Truncating Variants Contribute to Hearing Loss and Severe Retinopathy in USH2A-Associated Retinitis Pigmentosa in Japanese Patients

Abstract
:1. Introduction
2. Results
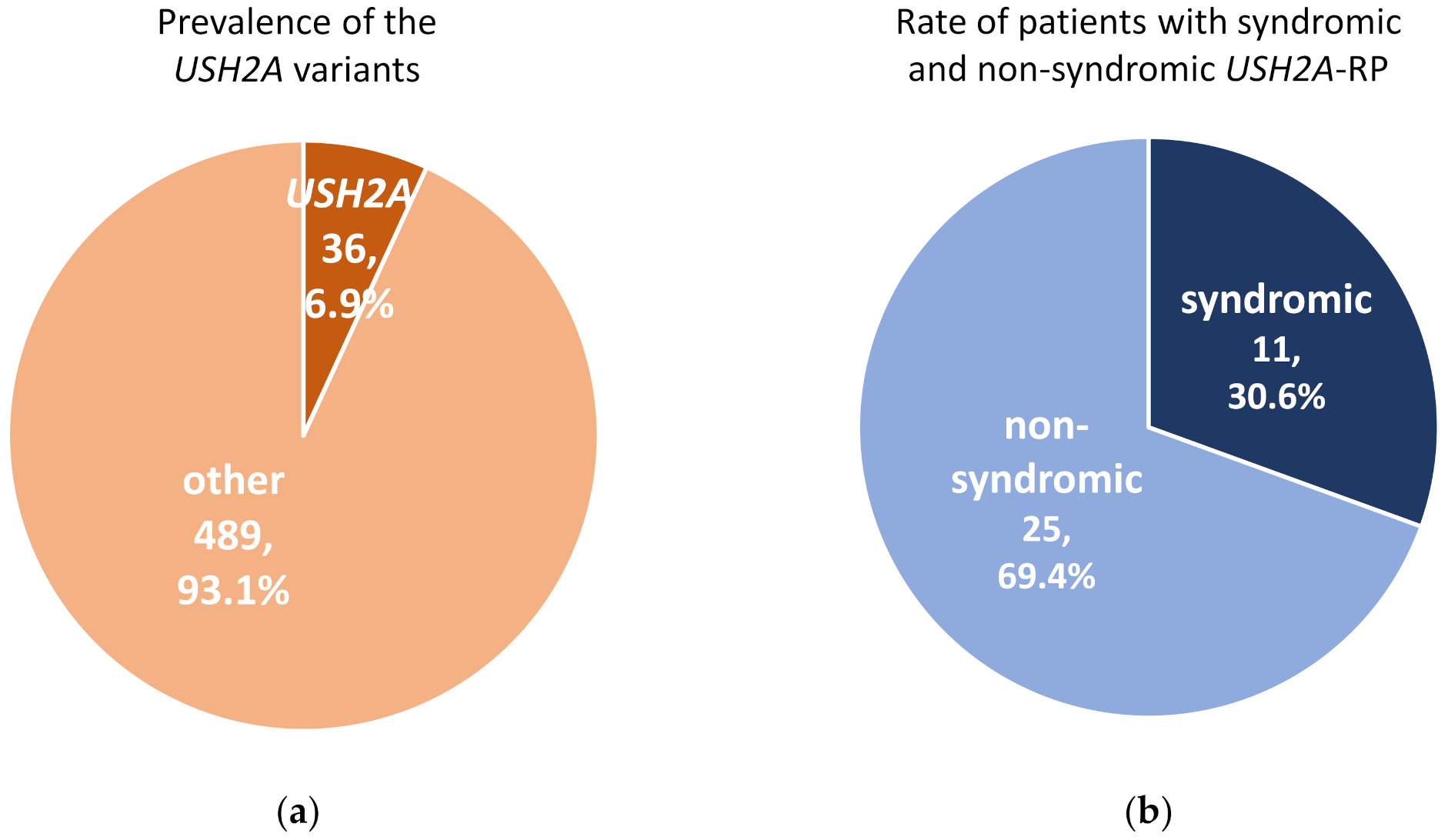
2.1. Syndromic and Non-Syndromic USH2A-RP
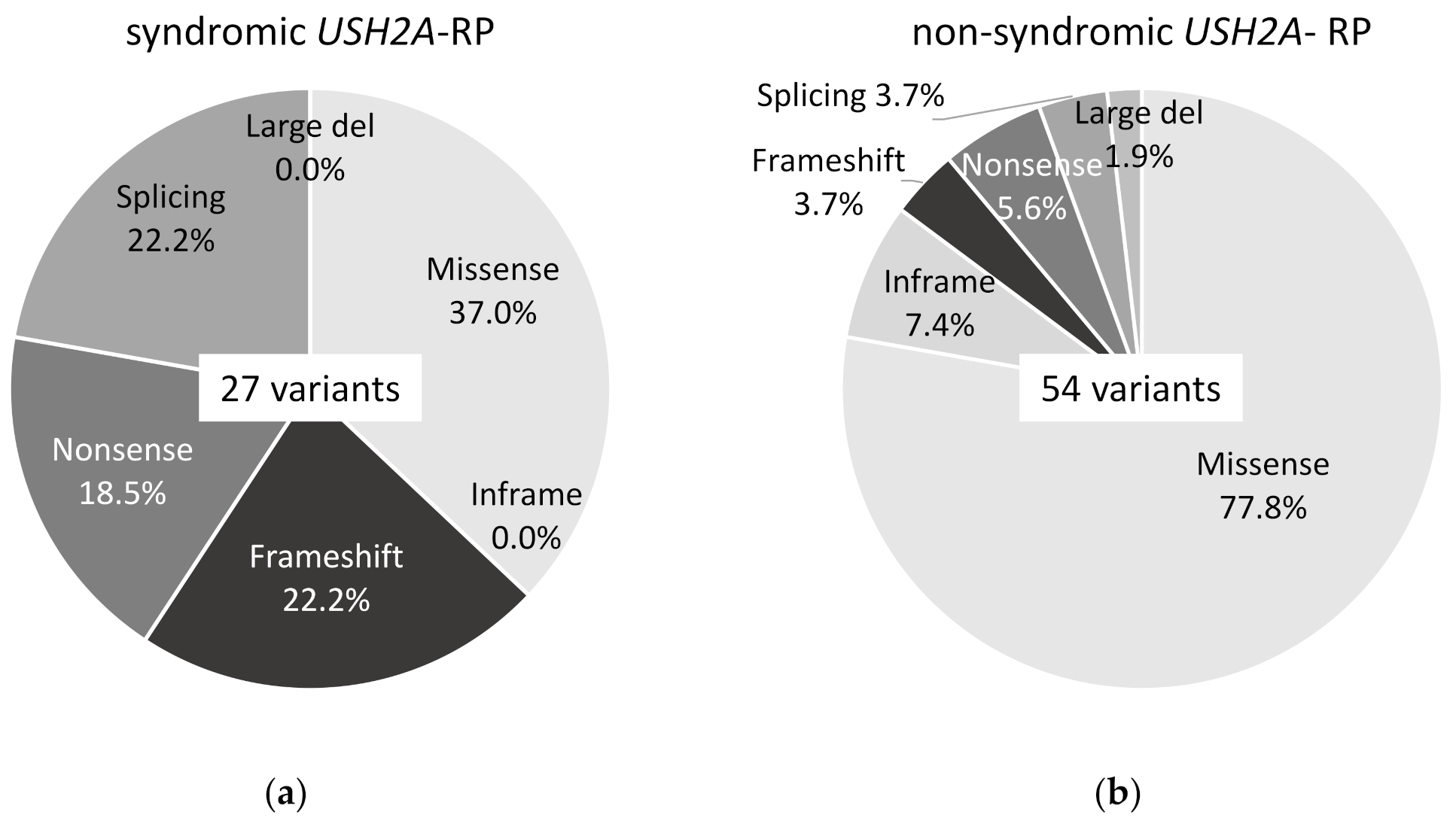
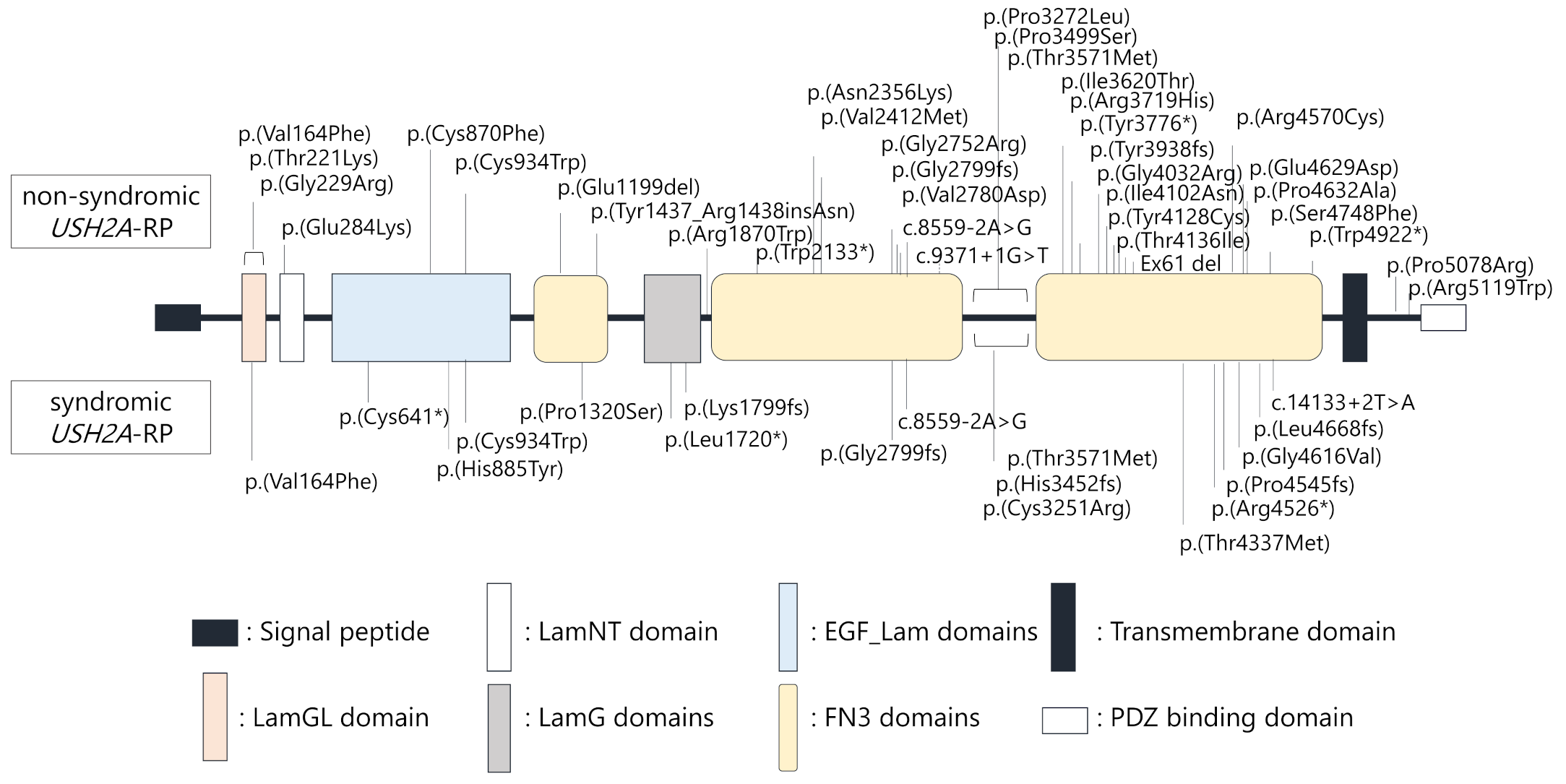
2.2. Truncating USH2A Variants were More Frequently Detected in Syndromic than Non-Syndromic USH2A-RP Patients
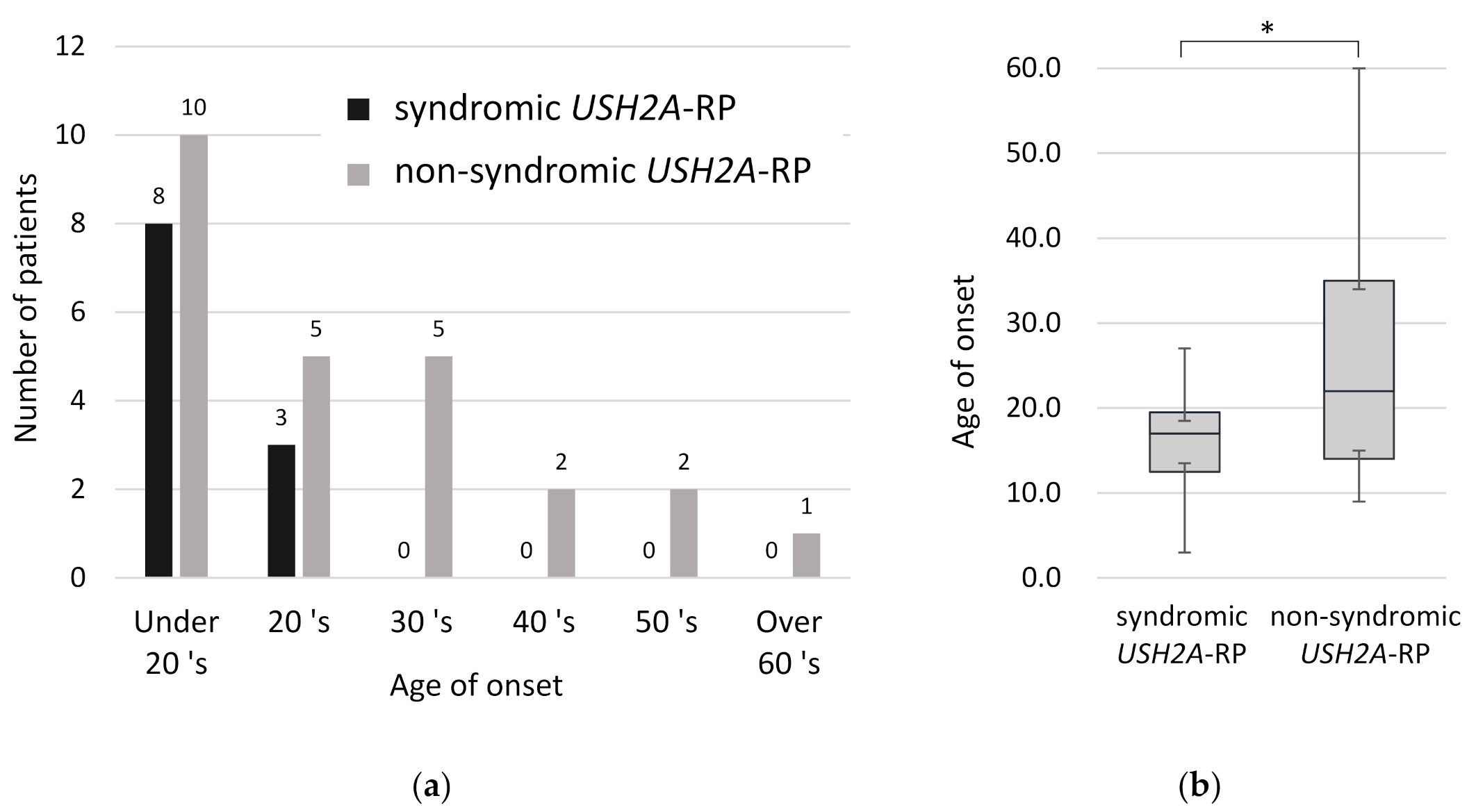
2.3. Earlier Onset of RP in Syndromic Patients than Non-Syndromic USH2A-RP Patients
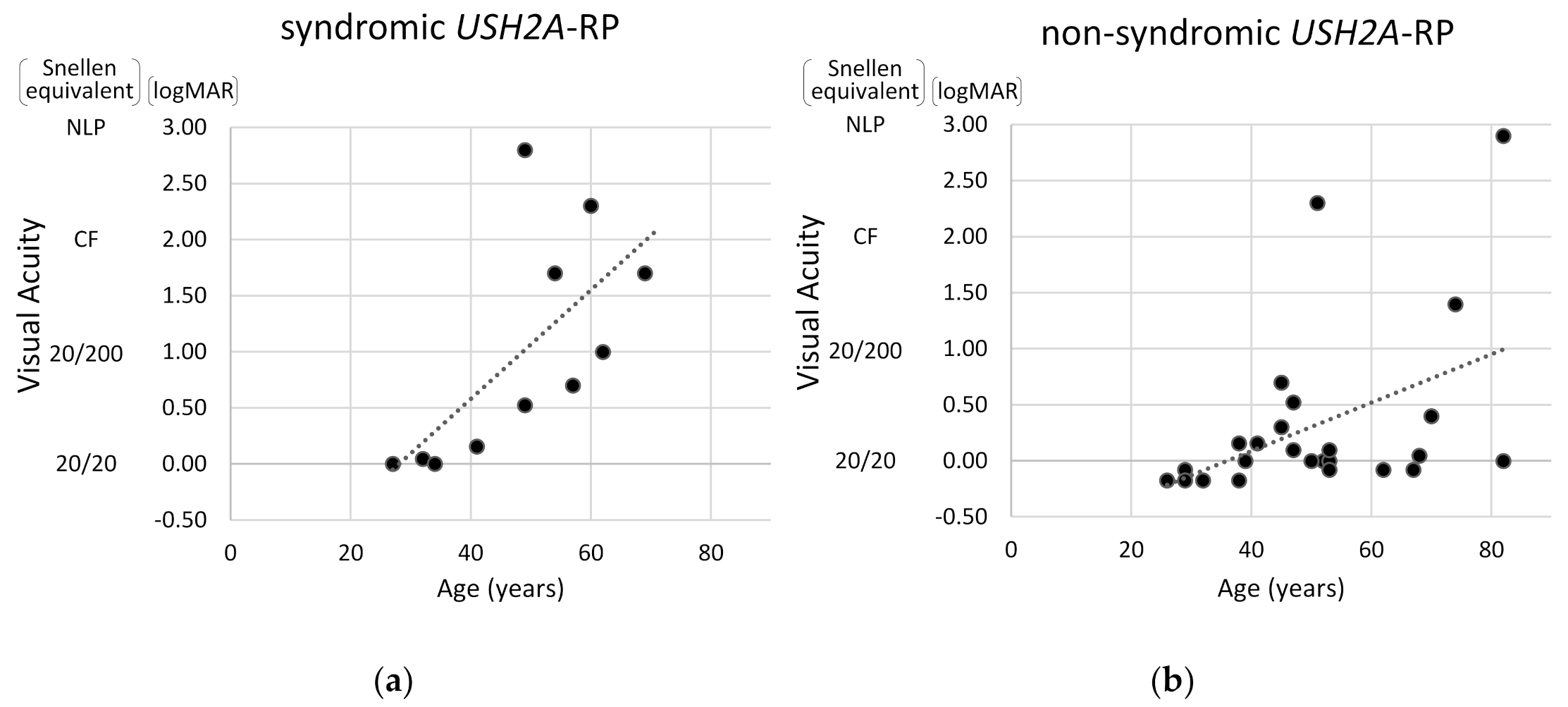
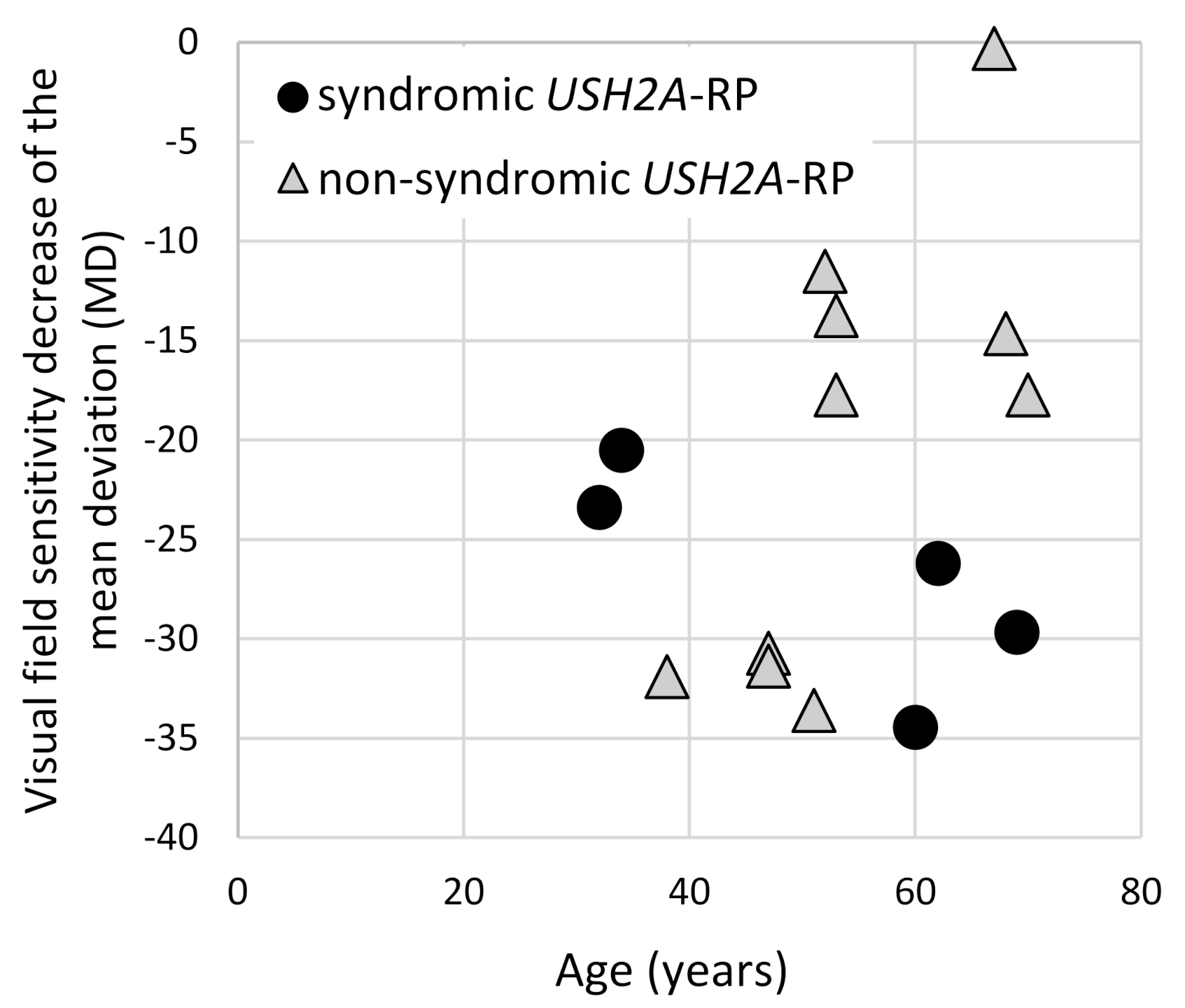
2.4. Visual Acuity and Visual Field Constriction in Syndromic and Non-Syndromic USH2A-RP Patients
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Patients Recruitment or Inclusion Criteria
4.3. Genetic Analysis
4.4. Clinical Evaluations
4.5. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RP | Retinitis Pigmentosa |
| IRD | Inherited Retinal Degenerative Disease |
| USH2A-RP | USH2A-associated RP |
| HFA | Humphrey field analyzer |
| MD | Mean Deviation |
References
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology 2020. [Google Scholar] [CrossRef]
- Koyanagi, Y.; Akiyama, M.; Nishiguchi, K.M.; Momozawa, Y.; Kamatani, Y.; Takata, S.; Inai, C.; Iwasaki, Y.; Kumano, M.; Murakami, Y.; et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. J. Med. Genet. 2019, 56, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Aparisi, M.J.; Aller, E.; Fuster-García, C.; García-García, G.; Rodrigo, R.; Vázquez-Manrique, R.P.; Blanco-Kelly, F.; Ayuso, C.; Roux, A.F.; Jaijo, T.; et al. Targeted next generation sequencing for molecular diagnosis of Usher syndrome. Orphanet J. Rare Dis. 2014, 9, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eudy, J.D.; Weston, M.D.; Yao, S.; Hoover, D.M.; Rehm, H.L.; Ma-Edmonds, M.; Yan, D.; Ahmad, I.; Cheng, J.J.; Ayuso, C.; et al. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 1998, 280, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Le Quesne Stabej, P.; Saihan, Z.; Rangesh, N.; Steele-Stallard, H.B.; Ambrose, J.; Coffey, A.; Emmerson, J.; Haralambous, E.; Hughes, Y.; Steel, K.P.; et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J. Med. Genet. 2012, 49, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, C.; Riahi, Z.; Chantot-Bastaraud, S.; Smagghe, L.; Letexier, M.; Marcaillou, C.; Lefèvre, G.M.; Hardelin, J.P.; El-Amraoui, A.; Singh-Estivalet, A.; et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 2016, 24, 1730–1738. [Google Scholar] [CrossRef] [Green Version]
- Rivolta, C.; Sweklo, E.A.; Berson, E.L.; Dryja, T.P. Missense mutation in the USH2A gene: Association with recessive retinitis pigmentosa without hearing loss. Am. J. Hum. Genet. 2000, 66, 1975–1978. [Google Scholar] [CrossRef] [Green Version]
- Lenassi, E.; Vincent, A.; Li, Z.; Saihan, Z.; Coffey, A.J.; Steele-Stallard, H.B.; Moore, A.T.; Steel, K.P.; Luxon, L.M.; Héon, E.; et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 2015, 23, 1318–1327. [Google Scholar] [CrossRef] [Green Version]
- Nishio, S.Y.; Usami, S. Deafness gene variations in a 1120 nonsyndromic hearing loss cohort: Molecular epidemiology and deafness mutation spectrum of patients in Japan. Ann. Otol. Rhinol. Laryngol. 2015, 124, 49s–60s. [Google Scholar] [CrossRef]
- van Wijk, E.; Pennings, R.J.; te Brinke, H.; Claassen, A.; Yntema, H.G.; Hoefsloot, L.H.; Cremers, F.P.; Cremers, C.W.; Kremer, H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am. J. Hum. Genet. 2004, 74, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Reiners, J.; Nagel-Wolfrum, K.; Jürgens, K.; Märker, T.; Wolfrum, U. Molecular basis of human Usher syndrome: Deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 2006, 83, 97–119. [Google Scholar] [CrossRef] [PubMed]
- Dona, M.; Slijkerman, R.; Lerner, K.; Broekman, S.; Wegner, J.; Howat, T.; Peters, T.; Hetterschijt, L.; Boon, N.; de Vrieze, E.; et al. Usherin defects lead to early-onset retinal dysfunction in zebrafish. Exp. Eye Res. 2018, 173, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; El-Amraoui, A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): Pathogenesis, molecular diagnosis and therapeutic approaches. Curr. Opin. Neurol. 2012, 25, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.; Zallocchi, M. Usher protein functions in hair cells and photoreceptors. Int. J. Biochem. Cell Biol. 2014, 46, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Bulgakov, O.V.; Darrow, K.N.; Pawlyk, B.; Adamian, M.; Liberman, M.C.; Li, T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4413–4418. [Google Scholar] [CrossRef] [Green Version]
- Bujakowska, K.M.; Liu, Q.; Pierce, E.A. Photoreceptor Cilia and Retinal Ciliopathies. Cold Spring Harb Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Usher Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Ohtsubo, M.; Iwasaki, S.; Hotta, Y.; Usami, S.; Mizuta, K.; Mineta, H.; Minoshima, S. Novel USH2A mutations in Japanese Usher syndrome type 2 patients: Marked differences in the mutation spectrum between the Japanese and other populations. J. Hum. Genet. 2011, 56, 484–490. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hosono, K.; Suto, K.; Ishigami, C.; Arai, Y.; Hikoya, A.; Hirami, Y.; Ohtsubo, M.; Ueno, S.; Terasaki, H.; et al. The first USH2A mutation analysis of Japanese autosomal recessive retinitis pigmentosa patients: A totally different mutation profile with the lack of frequent mutations found in Caucasian patients. J. Hum. Genet. 2014, 59, 521–528. [Google Scholar] [CrossRef]
- Hartel, B.P.; Löfgren, M.; Huygen, P.L.; Guchelaar, I.; Lo, A.N.K.N.; Sadeghi, A.M.; van Wijk, E.; Tranebjærg, L.; Kremer, H.; Kimberling, W.J.; et al. A combination of two truncating mutations in USH2A causes more severe and progressive hearing impairment in Usher syndrome type IIa. Hear. Res. 2016, 339, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Pierrache, L.H.; Hartel, B.P.; van Wijk, E.; Meester-Smoor, M.A.; Cremers, F.P.; de Baere, E.; de Zaeytijd, J.; van Schooneveld, M.J.; Cremers, C.W.; Dagnelie, G.; et al. Visual Prognosis in USH2A-Associated Retinitis Pigmentosa Is Worse for Patients with Usher Syndrome Type IIa Than for Those with Nonsyndromic Retinitis Pigmentosa. Ophthalmology 2016, 123, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Chen, D.F.; Wang, L.; Wu, S.; Wei, X.; Li, H.; Jin, Z.B.; Sui, R. USH2A variants in Chinese patients with Usher syndrome type II and non-syndromic retinitis pigmentosa. Br. J. Ophthalmol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Joo, K.; Oh, J.; Han, J.H.; Park, H.R.; Lee, S.; Oh, D.Y.; Woo, S.J.; Choi, B.Y. Severe or Profound Sensorineural Hearing Loss Caused by Novel USH2A Variants in Korea: Potential Genotype-Phenotype Correlation. Clin. Exp. Otorhinolaryngol 2020, 13, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandberg, M.A.; Rosner, B.; Weigel-DiFranco, C.; McGee, T.L.; Dryja, T.P.; Berson, E.L. Disease course in patients with autosomal recessive retinitis pigmentosa due to the USH2A gene. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5532–5539. [Google Scholar] [CrossRef] [Green Version]
- Nagase, Y.; Kurata, K.; Hosono, K.; Suto, K.; Hikoya, A.; Nakanishi, H.; Mizuta, K.; Mineta, H.; Minoshima, S.; Hotta, Y. Visual Outcomes in Japanese Patients with Retinitis Pigmentosa and Usher Syndrome Caused by USH2A Mutations. Semin. Ophthalmol. 2018, 33, 560–565. [Google Scholar] [CrossRef] [Green Version]
- Sayo, A.; Ueno, S.; Kominami, T.; Nishida, K.; Inooka, D.; Nakanishi, A.; Yasuda, S.; Okado, S.; Takahashi, K.; Matsui, S.; et al. Longitudinal study of visual field changes determined by Humphrey Field Analyzer 10-2 in patients with Retinitis Pigmentosa. Sci. Rep. 2017, 7, 16383. [Google Scholar] [CrossRef]
- Haque, M.N.; Kurata, K.; Hosono, K.; Ohtsubo, M.; Ohishi, K.; Sato, M.; Minoshima, S.; Hotta, Y. A Japanese family with cone-rod dystrophy of delayed onset caused by a compound heterozygous combination of novel CDHR1 frameshift and known missense variants. Hum. Genome Var. 2019, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Di Iorio, V.; Orrico, A.; Esposito, G.; Melillo, P.; Rossi, S.; Sbordone, S.; Auricchio, A.; Testa, F.; Simonelli, F. ASSOCIATION BETWEEN GENOTYPE AND DISEASE PROGRESSION IN ITALIAN STARGARDT PATIENTS: A Retrospective Natural History Study. Retina 2019, 39, 1399–1409. [Google Scholar] [CrossRef]
- Astuto, L.M.; Bork, J.M.; Weston, M.D.; Askew, J.W.; Fields, R.R.; Orten, D.J.; Ohliger, S.J.; Riazuddin, S.; Morell, R.J.; Khan, S.; et al. CDH23 mutation and phenotype heterogeneity: A profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am. J. Hum. Genet. 2002, 71, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Schultz, J.M.; Bhatti, R.; Madeo, A.C.; Turriff, A.; Muskett, J.A.; Zalewski, C.K.; King, K.A.; Ahmed, Z.M.; Riazuddin, S.; Ahmad, N.; et al. Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes. J. Med. Genet. 2011, 48, 767–775. [Google Scholar] [CrossRef] [PubMed]
- DuPont, M.; Jones, E.M.; Xu, M.; Chen, R. Investigating the disease association of USH2A p.C759F variant by leveraging large retinitis pigmentosa cohort data. Ophthalmic. Genet. 2018, 39, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Pozo, M.G.; Bravo-Gil, N.; Méndez-Vidal, C.; Montero-de-Espinosa, I.; Millán, J.M.; Dopazo, J.; Borrego, S.; Antiñolo, G. Re-evaluation casts doubt on the pathogenicity of homozygous USH2A p.C759F. Am. J. Med. Genet. A 2015, 167, 1597–1600. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, X.; Xie, S.; Gao, M.; Liu, F.; Yu, S.; Sun, P.; Wang, C.; Archacki, S.; Lu, Z.; et al. Knockout of ush2a gene in zebrafish causes hearing impairment and late onset rod-cone dystrophy. Hum. Genet. 2018, 137, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Pendse, N.D.; Lamas, V.; Pawlyk, B.S.; Maeder, M.L.; Chen, Z.Y.; Pierce, E.A.; Liu, Q. In Vivo Assessment of Potential Therapeutic Approaches for USH2A-Associated Diseases. Adv. Exp. Med. Biol. 2019, 1185, 91–96. [Google Scholar] [CrossRef]
- Zou, J.; Zheng, T.; Ren, C.; Askew, C.; Liu, X.P.; Pan, B.; Holt, J.R.; Wang, Y.; Yang, J. Deletion of PDZD7 disrupts the Usher syndrome type 2 protein complex in cochlear hair cells and causes hearing loss in mice. Hum. Mol. Genet. 2014, 23, 2374–2390. [Google Scholar] [CrossRef] [Green Version]
- Jouret, G.; Poirsier, C.; Spodenkiewicz, M.; Jaquin, C.; Gouy, E.; Arndt, C.; Labrousse, M.; Gaillard, D.; Doco-Fenzy, M.; Lebre, A.S. Genetics of Usher Syndrome: New Insights from a Meta-analysis. Otol. Neurotol. 2019, 40, 121–129. [Google Scholar] [CrossRef]
- Oishi, M.; Oishi, A.; Gotoh, N.; Ogino, K.; Higasa, K.; Iida, K.; Makiyama, Y.; Morooka, S.; Matsuda, F.; Yoshimura, N. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7369–7375. [Google Scholar] [CrossRef]
- Arai, Y.; Maeda, A.; Hirami, Y.; Ishigami, C.; Kosugi, S.; Mandai, M.; Kurimoto, Y.; Takahashi, M. Retinitis Pigmentosa with EYS Mutations Is the Most Prevalent Inherited Retinal Dystrophy in Japanese Populations. J. Ophthalmol. 2015, 2015, 819760. [Google Scholar] [CrossRef] [Green Version]
- Maeda, A.; Yoshida, A.; Kawai, K.; Arai, Y.; Akiba, R.; Inaba, A.; Takagi, S.; Fujiki, R.; Hirami, Y.; Kurimoto, Y.; et al. Development of a molecular diagnostic test for Retinitis Pigmentosa in the Japanese population. Jpn. J. Ophthalmol. 2018, 62, 451–457. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze-Bonsel, K.; Feltgen, N.; Burau, H.; Hansen, L.; Bach, M. Visual acuities “hand motion” and “counting fingers” can be quantified with the freiburg visual acuity test. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.; Fishman, G.A.; Anderson, R.J.; Tozatti, M.S.; Heckenlively, J.R.; Weleber, R.G.; Edwards, A.O.; Brown, J., Jr. Visual acuity impairment in patients with retinitis pigmentosa at age 45 years or older. Ophthalmology 1999, 106, 1780–1785. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Nucleotide Change | Protein Change | Zygosity |
|---|---|---|---|
| P1 | c.490G>T;c.13631dupG | p.(Val164Phe);p.(Pro4545Serfs*17) | Het;Het |
| P2 | c.1923T>A;c.3958C>T;c.5396delA | p.(Cys641*);p.(Pro1320Ser) 1;p.(Lys1799Serfs*18) | Het;Het;Het |
| P3 | c.13576C>T;c.13847G>T | p.(Arg4526*);p.(Gly4616Val) | Homo;Homo |
| P4 | c.2653C>T;c.9751T>C; c.13576C>T;c.13847G>T | p.(His885Tyr) 2; p.(Cys3251Arg) 2;p.(Arg4526*)2;p.(Gly4616Val)2 | Het;Het; Het;Het |
| P5 | c.8559-2A>G;c.14133+2T>A | p.(?);p.(?) | Het;Het |
| P6 | c.8396delG | p.(Gly2799Valfs*31) | Homo |
| P7 | c.10353_10356delTCAT;c.13010C>T | p.(His3452Glnfs*4);p.(Thr4337Met) | Het;Het |
| P8 | c.2802T>G;c.5158delC | p.(Cys934Trp);p.(Leu1720*) | Het;Het |
| P9 | c.8559-2A>G;c.10712C>T | p.(?);p.(Thr3571Met) | Het;Het |
| P10 | c.8559-2A>G;c.14004delG | p.(?);p.(Leu4668Phefs*10) | Het;Het |
| P11 | c.8559-2A>G | p.(?) | Homo |
| P12 | c.10859T>C;c.11328T>G | p.(Ile3620Thr);p.(Tyr3776*) | Het;Het |
| P13 | c.14243C>T | p.(Ser4748Phe) | Homo |
| P14 | c.3596_3598delAAG;c.8254G>A | p.(Glu1199del);p.(Gly2752Arg) | Het;Het |
| P15 | c.662C>A;c.7068T>G;c.7234G>A | p.(Thr221Lys) 1;p.(Asn2356Lys);p.(Val2412Met) 1 | Het;Het;Het |
| P16 | c.490G>T;c.3595_3597delGAA | p.(Val164Phe);p.(Glu1199del) | Het;Het |
| P17 | c.10859T>C;c.14766G>A | p.(Ile3620Thr);p.(Trp4922*) | Het;Het |
| P18 | c.8559-2A>G;c.14243C>T | p.(?);p.(Ser4748Phe) | Het;Het |
| P19 | c.11156G>A;c.13010C>T | p.(Arg3719His);p.(Thr4337Met) | Het;Het |
| P20 | c.2802T>G;c.13847G>T | p.(Cys934Trp);p.(Gly4616Val) | Het;Het |
| P21 | c.(11712_12066)del;c.15233C>G | p.(?);p.(Pro5078Arg) | Het;Het |
| P22 | c.8254G>A | p.(Gly2752Arg) | Homo |
| P23 | c.850G>A;c.2802T>G | p.(Glu284Lys);p.(Cys934Trp) | Het;Het |
| P24 | c.4310_4312dupATA;c.8254G>A | p.(Tyr1437_Arg1438insAsn);p.(Gly2752Arg) | Het;Het |
| P25 | c.6399G>A;c.13887G>T | p.(Trp2133*);p.(Glu4629Asp) | Het;Het |
| P26 | c.2802T>G;c.9815C>T | p.(Cys934Trp);p.(Pro3272Leu) | Het;Het |
| P27 | c.14243C>T;c.15233C>G | p.(Ser4748Phe);p.(Pro5078Arg) | Het;Het |
| P28 | c.9371+1G>T;c.12094G>A | p.(?);p.(Gly4032Arg) | Het;Het |
| P29 | c.3596_3598delAAG;c.8254G>A;c.13894C>G | p.(Glu1199del);p.(Gly2752Arg);p.(Pro4632Ala)1 | Het;Het;Het |
| P30 | c.685G>C;c.13708C>T | p.(Gly229Arg);p.(Arg4570Cys) | Het;Het |
| P31 | c.8254G>A;c.8396delG | p.(Gly2752Arg);p.(Gly2799Valfs*31) | Het;Het |
| P32 | c.2802T>G;c.11811_11812delCT | p.(Cys934Trp);p.(Tyr3938Argfs*8) | Het;Het |
| P33 | c.490G>T;c.12383A>G | p.(Val164Phe);p.(Tyr4128Cys) | Het;Het |
| P34 | c.2609G>T;c.5608C>T; c.12305T>A;c.15355C>T | p.(Cys870Phe) 1;p.(Arg1870Trp); p.(Ile4102Asn) 1;p.(Arg5119Trp)1 | Het;Het; Het;Het |
| P35 | c.10495C>T;c.10712C>T | p.(Pro3499Ser);p.(Thr3571Met) | Het;Het |
| P36 | c.8339T>A;c.12407C>T | p.(Val2780Asp);p.(Thr4136Ile) | Het;Het |
| Characteristics | Syndromic RP 1 | Non-Syndromic RP 2 | |
|---|---|---|---|
| Age (years, mean ± SD, range) | 48.5 ± 12.9, 27–69 | 50.9 ± 15.7, 26–82 | |
| Gender (n, %) | Male | 6 (54.5) | 13 (52.0) |
| Family History (n, %) | Yes | 5 (45.5) | 6 (24.0) |
| Consanguineous Marriage (n, %) | Yes | 1 (9.1) | 2 (8.0) |
| Type of Variant | Syndromic RP 1 | Non-Syndromic RP 2 |
|---|---|---|
| Truncating / Truncating (n, %) | 6 (54.5) | 0 (0.0) |
| Truncating / Missense (n, %) | 5 (45.5) | 8 (32.0) |
| Missense / Missense (n, %) | 0 (0.0) | 17 (68.0) |
| USH2A Variants | Syndromic RP 1 | Non-Syndromic RP 2 |
|---|---|---|
| p.(Val164Phe) | 1 | 2 |
| p.(Cys934Trp) | 1 | 4 |
| p.(Glu1199del) | 0 | 3 |
| p.(Gly2752Arg) | 0 | 5 |
| p.(Gly2799Valfs*31) | 1 | 1 |
| c.8559-2A>G | 4 | 1 |
| p.(Thr3571Met) | 1 | 1 |
| p.(Ile3620Thr) | 0 | 2 |
| p.(Ser4748Phe) | 0 | 3 |
| p.(Thr4337Met) | 1 | 1 |
| p.(Arg4526*) | 2 | 0 |
| p.(Gly4616Val) | 2 | 1 |
| p.(Pro5078Arg) | 0 | 2 |
| Phenotype | Patient | Rate of Changes in MD Values | Observation Period (Years) |
|---|---|---|---|
| syndromic RP | P8 | −1.54 | 1 |
| non-syndromic RP | P12 | −0.67 | 8 |
| P25 | −0.88 | 1 | |
| P27 | −0.65 | 6 | |
| P33 | −0.48 | 2 | |
| P36 | −0.34 * | 6 |
| ABCA4 | BEST1 | BBS1* | C2orf71 | CEP290* |
| CDH23 * | CDHR1 * | CHM * | CNGA1 | CNGB1 |
| CNGB3 | CRB1 | CRX | CYP4V2 | EYS |
| FAM161A * | GPR98 * | GUCA1A * | GUCY2D | IMPDH1 |
| IMPG2 | KLHL7 * | LRAT | MAK | MERTK |
| MYO7A * | NR2E3 | NRL | PCDH15 * | PDE6B |
| PRCD | PROM1 | PRPF31 | PRPF6 | PRPH2 |
| RDH5 | RDH12 | RHO | ROM1 | RP1 |
| RP1L1 | RP2 | RP9 | RPE65 | RPGR |
| RS1 * | SNRNP200 | TOPORS | TULP1 | USH2A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inaba, A.; Maeda, A.; Yoshida, A.; Kawai, K.; Hirami, Y.; Kurimoto, Y.; Kosugi, S.; Takahashi, M. Truncating Variants Contribute to Hearing Loss and Severe Retinopathy in USH2A-Associated Retinitis Pigmentosa in Japanese Patients. Int. J. Mol. Sci. 2020, 21, 7817. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217817
Inaba A, Maeda A, Yoshida A, Kawai K, Hirami Y, Kurimoto Y, Kosugi S, Takahashi M. Truncating Variants Contribute to Hearing Loss and Severe Retinopathy in USH2A-Associated Retinitis Pigmentosa in Japanese Patients. International Journal of Molecular Sciences. 2020; 21(21):7817. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217817
Chicago/Turabian StyleInaba, Akira, Akiko Maeda, Akiko Yoshida, Kanako Kawai, Yasuhiko Hirami, Yasuo Kurimoto, Shinji Kosugi, and Masayo Takahashi. 2020. "Truncating Variants Contribute to Hearing Loss and Severe Retinopathy in USH2A-Associated Retinitis Pigmentosa in Japanese Patients" International Journal of Molecular Sciences 21, no. 21: 7817. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217817