Role of Cadherins in Cancer—A Review

 , and
, and 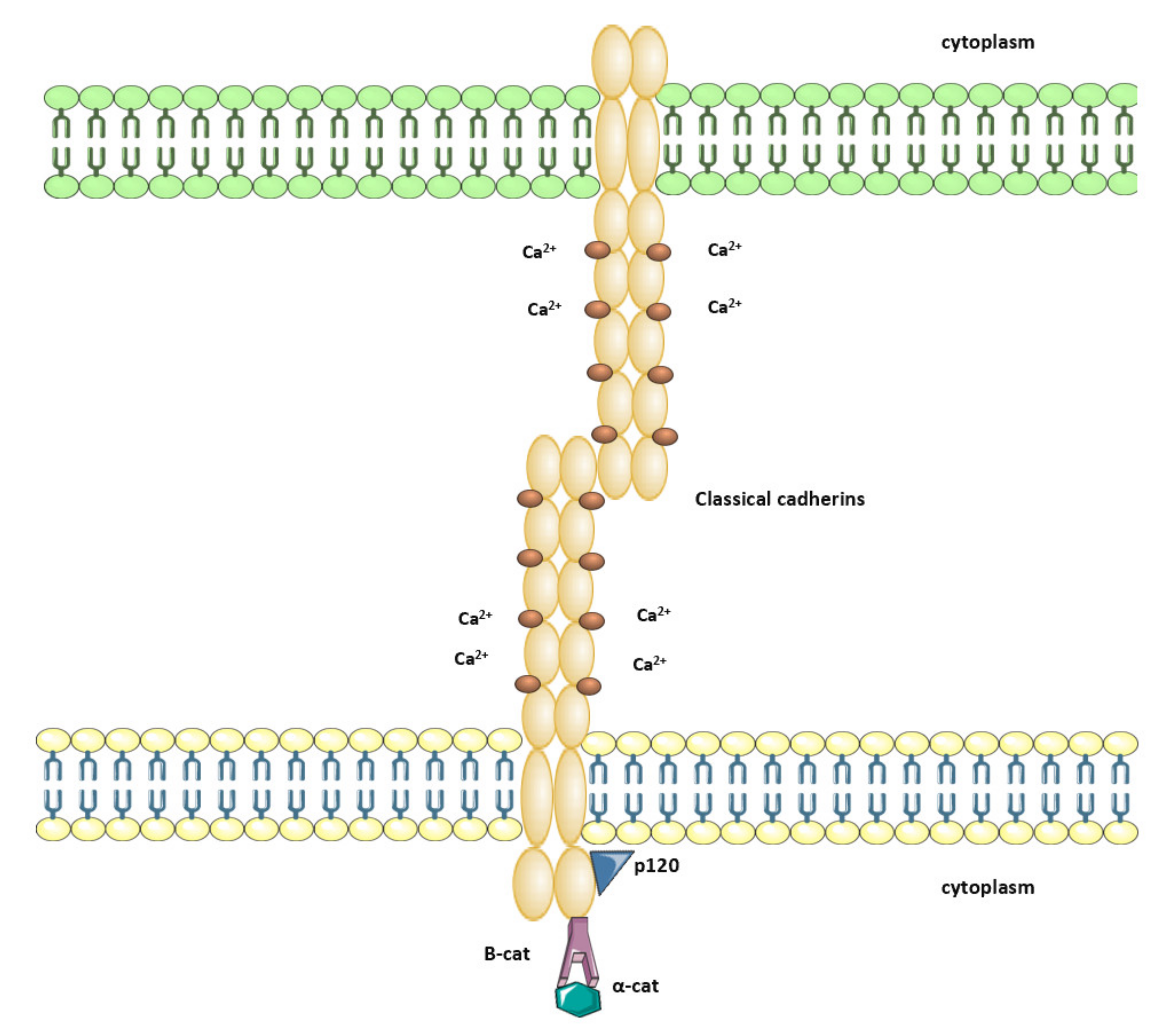{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cadherin-Catenin Complex
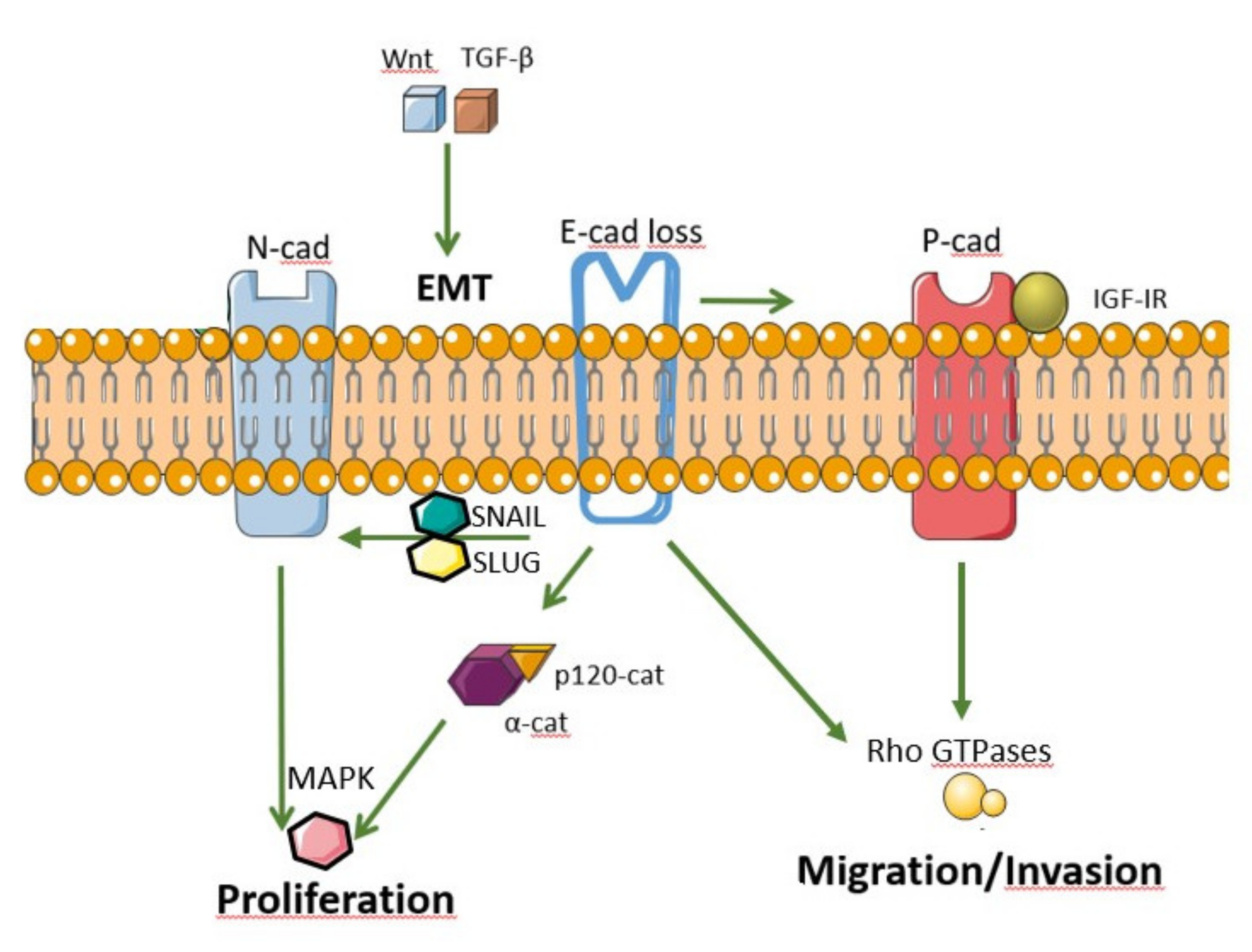
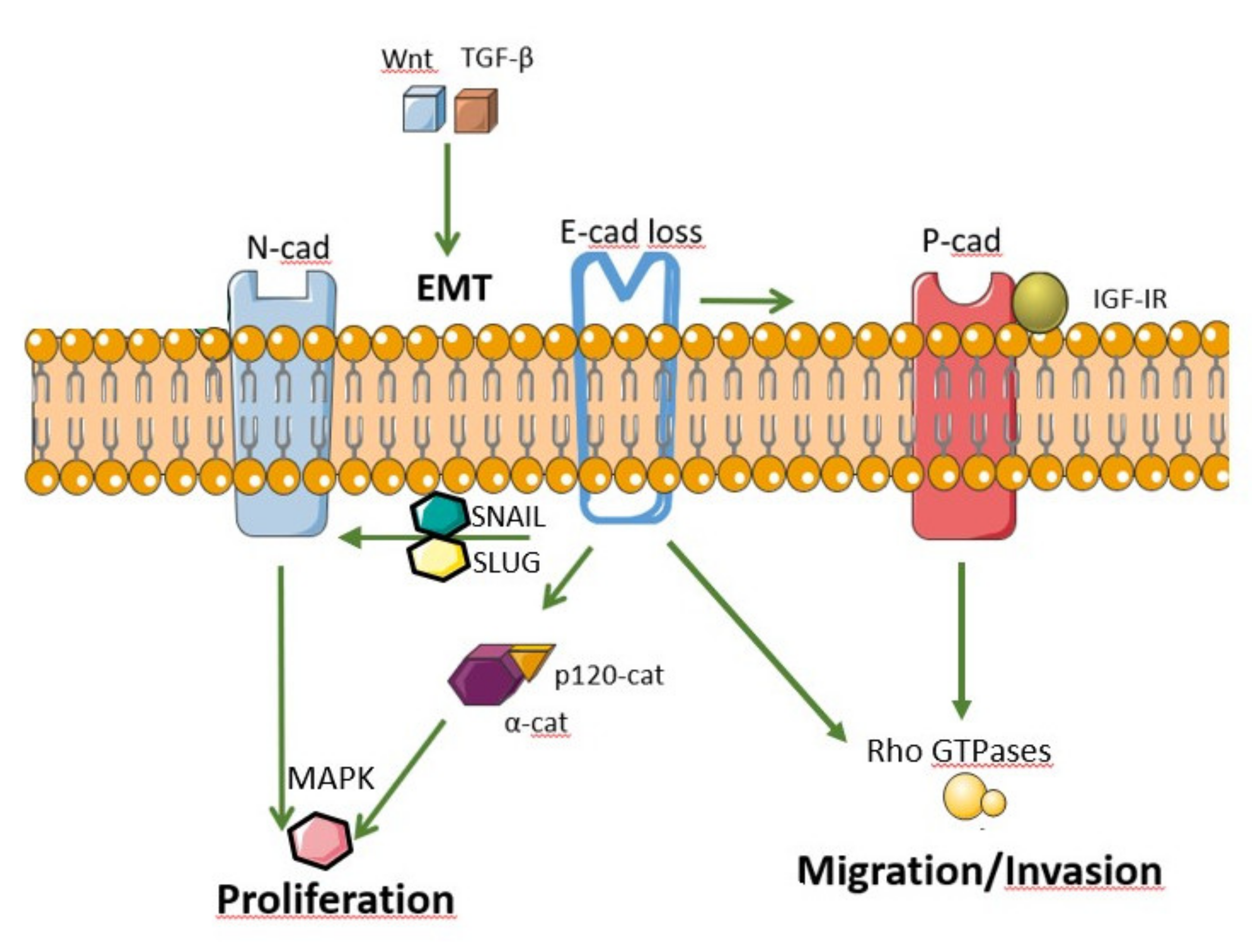
3. Cadherin and Catenin Signaling in Cancer
4. Epithelial Mesenchymal Transition
5. Cadherins in Human and Animal Cancer as a Prognostic Factor
6. Therapeutic Targets Associated with Cadherin Dysfunction
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | Adenomatous polyposis coli |
| ARVCF | Armadillo repeat protein deleted in velo-cardio-facial syndrome |
| CCH | Canine cutaneous histiocytoma |
| CSS | Cause specific survival |
| EMT | Epithelial mesenchymal transition |
| E-cadherin | Epithelial cadherin |
| EC | Extracellular domain |
| GSK-3β | Kinase glycogen synthase 3β |
| LEF | Lymphoid enhancer factor |
| MAPK | Mitogen activated kinase |
| MCRC | Metastatic colorectal cancer |
| MMP | Matrix metaloproteinase |
| N-cadherin | Neural cadherin |
| NPRAP/δ-catenin | Neural plakophilin-related armadillo protein, CTNND2 |
| OS | Overall survival |
| OSCC | Squamous cell carcinoma of the oral cavity |
| p0071 | Plakophilin 4 |
| p120 | Phosphorylated CREB1 |
| p-CREB | p120 catenin, also known as CTNND1 |
| PDAC | pancreatic ductal adenocarcinoma |
| PI3K | Phosphoinositide 3-kinase |
| P-cadherin | Placental cadherin |
| Ras | Rat sarcoma viral oncogene |
| Rac1 | Rhomboid-like-2 |
| RCC | Ras-related C3 botulinum toxin substrate |
| RHBDL2 | Renal cell carcinoma |
| RTK | Receptor tyrosine kinase |
| SNP | Single nucleotide pleomorphism |
| Snail | Transcription factor Snail, also known as SNAI1 |
| Slug | Transcription factor Slug, also known as SNAI2 |
| TCF | T-cell factor |
| TDG | Thymine-DNA-glycosylase |
| TGF-β | Transforming growth factor β |
| TT | Tumor thrombus |
| VEGF-A | Vascular endothelial growth factor A |
| VC | Vena cava |
References
- Colás-Algora, N.; Millán, J. How many cadherins do human endothelial cells express? Cell Mol. Life Sci. 2019, 76, 1299–1317. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Takeichi, M. Evolution: Structural and functional diversity of cadherin at the adherens junction. J. Cell Biol. 2011, 193, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, L.; Weis, W.I. Structure and biochemistry of cadherins and catenins. Cold Spring Harb. Perspect. Biol. 2009, 1, a003053. [Google Scholar] [CrossRef] [PubMed]
- Casal, J.I.; Bartolome, R.A. Beyond N-cadherin, relevance of cadherins 5,6 and 17 in cancer progression and metastasis. Int. J. Mol. Sci. 2019, 20, 3373. [Google Scholar] [CrossRef] [Green Version]
- Weis, W.I. Cadherin structure: A revealing zipper. Structure 1995, 3, 425–427. [Google Scholar] [CrossRef] [Green Version]
- Bruner, H.C.; Derksen, P.W.B. Loss of E-Cadherin-Dependent Cell-Cell Adhesion and the Development and Progression of Cancer. Cold Spring Harb. Perspect. Biol. 2018, 10, a029330. [Google Scholar] [CrossRef] [Green Version]
- Troyanovsky, R.B.; Chitaev, N.A.; Troyanovsky, S.M. Cadherin binding sites of plakoglobin: Localization, specificity and role in targeting to adhering junctions. J. Cell Sci. 1996, 109, 3069–3078. [Google Scholar]
- Grigorian, I.Y.; Linkova, N.S.; Polyakova, V.O.; Paltseva, E.M.; Kozlov, K.L. Signaling molecules of the endometrium: Gerontological and general pathological aspects. Adv. Gerontol. 2016, 6, 36–43. [Google Scholar] [CrossRef]
- Sousa, B.; Pereira, J.; Paredes, J. The Crosstalk Between Cell Adhesion and Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 1933. [Google Scholar] [CrossRef] [Green Version]
- Baranwal, S.; Alahari, S.K. Molecular mechanisms controlling E-cadherin expression in breast cancer. Biochem. Biophys. Res. Commun. 2009, 384, 6–11. [Google Scholar] [CrossRef] [Green Version]
- Roggiani, F.; Mezzanzanica, D.; Rea, K.; Tomassetti, A. Guidance of Signaling Activations by Cadherins and Integrins in Epithelial Ovarian Cancer Cells. Int. J. Mol. Sci. 2016, 17, 1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalle Vedove, A.; Falchi, F.; Donini, S.; Dobric, A.; Germain, S.; Di Martino, G.P.; Prosdocimi, T.; Vettraino, C.; Torretta, A.; Cavalli, A.; et al. Structure-Based Virtual Screening Allows the Identification of Efficient Modulators of E-Cadherin-Mediated Cell-Cell Adhesion. Int. J. Mol. Sci. 2019, 20, 3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, J.M.; Ting, A.H.; Vilardell, F.; Gallmeier, E.; Baylin, S.B.; Hruban, R.H.; Kern, S.E.; Iacobuzio-Donahue, C.A. Absence of E-cadherin expression distinguishes noncohesive from cohesive pancreatic cancer. Clin. Cancer Res. 2008, 14, 412–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, J.L.; Kim, A.C.; Henscor, J.R. The role and function of cadherins in the mammary gland. Breast Cancer Res. 2012, 14, 203. [Google Scholar] [CrossRef] [Green Version]
- Gruss, C.; Herlyn, M. Role of cadherins and matrixins in melanoma. Curr. Opin. Oncol. 2001, 13, 117–123. [Google Scholar] [CrossRef]
- Rossi, T.; Tedaldi, G.; Petracci, E.; Khouzam, R.A.; Ranzani, G.N.; Morgagni, P.; Saragoni, L.; Monti, M.; Calistro, D.; Ulivi, P.; et al. E-cadherin downregulation and microRNAs in sporadic intestinal-type gastric cancer. Int. J. Mol. Sci. 2019, 20, 4452. [Google Scholar] [CrossRef] [Green Version]
- Kourtidis, A.; Lu, R.; Pence, L.J.; Anastasiadis, P.Z. A central role for cadherin signaling in cancer. Exp. Cell Res. 2017, 358, 78–85. [Google Scholar] [CrossRef]
- Ceresa, D.; Alessandrini, F.; Bosio, L.; Marubbi, D.; Reverberi, D.; Malatesta, P.; Appolloni, I. Cdh4 Down-Regulation Impairs in Vivo Infiltration and Malignancy in Patients Derived Glioblastoma Cells. Int. J. Mol. Sci. 2019, 20, 4028. [Google Scholar] [CrossRef] [Green Version]
- Shimada, S.; Mimata, A.; Sekine, M.; Mogushi, K.; Akiyama, Y.; Fukamachi, H.; Jonkers, J.; Tanaka, H.; Eishi, Y.; Yuasa, Y. Synergistic tumour suppressor activity of E-cadherin and p53 in a conditional mouse model for metastatic diffuse-type gastric cancer. Gut 2012, 61, 344–353. [Google Scholar] [CrossRef]
- Daulagala, A.C.; Bridges, M.C.; Kourtidis, A. E-cadherin Beyond Structure: A signaling hub in colon homeostasis and disease. Int. J. Mol. Sci. 2019, 20, 2756. [Google Scholar] [CrossRef] [Green Version]
- Shamir, E.R.; Ewald, A.J. Adhesion in mammary development: Novel roles forE-cadherin in individul and collective cell migration. Curr. Top. Dev. Biol. 2015, 112, 353–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labernadie, A.; Kato, T.; Brugues, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; Gonzalez-Tarrago, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin- N-cadherin adhesion enables fibroblast to drive cancer cell invasion. Nat. Cell. Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Dohn, M.R.; Brown, M.V.; Reynolds, A.B. An essential role for p120-catenin in Src- and Rac1-mediated anchorage-independent cell growth. J. Cell Biol. 2009, 184, 437–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venhuizen, J.H.; Span, P.N.; van den Dries, K.; Sommer, S.; Friedl, P.; Zegers, M.M. P120 catenin isoforms differentially associate with breast cancer invasion and metastasis. Cancers 2019, 11, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, Y.I.; Schecterson, L.; Gumbiner, B.M. Roles for E- cadherin cell surface regulation in cancer. Mol. Biol. Cell. 2016, 27, 3233–3244. [Google Scholar] [CrossRef] [PubMed]
- Gurrapu, S.; Franzolin, G.; Fard, D.; Accardo, M.; Medico, E.; Sarotto, I.; Sapino, A.; Isella, C.; Tamagnone, L. Reverse signaling by semaphorin 4 C elicits SMAD1/5- and ID1/3- dependent invasive reprogramming in cancer cells. Sci. Signal. 2019, 12, eaav2041. [Google Scholar] [CrossRef] [PubMed]
- Padmanaban, V.; Krol, I.; Suhali, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Yamada, S.; Pokutta, S.; Drees, F.; Weis, W.I.; Nelson, W.J. Deconstructing the cadherin-catenin-actin complex. Cell 2005, 123, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Gama, A.; Schmitt, F. Cadherin cell adhesion system in canine mammary cancer: A review. Vet. Med. Int. 2012, 2012, 357187. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Iino, M.; Goto, K. Knockdown of Sec6 improves cell–cell adhesion by increasing α-E-catenin in oral cancer cells. FEBS Lett. 2012, 586, 924–933. [Google Scholar] [CrossRef] [Green Version]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef] [Green Version]
- Chamorro, M.N.; Schwartz, D.R.; Vonica, A.; Brivanlou, A.H.; Cho, K.R.; Varmus, H.E. FGF-20 and DKK1 are transcriptional targets of beta-catenin and FGF-20 is implicated in cancer and development. EMBO J. 2005, 24, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef]
- Kazanskaya, O.; Glinka, A.; del Barco Barrantes, I.; Stannek, P.; Niehrs, C.; Wu, W. R-Spondin2 Is a Secreted Activator of Wnt/β-Catenin Signaling and Is Required for Xenopus Myogenesis. Dev. Cell 2004, 7, 525–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, Z.; Vijayakumar, S.; de la Torre, T.V.; Rotolo, S.; Bafico, A. Analysis of endogenous LRP6 function reveals a novel feedback mechanism by which Wnt negatively regulates its receptor. Mol. Cell Biol. 2007, 27, 7291–7301. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Kourtidis, A.; Yanagisawa, M.; Huveldt, D.; Copland, J.A.; Anastasiadis, P.Z. Pro-tumorigenic phosphorylation of p 120 catenin in renal and breast cancer. PLoS ONE 2015, 10, e0129964. [Google Scholar] [CrossRef] [Green Version]
- Andl, C.D.; Fargnoli, B.B.; Okawa, T.; Bowser, M.; Takaoka, M.; Nakagawa, H.; Klein-Szanto, A.; Herlyn, M.; Rustgi, A.K. Coordinated functions of E-cadherin and transforming Growth Factor β receptor II in vitro and in vivo. Cancer Res. 2006, 66, 9878–9885. [Google Scholar] [CrossRef] [Green Version]
- Pece, S.; Gutkind, J.S. Signaling from E-cadherins to the MAPK pathway by the recuitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J. Biol. Chem. 2000, 275, 41227–41233. [Google Scholar] [CrossRef] [Green Version]
- Motti, M.L.; Califano, D.; Baldassarre, G.; Celetti, A.; Merolla, F.; Forzati, F.; Napolitano, M.; Tavernise, B.; Fusco, A.; Viglietto, G. Reduced E-cadherin expression contributes to the loss of p27 kip1 -mediated mechanism of contact inhibition in thyroid anaplastic carcinomas. Carcinogenesis 2005, 26, 1021–1034. [Google Scholar] [CrossRef]
- St Croix, B.; Sheehan, C.; Rak, J.W.; Flørenes, V.A.; Slingerland, J.M.; Kerbel, R.S. E-Cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1). J. Cell Biol. 1998, 142, 557–571. [Google Scholar] [CrossRef] [PubMed]
- De Santis, G.; Miotti, S.; Mazzi, M.; Canevari, S.; Tomassetti, A. E-cadherin directly contributes to PI3K/AKT activation by engaging the PI3K-p85 regulatory subunit to adherens junctions of ovarian carcinoma cells. Oncogene 2009, 28, 1206–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, J.H.; Kraemer, A.; Stehbens, S.J.; Frame, M.C.; Yap, A.S. Recruitment of Phosphoinositide 3-Kinase Defines a Positive Contribution of Tyrosine Kinase Signaling to E-cadherin Function. J. Biol. Chem. 2005, 280, 3043–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pece, S.; Chiariello, M.; Murga, C.; Gutkind, J.S. Activation of the Protein Kinase Akt/PKB by the Formation of E-cadherin-mediated Cell-Cell Junctions: Evidence for the association of phosphatidylinositol 3-kinase with the e-cadherin adhesion complex. J. Biol. Chem. 1999, 274, 19347–19351. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [Green Version]
- McCrea, P.D.; Maher, M.T.; Gottardi, C.J. Nuclear signaling from cadherin adhesion complexes. Curr. Top. Dev. Biol. 2015, 112, 129–196. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Wang, X.; Zhang, H.; Wang, Z.; Nan, G.; Li, Y.; Zhang, F.; Mohammed, M.K.; Haydon, R.C.; Luu, H.H.; et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: Implications in targeted cancer therapies. Lab. Investig. 2016, 96, 116–136. [Google Scholar] [CrossRef] [Green Version]
- Gloushankova, N.A.; Zhitnyak, I.Y.; Rubtsova, S.N. Role of Epithelial-Mesenchymal Transition in Tumor Progression. Biochemistry 2018, 83, 1469–1476. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazan, R.B.; Qiao, R.; Keren, R.; Badano, I.; Suyama, K. Cadherin Switch in Tumor Progression. Ann. N. Y. Acad. Sci. 2004, 1014, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.S.T.; Gumbiner, B.M. Adhesion-independent mechanism for suppression of tumor cell invasion by E-cadherin. J. Cell Biol. 2003, 161, 1191–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, E.; Yanagisawa, M.; Marlow, L.A.; Copland, J.A.; Perez, E.A.; Anastasiadis, P.Z. p120 catenin induces opposing effects on tumor cell growth depending on E-cadherin expression. J. Cell Biol. 2008, 183, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Pećina-Slaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowell, C.F.; Yan, J.K.; Eiseler, T.; Leightner, A.C.; Döppler, H.; Storz, P. Loss of cell-cell contacts induces NF-κB via RhoA-mediated activation of protein kinase D1. J. Cell Biochem. 2009, 106, 714–728. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.S.; Gao, W.; Chan, J.Y. Interactions between E-cadherin and microRNA deregulation in head and neck cancers: The potential interplay. Biomed. Res. Int. 2014, 2014, 126038. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tan, Z.; Guan, F. Tumor-derived exosomes mediate the instability of cadherins and promote tumor progression. Int. J. Mol. Sci. 2019, 20, 3652. [Google Scholar] [CrossRef] [Green Version]
- Kourtidis, A.; Anastasiadis, P.Z. Close encounters of the RNAi kind: The silencing life of the adherens junctions. Curr. Opin. Cell Biol. 2018, 54, 30–36. [Google Scholar] [CrossRef]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Weinberg, R.A. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, Y.; Katoh, M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review). Int. J. Mol. Med. 2008, 22, 271–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheel, C.; Eaton, E.N.; Li, S.H.-J.; Chaffer, C.L.; Reinhardt, F.; Kah, K.-J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [Green Version]
- Cichon, M.A.; Radisky, D.C. Extracellular matrix as a contextual determinant of transforming growth factor-β signaling in epithelial-mesenchymal transition and in cancer. Cell Adhes. Migr. 2014, 8, 588–594. [Google Scholar] [CrossRef] [Green Version]
- Boyer, B.; Tucker, G.C.; Vallés, A.M.; Franke, W.W.; Thiery, J.P. Rearrangements of desmosomal and cytoskeletal proteins during the transition from epithelial to fibroblastoid organization in cultured rat bladder carcinoma cells. J. Cell Biol. 1989, 109, 1495–1509. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, K.; Shirakihara, T.; Nakano, A.; Imamura, T.; Miyazono, K.; Saitoh, M. Role of Ras Signaling in the Induction of Snail by Transforming Growth Factor-β. J. Biol. Chem. 2009, 284, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Ghosh, S.; Wang, Z.; Hunter, T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of β-catenin, and enhanced tumor cell invasion. Cancer Cell 2003, 4, 499–515. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; He, W.; Tulley, S.; Gupta, G.P.; Serganova, I.; Chen, C.-R.; Manova-Todorova, K.; Blasberg, R.; Gerald, W.L.; Massagué, J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13909–13914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourtidis, A.; Ngok, S.P.; Anastasiadis, P.Z. p120 catenin: An essential regulator of cadherin stability, adhesion-induced signaling, and cancer progression. Prog. Mol. Biol. Transl. Sci. 2013, 116, 409–432. [Google Scholar] [CrossRef] [Green Version]
- Bellovin, D.I.; Bates, R.C.; Muzikansky, A.; Rimm, D.L.; Mercurio, A.M. Altered Localization of p120 Catenin During Epithelial to Mesenchymal Transition of Colon Carcinoma Is Prognostic for Aggressive Disease. Cancer Res. 2005, 65, 10938–10945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paredes, J.; Correia, A.L.; Ribeiro, A.S.; Milanezi, F.; Cameselle-Teijeiro, J.; Schmitt, F.C. Breast carcinomas that co-express E- and P-cadherin are associated with p120-catenin cytoplasmic localisation and poor patient survival. J. Clin. Pathol. 2008, 61, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Dohn, M.R.; Brown, M.V.; Reynolds, A.B. Association of Rho-associated protein kinase 1 with E-cadherin complexes is mediated by p120-catenin. Mol. Biol. Cell 2012, 23, 99–110. [Google Scholar] [CrossRef]
- Cheung, L.W.T.; Leung, P.C.K.; Wong, A.S.T. Cadherin switching and activation of p120 catenin signaling are mediators of gonadotropin-releasing hormone to promote tumor cell migration and invasion in ovarian cancer. Oncogene 2010, 29, 2427–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schackmann, R.C.J.; van Amersfoort, M.; Haarhuis, J.H.I.; Vlug, E.J.; Halim, V.A.; Roodhart, J.M.L.; Vermaat, J.S.; Voest, E.E.; van der Groep, P.; van Diest, P.J.; et al. Cytosolic p120-catenin regulates growth of metastatic lobular carcinoma through Rock1-mediated anoikis resistance. J. Clin. Investig. 2011, 121, 3176–3188. [Google Scholar] [CrossRef] [PubMed]
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J. Cell Biol. 1999, 147, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Mackowiak, I.I.; Gentile, L.B.; Chaible, L.M.; Nagamine, M.K.; Guerra, J.M.; Mota, E.F.F.; Matera, J.M.; Mennecier, G.; Sanches, D.S.; Dagli, M.L.Z. E-cadherin in canine mast cell tumors: Decreased expression and altered subcellular localization in Grade 3 tumors. Vet. J. 2012, 194, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Gottardi, C.J.; Wong, E.; Gumbiner, B.M. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J. Cell Biol. 2001, 153, 1049–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, L.V.; Green, K.J.; Dubash, A.D. Cadherins in Cancer. In The Cadherin Superfamily; Suzuki, S., Hirano, S., Eds.; Springer: Tokyo, Japan, 2016. [Google Scholar] [CrossRef]
- Figueira, A.C.; Teodósio, A.S.; Carvalheira, J.; Lacerda, M.; de Matos, A.; Gärtner, F. P-cadherin expression in feline mammary tissues. Vet. Med. Int. 2012, 2012, 687424. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.S.; Albergaria, A.; Sousa, B.; Correia, A.L.; Bracke, M.; Seruca, R.; Schmitt, F.C.; Paredes, J. Extracellular cleavage and shedding of P-cadherin: A mechanism underlying the invasive behaviour of breast cancer cells. Oncogene 2010, 29, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Guerra, M.F.; Marazuela, E.G.; Fernández-Contreras, M.E.; Gamallo, C. P-cadherin expression reduced in squamous cell carcinoma of the oral cavity. Cancer 2005, 103, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.A.; Jung, E.J.; Lee, H.S.; Lee, H.E.; Yang, H.-K.; Oh, D.-Y.; Bang, Y.-J.; Kim, W.H. P-cadherin expression in gastric carcinoma: Its regulation mechanism and prognostic significance. Hum. Pathol. 2010, 41, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, K.; Nakagawa, H.; Hosokawa, M.; Nakamura, T.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Katagiri, T.; Nakamura, Y. Overexpressed P-Cadherin/CDH3 Promotes Motility of Pancreatic Cancer Cells by Interacting with p120ctn and Activating Rho-Family GTPases. Cancer Res. 2005, 65, 3092–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jager, T.; Becker, M.; Eisenhardt, A.; Tilki, D.; Totsch, M.; Schmid, K.W.; Romics, I.; Rubben, H.; Ergun, S.; Szarvas, T. The prognostic value of cadherin switch in bladder cancer. Oncol. Rep. 2010, 23, 1125–1132. [Google Scholar] [CrossRef]
- Riener, M.-O.; Vogetseder, A.; Pestalozzi, B.C.; Clavien, P.-A.; Probst-Hensch, N.; Kristiansen, G.; Jochum, W. Cell adhesion molecules P-cadherin and CD24 are markers for carcinoma and dysplasia in the biliary tract. Hum. Pathol. 2010, 41, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.; Valletta, D.; Bauer, K.; Thasler, W.E.; Hartmann, A.; Müller, M.; Reichert, T.E.; Hellerbrand, C. Downregulation of P-cadherin expression in hepatocellular carcinoma induces tumorigenicity. Int. J. Clin. Exp. Pathol. 2014, 7, 6125–6132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.-Q.; Wang, Z.; Leng, P. Aberrant N-cadherin expression in cancer. Biomed. Pharmacother. 2019, 118, 109320. [Google Scholar] [CrossRef]
- Saadatmand, S.; de Kruijf, E.M.; Sajet, A.; Dekker-Ensink, N.G.; van Nes, G.H.; Putter, H. Expression of cell adhesion molecules and prognosis in breast cancer. Br. J. Surg. 2013, 100, 252–260. [Google Scholar] [CrossRef]
- Drivalos, A.; Chrisofos, M.; Efstathiou, E.; Kapranou, A.; Kollaitis, G.; Koutlis, G.; Antoniou, N.; Karanastasis, D.; Dimopoulos, M.A.; Bamias, A. Expression of alpha5-integrin, alpha7-integrin, Epsilon-cadherin, and N-cadherin in localized prostate cancer. Urol. Oncol. 2016, 34, e11–e18. [Google Scholar] [CrossRef]
- Hui, L.; Zhang, S.; Dong, X.; Tian, D.; Cui, Z.; Qiu, X. Prognostic significance of twist and N-cadherin expression in NSCLC. PLoS ONE 2013, 23, e62171. [Google Scholar] [CrossRef] [Green Version]
- Muramaki, M.; Miyake, H.; Terakawa, T.; Kusuda, Y.; Fujisawa, M. Expression profile of E-cadherin and N-cadherin in urothelial carcinoma of the upper urinary tract is associated with disease recurrence in patients undergoing nephroureterectomy. Urology 2011, 78, e7–e12. [Google Scholar] [CrossRef]
- Seo, D.D.; Lee, H.C.; Kim, H.J.; Min, H.J.; Kim, K.M.; Lim, Y.S.; Chung, Y.H.; Lee, Y.S.; Suh, D.J.; Yu, E.; et al. Neural cadherin overexpression is a predictive marker for early postoperative recurrence in hepatocellular carcinoma patients. J. Gastroenterol. Hepatol. 2008, 23, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Reginato, A.; Girolami, D.; Menchetti, L.; Foiani, G.; Mandara, M.T. E-cadherin, N-cadherin Expression and Histologic Characterization of Canine Choroid Plexus Tumors. Vet. Pathol. 2016, 53, 788–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buendia, A.J.; Peñafiel-Verdu, C.; Navarro, J.A.; Vilafranca, M.; Sanchez, J. N-cadherin Expression in Feline Mammary Tumors Is Associated with a Reduced E-cadherin Expression and the Presence of Regional Metastasis. Vet. Pathol. 2014, 51, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Yan, L.; Liu, S.; Shan, Z.; Tian, Y.; Jin, Z. N-cadherin, a novel prognostic biomarker, drives malignant progression of colorectal cancer. Mol. Med. Rep. 2015, 12, 2999–3006. [Google Scholar] [CrossRef] [Green Version]
- Lascombe, I.; Clairotte, A.; Fauconnet, S.; Bernardini, S.; Wallerand, H.; Kantelip, B.; Bittard, H. N-Cadherin as a Novel Prognostic Marker of Progression in Superficial Urothelial Tumors. Clin. Cancer Res. 2006, 12, 2780–2787. [Google Scholar] [CrossRef] [Green Version]
- Shimazui, T.; Kojima, T.; Onozawa, M.; Suzuki, M.; Asano, T.; Akaza, H. Expression profile of N-cadherin differs from other classical cadherins as a prognostic marker in renal cell carcinoma. Oncol. Rep. 2006, 15, 1181–1184. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, B.; Sarli, G.; Preziosi, R.; Leprotti, S.; Benazzi, C. E-cadherin Expression in Canine Mammary Carcinomas with Regional Lymph Node Metastases. J. Vet. Med. Ser. A 2003, 50, 496–500. [Google Scholar] [CrossRef]
- De Matos, A.J.F.; Lopes, C.C.C.; Faustino, A.M.R.; Carvalheira, J.G.V.; Rutteman, G.R.; Gärtner, M.D.F.R.M. E-cadherin, β-catenin, invasion and lymph node metastases in canine malignant mammary tumours. APMIS 2007, 115, 327–334. [Google Scholar] [CrossRef]
- Li, Z.; Yin, S.; Zhang, L.; Liu, W.; Chen, B. Prognostic value of reduced E-cadherin expression in breast cancer: A meta-analysis. Oncotarget 2017, 8, 16445–16455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaszak, I.; Ruszczak, A.; Kanafa, S.; Kacprzak, K.; Król, M.; Jurka, P. Current biomarkers of canine mammary tumors. Acta Vet. Scand. 2018, 60, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gama, A.; Paredes, J.; Gärtner, F.; Alves, A.; Schmitt, F. Expression of E-cadherin, P-cadherin and β-catenin in canine malignant mammary tumours in relation to clinicopathological parameters, proliferation and survival. Vet. J. 2008, 177, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccari, D.A.P.C.; Pavam, M.V.; Terzian, C.B.; Pereira, R.S.; Ruiz, C.M.; Andrade, J.C. Immunohistochemical evaluation of e-cadherin, Ki-67 and PCNA in canine mammary neoplasias: Correlation of prognostic factors and clinical outcome. Pesqui. Vet. Bras. 2008, 28, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Pires, I.; Queiroga, F.L.; Alves, A.; Silva, F.; Lopes, C. Decrease of E-Cadherin Expression in Canine Cutaneous Histiocytoma Appears to be Related to its Spontaneous Regression. Anticancer Res. 2009, 29, 2713–2717. [Google Scholar]
- Fonseca-Alves, C.E.; Rodrigues, M.M.P.; de Moura, V.M.B.D.; Rogatto, S.R.; Laufer-Amorim, R. Alterations of C-MYC, NKX3.1, and E-cadherin expression in canine prostate carcinogenesis. Micro Res. Tech. 2013, 76, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Vara, J.A.; Miller, M.A.; Gilbreath, E.; Patterson, J.S. Immunohistochemical Detection of CD34, E-cadherin, Claudin-1, Glucose Transporter 1, Laminin, and Protein Gene Product 9.5 in 28 Canine and 8 Feline Meningiomas. Vet. Pathol. 2010, 47, 725–737. [Google Scholar] [CrossRef]
- Munday, J.S.; Brennan, M.M.; Kiupel, M. Altered Expression of β-catenin, E-cadherin, Cycloxygenase-2, and p53 Protein by Ovine Intestinal Adenocarcinoma Cells. Vet. Pathol. 2006, 43, 613–621. [Google Scholar] [CrossRef]
- Zappulli, V.; De Cecco, S.; Trez, D.; Caliari, D.; Aresu, L.; Castagnaro, M. Immunohistochemical Expression of E-Cadherin and β-Catenin in Feline Mammary Tumours. J. Comp. Pathol. 2012, 147, 161–170. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ware, K.E.; Xu, S.; Gilja, S.; Shetler, S.; Yang, Y.; Wang, X.; Austin, R.G.; Runyambo, D.; Hish, A.J.; et al. E-Cadherin Represses Anchorage-Independent Growth in Sarcomas through Both Signaling and Mechanical Mechanisms. Mol. Cancer Res. 2019, 17, 1391–1402. [Google Scholar] [CrossRef] [Green Version]
- Bendardaf, R.; Sharif-Askari, F.S.; Sharif-Askari, N.S.; Syrjanen, K.; Pyrhonen, S. Cytoplasmic E-Cadherin Expression Is Associated with Higher Tumour Level of VEGFA, Lower Response Rate to Irinotecan-based Treatment and Poorer Prognosis in Patients with Metastatic Colorectal Cancer. Anticancer Res. 2019, 39, 1953–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canadas, A.; Santos, M.; Medeiros, R.; Dias-Pereira, P. Influence of E-cadherin genetic variation in canine mammary tumour risk, clinicopathological features and prognosis. Vet. Comp. Oncol. 2019, 17, 489–496. [Google Scholar] [CrossRef]
- Pal, M.; Bhattacharya, S.; Kalyan, G.; Hazra, S. Cadherin profiling for therapeutic interventions in Epithelial Mesenchymal Transition (EMT) and tumorigenesis. Exp. Cell Res. 2018, 368, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.-H.; Liao, A.C.-H.; Hung, J.-H.; Lee, W.-J.; Hu, K.-C.; Lin, P.-T.; Liao, R.-F.; Chen, P.-S. α-Solanine inhibits invasion of human prostate cancer cell by suppressing epithelial-mesenchymal transition and MMPs expression. Molecules 2014, 19, 11896–11914. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Liu, J.; Li, C.; Zhao, Y. Simvastatin blocks TGF-β1-induced epithelial-mesenchymal transition in human prostate cancer cells. Oncol. Lett. 2016, 11, 3377–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Shen, C.; Wang, L.; Ma, Q.; Xia, P.; Qi, M.; Yang, M.; Han, B. Metformin inhibits epithelial-mesenchymal transition in prostate cancer cells: Involvement of the tumor suppressor miR30a and its target gene SOX4. Biochem. Biophys. Res. Commun. 2014, 452, 746–752. [Google Scholar] [CrossRef]
- Blaschuk, O.W. N-cadherin antagonists as oncology therapeutics. Philos. Trans. R Soc. Lond. B Biol. Sci. 2015, 370, 20140039. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.; Williams, G.; Gour, B.J.; Blaschuk, O.W.; Doherty, P. A Novel Family of Cyclic Peptide Antagonists Suggests That N-cadherin Specificity Is Determined by Amino Acids That Flank the HAV Motif. J. Biol. Chem. 2000, 275, 4007–4012. [Google Scholar] [CrossRef] [Green Version]
- Shintani, Y.; Fukumoto, Y.; Chaika, N.; Grandgenett, P.M.; Hollingsworth, M.A.; Wheelock, M.J.; Johnson, K.R. ADH-1 suppresses N-cadherin-dependent pancreatic cancer progression. Int. J. Cancer 2008, 122, 71–77. [Google Scholar] [CrossRef]
- Perotti, A.; Sessa, C.; Mancuso, A.; Noberasco, C.; Cresta, S.; Locatelli, A.; Carcangiu, M.L.; Passera, K.; Braghetti, A.; Scaramuzza, D.; et al. Clinical and pharmacological phase I evaluation of Exherin™ (ADH-1), a selective anti-N-cadherin peptide in patients with N-cadherin-expressing solid tumours. Ann. Oncol. 2009, 20, 741–745. [Google Scholar] [CrossRef]
- Devemy, E.; Blaschuk, O.W. Identification of a novel N-cadherin antagonist. Peptides 2008, 29, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, X.; Qin, S.; Wang, H.; Du, N.; Li, Y.; Pang, Y.; Wang, C.; Xu, C.; Ren, H. CDH1 promoter methylation correlates with decreased gene expression and poor prognosis in patients with breast cancer. Oncol. Lett. 2016, 11, 2635–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Yao, X.; Cao, Q.; Wu, Z.; Wang, Z.; Liu, F.; Shen, L. Clinicopathological and prognostic significance of CDH1 hypermethylation in hepatocellular carcinoma: A meta-analysis. Cancer Manag. Res. 2019, 11, 857–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buda, A.; Pignatelli, M. E-Cadherin and the Cytoskeletal Network in Colorectal Cancer Development and Metastasis. Cell Commun. Adhes. 2011, 18, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, D.J.; Mikhaylova, L.; Fedulov, A.V. Selective DNA demethylation by fusion of TDG with a sequence-specific DNA-binding domain. Epigenetics 2012, 7, 344–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battistini, C.; Rehman, M.; Avolio, M.; Arduin, A.; Valdembri, D.; Serini, G.; Tamagnone, L. Rhomboid-Like-2 Intramembrane Protease Mediates Metalloprotease-Independent Regulation of Cadherins. Int. J. Mol. Sci. 2019, 20, 5958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, A.; Kolligs, F.T. Wnt Signaling as a Therapeutic Target for Cancer. In Target Discovery and Validation Reviews and Protocols: Volume 2: Emerging Molecular Targets and Treatment Options; Sioud, M., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 63–91. [Google Scholar] [CrossRef]
- Liu, L.X.; Lee, N.P.; Chan, V.W.; Xue, W.; Zender, L.; Zhang, C.; Mao, M.; Dai, H.; Wang, X.L.; Xu, M.Z. Targeting cadherin-17 inactivates Wnt signaling and inhibits tumor growth in liver carcinoma. Hepatology 2009, 50, 1453–1463. [Google Scholar] [CrossRef]
- Ciołczyk-Wierzbicka, D.; Laidler, P. The inhibition of invasion of human melanoma cells through N-cadherin knock-down. Med. Oncol. (Northwood Lond. Engl.) 2018, 35, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; He, W.; Lin, N.; Wang, X.; Fan, Q.-X. N-cadherin knock-down decreases invasiveness of esophageal squamous cell carcinoma in vitro. World J. Gastroenterol. 2009, 15, 697–704. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaszak, I.; Witkowska-Piłaszewicz, O.; Niewiadomska, Z.; Dworecka-Kaszak, B.; Ngosa Toka, F.; Jurka, P. Role of Cadherins in Cancer—A Review. Int. J. Mol. Sci. 2020, 21, 7624. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207624
Kaszak I, Witkowska-Piłaszewicz O, Niewiadomska Z, Dworecka-Kaszak B, Ngosa Toka F, Jurka P. Role of Cadherins in Cancer—A Review. International Journal of Molecular Sciences. 2020; 21(20):7624. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207624
Chicago/Turabian StyleKaszak, Ilona, Olga Witkowska-Piłaszewicz, Zuzanna Niewiadomska, Bożena Dworecka-Kaszak, Felix Ngosa Toka, and Piotr Jurka. 2020. "Role of Cadherins in Cancer—A Review" International Journal of Molecular Sciences 21, no. 20: 7624. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207624