Precise Regulation of the Basal PKCγ Activity by DGKγ Is Crucial for Motor Coordination

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
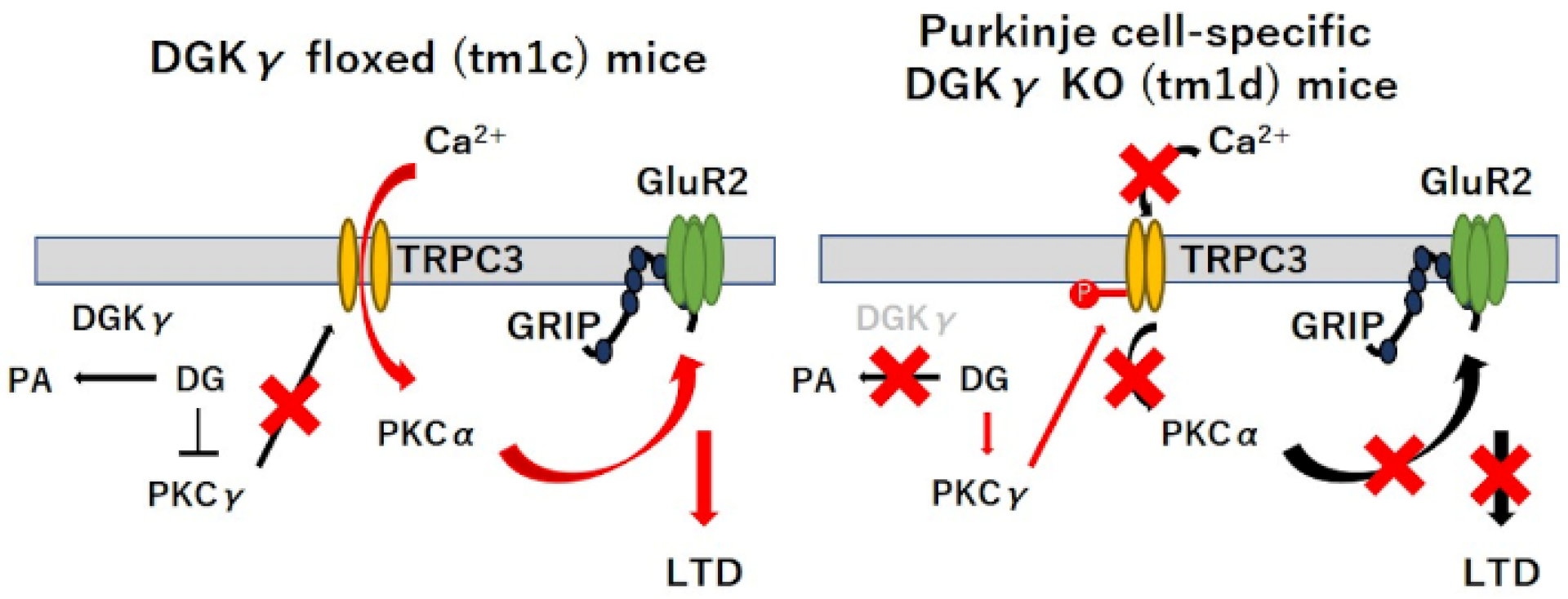
Abstract
:1. Introduction
2. Results
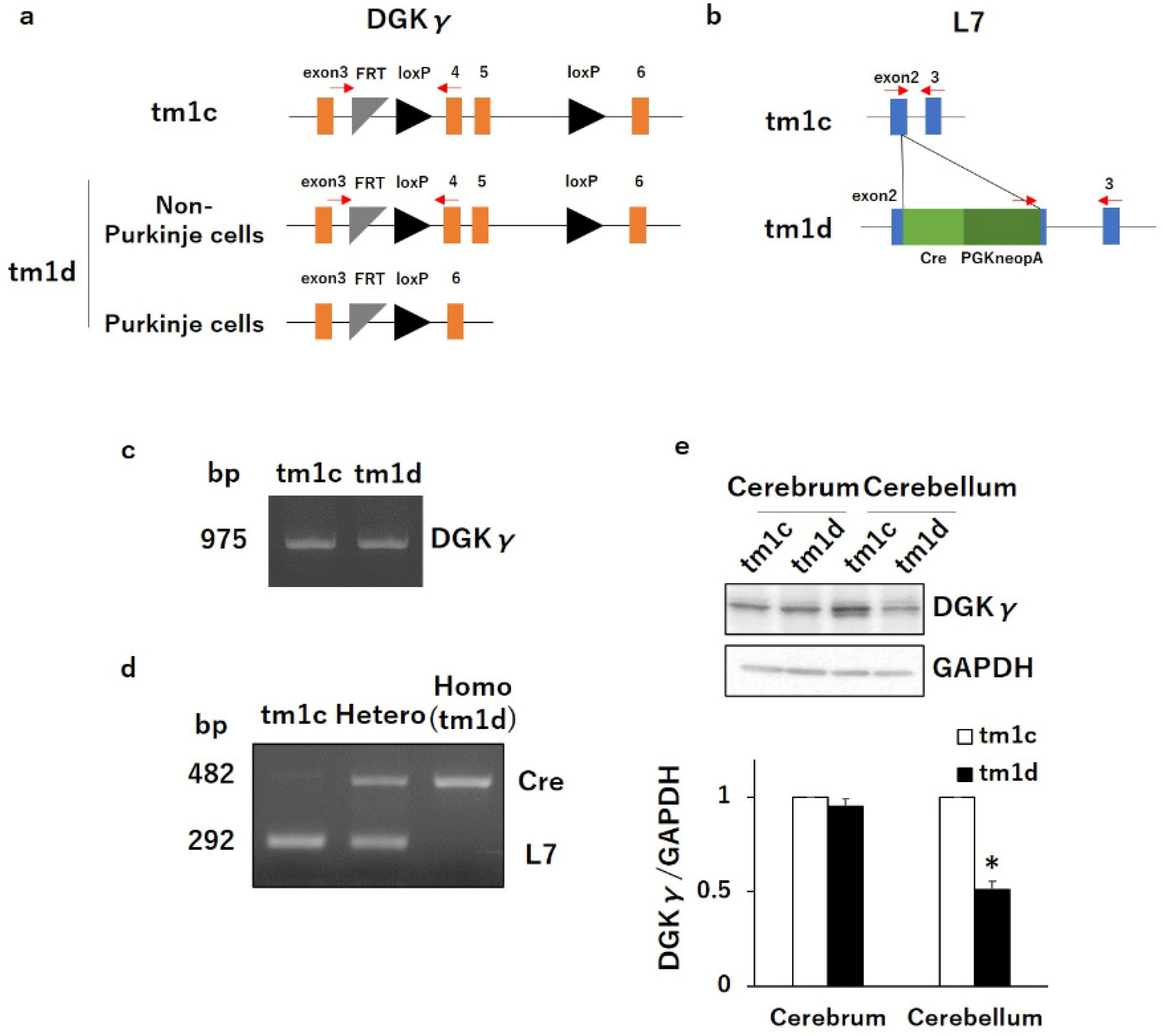
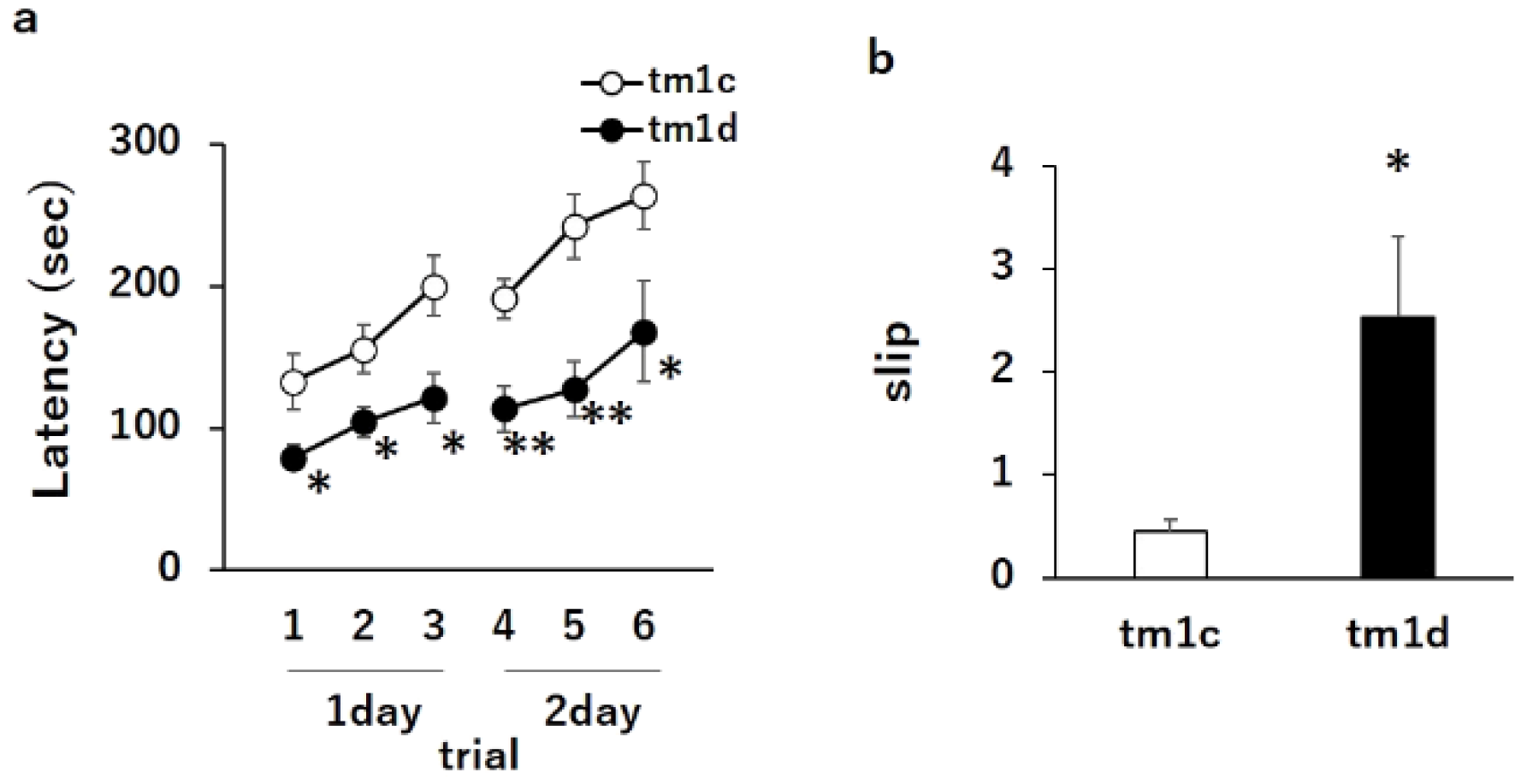
2.1. Motor Dyscoordination in Purkinje Cell-Specific DGKγ KO (tm1d) Mice
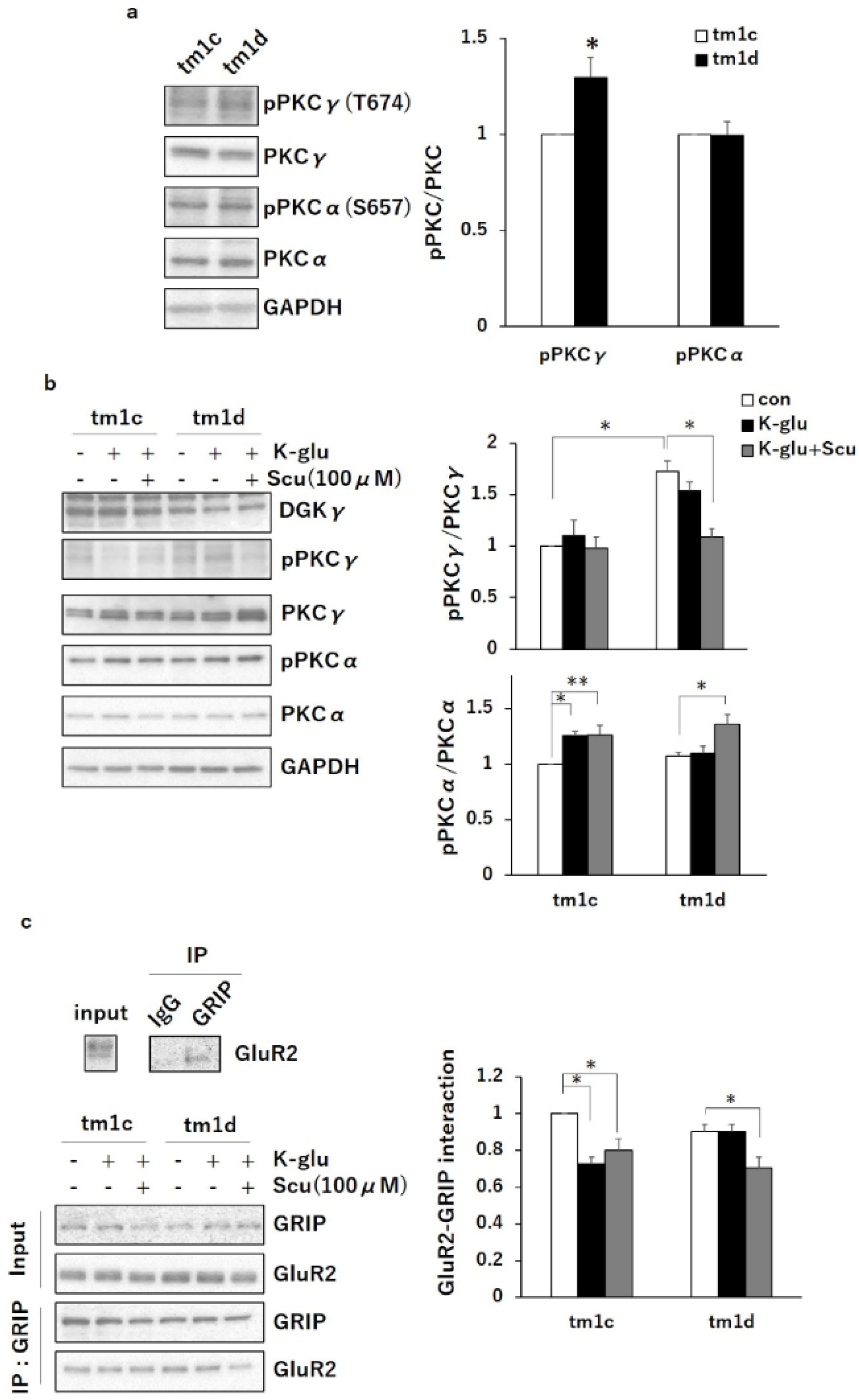
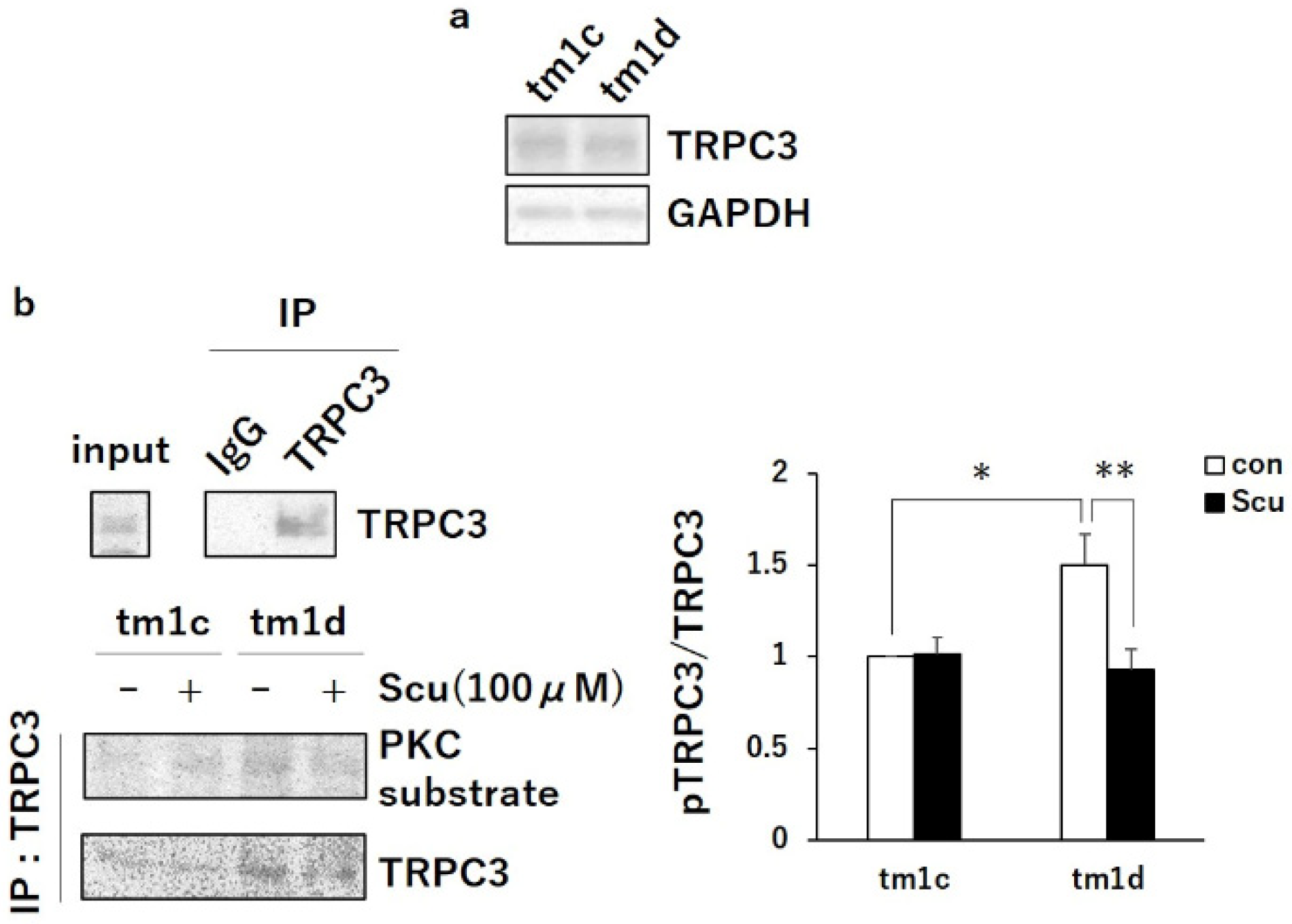
2.2. PKCα Inactivation during LTD by Upregulated PKCγ Mediated Negative Regulation of TRPC3
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Mice
4.3. Genotyping
4.4. Western Blotting
4.5. Rotarod Test
4.6. Beam Test
4.7. Acute Cerebellar Slice
4.8. Experimental Design and Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid |
| CF | Climbing fiber |
| CNS | Central nervous system |
| DGK | Diacylglycerol kinase |
| DGL | Diacylglycerol lipase |
| EPSC | Excitatory postsynaptic current |
| GRIP | Glutamate receptor interacting protein |
| K-glu | KCl-L-glutamate |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| mGluR1 | Metabotropic glutamate receptor 1 |
| mTOR | Mammalian of target of rapamycin |
| PA | Phosphatidic acid |
| PFs | Parallel fibers |
| PKC | Protein kinase C |
| SC | Schaffer-collateral |
| TRPC3 | Nonselective transient receptor potential cation channel type 3 |
| VDCCs | Voltage dependent calcium channels |
References
- Ito, M. The molecular organization of cerebellar long-term depression. Nat. Rev. Neurosci. 2002, 3, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Hashimoto, K.; Tabata, T. Type-1 metabotropic glutamate receptor in cerebellar Purkinje cells: A key molecule responsible for long-term depression, endocannabinoid signalling and synapse elimination. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 2173–2186. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, C.; Huber, K.M. Group 1 mGluR-dependent synaptic long-term depression: Mechanisms and implications for circuitry and disease. Neuron 2010, 65, 445–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.J.; Steinberg, J.P.; Huganir, R.L.; Linden, D.J. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science 2003, 300, 1751–1755. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Chung, H.J.; Wihler, C.; Huganir, R.L.; Linden, D.J. Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain–containing proteins. Neuron 2000, 28, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Leitges, M.; Kovac, J.; Plomann, M.; Linden, D.J. A unique PDZ ligand in PKCα confers induction of cerebellar long-term synaptic depression. Neuron 2004, 44, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Yamamoto, Y.; Kim, E.; Tanaka-Yamamoto, K. Functional and physical interaction of diacylglycerol kinase zeta with protein kinase Calpha Is required for cerebellar long-term depression. J. Neurosci. 2015, 35, 15453–15465. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Kano, M.; Abeliovich, A.; Chen, L.; Bao, S.; Kim, J.J.; Hashimoto, K.; Thompson, R.F.; Tonegawa, S. Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKCγ mutant mice. Cell 1995, 83, 1233–1242. [Google Scholar] [CrossRef] [Green Version]
- Saito, N.; Shirai, Y. Protein kinase C γ (PKC γ): Function of neuron specific isotype. J. Biochem. 2002, 132, 683–687. [Google Scholar] [CrossRef] [Green Version]
- Shuvaev, A.N.; Horiuchi, H.; Seki, T.; Goenawan, H.; Irie, T.; Iizuka, A.; Sakai, N.; Hirai, H. Mutant PKCγ in spinocerebellar ataxia type 14 disrupts synapse elimination and long-term depression in Purkinje cells in vivo. J. Neurosci. 2011, 31, 14324–14334. [Google Scholar] [CrossRef] [Green Version]
- Sakane, F.; Imai, S.I.; Kai, M.; Yasuda, S.; Kanoh, H. Diacylglycerol kinases: Why so many of them? Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Almena, M.; Mérida, I. Shaping up the membrane: Diacylglycerol coordinates spatial orientation of signaling. Trends Biochem. Sci. 2011, 36, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Oyasu, M.; Taniguchi, T.; Yamaguchi, Y.; Takenaka, R.; Shirai, Y.; Saito, N. Immunocytochemical localization of a neuron-specific diacylglycerol kinase β and γ in the developing rat brain. Mol. Brain Res. 2005, 139, 288–299. [Google Scholar] [CrossRef]
- Tsumagari, R.; Kakizawa, S.; Kikunaga, S.; Fujihara, Y.; Ueda, S.; Yamanoue, M.; Saito, N.; Ikawa, M.; Shirai, Y. DGKγ knockout mice show impairments in cerebellar motor coordination, LTD and the dendritic development of Purkinje cells through the activation of PKCγ. Eneuro 2020, 7. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Shirai, Y.; Matsubara, T.; Sanse, K.; Kuriyama, M.; Oshiro, N.; Yoshino, K.; Yonezawa, K.; Ono, Y.; Saito, N. Phosphorylation and up-regulation of diacylglycerol kinase γ via its interaction with protein kinase C γ. J. Biol. Chem. 2006, 281, 31627–31637. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H. Protein kinase C in the cerebellum: Its significance and remaining conundrums. Cerebellum 2018, 17, 23–27. [Google Scholar] [CrossRef]
- Matsuda, S.; Mikawa, S.; Hirai, H. Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J. Neurochem. 1999, 73, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Gallimore, A.R.; Kim, T.; Tanaka-Yamamoto, K.; De Schutter, E. Switching on depression and potentiation in the cerebellum. Cell Rep. 2018, 22, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J. TRPC3 channel underlies cerebellar long-term depression. Cerebellum 2013, 12, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Kobayashi, T.; Takahashi, H.; Kawasaki, T.; Shirai, Y.; Ueyama, T.; Matsuda, T.; Seki, T.; Sakai, N.; Saito, N. Enzymological analysis of mutant protein kinase Cgamma causing spinocerebellar ataxia type 14 and dysfunction in Ca2+ homeostasis. J. Biol. Chem. 2008, 283, 19854–19863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trebak, M.; Hempel, N.; Wedel, B.J.; Smyth, J.T.; Bird, G.S.J.; Putney, J.W. Negative regulation of TRPC3 channels by protein kinase C-mediated phosphorylation of serine 712. Mol. Pharmacol. 2005, 67, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Daniel, H.; Hemart, N.; Jaillard, D.; Crepel, F. Coactivation of metabotropic glutamate receptors and of voltage-gated calcium channels induces long-term depression in cerebellar Purkinje cells in vitro. Exp. Brain Res. 1992, 90, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Henning, H.A.; Konnerth, A. mGluR1/TRPC3-mediated synaptic transmission and calcium signaling in mammalian central neurons. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, L.; Trebak, M.; Putney, J.W. Complex regulation of the TRPC3, 6 and 7 channel subfamily by diacylglycerol and phosphatidylinositol-4,5-bisphosphate. Cell Calcium 2008, 43, 506–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, E.B.E.; Oliver, P.L.; Glitsch, M.D.; Banks, G.T.; Achillic, F.; Hardy, A.; Nolan, P.M.; Fisher, E.M.C.; Davies, K.E. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6706–6711. [Google Scholar] [CrossRef] [Green Version]
- Canepari, M.; Papageorgiou, G.; Corrie, J.E.T.; Watkins, C.; Ogden, D. The conductance underlying the parallel fibre slow EPSP in rat cerebellar Purkinje neurones studied with photolytic release of L-glutamate. J. Physiol. 2001, 533, 765–772. [Google Scholar] [CrossRef]
- Andrikopoulos, P.; Eccles, S.A.; Yaqoob, M.M. Coupling between the TRPC3 ion channel and the NCX1 transporter contributed to VEGF-induced ERK1/2 activation and angiogenesis in human primary endothelial cells. Cell. Signal. 2017, 37, 12–30. [Google Scholar] [CrossRef]
- Lee, D.; Kim, E.; Tanaka-Yamamoto, K. Diacylglycerol kinases in the coordination of synaptic plasticity. Front. Cell Dev. Biol. 2016, 4, 92. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Chen, C.; Goda, Y.; Silva, A.J.; Stevens, C.F.; Tonegawa, S. Modified hippocampal long-term potentiation in PKCγ-mutant mice. Cell 1993, 75, 1253–1262. [Google Scholar] [CrossRef]
- Abeliovich, A.; Paylor, R.; Chen, C.; Kim, J.J.; Wehner, J.M.; Tonegawa, S. PKCγ mutant mice exhibit mild deficits in spatial and contextual learning. Cell 1993, 75, 1263–1271. [Google Scholar] [CrossRef]
- Barber, C.N.; Raben, D.M. Roles of DGKs in neurons: Postsynaptic functions? Adv. Biol. Regul. 2020, 75, 100688. [Google Scholar] [CrossRef] [PubMed]
- Topham, M.K.; Prescott, S.M. Mammalian diacylglycerol kinases, a family of lipid kinases with signaling functions. J. Biol. Chem. 1999, 274, 11447–11450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanoh, H.; Yamada, K.; Sakane, F. Diacylglycerol kinases: Emerging downstream regulators in cell signaling systems. J. Biochem. 2002, 131, 629–633. [Google Scholar] [CrossRef]
- Van Blitterswijk, W.J.; Houssa, B. Properties and functions of diacylglycerol kinases. Cell. Signal. 2000, 12, 595–605. [Google Scholar] [CrossRef]
- Raben, D.M.; Tu-Sekine, B. Regulation and roles of neuronal diacylglycerol kinases: A lipid perspective. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 353–364. [Google Scholar] [CrossRef]
- Shirai, Y.; Saito, N. Diacylglycerol kinase as a possible therapeutic target for neuronal diseases. J. Biomed. Sci. 2014, 21, 28. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Prescott, S.M.; Topham, M.K. Association of diacylglycerol kinase zeta with protein kinase C alpha: Spatial regulation of diacylglycerol signaling. J. Cell Biol. 2003, 160, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Hozumi, Y.; Fujiwara, H.; Kaneko, K.; Fujii, S.; Topham, M.K.; Watanabe, M.; Goto, K. Diacylglycerol kinase ε localizes to subsurface cisterns of cerebellar Purkinje cells. Cell Tissue Res. 2017, 1–18. [Google Scholar] [CrossRef]
- Kano, T.; Kouzuki, T.; Mizuno, S.; Ueda, S.; Yamanoue, M.; Sakane, F.; Saito, N.; Shirai, Y. Both the C1 domain and a basic amino acid cluster at the C-terminus are important for the neurite and branch induction ability of DGKβ. Biochem. Biophys. Res. Commun. 2014, 447, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Kouzuki, T.; Kakefuda, K.; Moriguchi, S.; Oyagi, A.; Horie, K.; Morita, S.; Shimazawa, M.; Fukunaga, K.; Takeda, J.; et al. Essential role of neuron-enriched diacylglycerol kinase (DGK), DGKβ in neurite spine formation, contributing to cognitive function. PLoS ONE 2010, 5, e11602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishisaka, M.; Kakefuda, K.; Oyagi, A.; Ono, Y.; Tsuruma, K.; Shimazawa, M.; Kitaichi, K.; Hara, H. Diacylglycerol kinase β knockout mice exhibit attention-deficit behavior and an abnormal response on methylphenidate-induced hyperactivity. PLoS ONE 2012, 7, e37058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Seo, J.; Nair, R.; Han, S.; Jang, S.; Kim, K.; Han, K.; Paik, S.K.; Choi, J.; Lee, S.; et al. DGKι regulates presynaptic release during mGluR-dependent LTD. EMBO J. 2011, 30, 165–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.; Kim, K.; Jang, S.; Han, S.; Choi, S.Y.; Kim, E. Regulation of hippocampal long-term potentiation and long-term depression by diacylglycerol kinaseζ. Hippocampus 2012, 22, 1018–1026. [Google Scholar] [CrossRef]
- Tabet, R.; Moutin, E.; Becker, J.A.J.; Heintz, D.; Fouillen, L.; Flatter, E.; Krȩzel, W.; Alunni, V.; Koebel, P.; Dembélé, D.; et al. Fragile X mental retardation protein (FMRP) controls diacylglycerol kinase activity in neurons. Proc. Natl. Acad. Sci. USA 2016, 113, E3619–E3628. [Google Scholar] [CrossRef] [Green Version]
- Saito, H.; Tsumura, H.; Otake, S.; Nishida, A.; Furukawa, T.; Suzuki, N. L7/Pcp-2-specific expression of Cre recombinase using knock-in approach. Biochem. Biophys. Res. Commun. 2005, 331, 1216–1221. [Google Scholar] [CrossRef]
- Edwards, F.A.; Konnerth, A.; Sakmann, B.; Takahashi, T. A thin slice preparation for patch clamp recordings from neurones of the mammalian central nervous system. Pflügers Arch. Eur. J. Physiol. 1989, 414, 600–612. [Google Scholar] [CrossRef]
- Kakizawa, S.; Yamasaki, M.; Watanabe, M.; Kano, M. Critical period for activity-dependent synapse elimination in developing cerebellum. J. Neurosci. 2000, 20, 4954–4961. [Google Scholar] [CrossRef]
- Kakizawa, S.; Yamazawa, T.; Chen, Y.; Ito, A.; Murayama, T.; Oyamada, H.; Kurebayashi, N.; Sato, O.; Watanabe, M.; Mori, N.; et al. Nitric oxide-induced calcium release via ryanodine receptors regulates neuronal function. EMBO J. 2012, 31, 417–428. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsumagari, R.; Maruo, K.; Kakizawa, S.; Ueda, S.; Yamanoue, M.; Saito, H.; Suzuki, N.; Shirai, Y. Precise Regulation of the Basal PKCγ Activity by DGKγ Is Crucial for Motor Coordination. Int. J. Mol. Sci. 2020, 21, 7866. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217866
Tsumagari R, Maruo K, Kakizawa S, Ueda S, Yamanoue M, Saito H, Suzuki N, Shirai Y. Precise Regulation of the Basal PKCγ Activity by DGKγ Is Crucial for Motor Coordination. International Journal of Molecular Sciences. 2020; 21(21):7866. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217866
Chicago/Turabian StyleTsumagari, Ryosuke, Kenta Maruo, Sho Kakizawa, Shuji Ueda, Minoru Yamanoue, Hiromitsu Saito, Noboru Suzuki, and Yasuhito Shirai. 2020. "Precise Regulation of the Basal PKCγ Activity by DGKγ Is Crucial for Motor Coordination" International Journal of Molecular Sciences 21, no. 21: 7866. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217866