Ligand-Based Pharmacophore Modeling, Molecular Docking, and Molecular Dynamic Studies of Dual Tyrosine Kinase Inhibitor of EGFR and VEGFR2



Abstract
:1. Introduction
2. Results and Discussion
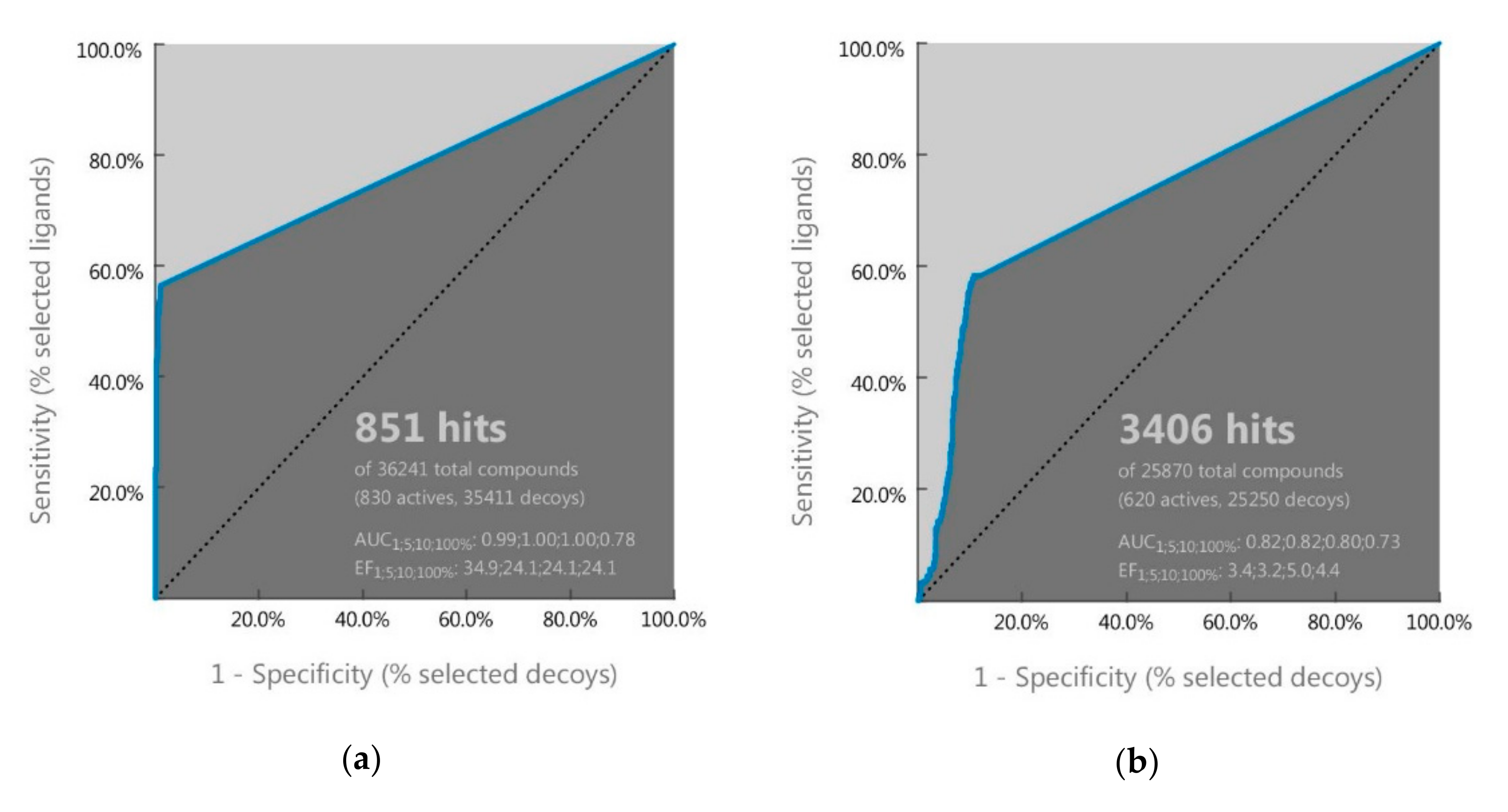
2.1. Ligand-Based Pharmacophore Screening
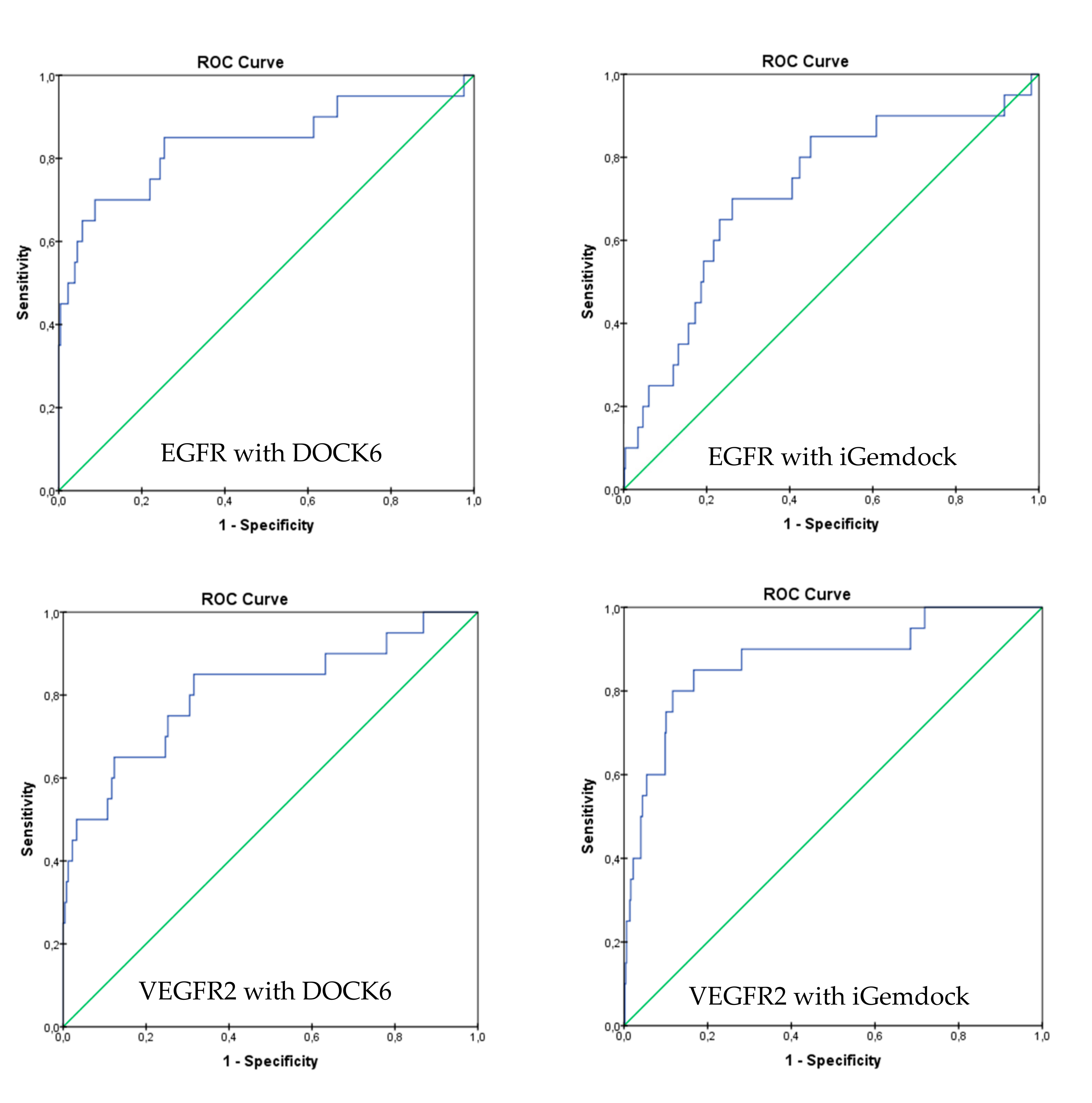
2.2. Molecular Docking Screening
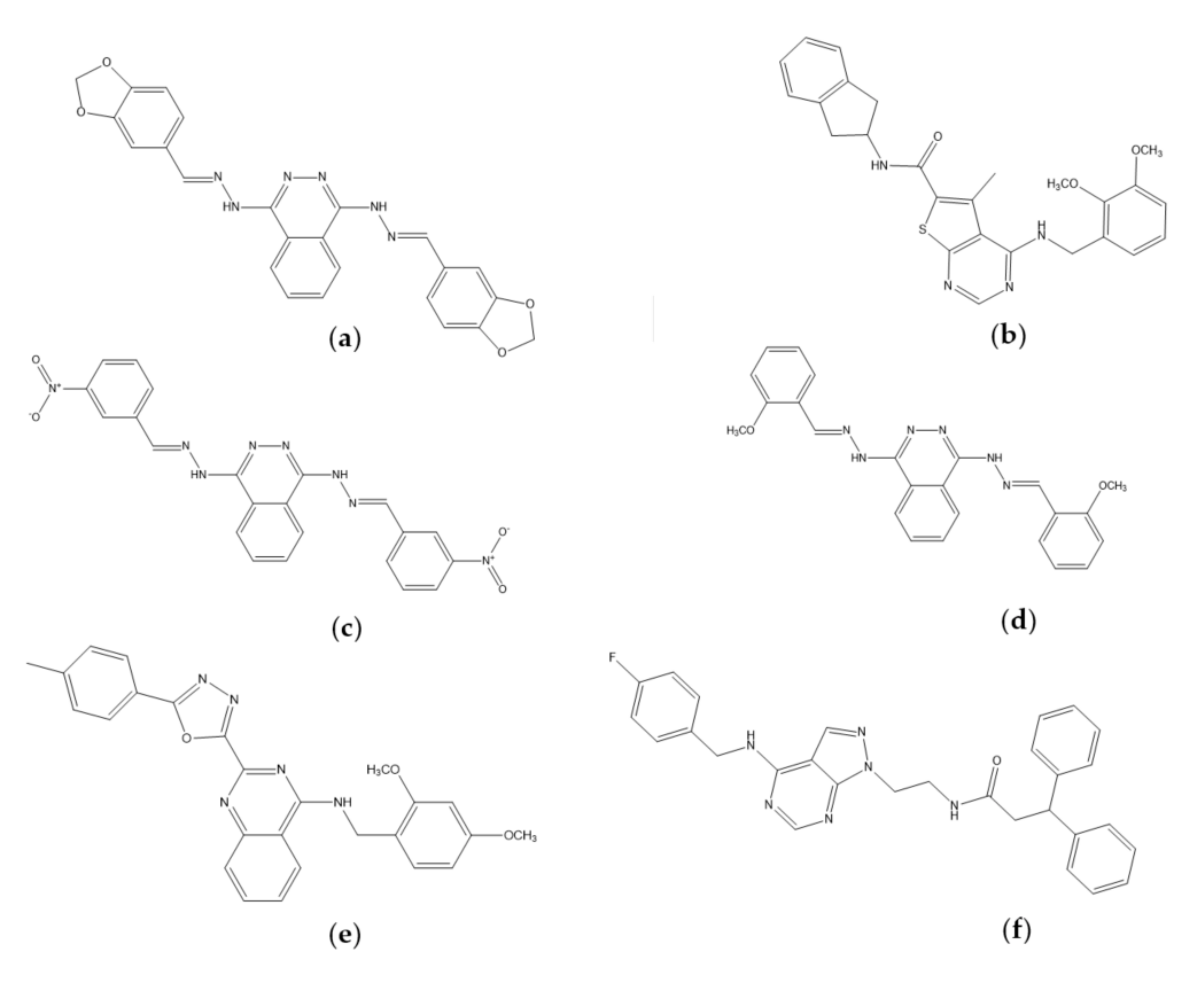
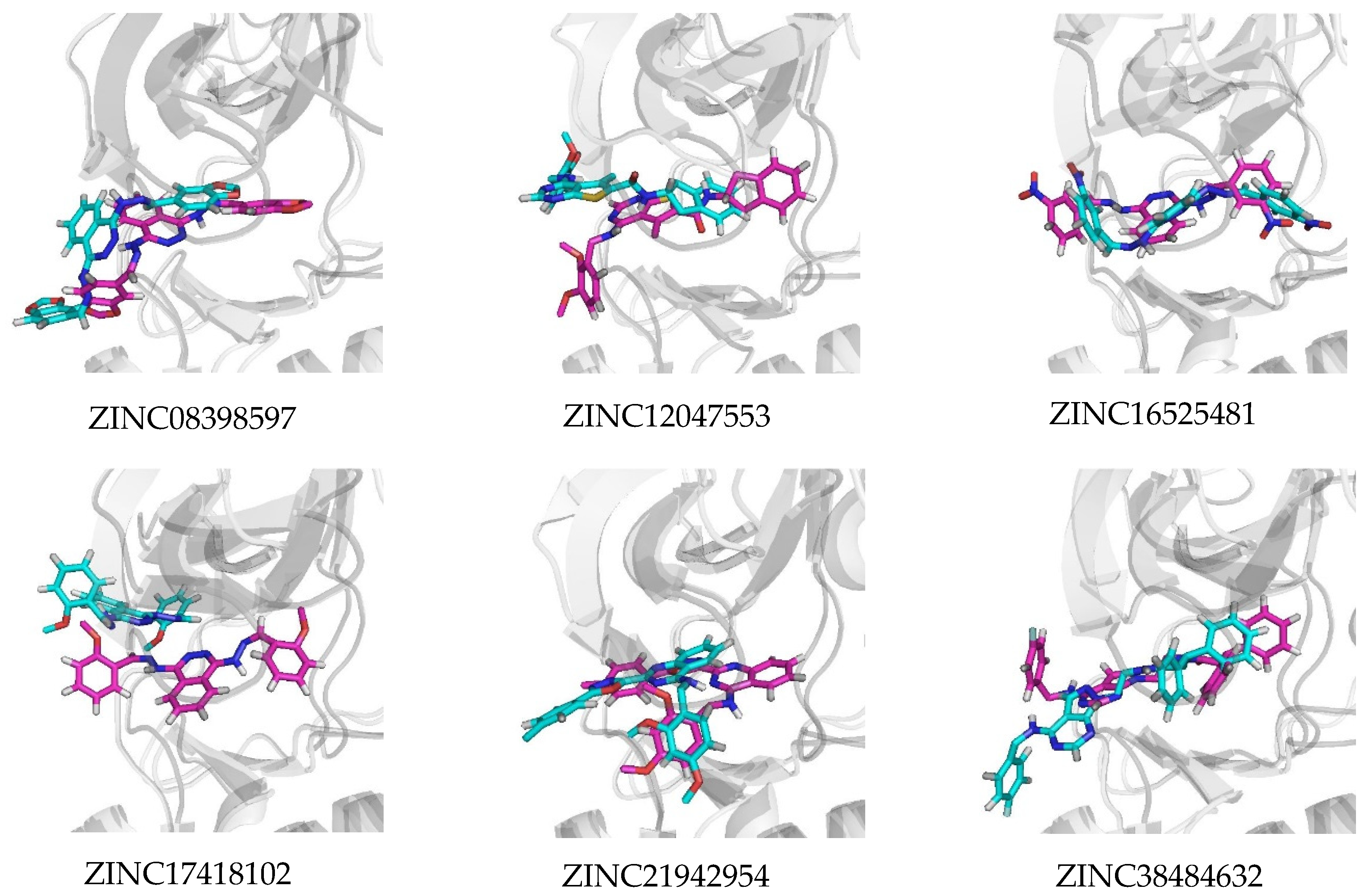
2.3. Molecular Docking Analysis
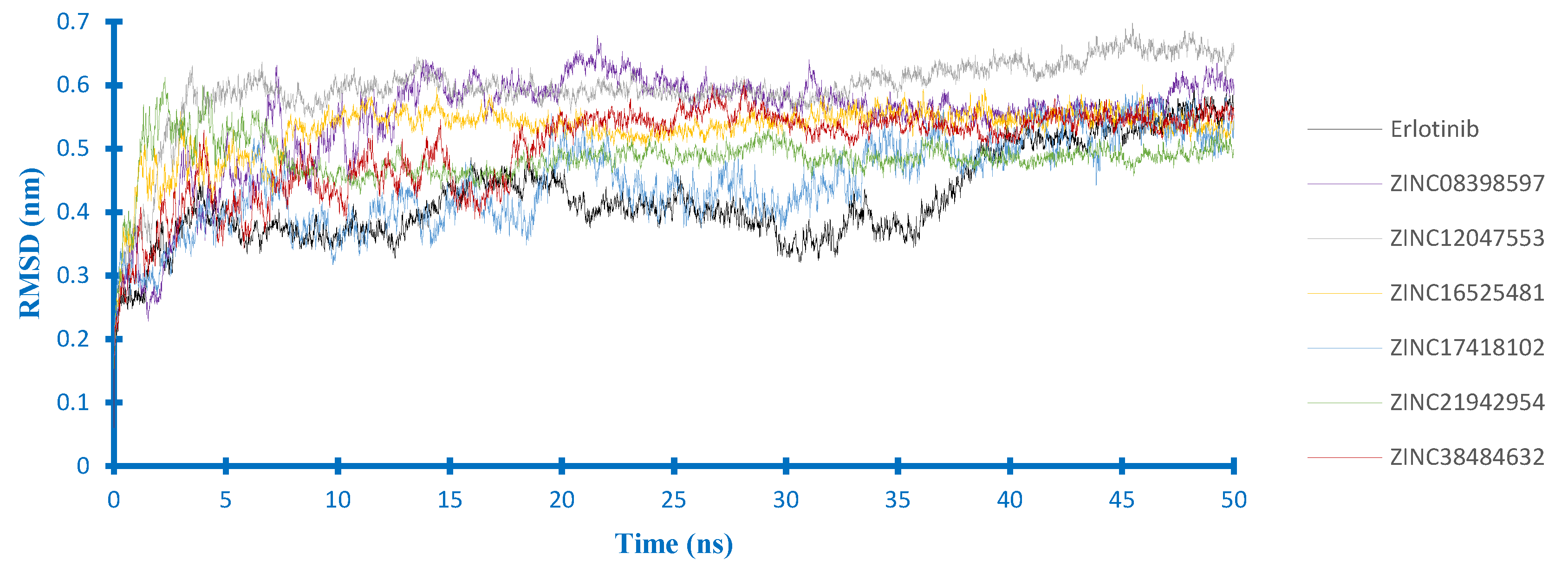
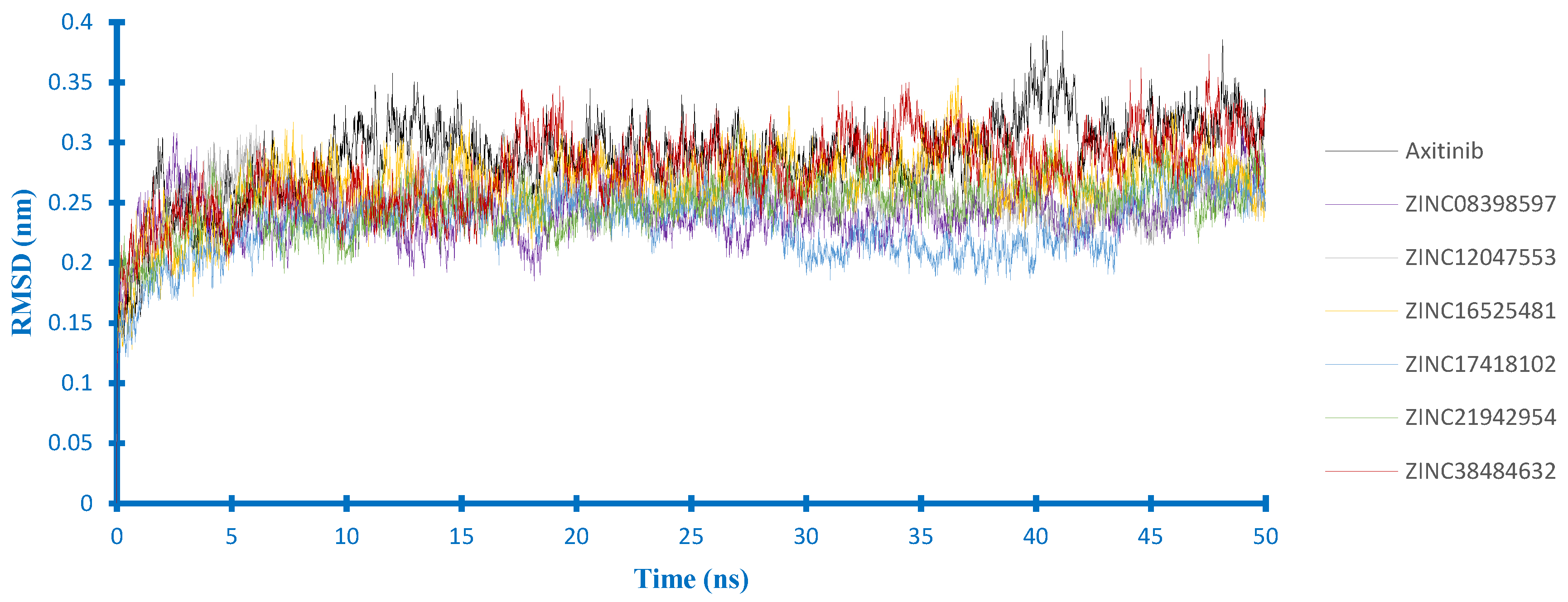
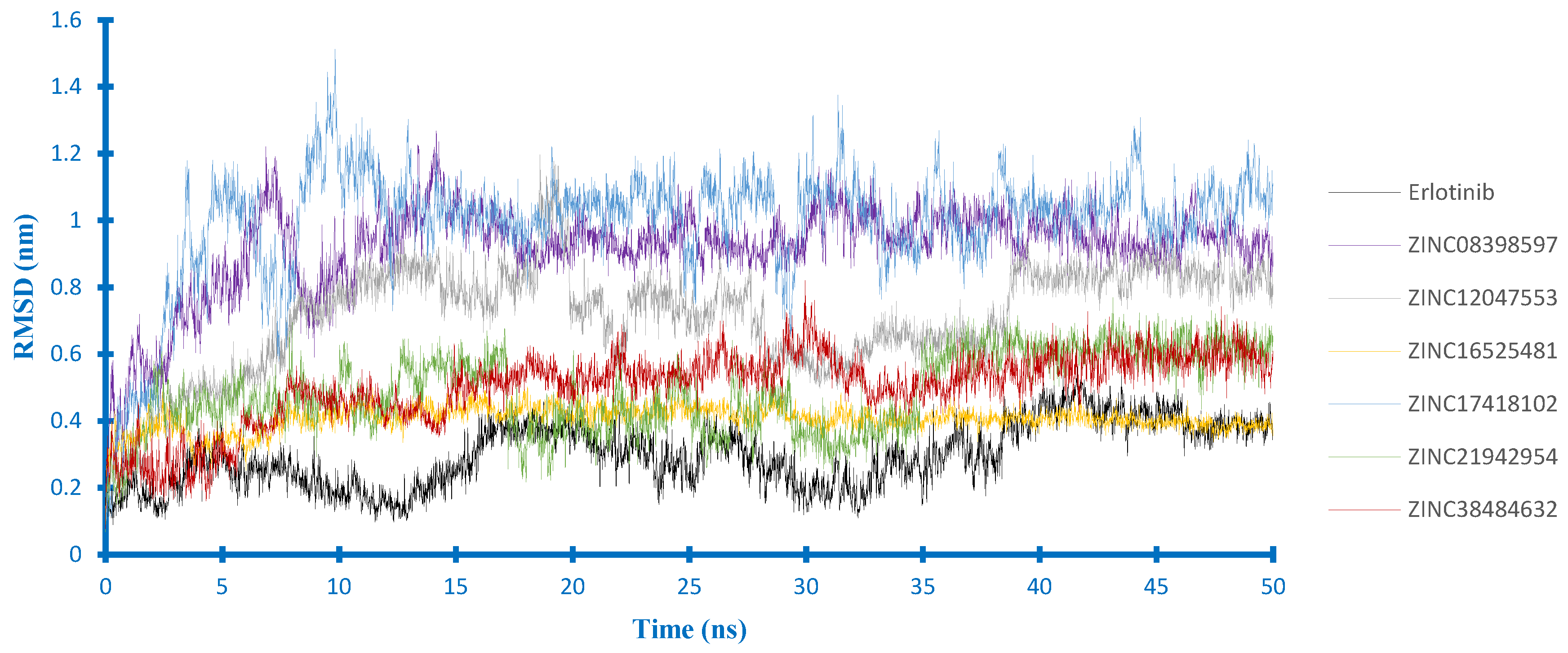
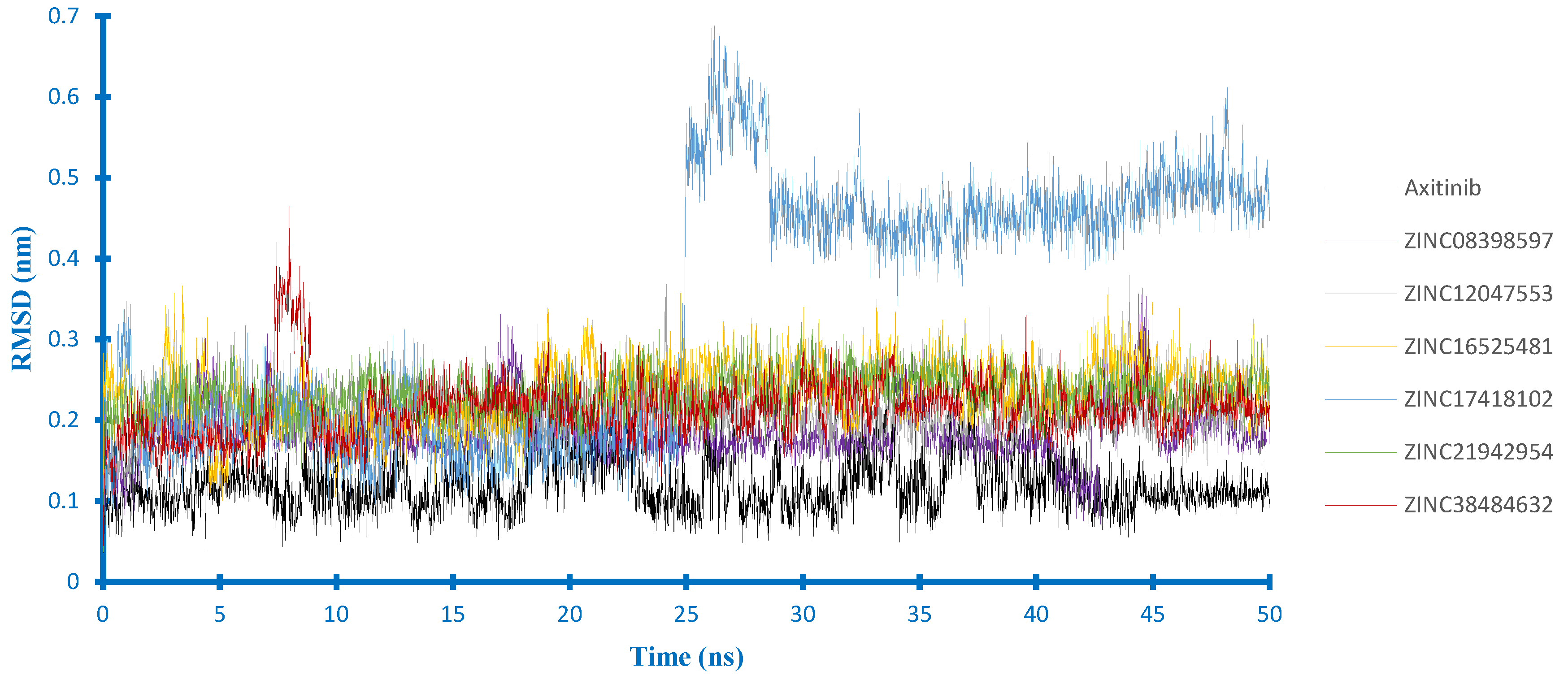
2.4. Molecular Dynamics Studies
2.4.1. Stability Analysis
2.4.2. Hydrogen Bond Analysis
2.4.3. MMPBSA Analysis
3. Materials and Methods
3.1. Ligand-Based Pharmacophore Modeling
3.2. Molecular Docking
3.3. Molecular Dynamics Simulations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Broekman, F. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J. Clin. Oncol. 2011, 2, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Harari, P.M. Epidermal growth factor receptor inhibition strategies in oncology. Endocr. Relat. Cancer 2004, 11, 689–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yewale, C.; Baradia, D.; Vhora, I.; Patil, S.; Misra, A. Epidermal growth factor receptor targeting in cancer: A review of trends and strategies. Biomaterials 2013, 34, 8690–8707. [Google Scholar] [CrossRef]
- Pennell, N.A.; Lynch, T.J. Combined Inhibition of the VEGFR and EGFR Signaling Pathways in the Treatment of NSCLC. Oncologist 2009, 14, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J. The role of VEGF and EGFR inhibition: Implications for combining Anti-VEGF and Anti-EGFR Agents. Mol. Cancer Res. 2007, 5, 203–220. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Mazumdar, A.; Dash, R.; Sarkar, D.; Fisher, P.B.; Mandal, M. ZD6474, a dual tyrosine kinase inhibitor of EGFR and VEGFR-2, inhibits MAPK/ERK and AKT/PI3-K and induces apoptosis in breast cancer cells. Cancer Biol. Ther. 2010, 9, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Grande, E.; Kreissl, M.C.; Filetti, S.; Newbold, K.; Reinisch, W.; Robert, C.; Schlumberger, M.; Tolstrup, L.K.; Zamorano, J.L.; Capdevila, J. Vandetanib in advanced medullary thyroid cancer: Review of adverse event management strategies. Adv. Ther. 2013, 30, 945–966. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.L.; Lima, L.M.; Tesch, R.; Sant’Anna, C.M.; Totzke, F.; Kubbutat, M.H.; Schächtele, C.; Laufer, S.A.; Barreiro, E.J. Novel 2-chloro-4-anilino-quinazoline derivatives as EGFR and VEGFR-2 dual inhibitors. Eur. J. Med. Chem. 2014, 71, 1–14. [Google Scholar] [CrossRef]
- Zhang, H.Q.; Gong, F.H.; Li, C.G.; Zhang, C.; Wang, Y.J.; Xu, Y.G.; Sun, L.P. Design and discovery of 4-anilinoquinazoline-acylamino derivatives as EGFR and VEGFR-2 dual TK inhibitors. Eur. J. Med. Chem. 2016, 109, 371–379. [Google Scholar] [CrossRef]
- Li, Y.; Tan, C.; Gao, C.; Zhang, C.; Luan, X.; Chen, X.; Liu, H.; Chen, Y.; Jiang, Y. Discovery of benzimidazole derivatives as novel multi-target EGFR, VEGFR-2 and PDGFR kinase inhibitors. Bioorg. Med. Chem. 2011, 19, 4529–4535. [Google Scholar] [CrossRef]
- Amin, K.M.; Barsoum, F.F.; Awadallah, F.M.; Mohamed, N.E. Identification of new potent phthalazine derivatives with VEGFR-2 and EGFR kinase inhibitory activity. Eur. J. Med. Chem. 2016, 123, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Bhunia, S.S.; Balaramnavar, V.M.; Saxena, A.K. Pharmacophore modelling, molecular docking and virtual screening for EGFR (HER 1) tyrosine kinase inhibitors. SAR QSAR Environ. Res. 2011, 22, 239–263. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sun, X.; Zhao, H.; Tang, Y.; Lan, M. Discovery of novel EGFR tyrosine kinase inhibitors by structure-based virtual screening. Bioorg. Med. Chem. Lett. 2012, 22, 4004–4009. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Jeong, K.W.; Lee, Y.; Song, J.Y.; Kim, M.S.; Lee, G.S.; Kim, Y. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur. J. Med. Chem. 2010, 45, 5420–5427. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, N.; Luo, K.; Zhang, W.; Li, X.; Wu, C.; Bao, J. In silico discovery of potential VEGFR-2 inhibitors from natural derivatives for anti-angiogenesis therapy. Int. J. Mol. Sci. 2014, 15, 15994–16011. [Google Scholar] [CrossRef] [Green Version]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef]
- Hosmer, D.W.; Lemeshow, S.; Sturdivant, R.X. Applied Logistic Regression, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; p. 177. [Google Scholar]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Nasab, R.R.; Mansourian, M.; Hassanzadeh, F.; Shahlaei, M. Exploring the interaction between epidermal growth factor receptor tyrosine kinase and some of the synthesized inhibitors using combination of in-silico and in-vitro cytotoxicity methods. Res. Pharm. Sci. 2018, 13, 509–522. [Google Scholar]
- Yang, Y.A.; Tang, W.J.; Zhang, X.; Yuan, J.W.; Liu, X.H.; Zhu, H.L. Synthesis, molecular docking and biological evaluation of Glycyrrhizin analogs as anticancer agents targeting EGFR. Molecules 2014, 19, 6368–6381. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.H.; Shiao, H.Y.; Tu, C.H.; Liu, P.M.; Hsu, J.T.; Amancha, P.K.; Wu, J.S.; Coumar, M.S.; Chen, C.H.; Wang, S.Y.; et al. Protein kinase inhibitor design by targeting the Asp-Phe-Gly (DFG) motif: The role of the DFG motif in the design of epidermal growth factor receptor inhibitors. J. Med. Chem. 2013, 56, 3889–3903. [Google Scholar] [CrossRef] [PubMed]
- Sanphanya, K.; Wattanapitayakul, S.K.; Phowichit, S.; Fokin, V.V.; Vajragupta, O. Novel VEGFR-2 kinase inhibitors identified by the back-to-front approach. Bioorg. Med. Chem. Lett. 2013, 23, 2962–2967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Structures, C.; Indices, P.; Studies, E.; Parade, C. Selecting protein structure/s for docking-based virtual screening: A case study on type II inhibitors of VEGFR-2 Kinase. Int. J. Pharm. Sci. Res. 2019, 10, 2998–3011. [Google Scholar]
- Desheng, L.; Jian, G.; Yuanhua, C.; Wei, C.; Huai, Z.; Mingjuan, J. Molecular dynamics simulations and MM/GBSA methods to investigate binding mechanisms of aminomethylpyrimidine inhibitors with DPP-IV. Bioorg. Med. Chem. Lett. 2011, 21, 6630–6635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, S.; Jiao, Y.; Liu, H.; Yuan, H.; Lu, S.; Ran, T.; Yao, S.; Ke, Z.; Xu, J.; et al. An integrated virtual screening approach for VEGFR-2 inhibitors. J. Chem. Inf. Model. 2013, 53, 3163–3177. [Google Scholar] [CrossRef] [PubMed]
- Froimowitz, M. HyperChem: A software package for computational chemistry and molecular modeling. Biotechniques 1993, 14, 1010–1013. [Google Scholar]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, 409–414. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC-A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.M.; Chen, C.C. GEMDOCK: A Generic Evolutionary Method for Molecular Docking. Proteins 2004, 55, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modeling of protein structures and complexes. Nucleic Acids Res. 2018, 46, 296–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated Topology Builder Version 3.0: Prediction of Solvation Free Enthalpies in Water and Hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A. g-mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | DOCK6 Score | iGemdock Score | H-Bond | Hydrophobic interaction |
|---|---|---|---|---|
| Erlotinib | −64.26 | −107.61 | Met769 | Leu694, Ala719, Lys721, Leu764, Thr766, Leu768, Gly772, Leu820, Asp831 |
| ZINC08398597 | −68.76 | −129.93 | Met769 | Leu694, Ala719, Val702, Leu768, Pro770, Gly772, Asp776, Tyr777, Leu820 |
| ZINC12047553 | −72.76 | −125.93 | Met769 | Leu694, Ala719, Val702, Lys721, Met742, Leu764, Pro770, Gly772, Asp776, Leu820 |
| ZINC16525481 | −73.03 | −128.81 | Lys692, Lys721, Asp831 | Leu694, Ala719, Lys721, Met742, Pro770, Gly772, Cys773, Leu820 |
| ZINC17418102 | −65.89 | −122.16 | - | Lys692, Leu694, Ala719, Val702, Lys704, Lys721, Leu764, Thr766, Pro770, Gly772, Leu820, Asp831 |
| ZINC21942954 | −72.93 | −117.69 | Met769, Cys773, Asp831 | Leu694, Gly695, Lys721, Leu768, Pro770, Gly772, Leu820 |
| ZINC38484632 | −72.97 | −111.43 | Met769, Pro770 | Lys692, Leu694, Ala719, Val702, Lys721, Met742, Thr766, Leu768, Gly772, Leu820 |
| Ligand | DOCK6 Score | iGemdock Score | H-Bond | Hydrophobic interaction |
|---|---|---|---|---|
| Axitinib | −83.00 | −149.07 | Glu885, Glu917, Cys919, Asp1046 | Leu840, Ala866, Lys868, Val916, Phe918, Gly992, Leu1035, Phe1047 |
| ZINC08398597 | −89.65 | −156.25 | Glu885, Asp1046 | Leu840, Val848, Ala866, Val867, Lys868, Leu889, Val914, Val916, Gly992, Leu1019, His1026, Leu1035, Phe1047 |
| ZINC12047553 | −85.20 | −168.46 | Asp1046 | Leu840, Val848, Ala866, Lys868, Glu885, Val899, Val914, Val916, Phe918, Cys919, Gly992, His1026, Leu1035, Ile1044, Cys1045, Phe1047 |
| ZINC16525481 | −94.84 | −162.89 | Glu885, Asp1046 | Leu840, Val848, Ala866, Val867, Lys868, Leu889, Val914, Val916, Phe918, His1026, Leu1035, Cys1045, Phe1047 |
| ZINC17418102 | −89.39 | −155.33 | Glu885, Asp1046 | Leu840, Val848, Ala866, Val867, Lys868, Leu889, Val914, Val916, Glu917, Phe918, His1026, Leu1035, Ile1044, Phe1047 |
| ZINC21942954 | −84.25 | −152.80 | Cys1045, Asp1046 | Leu840, Lys868, Leu889, Val899, Val914, Val916, Phe918, Cys919, Leu1035, Ile1044, Phe1047 |
| ZINC38484632 | −85.05 | −151.26 | Glu885, Cys1045, Asp1046 | Leu840, Val848, Ala866, Lys868, Leu889, Ile892, Val916, Cys1024, Leu1019, Ile1025, His1026, Leu1035, Phe1047 |
| Ligand | Target | |||
|---|---|---|---|---|
| EGFR | VEGFR2 | |||
| Donor-Acceptor | Occupancy (%) | Donor-Acceptor | Occupancy (%) | |
| Erlotinib | Met769 (H)---(N2) | 28.1 | - | - |
| Axitinib | - | - | (H12)---Glu917 (O) | 76.5 |
| (H1)---Glu885 (OE2) | 30.3 | |||
| (H1)---Glu885 (OE1) | 27.7 | |||
| Asp1046 (H)---(O81) | 69.5 | |||
| Cys919 (H)---(N14) | 88.8 | |||
| ZINC08398597 | Cys773 (H)---(N1) | 21.1 | (H12)---Glu885 (OE2) | 41.5 |
| Cys773 (H)---(N2) | 13.4 | (H12)---Glu885 (OE1) | 34.5 | |
| Met769 (H)---(O4) | 10.2 | Asp1046 (H)---(N6) | 32.5 | |
| Asp1046 (H)---(N1) | 45.6 | |||
| ZINC12047553 | Met769 (H)---(O1) | 25.1 | (H15)---Asp1046 (O) | 11.5 |
| ZINC16525481 | (H5)---Gln767 (O) | 74.3 | (H11)---Glu885 (OE2) | 17.6 |
| Phe832 (H)---(O) | 45.4 | (H11)---Glu885 (OE1) | 17.4 | |
| Met769 (H)---(N1) | 88 | Asp1046 (H)---(N1) | 70.2 | |
| Met769 (H)---(N2) | 73.7 | Asp1046 (H)---(N2) | 47.7 | |
| Cys919 (H)---(O) | 70.5 | |||
| ZINC17418102 | - | - | Asp1046(H)---(N6) | 41.3 |
| ZINC21942954 | Cys773 (H)---(O3) | 48.7 | (H12)---Glu885 (OE2) | 31.5 |
| (H12)---Glu885 (OE1) | 27.4 | |||
| Asp1046 (H)---(N4) | 37.4 | |||
| Asp1046 (H)---(N5) | 24.3 | |||
| ZINC38484632 | (H16)---Pro770 (O) | 10.5 | (H9)---Glu885 (OE2) | 16.8 |
| Cys773 (H)---(N4) | 16.8 | (H9)---Glu885 (OE1) | 19.8 | |
| Met769 (H)---(O1) | 92.1 | Asp1046 (H)---(O1) | 65.3 | |
| Ligands | Van der Waals Energy (ΔEvdW) | Electrostatic Energy (ΔEelec) | Polar Solvation Energy (ΔGpolar) | SASA Energy (ΔGnonpolar) | Binding Energy (ΔGbind) |
|---|---|---|---|---|---|
| Erlotinib | −232.64 ± 13.43 | −46.89 ± 10.03 | 176.32 ± 17.18 | −24.18 ± 0.92 | −127.38 ± 17.58 |
| ZINC08398597 | −220.76 ± 10.87 | −24.78 ± 7.60 | 114.24 ± 14.70 | −23.37 ± 1.72 | −154.67 ± 13.85 |
| ZINC12047553 | −183.44 ± 16.81 | −23.33 ± 7.92 | 131.99 ± 29.16 | −19.71 ± 1.58 | −94.49 ± 19.15 |
| ZINC16525481 | −223.17 ± 12.92 | −43.01 ± 12.74 | 182.64 ± 14.30 | −22.30 ± 0.99 | −105.84 ± 17.08 |
| ZINC17418102 | −131.71 ± 12.28 | −16.65 ± 10.30 | 85.25 ± 20.67 | −15.09 ± 1.43 | −78.21 ± 11.57 |
| ZINC21942954 | −200.15 ± 14.24 | −16.94 ± 12.14 | 143.44 ± 29.17 | −19.55 ± 1.2 | −93.20 ± 18.56 |
| ZINC38484632 | −179.60 ± 11.35 | −33.06 ± 8.88 | 118.60 ± 20.60 | −21.25 ± 1.24 | −115.31 ± 19.27 |
| Ligands | Van der Waals Energy (ΔEvdW) | Electrostatic Energy (ΔEelec) | Polar Solvation Energy (ΔGpolar) | SASA Energy (ΔGnonpolar) | Binding Energy (ΔGbind) |
|---|---|---|---|---|---|
| Axitinib | −202.38 ± 11.43 | −75.49 ± 10.19 | 163.49 ± 10.45 | −20.69 ± 0.63 | −135.08 ± 7.94 |
| ZINC08398597 | −267.39 ± 13.12 | −32.26 ± 8.51 | 154.24 ± 12.60 | −25.56 ± 0.76 | −170.96 ± 12.17 |
| ZINC12047553 | −258.36 ± 10.04 | −24.89 ± 8.73 | 163.99 ± 21.33 | −26.22 ± 1.16 | −145.48 ± 23.74 |
| ZINC16525481 | −272.50 ± 11.94 | −29.68 ± 8.44 | 184.00 ± 15.54 | −26.25 ± 1.03 | −144.43 ± 12.32 |
| ZINC17418102 | −252.86 ± 10.25 | −28.79 ± 6.46 | 166.57 ± 12.21 | −24.63 ± 1.13 | −139.71 ± 14.81 |
| ZINC21942954 | −268.09 ± 9.85 | −42.85 ± 6.59 | 199.97 ± 25.24 | −26.72 ± 0.73 | −137.69 ± 25.23 |
| ZINC38484632 | −260.64 ± 9.06 | −25.80 ± 7.40 | 167.18 ± 16.01 | −27.72 ± 1.15 | −146.97 ± 12.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sangande, F.; Julianti, E.; Tjahjono, D.H. Ligand-Based Pharmacophore Modeling, Molecular Docking, and Molecular Dynamic Studies of Dual Tyrosine Kinase Inhibitor of EGFR and VEGFR2. Int. J. Mol. Sci. 2020, 21, 7779. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207779
Sangande F, Julianti E, Tjahjono DH. Ligand-Based Pharmacophore Modeling, Molecular Docking, and Molecular Dynamic Studies of Dual Tyrosine Kinase Inhibitor of EGFR and VEGFR2. International Journal of Molecular Sciences. 2020; 21(20):7779. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207779
Chicago/Turabian StyleSangande, Frangky, Elin Julianti, and Daryono Hadi Tjahjono. 2020. "Ligand-Based Pharmacophore Modeling, Molecular Docking, and Molecular Dynamic Studies of Dual Tyrosine Kinase Inhibitor of EGFR and VEGFR2" International Journal of Molecular Sciences 21, no. 20: 7779. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207779