Monitoring Cannabinoid CB2 -Receptor Mediated cAMP Dynamics by FRET-Based Live Cell Imaging


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
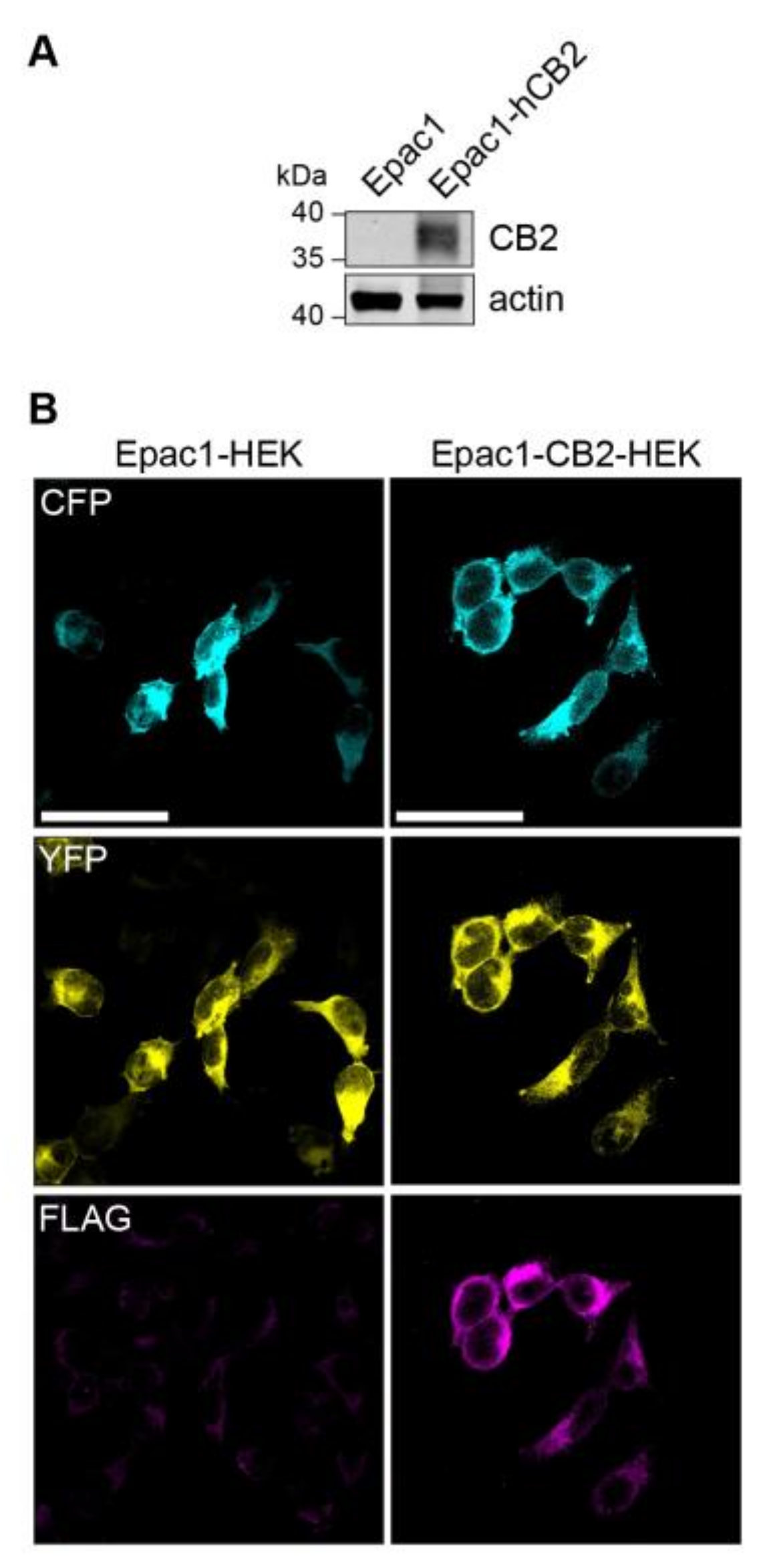
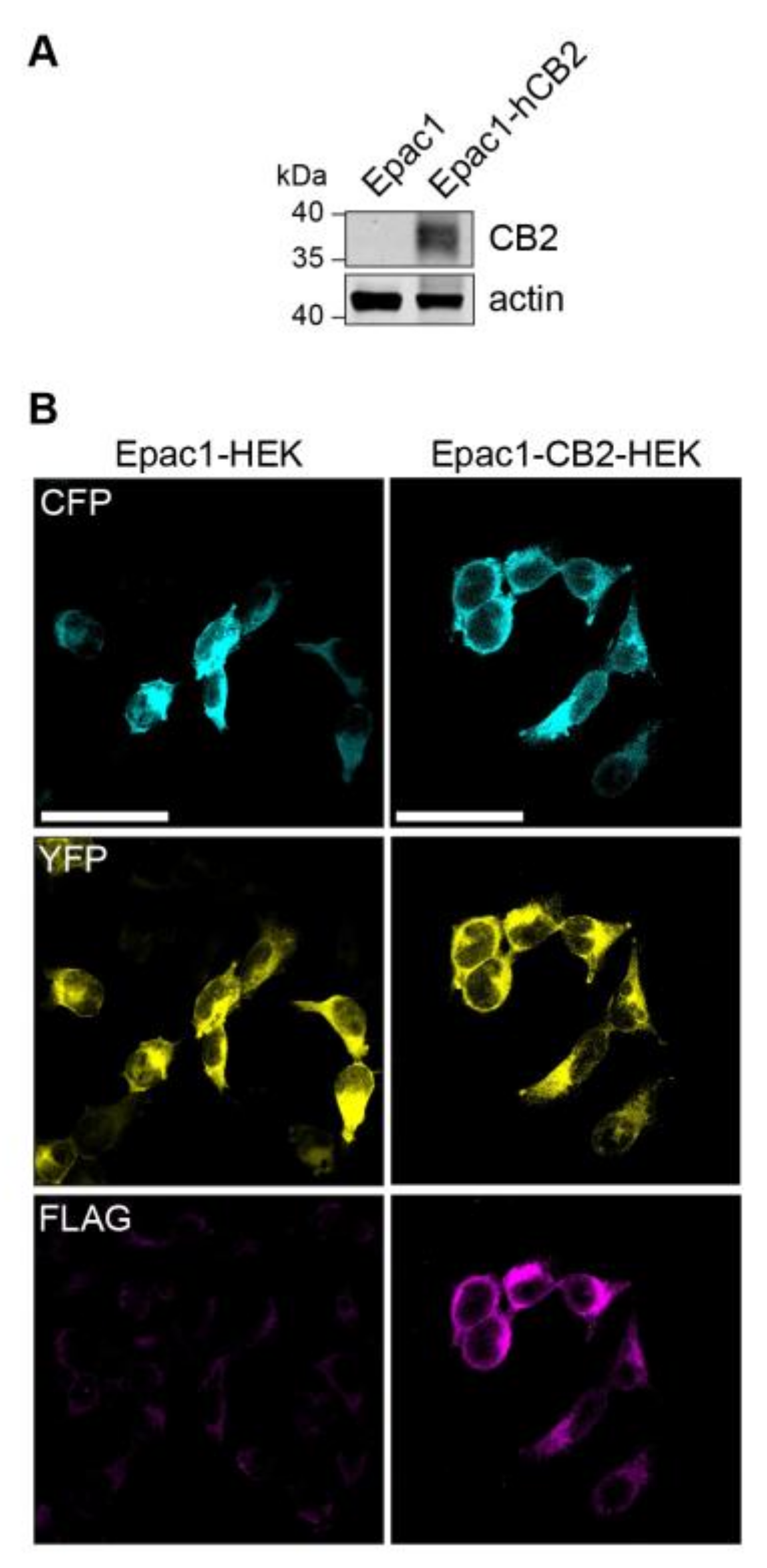
2.1. Stable Expression of FLAG-hCB2 in Epac1-HEK Cells
2.2. Evaluation of Live Cell FRET Imaging and cAMP Measurement in Epac1-HEK Cells
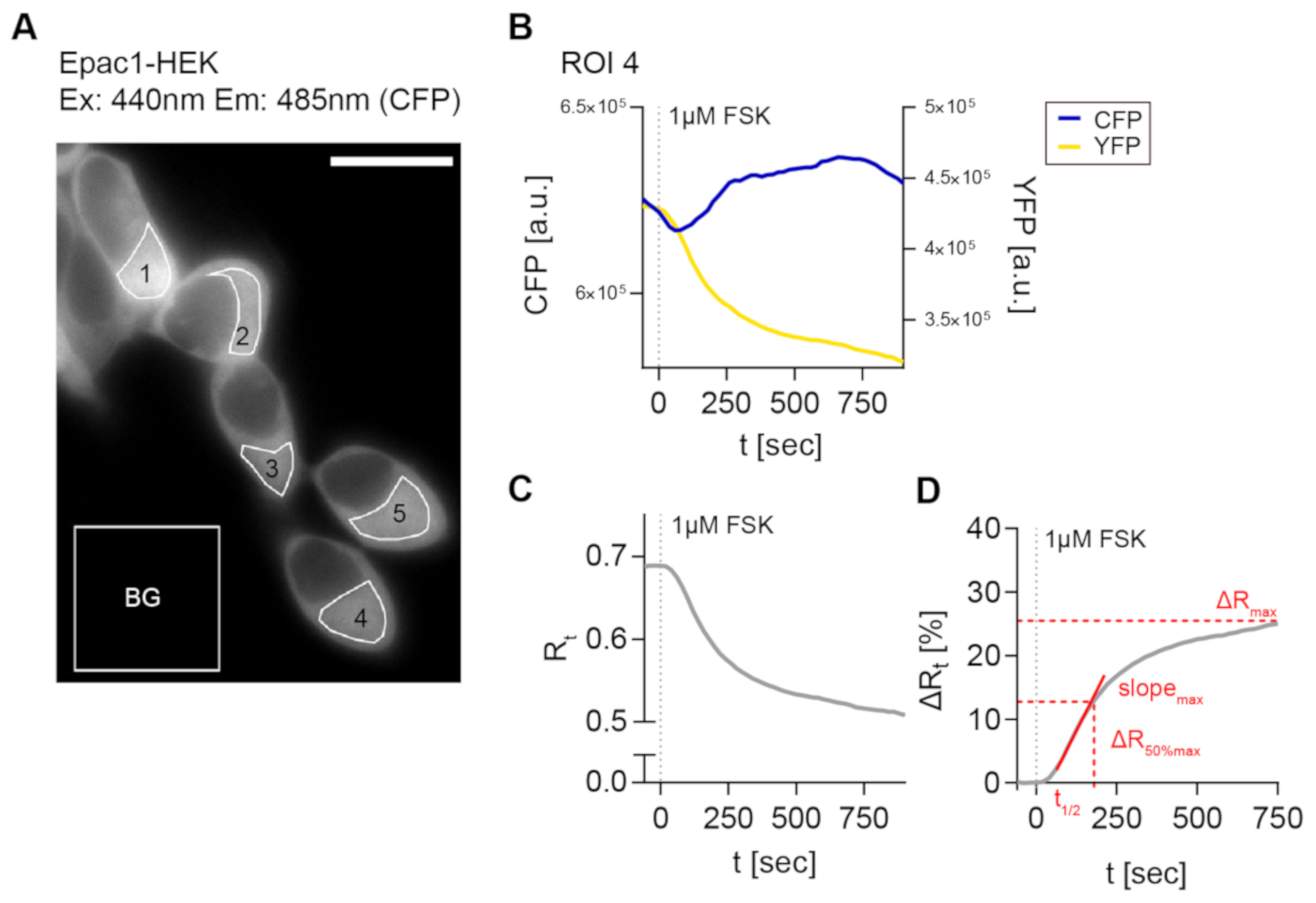
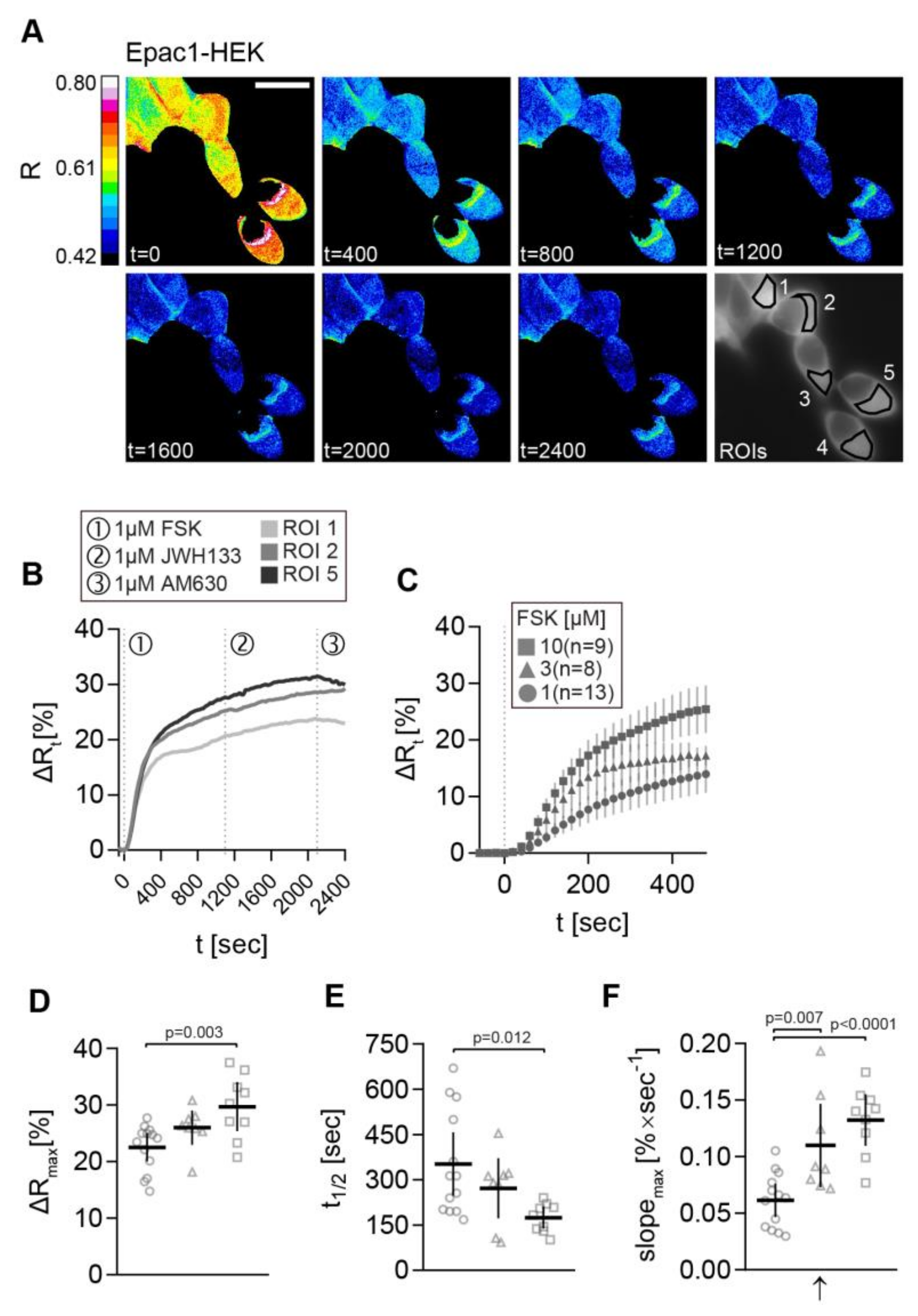
2.3. FRET Recordings from FSK-Stimulated Epac1-HEK Cells Showed the Feasibility of the Image Acquisition and Analysis Pipeline
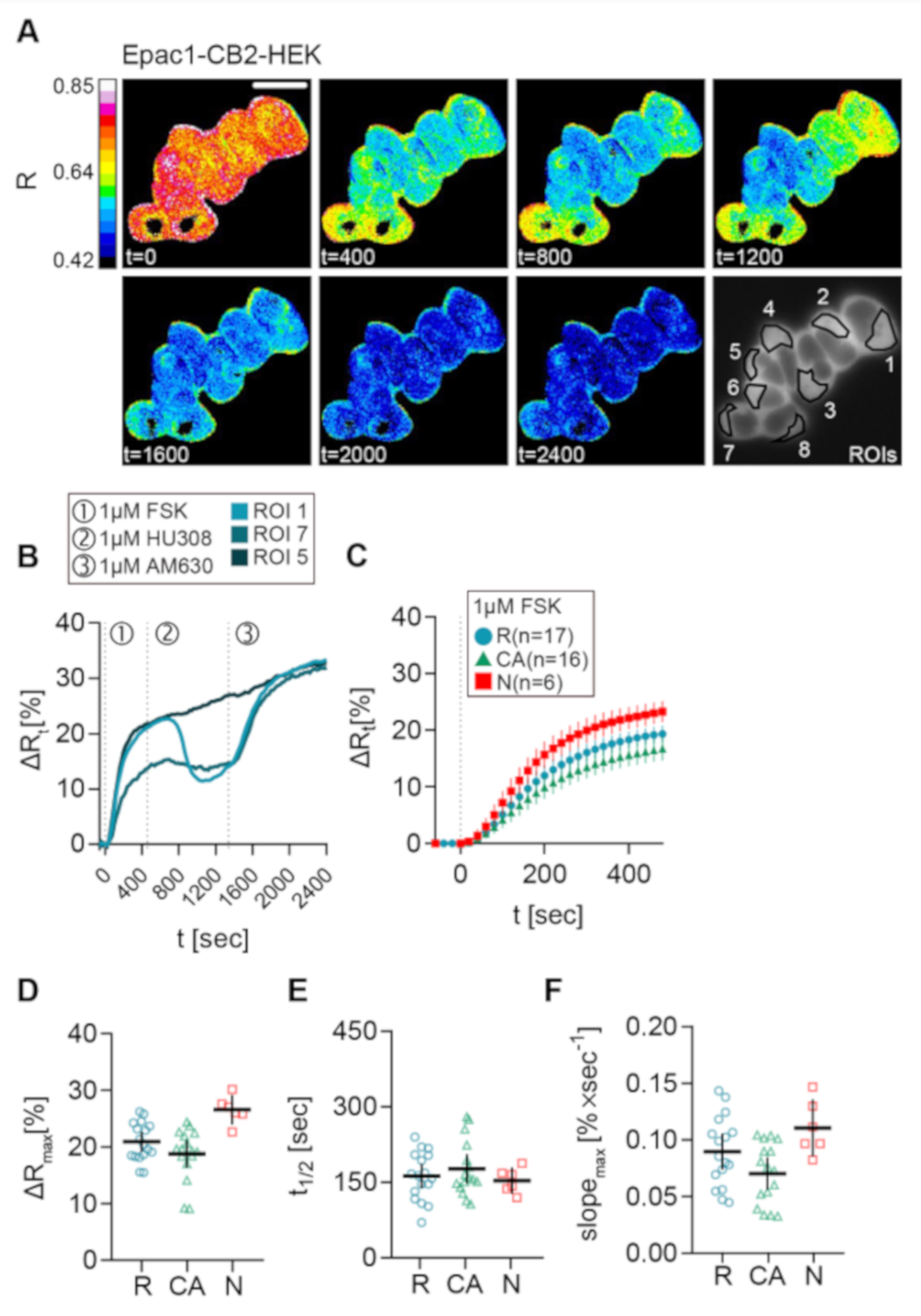
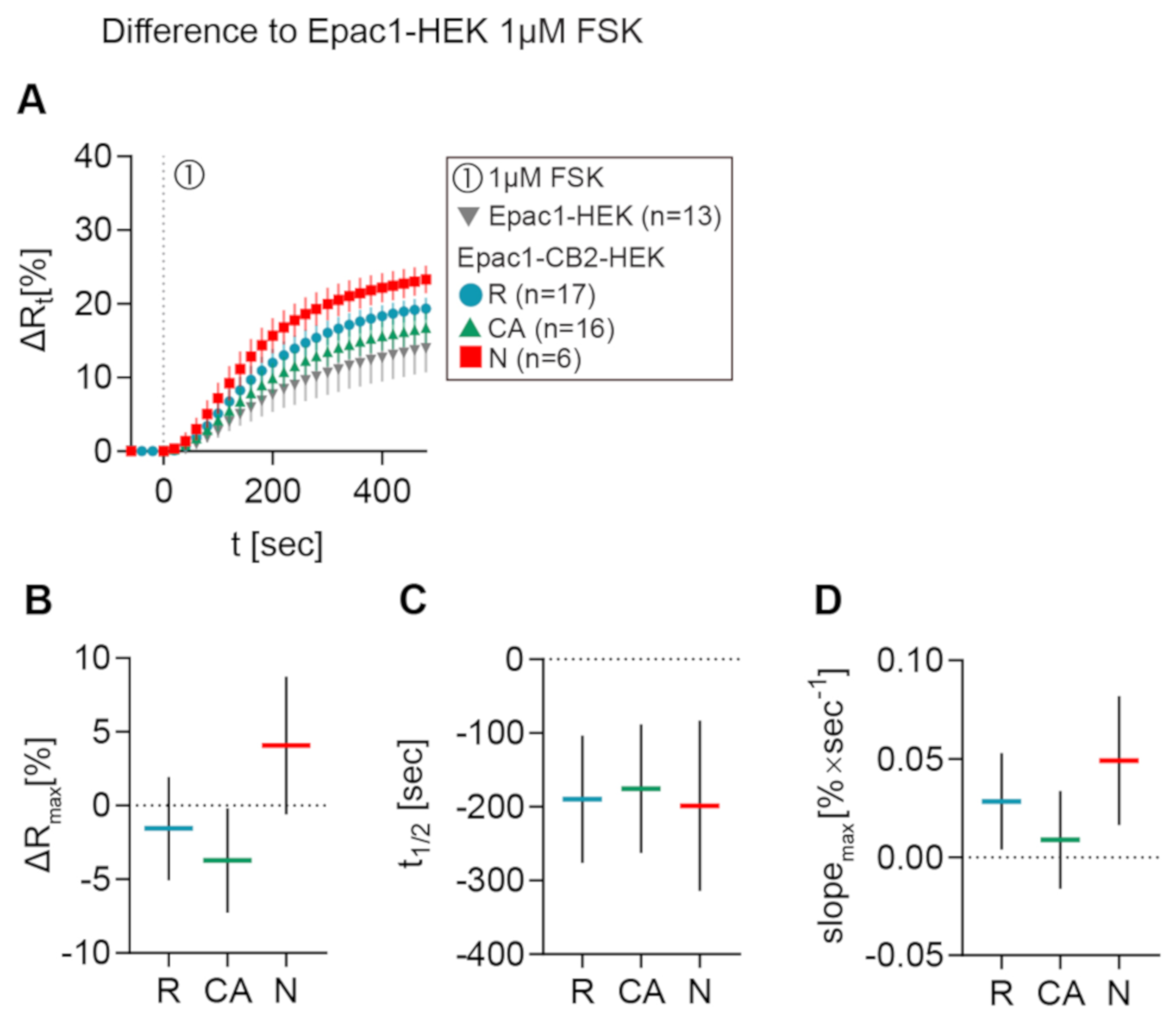
2.4. Live Measurement of CB2-Mediated cAMP Dynamics Revealed Different CB2-Mediated cAMP Response Patterns in Epac1-CB2-HEK Cells
2.5. Reduction of cAMP Levels After CB2 Activation with Different Agonists in Epac1-CB2-HEK Cells
3. Discussion
4. Materials and Methods
4.1. Cells, Reagents and Software
4.2. HEK293 Cell Culture and Preparation for Downstream Assays
4.3. SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Immunoblotting
4.4. Immunocytochemistry
4.5. Live Cell FRET Imaging
4.6. FRET Data Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moreno, E.; Cavic, M.; Krivokuca, A.; Casado, V.; Canela, E. The Endocannabinoid System as a Target in Cancer Diseases: Are We There Yet? Front. Pharmacol. 2019, 10, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallat, A.; Teixeira-Clerc, F.; Deveaux, V.; Manin, S.; Lotersztajn, S. The endocannabinoid system as a key mediator during liver diseases: New insights and therapeutic openings. Br. J. Pharmacol. 2011, 163, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.S.V.; Kelly, M.E.M. Cannabinoids in the Cardiovascular System. Adv. Pharmacol. 2017, 80, 329–366. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Sledzinski, P.; Zeyland, J.; Slomski, R.; Nowak, A. The current state and future perspectives of cannabinoids in cancer biology. Cancer Med. 2018, 7, 765–775. [Google Scholar] [CrossRef]
- Sarfaraz, S.; Adhami, V.M.; Syed, D.N.; Afaq, F.; Mukhtar, H. Cannabinoids for cancer treatment: Progress and promise. Cancer Res. 2008, 68, 339–342. [Google Scholar] [CrossRef] [Green Version]
- Bouaboula, M.; Desnoyer, N.; Carayon, P.; Combes, T.; Casellas, P. Gi protein modulation induced by a selective inverse agonist for the peripheral cannabinoid receptor CB2: Implication for intracellular signalization cross-regulation. Mol. Pharmacol. 1999, 55, 473–480. [Google Scholar]
- Saroz, Y.; Kho, D.T.; Glass, M.; Graham, E.S.; Grimsey, N.L. Cannabinoid Receptor 2 (CB2) Signals via G-alpha-s and Induces IL-6 and IL-10 Cytokine Secretion in Human Primary Leukocytes. ACS Pharmacol. Transl. Sci. 2019, 2, 414–428. [Google Scholar] [CrossRef] [Green Version]
- Sharaf, A.; Mensching, L.; Keller, C.; Rading, S.; Scheffold, M.; Palkowitsch, L.; Djogo, N.; Rezgaoui, M.; Kestler, H.A.; Moepps, B.; et al. Systematic Affinity Purification Coupled to Mass Spectrometry Identified p62 as Part of the Cannabinoid Receptor CB2 Interactome. Front. Mol. Neurosci. 2019, 12, 224. [Google Scholar] [CrossRef] [Green Version]
- Mancini, I.; Brusa, R.; Quadrato, G.; Foglia, C.; Scandroglio, P.; Silverman, L.S.; Tulshian, D.; Reggiani, A.; Beltramo, M. Constitutive activity of cannabinoid-2 (CB2) receptors plays an essential role in the protean agonism of (+)AM1241 and L768242. Br. J. Pharmacol. 2009, 158, 382–391. [Google Scholar] [CrossRef]
- Bolognini, D.; Cascio, M.G.; Parolaro, D.; Pertwee, R.G. AM630 behaves as a protean ligand at the human cannabinoid CB2 receptor. Br. J. Pharmacol. 2012, 165, 2561–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soethoudt, M.; Grether, U.; Fingerle, J.; Grim, T.W.; Fezza, F.; de Petrocellis, L.; Ullmer, C.; Rothenhausler, B.; Perret, C.; van Gils, N.; et al. Cannabinoid CB2 receptor ligand profiling reveals biased signalling and off-target activity. Nat. Commun. 2017, 8, 13958. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Stella, N.; Schweitzer, P.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Gertsch, J.; Leonti, M.; Raduner, S.; Racz, I.; Chen, J.Z.; Xie, X.Q.; Altmann, K.H.; Karsak, M.; Zimmer, A. Beta-caryophyllene is a dietary cannabinoid. Proc. Natl. Acad. Sci. USA 2008, 105, 9099–9104. [Google Scholar] [CrossRef] [Green Version]
- Alberti, T.B.; Barbosa, W.L.; Vieira, J.L.; Raposo, N.R.; Dutra, R.C. (-)-beta-Caryophyllene, a CB2 Receptor-Selective Phytocannabinoid, Suppresses Motor Paralysis and Neuroinflammation in a Murine Model of Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 691. [Google Scholar] [CrossRef]
- Finlay, D.B.; Sircombe, K.J.; Nimick, M.; Jones, C.; Glass, M. Terpenoids From Cannabis Do Not Mediate an Entourage Effect by Acting at Cannabinoid Receptors. Front. Pharmacol. 2020, 11, 359. [Google Scholar] [CrossRef]
- Santiago, M.; Sachdev, S.; Arnold, J.C.; McGregor, I.S.; Connor, M. Absence of Entourage: Terpenoids Commonly Found in Cannabis sativa Do Not Modulate the Functional Activity of Delta(9)-THC at Human CB1 and CB2 Receptors. Cannabis Cannabinoid Res. 2019, 4, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Dhopeshwarkar, A.; Mackie, K. Functional Selectivity of CB2 Cannabinoid Receptor Ligands at a Canonical and Noncanonical Pathway. J. Pharmacol. Exp. Ther. 2016, 358, 342–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, M.R.; Finlay, D.B.; Macdonald, C.E.; Cawston, E.E.; Grimsey, N.L.; Glass, M. Real-Time Measurement of Cannabinoid Receptor-Mediated cAMP Signaling. Methods Enzymol. 2017, 593, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, S.; Boyd, R.; Grimsey, N.L.; Santiago, M.; Connor, M. Brodifacoum does not modulate human cannabinoid receptor-mediated hyperpolarization of AtT20 cells or inhibition of adenylyl cyclase in HEK 293 cells. PeerJ 2019, 7, e7733. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.; Nikolaev, V.O. Imaging alterations of cardiomyocyte cAMP microdomains in disease. Front. Pharmacol. 2015, 6, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaev, V.O.; Lohse, M.J. Monitoring of cAMP synthesis and degradation in living cells. Physiology (Bethesda) 2006, 21, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bunemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 2004, 279, 37215–37218. [Google Scholar] [CrossRef] [Green Version]
- Willoughby, D.; Cooper, D.M. Live-cell imaging of cAMP dynamics. Nat. Methods 2008, 5, 29–36. [Google Scholar] [CrossRef]
- Hill, S.J. G-protein-coupled receptors: Past, present and future. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S27–S37. [Google Scholar] [CrossRef] [Green Version]
- Storch, U.; Straub, J.; Erdogmus, S.; Gudermann, T.; Mederos, Y.S.M. Dynamic monitoring of Gi/o-protein-mediated decreases of intracellular cAMP by FRET-based Epac sensors. Pflug. Arch. 2017, 469, 725–737. [Google Scholar] [CrossRef] [Green Version]
- Insel, P.A.; Ostrom, R.S. Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell. Mol. Neurobiol. 2003, 23, 305–314. [Google Scholar] [CrossRef]
- Sato, J.; Makita, N.; Iiri, T. Inverse agonism: The classic concept of GPCRs revisited [Review]. Endocr. J. 2016, 63, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Melsom, C.B.; Orstavik, O.; Osnes, J.B.; Skomedal, T.; Levy, F.O.; Krobert, K.A. Gi proteins regulate adenylyl cyclase activity independent of receptor activation. PLoS ONE 2014, 9, e106608. [Google Scholar] [CrossRef] [Green Version]
- Tubio, M.R.; Fernandez, N.; Fitzsimons, C.P.; Copsel, S.; Santiago, S.; Shayo, C.; Davio, C.; Monczor, F. Expression of a G protein-coupled receptor (GPCR) leads to attenuation of signaling by other GPCRs: Experimental evidence for a spontaneous GPCR constitutive inactive form. J. Biol. Chem. 2010, 285, 14990–14998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klarenbeek, J.B.; Goedhart, J.; Hink, M.A.; Gadella, T.W.; Jalink, K. A mTurquoise-based cAMP sensor for both FLIM and ratiometric read-out has improved dynamic range. PLoS ONE 2011, 6, e19170. [Google Scholar] [CrossRef] [PubMed]
- Borner, C.; Smida, M.; Hollt, V.; Schraven, B.; Kraus, J. Cannabinoid receptor type 1- and 2-mediated increase in cyclic AMP inhibits T cell receptor-triggered signaling. J. Biol. Chem. 2009, 284, 35450–35460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Jansen, V.; Jikeli, J.F.; Hamzeh, H.; Alvarez, L.; Dombrowski, M.; Balbach, M.; Strunker, T.; Seifert, R.; Kaupp, U.B.; et al. A novel biosensor to study cAMP dynamics in cilia and flagella. Elife 2016, 5. [Google Scholar] [CrossRef]
- Borner, S.; Schwede, F.; Schlipp, A.; Berisha, F.; Calebiro, D.; Lohse, M.J.; Nikolaev, V.O. FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat. Protoc. 2011, 6, 427–438. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, J.U.; Perera, R.K.; Gotz, K.R.; Nikolaev, V.O. FRET microscopy for real-time monitoring of signaling events in live cells using unimolecular biosensors. J. Vis. Exp. 2012, e4081. [Google Scholar] [CrossRef] [Green Version]
- Nagler, M.; Palkowitsch, L.; Rading, S.; Moepps, B.; Karsak, M. Cannabinoid receptor 2 expression modulates Gbeta(1)gamma(2) protein interaction with the activator of G protein signalling 2/dynein light chain protein Tctex-1. Biochem. Pharmacol. 2016, 99, 60–72. [Google Scholar] [CrossRef]
- Kraft, A.E.; Nikolaev, V.O. FRET Microscopy for Real-Time Visualization of Second Messengers in Living Cells. Methods Mol. Biol. 2017, 1563, 85–90. [Google Scholar] [CrossRef] [PubMed]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mensching, L.; Rading, S.; Nikolaev, V.; Karsak, M. Monitoring Cannabinoid CB2 -Receptor Mediated cAMP Dynamics by FRET-Based Live Cell Imaging. Int. J. Mol. Sci. 2020, 21, 7880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217880
Mensching L, Rading S, Nikolaev V, Karsak M. Monitoring Cannabinoid CB2 -Receptor Mediated cAMP Dynamics by FRET-Based Live Cell Imaging. International Journal of Molecular Sciences. 2020; 21(21):7880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217880
Chicago/Turabian StyleMensching, Leonore, Sebastian Rading, Viacheslav Nikolaev, and Meliha Karsak. 2020. "Monitoring Cannabinoid CB2 -Receptor Mediated cAMP Dynamics by FRET-Based Live Cell Imaging" International Journal of Molecular Sciences 21, no. 21: 7880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217880