Serotonin Transporter and Plasma Membrane Monoamine Transporter Are Necessary for the Antidepressant-Like Effects of Ketamine in Mice


Abstract
:1. Introduction
2. Results
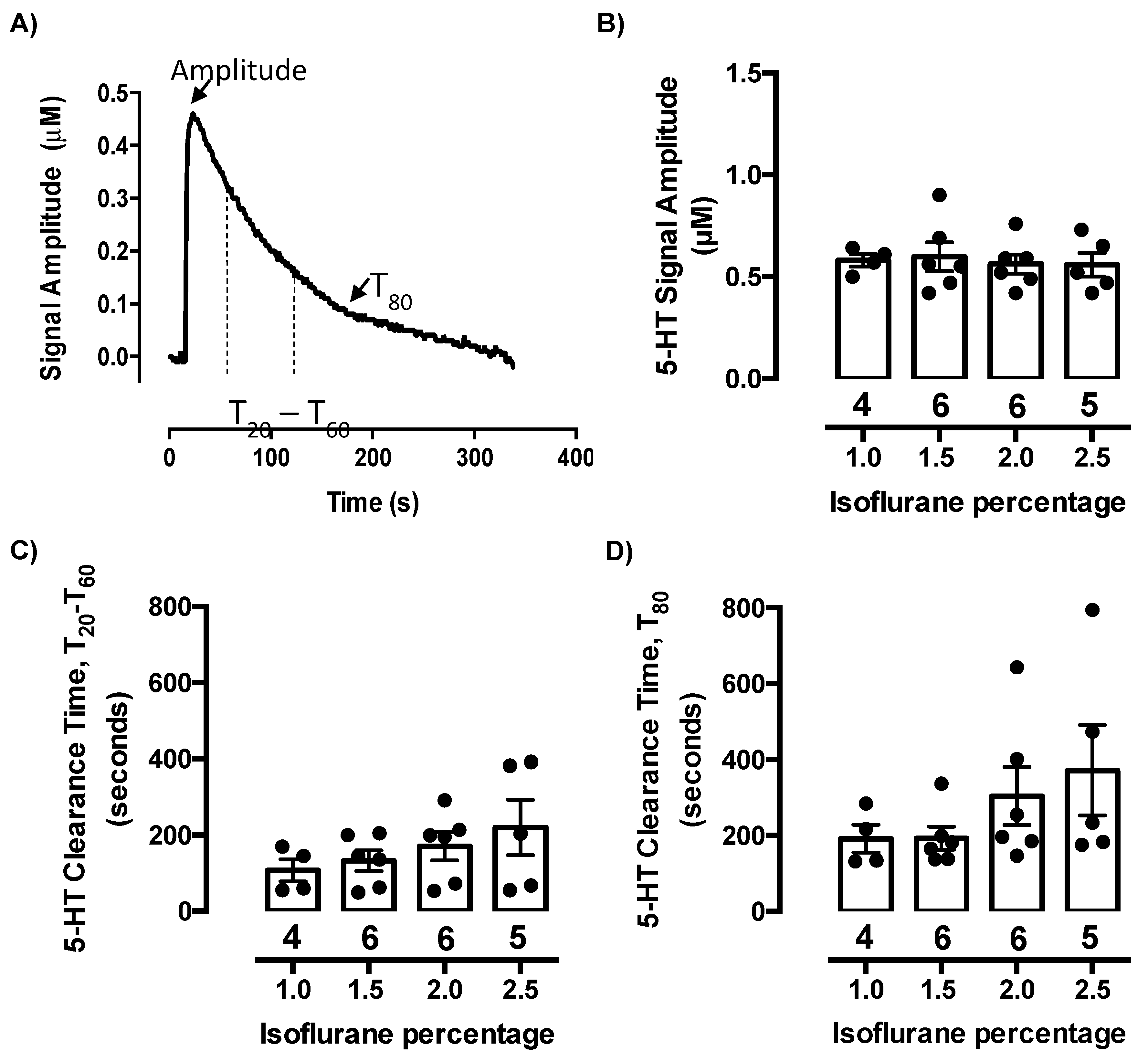
2.1. Isoflurane Does Not Evoke Serotonin Release or Inhibit Serotonin Clearance at Concentrations Needed to Maintain Anesthesia
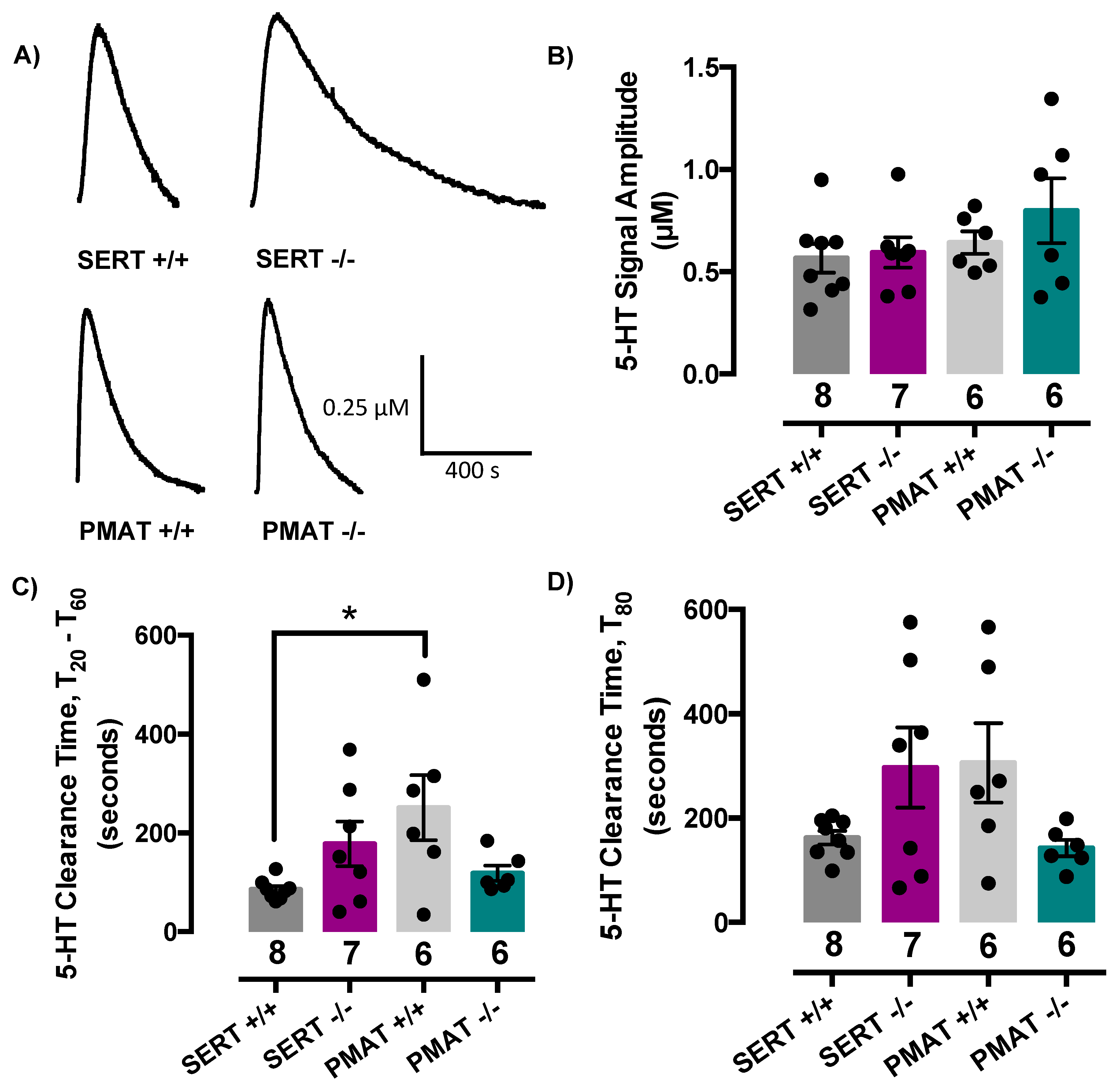
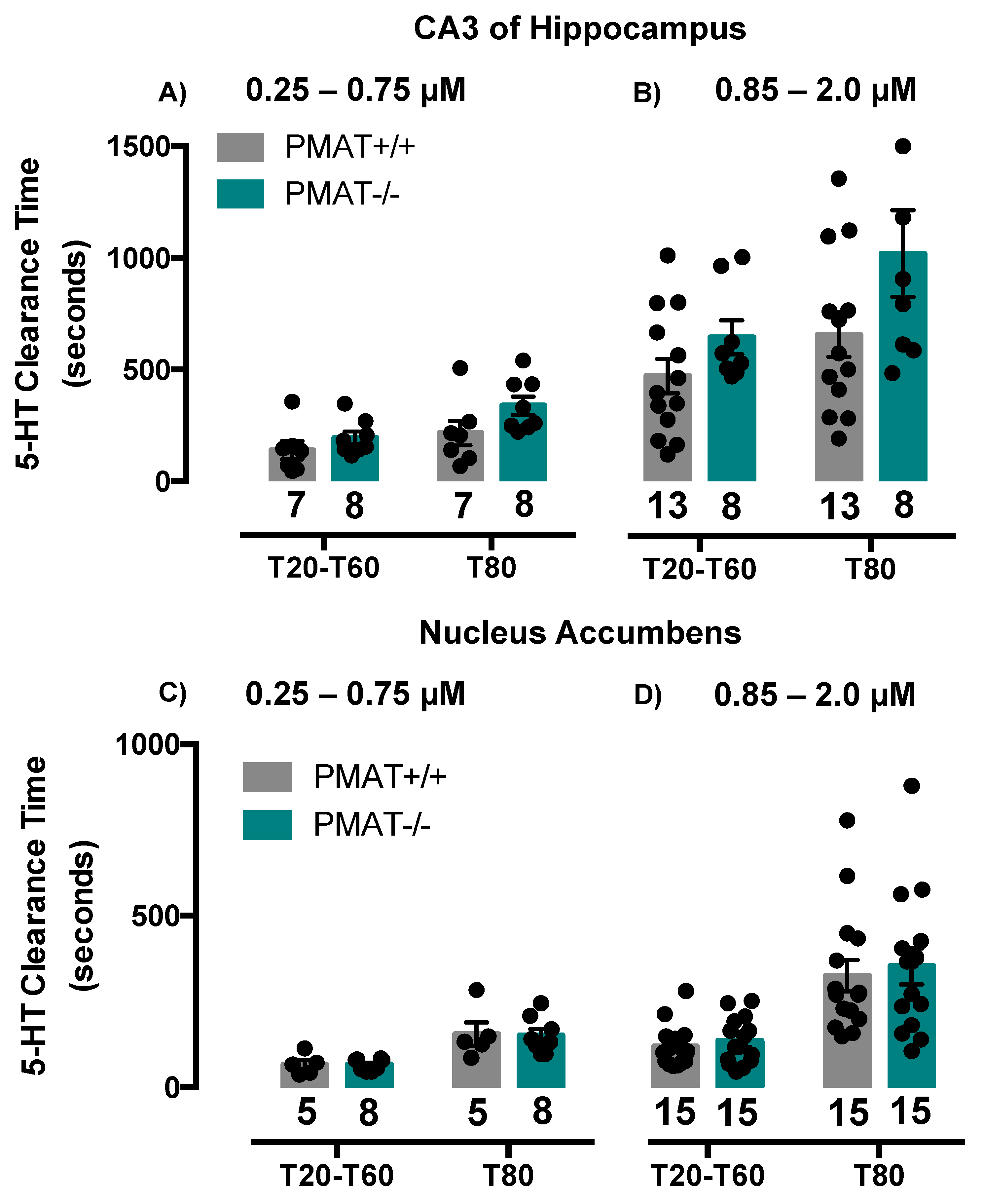
2.2. Comparison of Serotonin Clearance among SERT and PMAT Genotypes
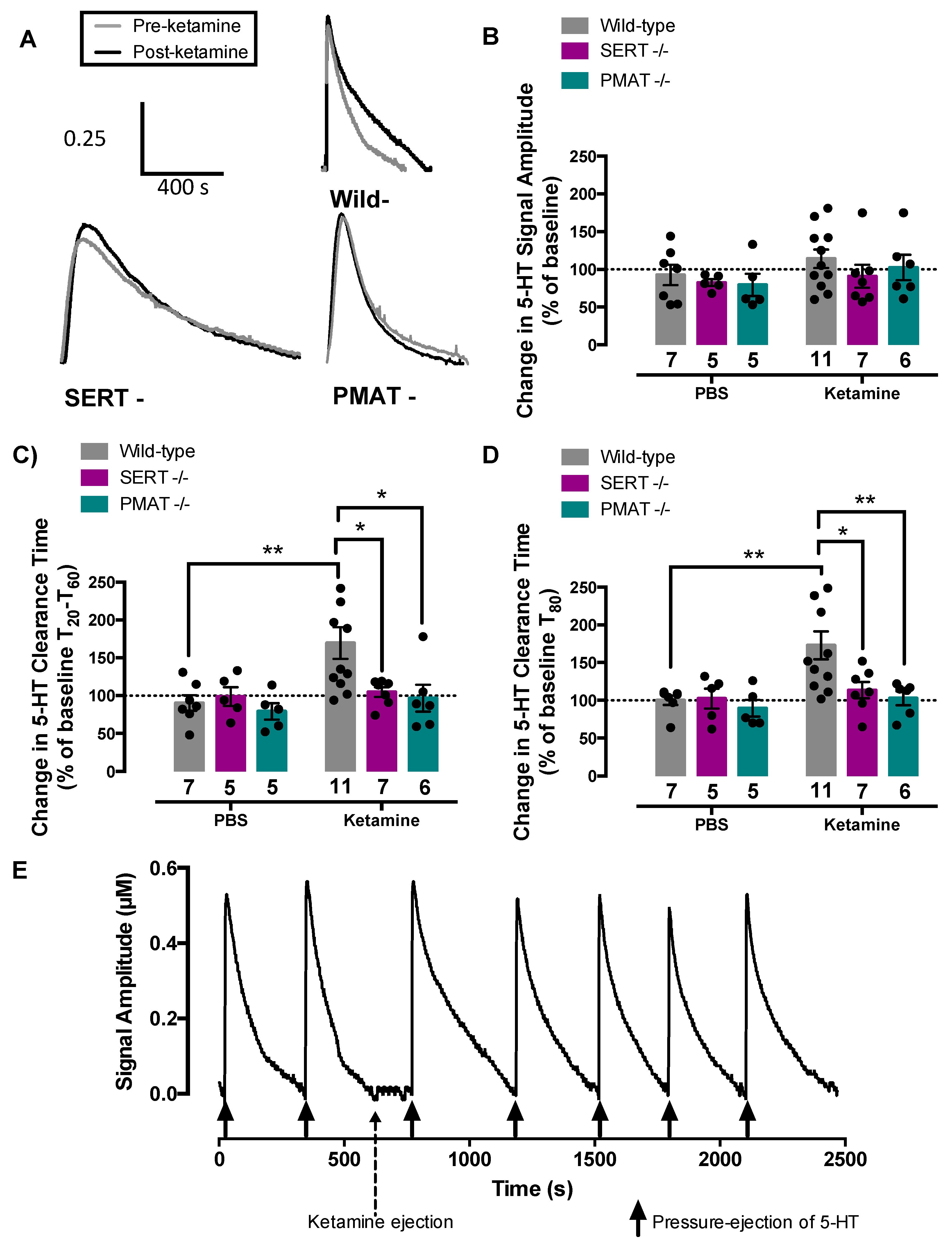
2.3. Ketamine Inhibits Serotonin Clearance in Wild-Type Mice, but Not in SERT−/− or PMAT−/− Mice
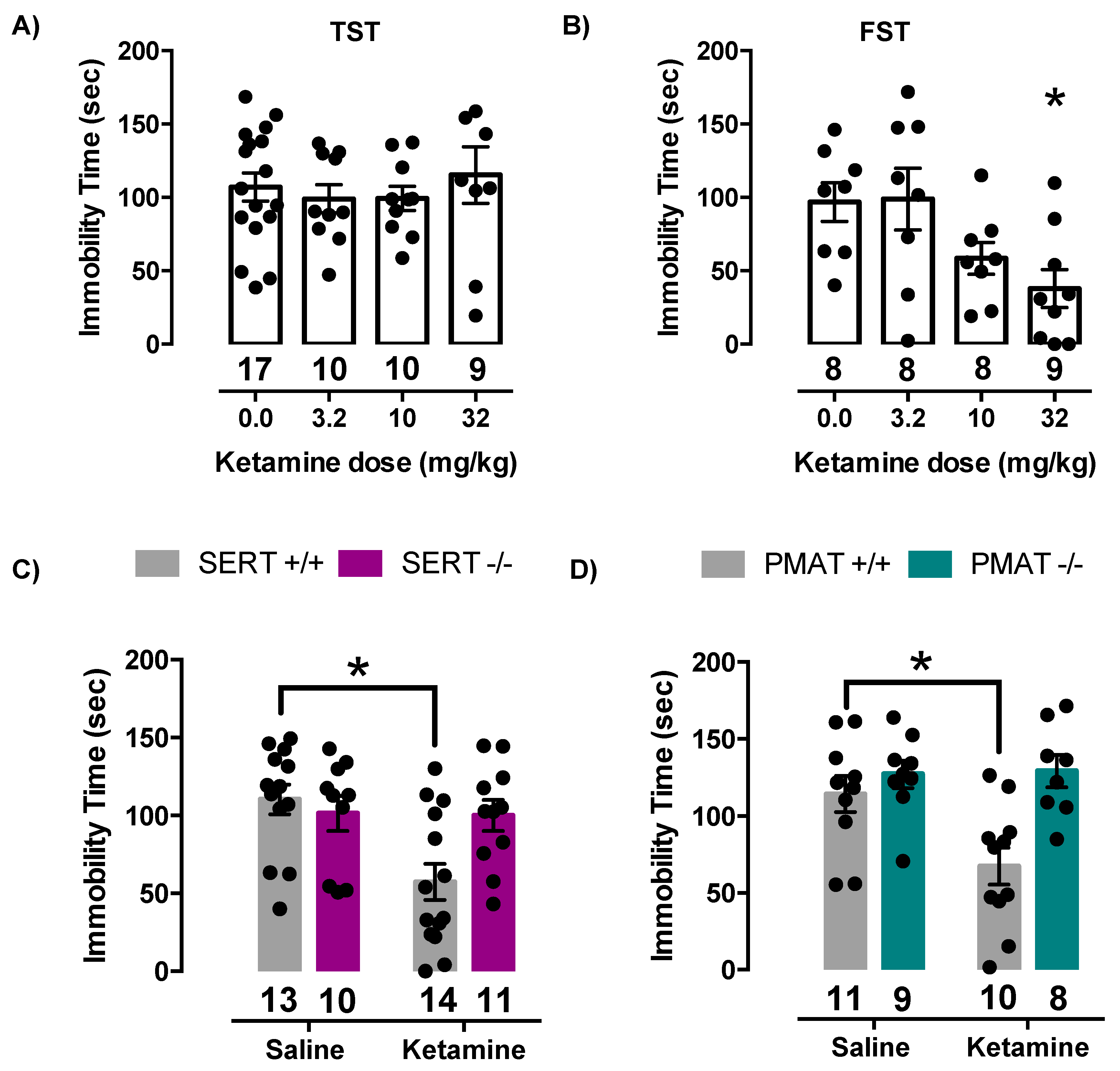
2.4. Antidepressant-Like Effects of Ketamine Are Lost in Mice Lacking SERT or PMAT
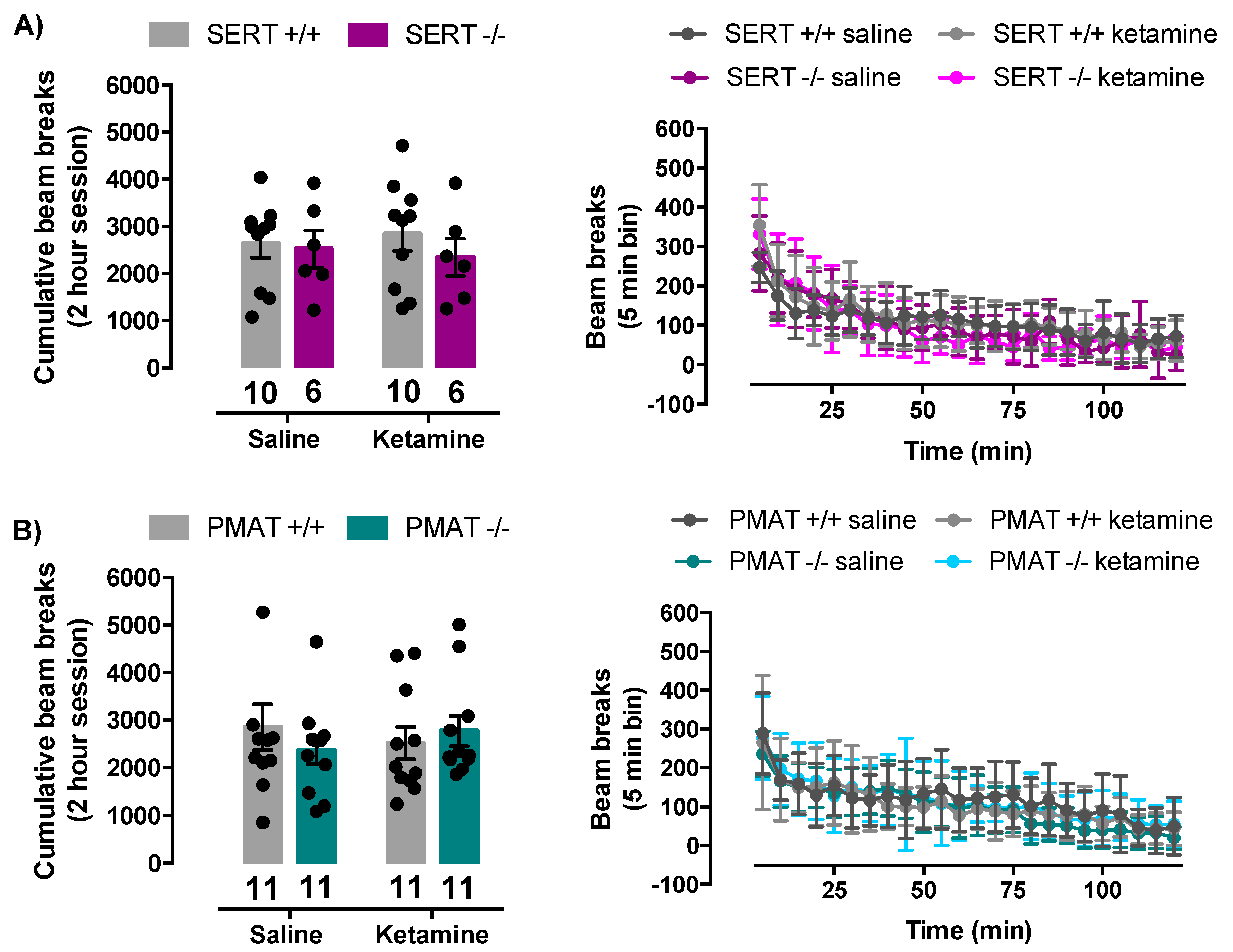
2.5. Ketamine Does Not Influence Locomotor Activity of SERT+/+, SERT−/−, PMAT+/+, or PMAT−/− Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. High-Speed Chronoamperometry
4.3. Effects of Isoflurane on Serotonin Clearance
4.4. Tail Suspension Test
4.5. Forced Swim Test
4.6. Locomotor Activity
4.7. Drugs
4.8. Data Analysis
4.8.1. High-Speed Chronoamperometry
4.8.2. Tail Suspension Test and Forced Swim Test
4.8.3. Locomotor Activity
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| +/+ | Wild-type |
| −/− | Knockout |
| [11C]DASB | [11C]-3-amino-4-(2-dimethylaminomethyl-phenylsulfanyl)benzonitrile |
| 5-HIAA | 5-hydroxyindoleacetic acid |
| ANOVA | Analysis of variance |
| BDNF | Brain derived neurotrophic factor |
| DAT | Dopamine transporter |
| FST | Forced swim test |
| HNK | (2R,6R)-hydroxynorketamine |
| NMDA | N-methyl-d-aspartate |
| NAcc | Nucleus accumbens |
| NET | Norepinephrine transporter |
| OCT1 | Organic cation transporter 1 |
| OCT2 | Organic cation transporter 2 |
| OCT3 | Organic cation transporter 3 |
| PBS | Phosphate buffered saline |
| PCA | p-Chloroamphetamine |
| PCPA | Parachlorophenylalanine |
| PET | Positron emission tomography |
| PMAT | Plasma membrane monoamine transporter |
| SERT | Serotonin transporter |
| SSRI | Selective serotonin reuptake inhibitor |
| TST | Tail suspension test |
References
- Center for Behavioral Health Statistics and Quality. Key Substance Use and Mental Health Indicators in the United States: Results from the 2015 National Survey on Drug Use and Health 2016, HHS Publication No. SMA 16-4984, NSDUH Series H-51. Available online: http://www.samhsa.gov/data (accessed on 28 April 2020).
- Mathys, M.; Mitchell, B.G. Targeting treatment-resistant depression. J. Pharm. Pract. 2011, 24, 520–533. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.S.; Filteau, M.-J.; Martin, L.; Patry, S.; Carvalho, A.; Cha, D.S.; Barakat, M.; Miguelez, M. Treatment-resistant depression: Definitions, review of the evidence, and algorithmic approach. J. Affect. Disord. 2014, 156, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.J.; Shah, A.; Lapidus, K.; Clark, C.; Jarun, N.; Ostermeyer, B.; Murrough, J.W. Ketamine for treatment-resistant unipolar depression. CNS Drugs 2012, 26, 189–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, D.C.; Henter, I.D.; Zarate, C.A., Jr. Targeting the glutamatergic system to treat major depressive disorder. Drugs 2012, 72, 1313–1333. [Google Scholar] [CrossRef] [PubMed]
- Monteggia, L.M.; Zarate, C., Jr. Antidepressant actions of ketamine: From molecular mechanisms to clinical practice. Curr. Opin. Neurol. 2015, 30, 139–143. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. FDA Approves New Nasal Spray Medication for Treatment-Resistant Depression; Available Only at Certified Doctor’s Office or Clinic [Press Release] 2019. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-nasal-spray-medication-treatment-resistant-depression-available-only-certified (accessed on 28 April 2020).
- Kavalali, E.T.; Monteggia, L.M. Targeting homeostatic synaptic plasticity for treatment of mood disorders. Neuron 2020, 106, 715–726. [Google Scholar] [CrossRef]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.-F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Lee, B.; Liu, R.-J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.-Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Angoa-Pérez, M.; Kane, M.J.; Briggs, D.I.; Herrera-Mundo, N.; Sykes, C.E.; Francescutti, D.M.; Kuhn, D.M. Mice genetically depleted of brain serotonin do not display a depression-like behavioral phenotype. ACS Chem. Neurosci. 2014, 5, 908–919. [Google Scholar] [CrossRef] [Green Version]
- Lucki, I. The spectrum of behaviors influenced by serotonin. Biol. Psychiatry 1998, 44, 151–162. [Google Scholar] [CrossRef]
- Lindefors, N.; Barati, S.; O’Connor, W.T. Differential effects of single and repeated ketamine administration on dopamine, serotonin, and GABA transmission in rat medial prefrontal cortex. Brain Res. 2014, 759, 205–212. [Google Scholar] [CrossRef]
- Iravani, M.M.; Muscat, R.; Kruk, Z.L. MK-801 interaction with the 5-HT transporter: A real-time study in brain slices using fast cyclic voltammetry. Synapse 1999, 32, 212–224. [Google Scholar] [CrossRef]
- Amargós-Bosch, M.; Lopez-Gil, X.; Artigas, F.; Adell, A. Clozapine and olanzapine, but not haloperidol, suppress serotonin efflux in the medial prefrontal cortex elicited by phencyclidine and ketamine. Int. J. Neuropsychopharmacol. 2006, 9, 565–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumoto, K.; Iijima, M.; Chaki, S. The antidepressant effects of an mGlu2/3 receptor antagonist and ketamine require AMPA receptor stimulation in the mPFC and subsequent activation of the 5-HT neurons in the DRN. Neuropsychopharmacology 2016, 41, 1046–1056. [Google Scholar] [CrossRef]
- Gigliucci, V.; O’Dowd, G.; Casey, S.; Egan, D.; Gibney, S.; Harkin, A. Ketamine elicits sustained antidepressant-like activity via a serotonin-dependent mechanism. Psychopharmacology 2013, 228, 157–166. [Google Scholar] [CrossRef]
- Pham, T.H.; Mendez-David, I.; Defaix, C.; Guiard, B.P.; Tritschler, L.; David, D.J.; Gardier, A.M. Ketamine treatment involves medial prefrontal cortex serotonin to induce a rapid antidepressant-like activity in BALB/cJ mice. Neuropharmacology 2017, 112, 198–209. [Google Scholar] [CrossRef]
- Gaarn du Jardin, K.; Liebenberg, N.; Müller, H.K.; Elfving, B.; Sanchez, C.; Wegener, G. Differential interaction with the serotonin system by S-ketamine, vortioxetine, and fluoxetine in a genetic rat model of depression. Psychopharmacology 2016, 233, 2813–2825. [Google Scholar] [CrossRef]
- Can, A.; Zanos, P.; Moaddel, R.; Kang, H.J.; Dossou, K.S.S.; Wainer, I.W.; Cheer, J.F.; Frost, D.O.; Huang, X.-P.; Gould, T.D. Effects of ketamine and ketamine metabolites on evoked striatal dopamine release, dopamine receptors, and monoamine transporters. J. Pharmacol. Exp. Ther. 2016, 359, 159–170. [Google Scholar] [CrossRef]
- Baganz, N.L.; Horton, R.E.; Calderon, A.S.; Owens, W.A.; Munn, J.L.; Watts, L.T.; Koldzic-Zivanovic, N.; Jeske, N.A.; Koek, W.; Toney, G.M.; et al. Organic cation transporter 3: Keeping the brake on extracellular serotonin in serotonin-transporter-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 18976–18981. [Google Scholar] [CrossRef] [Green Version]
- Baganz, N.; Horton, R.; Martin, K.; Holmes, A.; Daws, L.C. Repeated swim impairs serotonin clearance via a corticosterone-sensitive mechanism: Organic cation transporter 3, the smoking gun. J. Neurosci. 2010, 30, 15185–15195. [Google Scholar] [CrossRef] [Green Version]
- Daws, L.C. Unfaithful neurotransmitter transporters: Focus on serotonin uptake and implications for antidepressant efficacy. Pharmacol. Ther. 2009, 121, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, R.E.; Apple, D.M.; Owens, W.A.; Baganz, N.L.; Cano, S.; Mitchell, N.C.; Vitela, M.; Gould, G.G.; Koek, W.; Daws, L.C. Decynium-22 enhances SSRI-induced antidepressant-like effects in mice: Uncovering novel targets to treat depression. J. Neurosci. 2013, 33, 10534–10543. [Google Scholar] [CrossRef]
- Zhou, M.; Engel, K.; Wang, J. Evidence for significant contribution of a newly identified monoamine transporter (PMAT) to serotonin uptake in the human brain. Biochem. Pharmacol. 2007, 73, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, S.; MacQueen, G. The role of the hippocampus in the pathophysiology of major depression. J. Psychiatry Neurosci. 2004, 29, 417–426. [Google Scholar]
- Bowman, M.A.; Daws, L.C. Targeting serotonin transporters in the treatment of juvenile and adolescent depression. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Holmes, A. The ascent of mouse: Advances in modeling human depression and anxiety. Nat. Rev. Drug Discov. 2005, 4, 775–790. [Google Scholar] [CrossRef]
- Garris, P.A.; Budygin, E.A.; Phillips, P.E.M.; Venton, B.J.; Robinson, D.L.; Bergstrom, B.P.; Rebec, G.V.; Wightman, R.M. A role for presynaptic mechanisms in the actions of nomifensine and haloperidol. Neuroscience 2003, 118, 819–829. [Google Scholar] [CrossRef]
- Sabeti, J.; Gerhardt, G.A.; Zahniser, N.R. Chloral hydrate and ethanol, but not urethane, alter the clearance of exogenous dopamine recorded by chronoamperometry in striatum of unrestrained rats. Neurosci. Lett. 2003, 343, 9–12. [Google Scholar] [CrossRef]
- Brodnik, Z.D.; España, R.A. Dopamine uptake dynamics are preserved under isoflurane anesthesia. Neurosci. Lett. 2015, 606, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Johansen, S.L.; Iceman, K.E.; Iceman, C.R.; Taylor, B.E.; Harris, M.B. Isoflurane causes concentration-dependent inhibition of medullary raphe 5-HT neurons in situ. Auton. Neurosci. 2015, 193, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.C.; Adams, D.J.; Aronstam, R.S. The influence of isoflurane on the synaptic activity of 5-Hydroxytryptamine. Neurochem. Res. 1990, 15, 969–973. [Google Scholar] [CrossRef]
- Mukaida, K.; Shichino, T.; Koyanagi, S.; Himukashi, S.; Fukuda, K. Activity of the serotonergic system during isoflurane anesthesia. Anesth. Analg. 2007, 104, 836–839. [Google Scholar] [CrossRef] [Green Version]
- Whittington, R.A.; Virág, L. Isoflurane decreases extracellular serotonin in the mouse hippocampus. Anesth. Analg. 2006, 103, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daws, L.C.; Montañez, S.; Munn, J.L.; Owens, W.A.; Baganz, B.L.; Boyce-Rustay, J.M.; Millstein, R.A.; Wiedholz, L.M.; Murphy, D.L.; Holmes, A. Ethanol inhibits clearance of brain serotonin by a serotonin transporter-independent mechanism. J. Neurosci. 2006, 26, 6431–6438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montañez, S.; Owens, W.A.; Gould, G.G.; Murphy, D.L.; Daws, L.C. Exaggerated effect of fluvoxamine in heterozygote serotonin transporter knockout mice. J. Neurochem. 2003, 86, 210–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.C. Multiple Comparisons. In Theory and Methods; Chapman & Hall/CRC: Boca Raton, FL, USA, 1996. [Google Scholar]
- Maxwell, S.E.; Delaney, H.D.; Kelley, K. Designing Experiments and Analyzing Data. In A Model Comparison Perspective, 3rd ed.; Routledge: New York, NY, USA, 2018. [Google Scholar]
- Fukumoto, K.; Iijima, M.; Chaki, S. Serotonin-1A receptor stimulation mediates effects of a metabotropic glutamate 2/3 receptor antagonist, 2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl) propanoic acid (LY341495), and an N-methyl-D-aspartate receptor antagonist, ketamine, in the novelty-suppressed feeding test. Psychopharmacology 2014. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Georgiou, P.; Fischell, J.; Elmer, G.I.; Alkondon, M.; Yuan, P.; Pribut, H.J.; Singh, N.S.; et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 2016, 533, 481–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popik, P.; Kos, T.; Sow-Kucma, M.; Nowak, G. Lack of persistent effects of ketamine in rodent models of depression. Psychopharmacology 2008, 198, 421–430. [Google Scholar] [CrossRef]
- Polis, A.J.; Fitzgerald, P.J.; Hale, P.J.; Watson, B.O. Rodent ketamine depression-related research: Finding patterns in a literature of variability. Behav. Brain Res. 2019, 376, 112153. [Google Scholar] [CrossRef]
- Cryan, J.F.; Valentino, R.J.; Lucki, I. Assessing substrates underlying the behavioral effects of antidepressants using the modified rat forced swimming test. Neurosci. Biobehav. Rev. 2005, 29, 547–569. [Google Scholar] [CrossRef]
- Bogdanova, O.V.; Kanekar, S.; D’Anci, K.E.; Renshaw, P.F. Factors influencing behavior in the forced swim test. Physiol. Behav. 2013, 118, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ago, Y.; Tanabe, W.; Higuchi, M.; Tsukada, S.; Tanaka, T.; Yamaguchi, T.; Igarashi, H.; Yokoyama, R.; Seiriki, K.; Kasai, A.; et al. (R)-ketamine induces a greater increase in prefrontal 5-HT release than (S)-ketamine and ketamine metabolites via an AMPA receptor-independent mechanism. Int. J. Neuropsychopharmacol. 2019, 22, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Nishitani, N.; Nagai, Y.; Andoh, C.; Asaoka, N.; Kawai, H.; Shibui, N.; Nagayasu, K.; Shirakawa, H.; Nakagawa, T.; et al. Ketamine-induced prefrontal serotonin release is mediated by cholinergic neurons in the pedunculopontine tegmental nucleus. Int. J. Neuropharmacol. 2018, 21, 305–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Gil, X.; Jiménez-Sánchez, L.; Campa, L.; Castro, E.; Frago, C.; Adell, A. Role of serotonin and noradrenaline in the rapid antidepressant action of ketamine. ACS Chem. Neurosci. 2019, 10, 3318–3326. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.H.; Defaix, C.; Xu, X.; Deng, S.-X.; Fabresse, N.; Alvarez, J.-C.; Landry, D.W.; Brachman, R.A.; Denny, C.A.; Gardier, A.M. Common neurotransmission recruited in (R,S)-ketamine and (2R,2S)-hydroxynorketamine-induced sustained antidepressant-like effects. Biol. Psychiatry 2018, 84, e3–e6. [Google Scholar] [CrossRef]
- Yamamoto, S.; Ohba, H.; Nishiyama, S.; Harada, N.; Kakiuchi, T.; Tsukada, H.; Domino, E.F. Subanesthetic doses of ketamine transiently decrease serotonin transporter activity: A PET study in conscious monkeys. Neuropsychopharmacology 2013, 38, 2666–2674. [Google Scholar] [CrossRef] [Green Version]
- Azzaro, A.J.; Smith, D.J. The inhibitory action of ketamine hcl on [3H]5-hydroxytryptamine accumulation by rat brain synaptosomal-rich fractions: Comparison with [3H]catecholamine and [3H]gamma-aminobutyric acid uptake. Neuropharmacology 1977, 16, 349–356. [Google Scholar] [CrossRef]
- Nishimura, M.; Sato, K.; Okada, T.; Yoshiya, I.; Schloss, P.; Shimada, S.; Tohyama, M. Ketamine inhibits monoamine transporters expressed in human embryonic kidney 293 cells. Anesthesiology 1998, 88, 768–774. [Google Scholar] [CrossRef]
- Barann, M.; Stamer, U.M.; Lyutenska, M.; Stüber Bönisch, H.; Urban, B. Effects of opioids on human serotonin transporters. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 43–49. [Google Scholar] [CrossRef]
- Martin, L.L.; Bouchal, R.L.; Smith, D.J. Ketamine inhibits serotonin uptake in vivo. Neuropharmacology 1982, 21, 113–118. [Google Scholar] [CrossRef]
- Idvall, J.; Ahlgren, I.; Aronsen, K.R.; Stenberg, P. Ketamine infusions: Pharmacokinetics and clinical effects. Br. J. Anesth. 1979, 51, 1167–1173. [Google Scholar] [CrossRef]
- Zarate, C.A., Jr.; Brutsche, N.E.; Ibrahim, L.; Franco-Chaves, J.; Diazgranados, N.; Cravchik, A.; Selter, J.; Marquardt, C.A.; Liberty, V.; Luckenbaugh, D.A. Replication of ketamine’s antidepressant efficacy in bipolar depression: A randomized controlled add-on trial. Biol. Psychiatry 2012, 71, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, C.K.; Gálvez, V.; O’Keefe, E.; Mitchell, P.B.; Hadzi-Pavlovic, D.; Leyden, J.; Harper, S.; Somogyi, A.A.; Lai, R.; Weickert, C.S.; et al. Placebo-controlled pilot trial testing dose titration and intravenous, intramuscular and subcutaneous routes for ketamine in depression. Acta Psychiatr. Scand. 2016, 134, 48–56. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Riggs, L.M.; Highland, J.N.; Georgiou, P.; Pereira, E.F.R.; Albuquerque, E.X.; Thomas, C.J.; Zarate, C.A., Jr.; et al. Ketamine and ketamine metabolite pharmacology: Insights into therapeutic mechanisms. Pharmacol. Rev. 2018, 70, 621–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spies, M.; James, G.M.; Berroterán-Infante Ibeschitz, H.; Kranz, G.S.; Unterholzner, J.; Godbersen, M.; Gryglewski, G.; Hienert, M.; Jungwirth, J.; Pichler, V.; et al. Assessment of ketamine binding of the serotonin transporter in humans with positron emission tomography. Int. J. Neuropharmacol. 2018, 21, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, H.; Wang, J. Impaired monoamine and organic cation uptake in choroid plexus in mice with targeted disruption of the plasma membrane monoamine transporter (Slc29a4) gene. J. Biol. Chem. 2013, 288, 3535–3544. [Google Scholar] [CrossRef] [Green Version]
- Gilman, T.L.; George, C.M.; Vitela, M.; Herrera-Rosales, M.; Basiouny, M.S.; Koek, W.; Daws, L.C. Constitutive plasma membrane monoamine transporter (PMAT, Slc29a4) deficiency subtly affects anxiety-like and coping behaviours. Eur. J. Neurosci. 2018, 48, 1706–1716. [Google Scholar] [CrossRef]
- Fukumoto, K.; Toki, H.; Iijima, M.; Hashihayata, T.; Yamaguchi, J.-I.; Hashimoto, K.; Chaki, S. Antidepressant potential of (R)-ketamine in rodent models: Comparison with (S)-ketamine. J. Pharmacol. Exper. Ther. 2017, 361, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Lindholm, J.S.O.; Autio, H.; Vesa, L.; Antila, H.; Lindemann, L.; Hoener, M.C.; Skolnick, P.; Rantamaki, T.; Castren, E. The antidepressant-like effects of glutamatergic drugs ketamine and AMPA receptor potentiator LY 451646 are preserved in bdnf+/- heterozygous null mice. Neuropharmacology 2012, 62, 391–397. [Google Scholar] [CrossRef]
- Yamaguchi, J.I.; Toki, H.; Qu, Y.; Yang, C.; Koike, H.; Hashimoto, K.; Mizuno-Yasuhira, A.; Chaki, S. (2R,6R)-Hydroxynorketamine is not essential for the antidepressant actions of (R)-ketamine in mice. Neuropsychopharmacology 2018, 43, 1900–1907. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.E.; Chandra, J.; Sheriff, S.; Malinow, R. Opioid system is necessary but not sufficient for antidepressive actions of ketamine in rodents. Proc. Natl. Acad. Sci. USA 2020, 117, 2656–2662. [Google Scholar] [CrossRef] [PubMed]
- Carrier, N.; Kabbaj, M. Sex differences in the antidepressant-like effects of ketamine. Neuropharmacology 2013, 70, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Franceschelli, A.; Sens, J.; Herchick, S.; Thelen, C.; Pitychoutis, M. Sex differences in the rapid and the sustained antidepressant-like effects of ketamine in stress-naïve and “depressed” mice exposed to chronic mild stress. Neuroscience 2015, 290, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thelen, C.; Sens, J.; Mauch, J.; Pandit, R.; Pitychoutis, P.M. Repeated ketamine treatment induces sex-specific behavioral and neurochemical effects in mice. Behav. Brain Res. 2016, 312, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Daws, L.C.; Owens, W.A.; Toney, G.M. High-speed chronoamperometry to measure biogenic amine release and uptake in vivo. In Neurotransmitter Transporters–Investigate Methods; Sitte, H., Bonisch, H., Eds.; Humana Press: Totowa, NJ, USA, 2016; pp. 53–81. [Google Scholar]
- Daws, L.C.; Toney, G.M. High-speed chronoamperometry to study kinetics and mechanisms for serotonin clearance in vivo. In Electrochemical Methods for Neuroscience; Michael, A.C., Borland, L.M., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2007. [Google Scholar]
- Gerhardt, G.A. Rapid chronocoulometric measurements of norepinephrine overflow and clearance in CNS tissues. In Voltammetric Methods in Brain Systems, Neuromethods; Boulton, A.A., Baker, G.B., Adams, R.N., Eds.; Humana Press: Totowa, NJ, USA, 1995; Volume 27. [Google Scholar] [CrossRef]
- Perez, X.A.; Andrews, A.M. Chronoamperometry to determine differential reductions in uptake in brain synaptosomes from serotonin transporter knockout mice. Anal. Chem. 2005, 77, 818–826. [Google Scholar] [CrossRef]
- Williams, J.M.; Owens, W.A.; Turner, G.H.; Saunders, C.; Dipace, C.; Blakely, R.D.; France, C.P.; Gore, J.C.; Daws, L.C.; Avison, M.J.; et al. Hypoinsulinemia regulates amphetamine-induced reverse transport of dopamine. PLoS Biol. 2007, 5, e274. [Google Scholar] [CrossRef] [Green Version]
- Franklin, K.B.J.; Paxinos, G. The Mouse Brain in Stereotaxic Coordinates; Academic: Syndey, Australia, 1997. [Google Scholar]
- Steru, L.; Chermat, R.; Thierry, B.; Simon, P. The tail suspension test: A new method for screening antidepressants in mice. Psychopharmacology 1985, 85, 367–370. [Google Scholar] [CrossRef]
- Castagné, V.; Moser, P.; Roux, S.; Porsolt, R.D. Rodent models of depression: Forced swim and tail suspension behavioral despair tests in rats and mice. Curr. Protoc. Neurosci. 2011, 8. [Google Scholar] [CrossRef]
- Koike, H.; Iijima, M.; Chaki, S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav. Brain Res. 2011, 224, 107–111. [Google Scholar] [CrossRef]
- Lucki, I. The forced swimming test as a model for core and component behavioral effects of antidepressant drugs. Behav. Pharmacol. 1997, 8, 523–532. [Google Scholar] [CrossRef]
- Lucki, I.; Dalvi, A.; Mayorga, A.J. Sensitivity to the effects of pharmacologically selective antidepressants in different strains of mice. Psychopharmacology 2001, 155, 315–322. [Google Scholar] [CrossRef]
- Imre, G.; Fokkema, D.S.; Den Boer, J.A.; Ter Horst, G.J. Dose-response characteristics of ketamine effect on locomotion, cognitive function and central neuronal activity. Brain Res. Bull. 2005, 69, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, K.A.; Zamora, J.J.; Warmoth, K.P. Increased response to ketamine following treatment at long intervals: Implications for intermittent use. Biol. Psychiatry 2008, 63, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Parise, E.M.; Alcantara, L.F.; Warren, B.L.; Wright, K.N.; Hadad, R.; Sial, O.K.; Kroeck, K.G.; Iñiguez, S.D.; Bolaños-Guzmán, C.A. Repeated ketamine exposure induces an enduring resilient phenotype in adolescent and adult rats. Biol. Psychiatry 2013, 74, 750–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | SERT+/+ | SERT+/+ | SERT−/− | SERT−/− | PMAT+/+ | PMAT+/+ | PMAT−/− | PMAT−/− | |
|---|---|---|---|---|---|---|---|---|---|
| Drug | PBS | Ketamine | PBS | Ketamine | PBS | Ketamine | PBS | Ketamine | |
| N | 4 | 5 | 5 | 7 | 3 | 6 | 5 | 6 | |
| Amp | Pre | 0.44 ± 0.08 | 0.62 ± 0.09 | 0.46 ± 0.06 | 0.59 ± 0.07 | 0.56 ± 0.11 | 0.64 ± 0.06 | 0.74 ± 0.16 | 0.77 ± 0.15 |
| (µM) | Post | 0.47 ± 0.16 | 0.66 ± 0.18 | 0.37 ± 0.32 | 0.59 ± 0.19 | 0.48 ± 0.15 | 0.74 ± 0.08 | 0.62 ± 0.18 | 0.72 ± 0.11 |
| T20-T60 | Pre | 109 ± 13 | 83 ± 6 | 173 ± 39 | 178 ± 45 | 361 ± 99 | 251 ± 66 | 138 ± 54 | 171 ± 51 |
| (s) | Post | 114 ± 22 | 149 ± 28 # | 186 ± 56 | 188 ± 50 | 282 ± 102 | 326 ± 55 # | 109 ± 46 | 171 ± 60 |
| T80 | Pre | 194 ± 20 | 155 ± 18 | 273 ± 56 | 297 ± 77 | 475 ± 173 | 306 ± 76 | 183 ± 55 | 193 ± 49 |
| (s) | Post | 202 ± 19 | 278 ± 21 ** | 305 ± 86 | 321 ± 78 | 487 ± 221 | 422 ± 81 * | 165 ± 60 | 206 ± 67 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowman, M.A.; Vitela, M.; Clarke, K.M.; Koek, W.; Daws, L.C. Serotonin Transporter and Plasma Membrane Monoamine Transporter Are Necessary for the Antidepressant-Like Effects of Ketamine in Mice. Int. J. Mol. Sci. 2020, 21, 7581. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207581
Bowman MA, Vitela M, Clarke KM, Koek W, Daws LC. Serotonin Transporter and Plasma Membrane Monoamine Transporter Are Necessary for the Antidepressant-Like Effects of Ketamine in Mice. International Journal of Molecular Sciences. 2020; 21(20):7581. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207581
Chicago/Turabian StyleBowman, Melodi A., Melissa Vitela, Kyra M. Clarke, Wouter Koek, and Lynette C. Daws. 2020. "Serotonin Transporter and Plasma Membrane Monoamine Transporter Are Necessary for the Antidepressant-Like Effects of Ketamine in Mice" International Journal of Molecular Sciences 21, no. 20: 7581. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207581