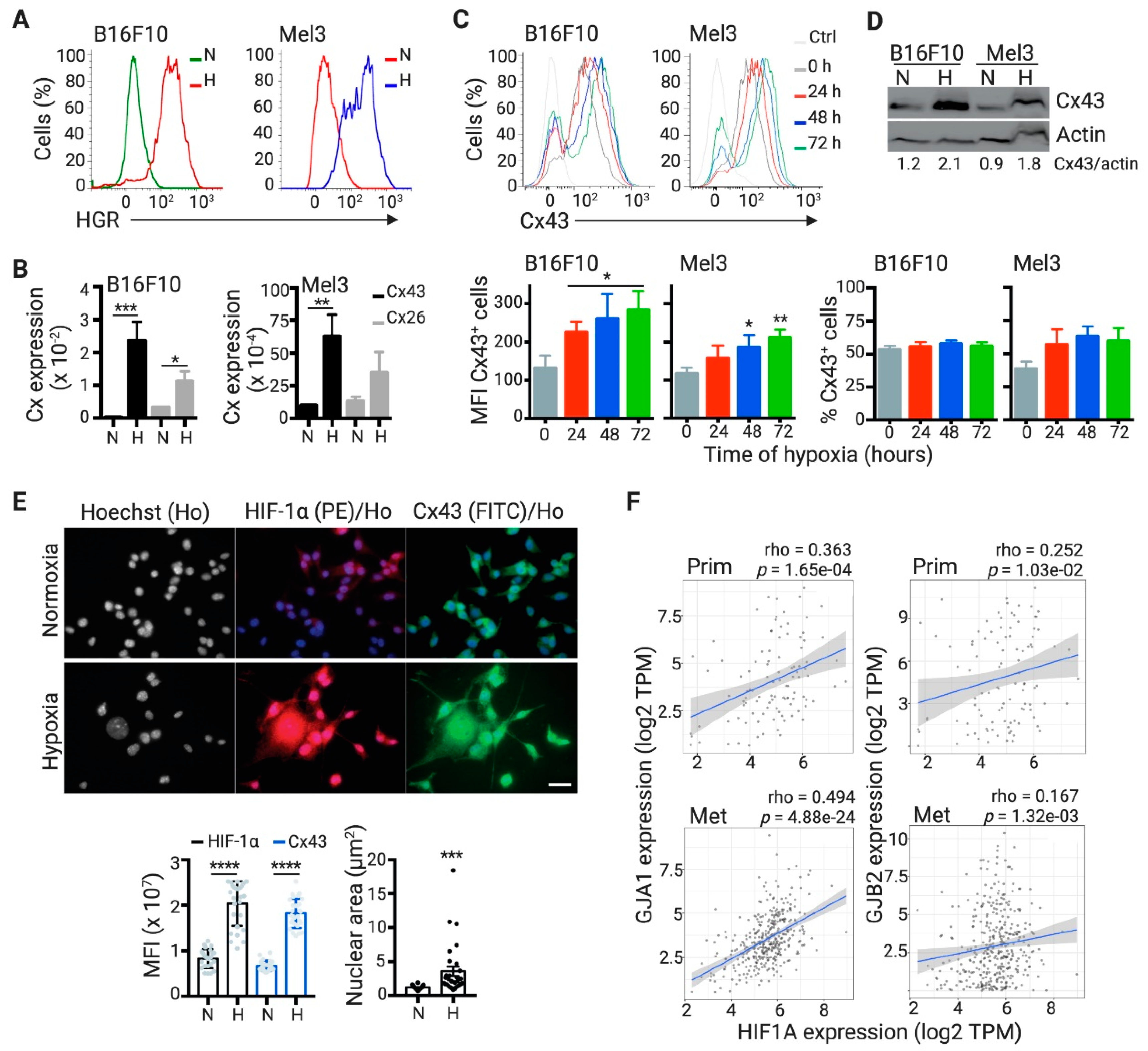
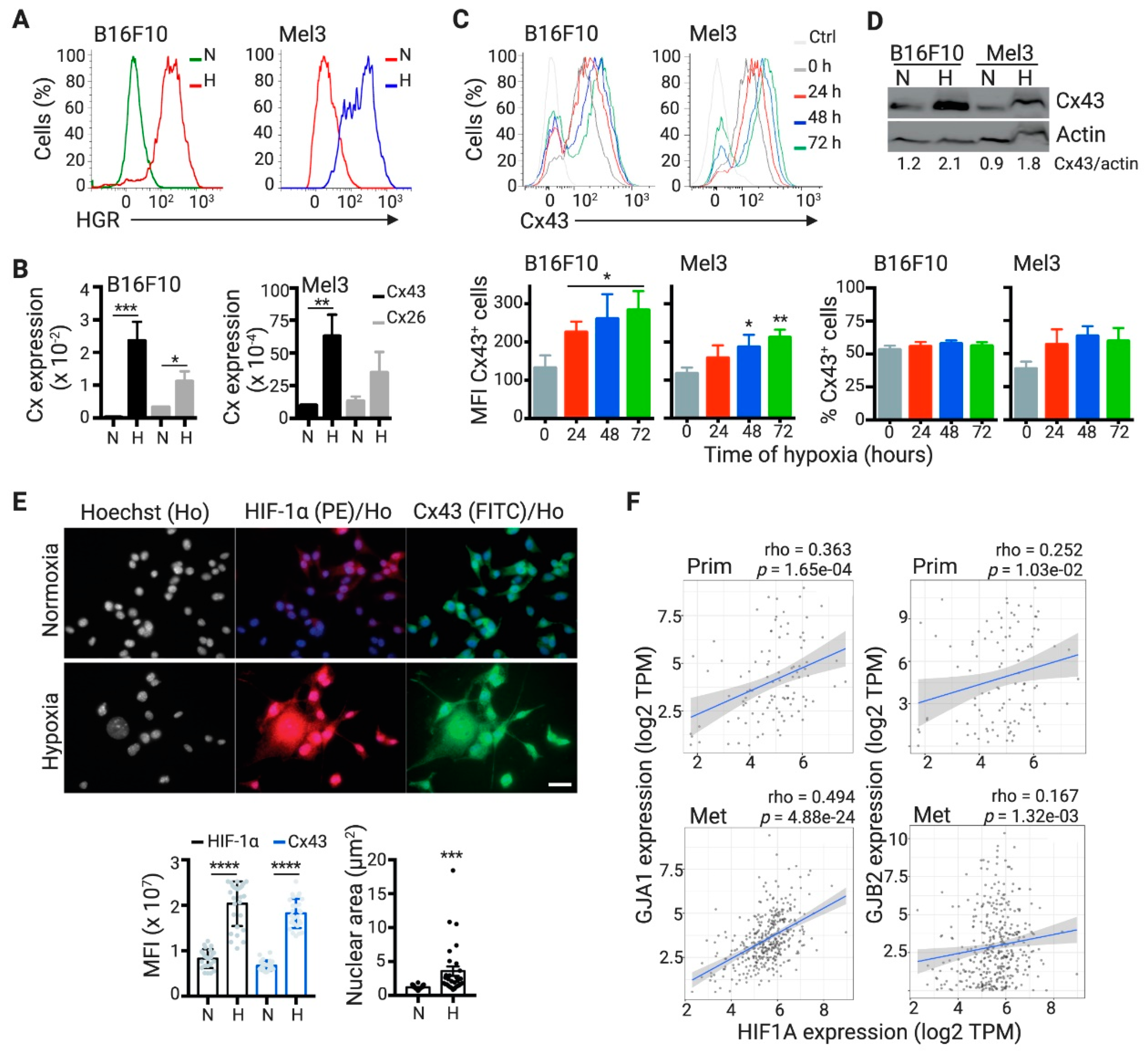
2.2. DCs Acquire Hypoxia-Induced miRNAs from Melanoma Cells by a Cx43-Dependent Mechanism
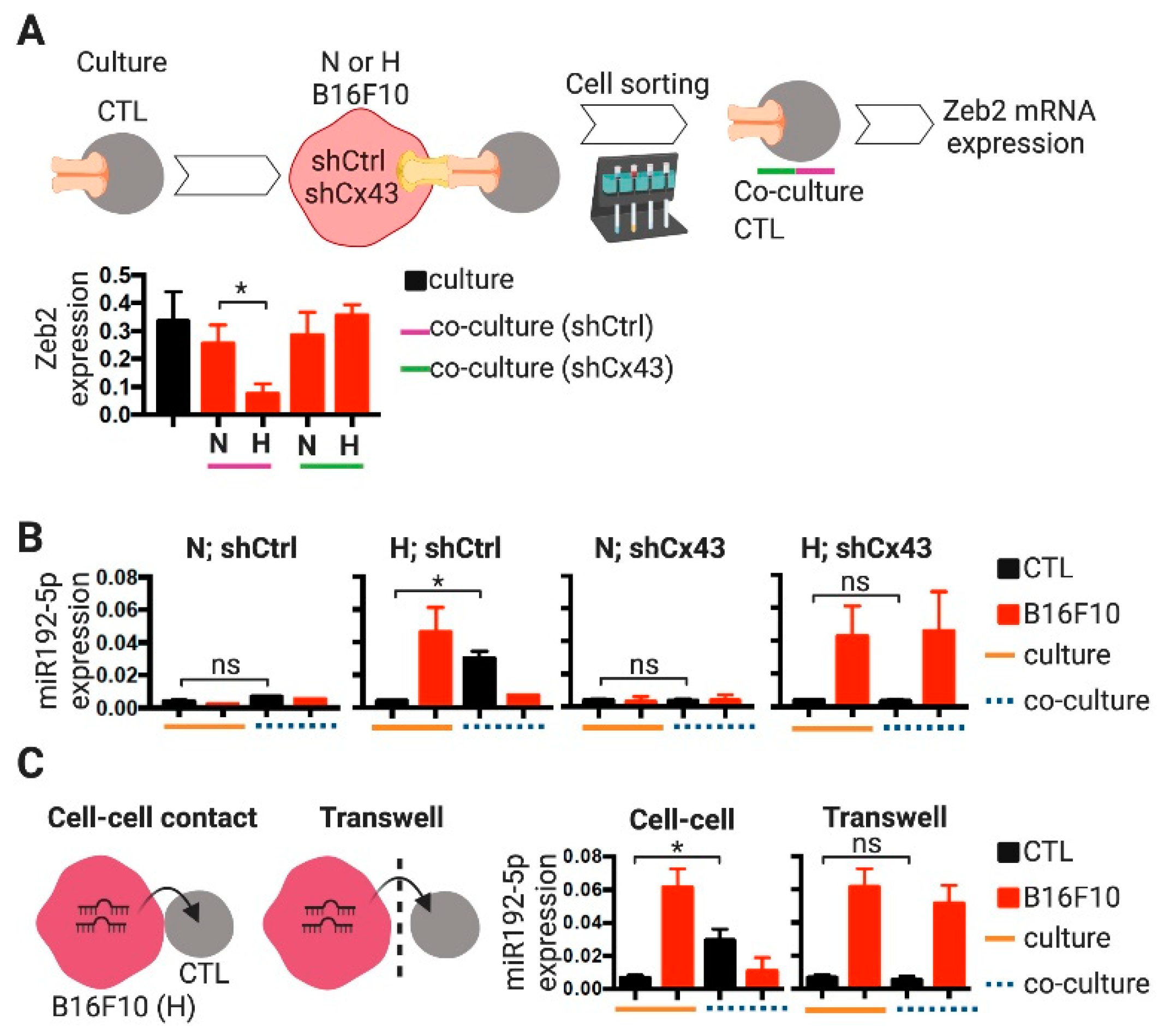
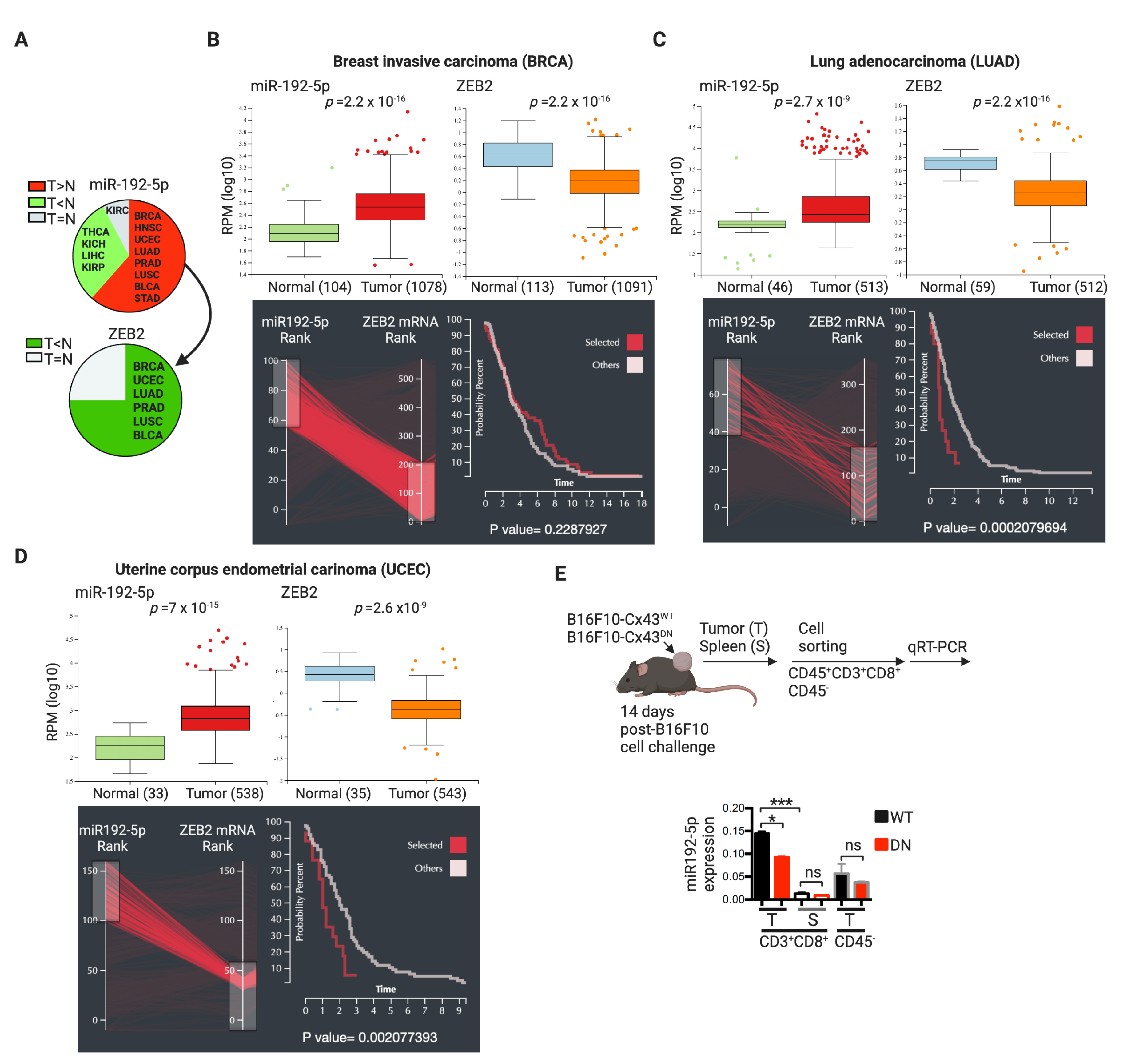
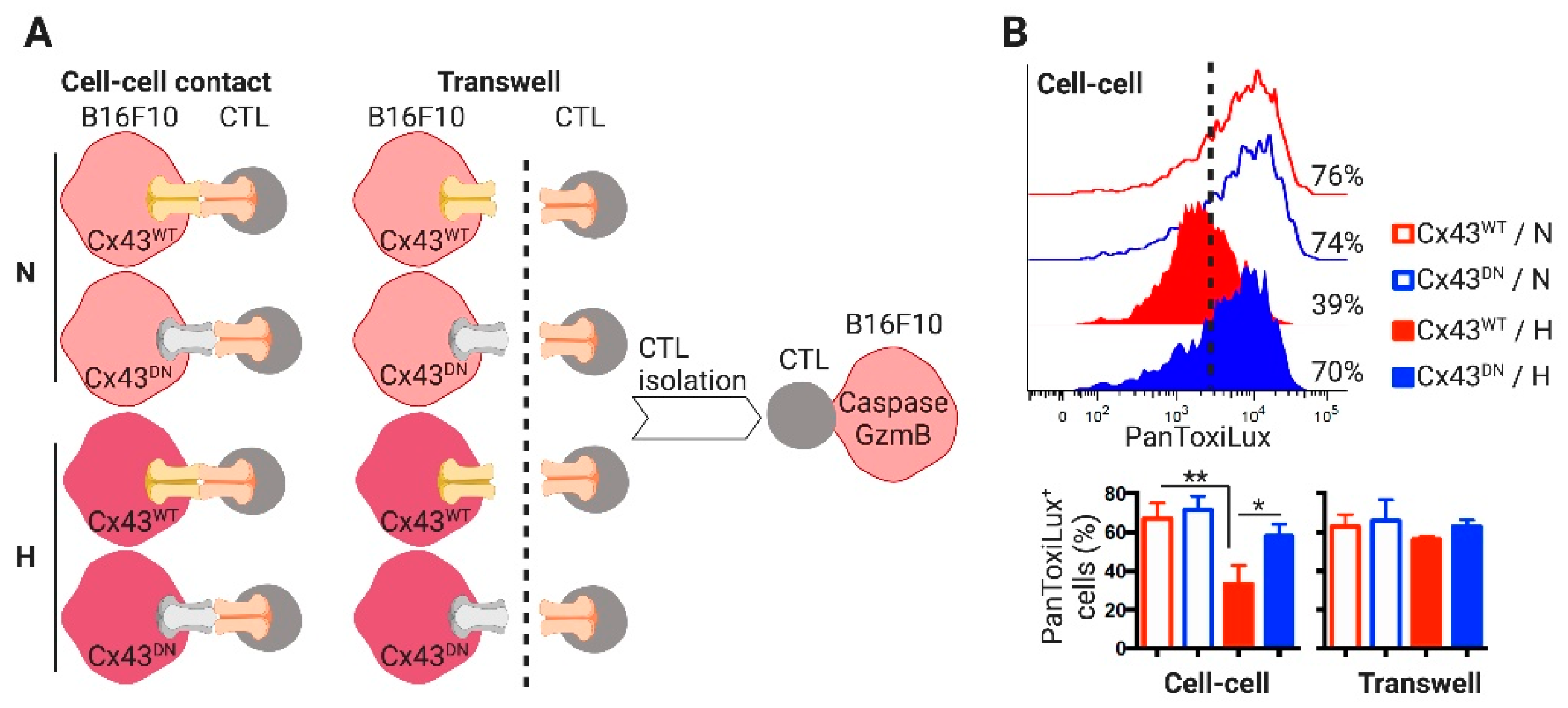
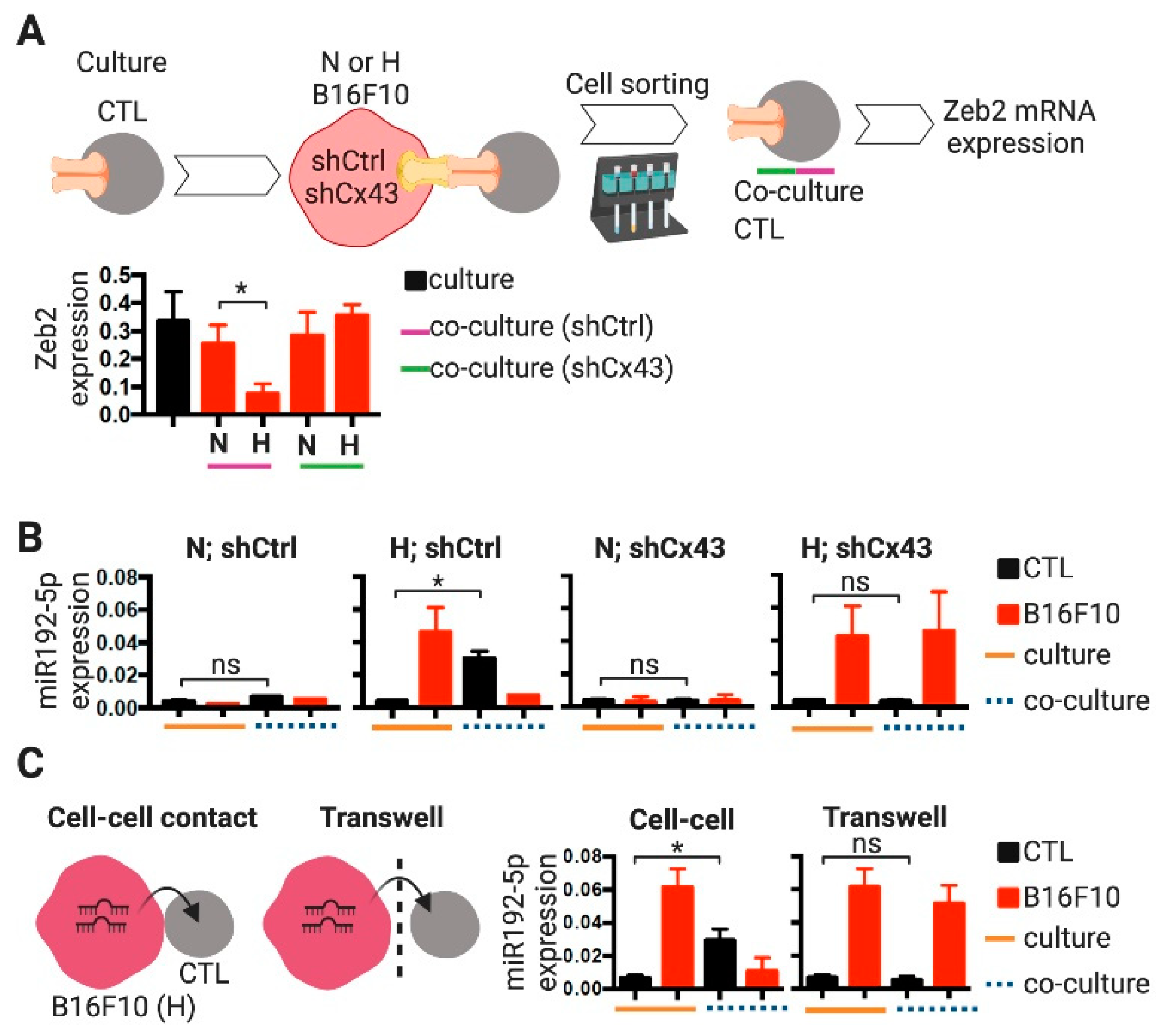
2.3. CTLs Acquire miR-192-5p from Hypoxic Melanoma Cells by a Cell Contact and Cx43-Dependent Mechanism
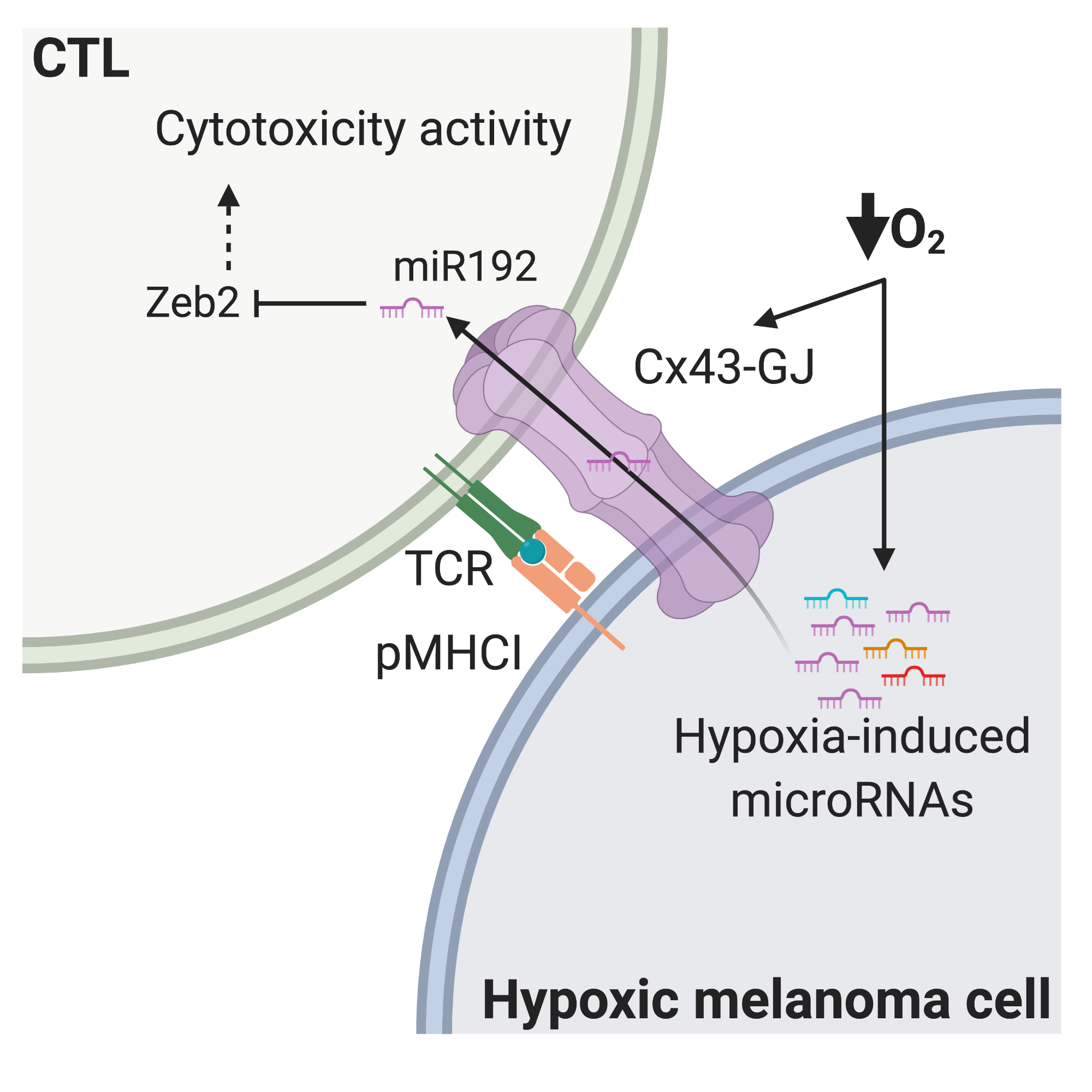
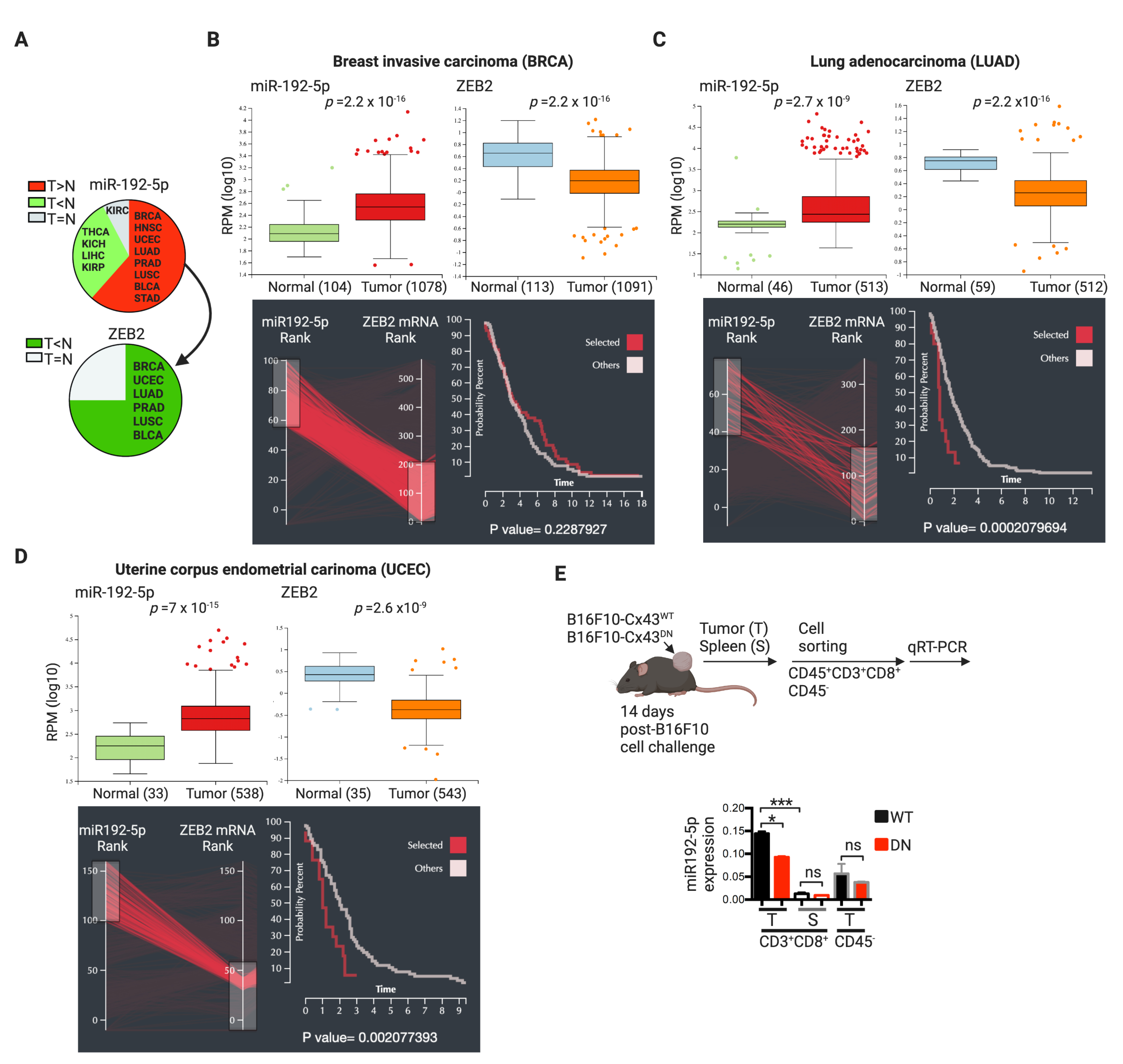
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cell Lines
4.3. Hypoxia Treatments
4.4. pMEL-1 CTL Differentiation
4.5. moDC and BM-DC Differentiation
4.6. Total RNA Transfer Assay
4.7. miRNA Transfer Assays
4.8. Pantoxilux Cytotoxic Assay
4.9. Flow Cytometry
4.10. Microscopy
4.11. Transfections
4.12. qRT-PCR
4.13. Western Blot
4.14. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BM-DC | Bone marrow-derived DC |
| CBX | Carbenoxolone |
| cGAMP | Cyclic guanosine monophosphate-adenosine monophosphate |
| CTL | Cytotoxic T lymphocyte |
| Cx | Connexin |
| DC | Dendritic cell |
| EV | Extracellular vesicles |
| H | Hypoxia |
| HGR | Hypoxia green reagent |
| HIF | Hypoxia induced factor |
| HRM | Hypoxia responsive miRNA |
| GJ | Gap junction |
| GJA1 | Gap junction protein alpha 1 (Cx43) |
| GJB2 | Gap junction protein beta 2 (Cx26) |
| GJIC | Gap junction-mediated intercellular communication |
| GZM | Granzyme |
| MFI | Mean fluorescence intensity |
| miRNA | microRNA |
| mo-DC | Monocyte-derived DC |
| N | Normoxia |
| NK | Natural killer |
| PD-1 | Programmed death-1 |
| PD-L1 | PD-ligand 1 |
| pMHC | Major histocompatibility-antigen peptide complex |
| shRNA | Short hairpin RNA |
| TAA | Tumor associated antigen |
| TCGA | The cancer genome atlas |
| TCR | T cell receptor |
| TME | Tumor microenvironment |
References
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yatim, N.; Cullen, S.; Albert, M.L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; Long, E.O. Cytotoxic immunological synapses. Immunol. Rev. 2010, 235, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Orlowski, R.J.; Xu, X.; Mick, R.; George, S.M.; Yan, P.K.; Manne, S.; Kraya, A.A.; Wubbenhorst, B.; Dorfman, L.; et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med. 2019, 25, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Chouaib, S.; Janji, B.; Tittarelli, A.; Eggermont, A.; Thiery, J.P. Tumor plasticity interferes with anti-tumor immunity. Crit. Rev. Immunol. 2014, 34, 91–102. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Najjar, Y.G.; Menk, A.V.; Sander, C.; Rao, U.; Karunamurthy, A.; Bhatia, R.; Zhai, S.; Kirkwood, J.M.; Delgoffe, G.M. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight. 2019, 4, e124989. [Google Scholar] [CrossRef]
- Omar, H.A.; El-Serafi, A.T.; Hersi, F.; Arafa, E.A.; Zaher, D.M.; Madkour, M.; Arab, H.H.; Tolba, M.F. Immunomodulatory MicroRNAs in cancer: Targeting immune checkpoints and the tumor microenvironment. FEBS J. 2019, 286, 3540–3557. [Google Scholar] [CrossRef] [Green Version]
- Romano, G.; Kwong, L.N. miRNAs, Melanoma and Microenvironment: An Intricate Network. Int. J. Mol. Sci. 2017, 18, 2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Buart, S.; Romero, P.; Ketari, S.; Janji, B.; Mari, B.; Mami-Chouaib, F.; Chouaib, S. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res. 2012, 72, 4629–4641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlhapp, F.J.; Mitra, A.K.; Lengyel, E.; Peter, M.E. MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 2015, 34, 5857–5868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichmüller, S.B.; Osen, W.; Mandelboim, O.; Seliger, B. Immune Modulatory microRNAs Involved in Tumor Attack and Tumor Immune Escape. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le Cam, E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-β and miR23a transfer. Oncoimmunology 2015, 5, e1062968. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.L.; Hung, J.Y.; Chang, W.A.; Jian, S.F.; Lin, Y.S.; Pan, Y.C.; Wu, C.Y.; Kuo, P.L. Hypoxic Lung-Cancer-Derived Extracellular Vesicle MicroRNA-103a Increases the Oncogenic Effects of Macrophages by Targeting PTEN. Mol. Ther. 2018, 26, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Lemcke, H.; Steinhoff, G.; David, R. Gap junctional shuttling of miRNA--A novel pathway of intercellular gene regulation and its prospects in clinical application. Cell Signal. 2015, 27, 2506–2514. [Google Scholar] [CrossRef]
- Lemcke, H.; David, R. Potential mechanisms of microRNA mobility. Traffic 2018, 19, 910–917. [Google Scholar] [CrossRef]
- Gleisner, M.A.; Navarrete, M.; Hofmann, F.; Salazar-Onfray, F.; Tittarelli, A. Mind the Gaps in Tumor Immunity: Impact of Connexin-Mediated Intercellular Connections. Front. Immunol. 2017, 8, 1067. [Google Scholar] [CrossRef] [Green Version]
- Elgueta, R.; Tobar, J.A.; Shoji, K.F.; De Calisto, J.; Kalergis, A.M.; Bono, M.R.; Rosemblatt, M.; Saez, J.C. Gap junctions at the dendritic cell-T cell interface are key elements for antigen-dependent T cell activation. J. Immunol. 2009, 183, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Mendoza-Naranjo, A.; Bouma, G.; Pereda, C.; Ramírez, M.; Webb, K.F.; Tittarelli, A.; López, M.N.; Kalergis, A.M.; Thrasher, A.J.; Becker, D.L.; et al. Functional gap junctions accumulate at the immunological synapse and contribute to T cell activation. J. Immunol. 2011, 187, 3121–3132. [Google Scholar] [CrossRef]
- Tittarelli, A.; Mendoza-Naranjo, A.; Farías, M.; Guerrero, I.; Ihara, F.; Wennerberg, E.; Riquelme, S.; Gleisner, A.; Kalergis, A.; Lundqvist, A.; et al. Gap junction intercellular communications regulate NK cell activation and modulate NK cytotoxic capacity. J. Immunol. 2014, 192, 1313–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tittarelli, A.; Navarrete, M.; Gleisner, M.A.; Gebicke-Haerter, P.; Salazar-Onfray, F. Connexin-Mediated Signaling at the Immunological Synapse. Int. J. Mol. Sci. 2020, 21, 3736. [Google Scholar] [CrossRef] [PubMed]
- Tittarelli, A.; Janji, B.; Van Moer, K.; Noman, M.Z.; Chouaib, S. The selective degradation of synaptic connexin 43 protein by hypoxia-induced autophagy impairs natural killer cell-mediated tumor cell killing. J. Biol. Chem. 2015, 290, 23670–23679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, F.; Navarrete, M.; Álvarez, J.; Guerrero, I.; Gleisner, M.A.; Tittarelli, A.; Salazar-Onfray, F. Cx43-Gap Junctions Accumulate at the Cytotoxic Immunological Synapse Enabling Cytotoxic T Lymphocyte Melanoma Cell Killing. Int. J. Mol. Sci. 2019, 20, 4509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccheri, F.; Pozzi, C.; Avogadri, F.; Barozzi, S.; Faretta, M.; Fusi, P.; Rescigno, M. Bacteria-induced gap junctions in tumors favor antigen cross-presentation and antitumor immunity. Sci. Transl. Med. 2010, 2, 44ra57. [Google Scholar] [CrossRef] [PubMed]
- Kiszner, G.; Balla, P.; Wichmann, B.; Barna, G.; Baghy, K.; Nemeth, I.B.; Varga, E.; Furi, I.; Toth, B.; Krenacs, T. Exploring Differential Connexin Expression across Melanocytic Tumor Progression Involving the Tumor Microenvironment. Cancers 2019, 11, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Sánchez, L.M.; Jimenez, C.; Valverde, A.; Hernandez, V.; Peñarando, J.; Martinez, A.; Lopez-Pedrera, C.; Muñoz-Castañeda, J.R.; De la Haba-Rodríguez, J.R.; Aranda, E.; et al. CoCl2, a mimic of hypoxia, induces formation of polyploid giant cells with stem characteristics in colon cancer. PLoS ONE 2014, 9, e99143. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.J.; Deng, Q.; Hu, D.P.; Wang, Z.Y.; Huang, B.H.; Du, Z.Y.; Fang, Y.X.; Wong, W.L.; Zhang, K.; Chow, C.F. A molecular fluorescent dye for specific staining and imaging of RNA in live cells: A novel ligand integration from classical thiazole orange and styryl compounds. Chem. Commun. (Camb). 2015, 51, 15241–15244. [Google Scholar] [CrossRef]
- Mendoza-Naranjo, A.; Sáez, P.J.; Johansson, C.C.; Ramírez, M.; Mandakovic, D.; Pereda, C.; López, M.N.; Kiessling, R.; Sáez, J.C.; Salazar-Onfray, F. Functional gap junctions facilitate melanoma antigen transfer and cross-presentation between human dendritic cells. J. Immunol. 2007, 178, 6949–6957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Wang, X.; Guo, Y.; Peng, F.; Zheng, N.; He, B.; Ge, H.; Tao, L.; Wang, Q. Pattern of cell-to-cell transfer of microRNA by gap junction and its effect on the proliferation of glioma cells. Cancer Sci. 2019, 110, 1947–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Le, Q.T.; Giaccia, A.J. MiR-210-micromanager of the hypoxia pathway. Trends Mol. Med. 2010, 16, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.Y.; Lin, Y.C.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Zhang, J.; Wang, M.; Lanting, L.; Yuan, H.; Rossi, J.J.; Natarajan, R. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc. Natl. Acad. Sci. USA 2007, 104, 3432–3437. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.L.; Omilusik, K.D. ZEBs: Novel Players in Immune Cell Development and Function. Trends Immunol. 2019, 40, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Omilusik, K.D.; Best, J.A.; Yu, B.; Goossens, S.; Weidemann, A.; Nguyen, J.V.; Seuntjens, E.; Stryjewska, A.; Zweier, C.; Roychoudhuri, R.; et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J. Exp. Med. 2015, 212, 2027–2039. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.X.; Amezquita, R.A.; Guan, T.; Marshall, H.D.; Joshi, N.S.; Kleinstein, S.H.; Kaech, S.M. The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J. Exp. Med. 2015, 212, 2041–2056. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhan, Z.; Xu, L.; Ma, F.; Li, D.; Guo, Z.; Li, N.; Cao, X. MicroRNA-148/152 impairs innate response and antigen presentation of TLR-triggered dendritic cells by targeting CaMKIIα. J. Immunol. 2010, 185, 7244–7251. [Google Scholar] [CrossRef]
- Wu, C.; Chen, W.; He, J.; Jin, S.; Liu, Y.; Yi, Y.; Gao, Z.; Yang, J.; Yang, J.; Cui, J.; et al. Interplay of m6A and H3K27 trimethylation restrains inflammation during bacterial infection. Sci. Adv. 2020, 6, eaba0647. [Google Scholar] [CrossRef]
- Gallo, R.M.; Khan, M.A.; Shi, J.; Kapur, R.; Wei, L.; Bailey, J.C.; Liu, J.; Brutkiewicz, R.R. Regulation of the actin cytoskeleton by Rho kinase controls antigen presentation by CD1d. J. Immunol. 2012, 189, 1689–1698. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yi, H.; Wang, C.; He, H.; Li, P.; Pan, H.; Sheng, N.; Ji, M.; Cai, L.; Ma, Y. Integrated Nanovaccine with MicroRNA-148a Inhibition Reprograms Tumor-Associated Dendritic Cells by Modulating miR-148a/DNMT1/SOCS1 Axis. J. Immunol. 2016, 197, 1231–1241. [Google Scholar] [CrossRef] [Green Version]
- Benitez-Ribas, D.; Adema, G.J.; Winkels, G.; Klasen, I.S.; Punt, C.J.; Figdor, C.G.; de Vries, I.J. Plasmacytoid dendritic cells of melanoma patients present exogenous proteins to CD4+ T cells after Fc gamma RII-mediated uptake. J. Exp. Med. 2006, 203, 1629–1635. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, A.; Hester, J.; Macklin, P.S.; Kawai, K.; Uchiyama, M.; Biggs, D.; Bishop, T.; Bull, K.; Cheng, X.; Cawthorne, E.; et al. Systemic silencing of PHD2 causes reversible immune regulatory dysfunction. J. Clin. Investig. 2019, 129, 3640–3656. [Google Scholar] [CrossRef]
- Soares, A.R.; Martins-Marques, T.; Ribeiro-Rodrigues, T.; Ferreira, J.V.; Catarino, S.; Pinho, M.J.; Zuzarte, M.; Isabel Anjo, S.; Manadas, B.; Sluijter, J.; et al. Gap junctional protein Cx43 is involved in the communication between extracellular vesicles and mammalian cells. Sci. Rep. 2015, 5, 13243. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.Y.; Lin, W.C. miR-TV: An interactive microRNA Target Viewer for microRNA and target gene expression interrogation for human cancer studies. Database (Oxford) 2020, 2020, baz148. [Google Scholar] [CrossRef] [Green Version]
- Zucker, S.N.; Bancroft, T.A.; Place, D.E.; Des Soye, B.; Bagati, A.; Berezney, R. A dominant negative Cx43 mutant differentially affects tumorigenic and invasive properties in human metastatic melanoma cells. J. Cell. Physiol. 2013, 228, 853–859. [Google Scholar] [CrossRef]
- Betof Warner, A.; Palmer, J.S.; Shoushtari, A.N.; Goldman, D.A.; Panageas, K.S.; Hayes, S.A.; Bajwa, R.; Momtaz, P.; Callahan, M.K.; Wolchok, J.D.; et al. Long-Term Outcomes and Responses to Retreatment in Patients with Melanoma Treated With PD-1 Blockade. J. Clin. Oncol. 2020, 38, 1655–1663. [Google Scholar] [CrossRef]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef]
- Kanno, Y.; Loewenstein, W.R. Cell-to-cell passage of large molecules. Nature 1966, 212, 629–630. [Google Scholar] [CrossRef]
- Alaga, K.C.; Crawford, M.; Dagnino, L.; Laird, D.W. Aberrant Cx43 Expression and Mislocalization in Metastatic Human Melanomas. J. Cancer 2017, 8, 1123–1128. [Google Scholar] [CrossRef] [Green Version]
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 2019, 29, 1236–1248. [Google Scholar] [CrossRef] [Green Version]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–1498. [Google Scholar] [CrossRef]
- Bohuslavova, R.; Cerychova, R.; Papousek, F.; Olejnickova, V.; Bartos, M.; Görlach, A.; Kolar, F.; Sedmera, D.; Semenza, G.L.; Pavlinkova, G. HIF-1α is required for development of the sympathetic nervous system. Proc. Natl. Acad. Sci. USA 2019, 116, 13414–13423. [Google Scholar] [CrossRef] [Green Version]
- Dovmark, T.H.; Saccomano, M.; Hulikova, A.; Alves, F.; Swietach, P. Connexin-43 channels are a pathway for discharging lactate from glycolytic pancreatic ductal adenocarcinoma cells. Oncogene 2017, 36, 4538–4550. [Google Scholar] [CrossRef]
- Dovmark, T.H.; Hulikova, A.; Niederer, S.A.; Vaughan-Jones, R.D.; Swietach, P. Normoxic cells remotely regulate the acid-base balance of cells at the hypoxic core of connexin-coupled tumor growths. FASEB J. 2018, 32, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Valiunas, V.; Polosina, Y.Y.; Miller, H.; Potapova, I.A.; Valiuniene, L.; Doronin, S.; Mathias, R.T.; Robinson, R.B.; Rosen, M.R.; Cohen, I.S.; et al. Connexin-specific cell-to-cell transfer of short interfering RNA by gap junctions. J. Physiol. 2005, 568, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Wolvetang, E.J.; Pera, M.F.; Zuckerman, K.S. Gap junction mediated transport of shRNA between human embryonic stem cells. Biochem. Biophys. Res. Commun. 2007, 363, 610–615. [Google Scholar] [CrossRef]
- Katakowski, M.; Buller, B.; Wang, X.; Rogers, T.; Chopp, M. Functional microRNA is transferred between glioma cells. Cancer Res. 2010, 70, 8259–8263. [Google Scholar] [CrossRef] [Green Version]
- Zong, L.; Zhu, Y.; Liang, R.; Zhao, H.B. Gap junction mediated miRNA intercellular transfer and gene regulation: A novel mechanism for intercellular genetic communication. Sci. Rep. 2016, 27, 19884. [Google Scholar] [CrossRef] [Green Version]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [Green Version]
- Aucher, A.; Rudnicka, D.; Davis, D.M. MicroRNAs transfer from human macrophages to hepato-carcinoma cells and inhibit proliferation. J. Immunol. 2013, 191, 6250–6260. [Google Scholar] [CrossRef]
- Segura, M.F.; Greenwald, H.S.; Hanniford, D.; Osman, I.; Hernando, E. MicroRNA and cutaneous melanoma: From discovery to prognosis and therapy. Carcinogenesis 2012, 33, 1823–1832. [Google Scholar] [CrossRef] [Green Version]
- Walbrecq, G.; Lecha, O.; Gaigneaux, A.; Fougeras, M.R.; Philippidou, D.; Margue, C.; Tetsi Nomigni, M.; Bernardin, F.; Dittmar, G.; Behrmann, I.; et al. Hypoxia-Induced Adaptations of miRNomes and Proteomes in Melanoma Cells and Their Secreted Extracellular Vesicles. Cancers (Basel) 2020, 12, 692. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.Z.; Cheng, W.C.; Chen, S.F.; Nieh, S.; O’Connor, C.; Liu, C.L.; Tsai, W.W.; Wu, C.J.; Martin, L.; Lin, Y.S.; et al. miR-25/93 mediates hypoxia-induced immunosuppression by repressing cGAS. Nat. Cell. Biol. 2017, 19, 1286–1296. [Google Scholar] [CrossRef] [Green Version]
- Martins-Marques, T.; Pinho, M.J.; Zuzarte, M.; Oliveira, C.; Pereira, P.; Sluijter, J.P.; Gomes, C.; Girao, H. Presence of Cx43 in extracellular vesicles reduces the cardiotoxicity of the anti-tumour therapeutic approach with doxorubicin. J. Extracell. Vesicles 2016, 5, 32538. [Google Scholar] [CrossRef]
- Varela-Eirin, M.; Varela-Vazquez, A.; Rodríguez-Candela Mateos, M.; Vila-Sanjurjo, A.; Fonseca, E.; Mascareñas, J.L.; Eugenio Vázquez, M.; Mayan, M.D. Recruitment of RNA molecules by connexin RNA-binding motifs: Implication in RNA and DNA transport through microvesicles and exosomes. Biochim. Biophys. Acta. Mol. Cell. Res. 2017, 1864, 728–736. [Google Scholar] [CrossRef]
- Banerjee, D.; Gakhar, G.; Madgwick, D.; Hurt, A.; Takemoto, D.; Nguyen, T.A. A novel role of gap junction connexin46 protein to protect breast tumors from hypoxia. Int. J. Cancer 2010, 127, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Acuña, R.A.; Varas-Godoy, M.; Berthoud, V.M.; Alfaro, I.E.; Retamal, M.A. Connexin-46 Contained in Extracellular Vesicles Enhance Malignancy Features in Breast Cancer Cells. Biomolecules 2020, 10, 676. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Kouwaki, T.; Oshiumi, H. Aging-Associated Extracellular Vesicles Contain Immune Regulatory microRNAs Alleviating Hyperinflammatory State and Immune Dysfunction in the Elderly. iScience. 2020, 23, 101520. [Google Scholar] [CrossRef]
- Zhang, D.; Wu, Y.; Sun, G. miR-192 suppresses T follicular helper cell differentiation by targeting CXCR5 in childhood asthma. Scand. J. Clin. Lab. Investig. 2018, 78, 236–242. [Google Scholar] [CrossRef]
- Overwijk, W.W.; Theoret, M.R.; Finkelstein, S.E.; Surman, D.R.; De Jong, L.A.; Vyth-Dreese, F.A.; Dellemijn, T.A.; Antony, P.A.; Spiess, P.J.; Palmer, D.C.; et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med. 2003, 198, 569–580. [Google Scholar] [CrossRef]
- Aguilera, R.; Saffie, C.; Tittarelli, A.; González, F.E.; Ramírez, M.; Reyes, D.; Pereda, C.; Hevia, D.; García, T.; Salazar, L.; et al. Heat-shock induction of tumor-derived danger signals mediates rapid monocyte differentiation into clinically effective dendritic cells. Clin. Cancer Res. 2011, 17, 2474–2483. [Google Scholar] [CrossRef] [Green Version]
- Gleisner, M.A.; Pereda, C.; Tittarelli, A.; Navarrete, M.; Fuentes, C.; Ávalos, I.; Tempio, F.; Araya, J.P.; Becker, M.I.; González, F.E.; et al. A heat-shocked melanoma cell lysate vaccine enhances tumor infiltration by prototypic effector T cells inhibiting tumor growth. J. Immunother. Cancer 2020, 8, e000999. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target 1 | Description | Relevant Immune Role | Ref. |
|---|---|---|---|
| miR-192-5p | |||
| ZEB2 | Zinc finger E-box binding homeobox 2 | Effector T cell and DC differentiation and function | [35] |
| miR-148a-3p | |||
| Camk2a | Calcium/calmodulin dependent protein kinase II alpha | DC maturation and function | [38] |
| Kdm6b | Lysine demethylase 6B | Proinflammatory cytokine production by DCs | [39] |
| Rock1 | Rho associated coiled-coil containing protein kinase 1 | DC antigen presentation | [40] |
| Dnmt1 | DNA methyltransferase 1 | Tumor associated-DC maturation | [41] |
| Fcgr2b | Fc fragment of IgG receptor IIb | Tumor antigen uptake and DC activation | [42] |
| Epas1 | Endothelial PAS domain protein 1 (HIF2A) | Regulatory T cell function | [43] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tittarelli, A.; Navarrete, M.; Lizana, M.; Hofmann-Vega, F.; Salazar-Onfray, F. Hypoxic Melanoma Cells Deliver microRNAs to Dendritic Cells and Cytotoxic T Lymphocytes through Connexin-43 Channels. Int. J. Mol. Sci. 2020, 21, 7567. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207567
Tittarelli A, Navarrete M, Lizana M, Hofmann-Vega F, Salazar-Onfray F. Hypoxic Melanoma Cells Deliver microRNAs to Dendritic Cells and Cytotoxic T Lymphocytes through Connexin-43 Channels. International Journal of Molecular Sciences. 2020; 21(20):7567. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207567
Chicago/Turabian StyleTittarelli, Andrés, Mariela Navarrete, Marcelo Lizana, Francisca Hofmann-Vega, and Flavio Salazar-Onfray. 2020. "Hypoxic Melanoma Cells Deliver microRNAs to Dendritic Cells and Cytotoxic T Lymphocytes through Connexin-43 Channels" International Journal of Molecular Sciences 21, no. 20: 7567. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207567