Interaction of Cucurbit[7]uril with Oxime K027, Atropine, and Paraoxon: Risky or Advantageous Delivery System?

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. In Silico Study
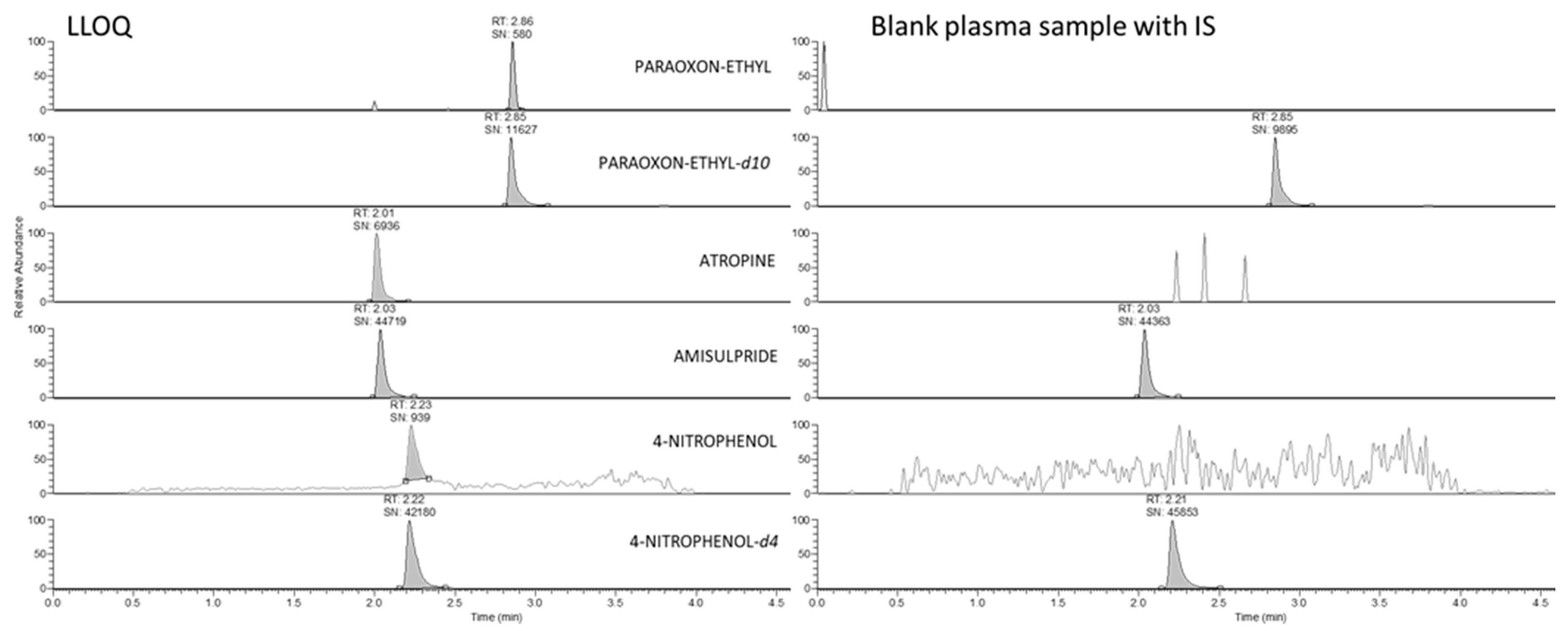
2.2. LC-HRMS Analysis and Method Validation
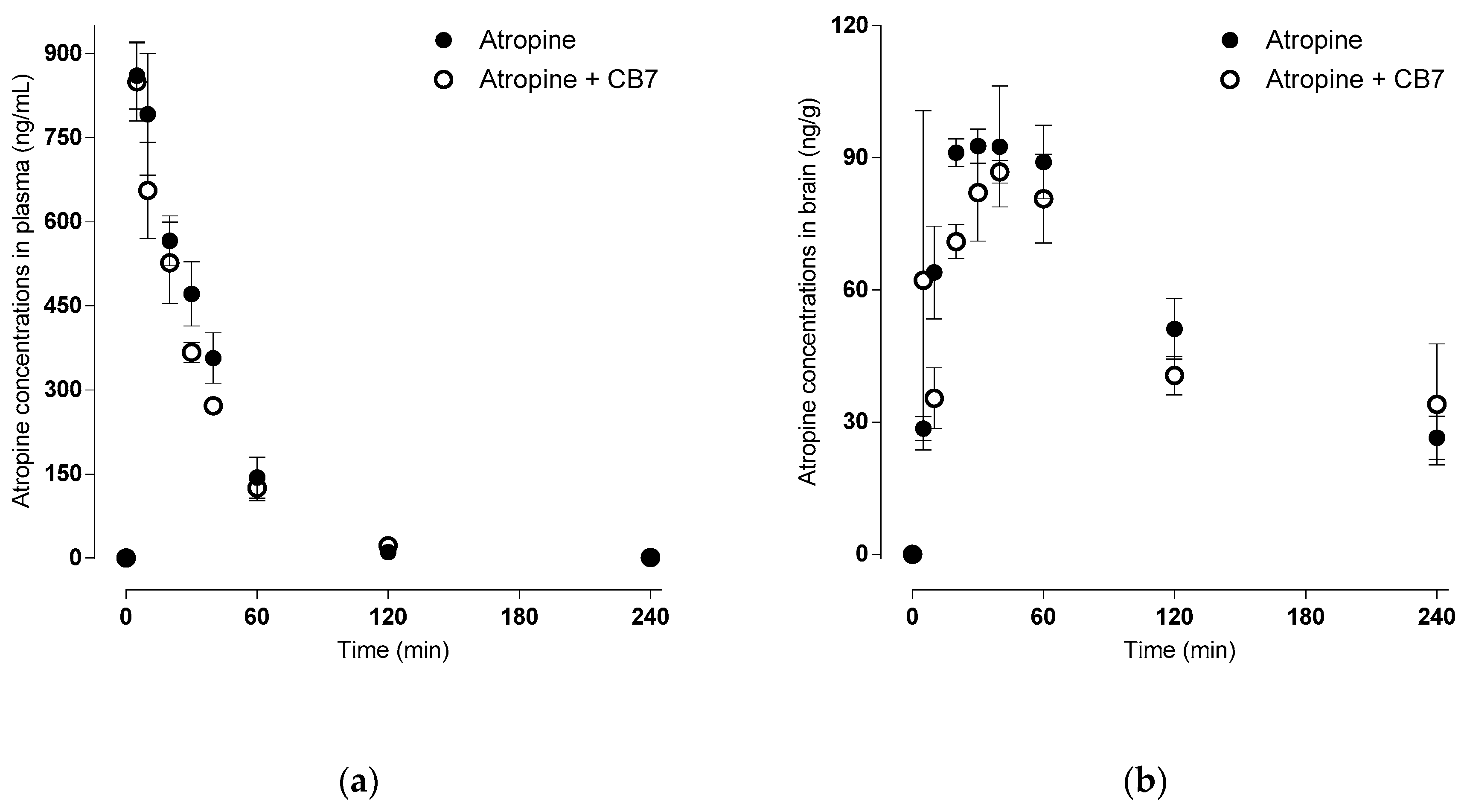
2.3. Pharmacokinetic Study of Atropine—Absorption and Brain Distribution
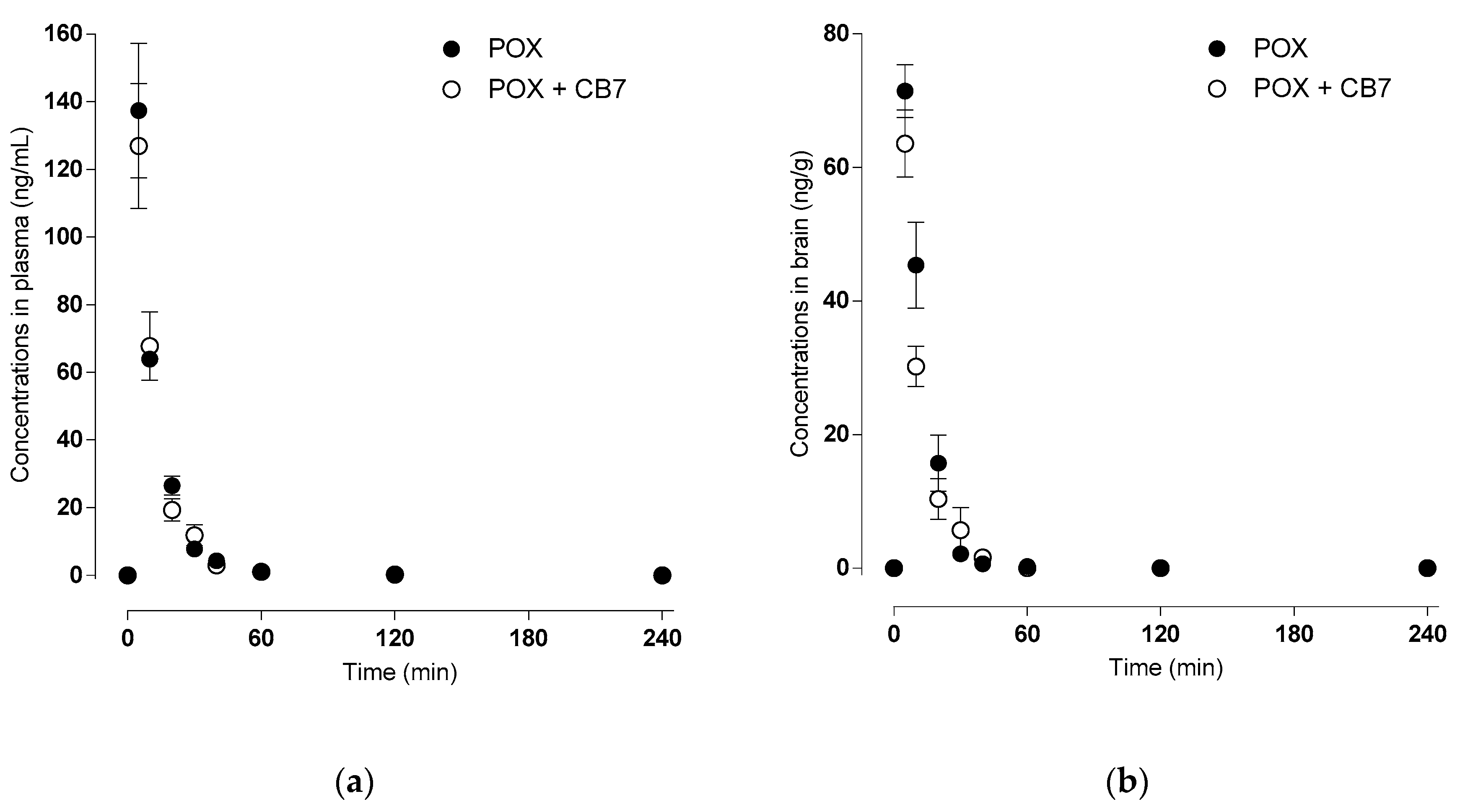
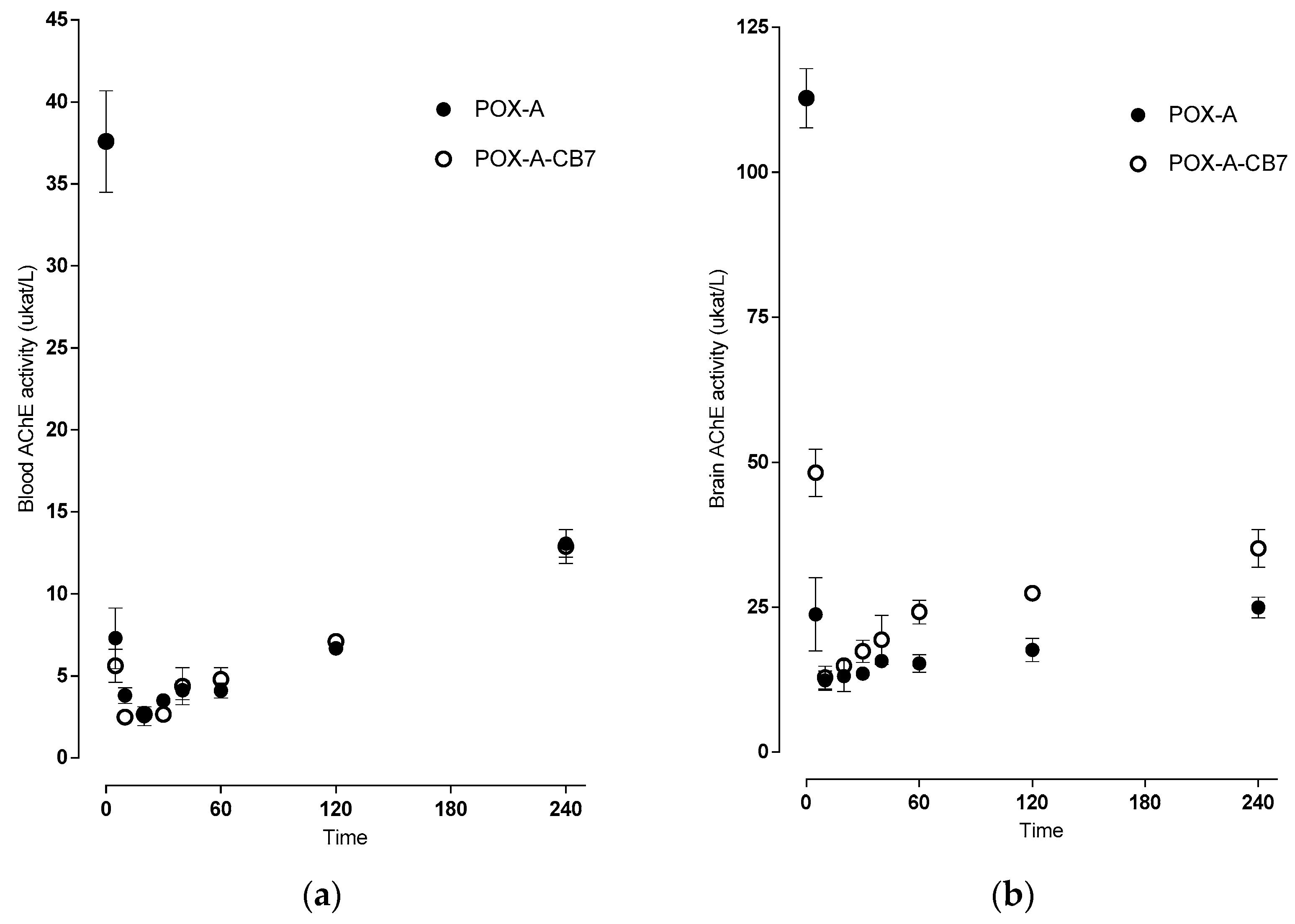
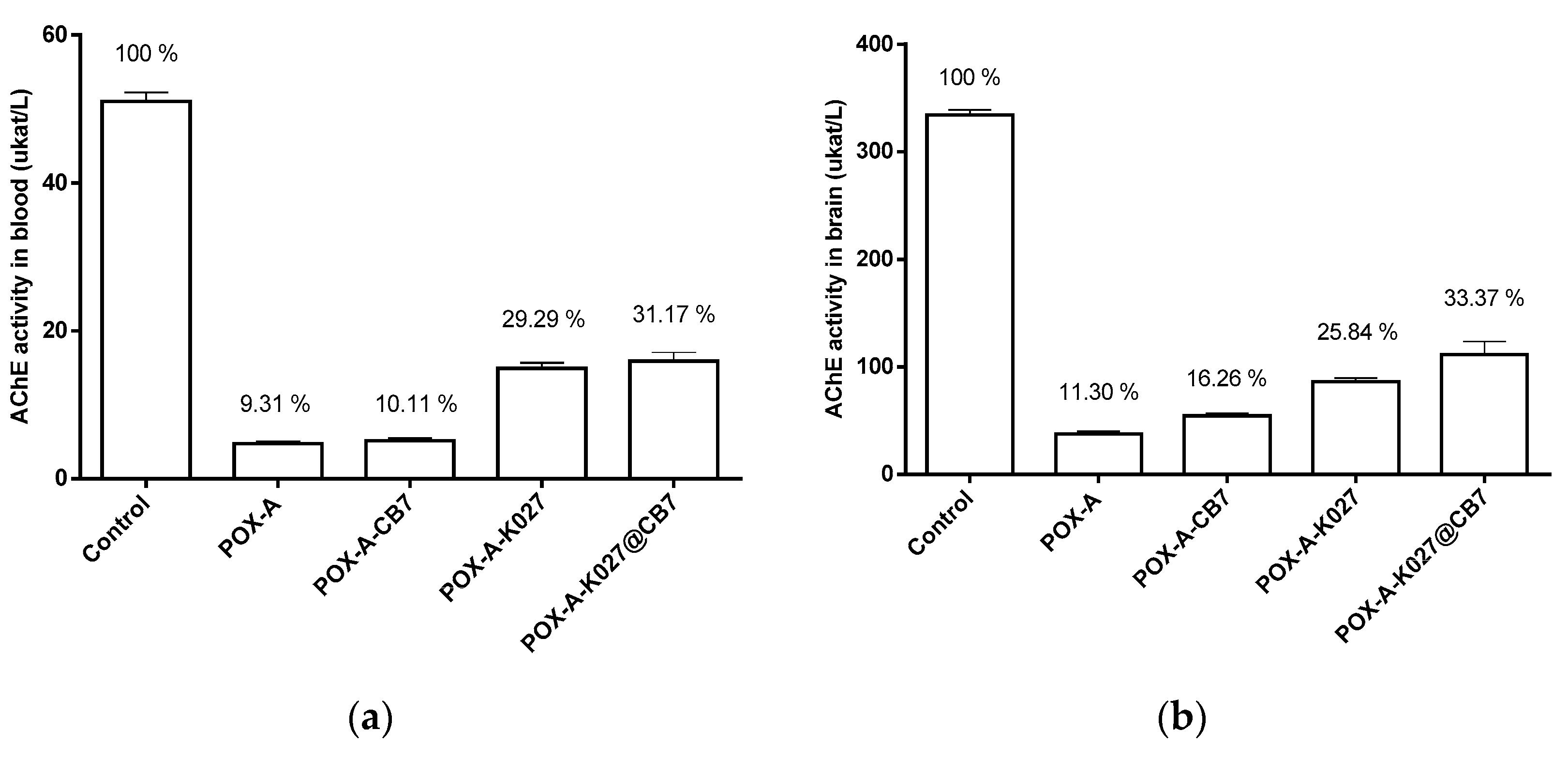
2.4. Toxicokinetics of Paraoxon versus Acetylcholinesterase Inhibition
2.5. Pharmacodynamic Effectiveness of K027—Reactivation Study
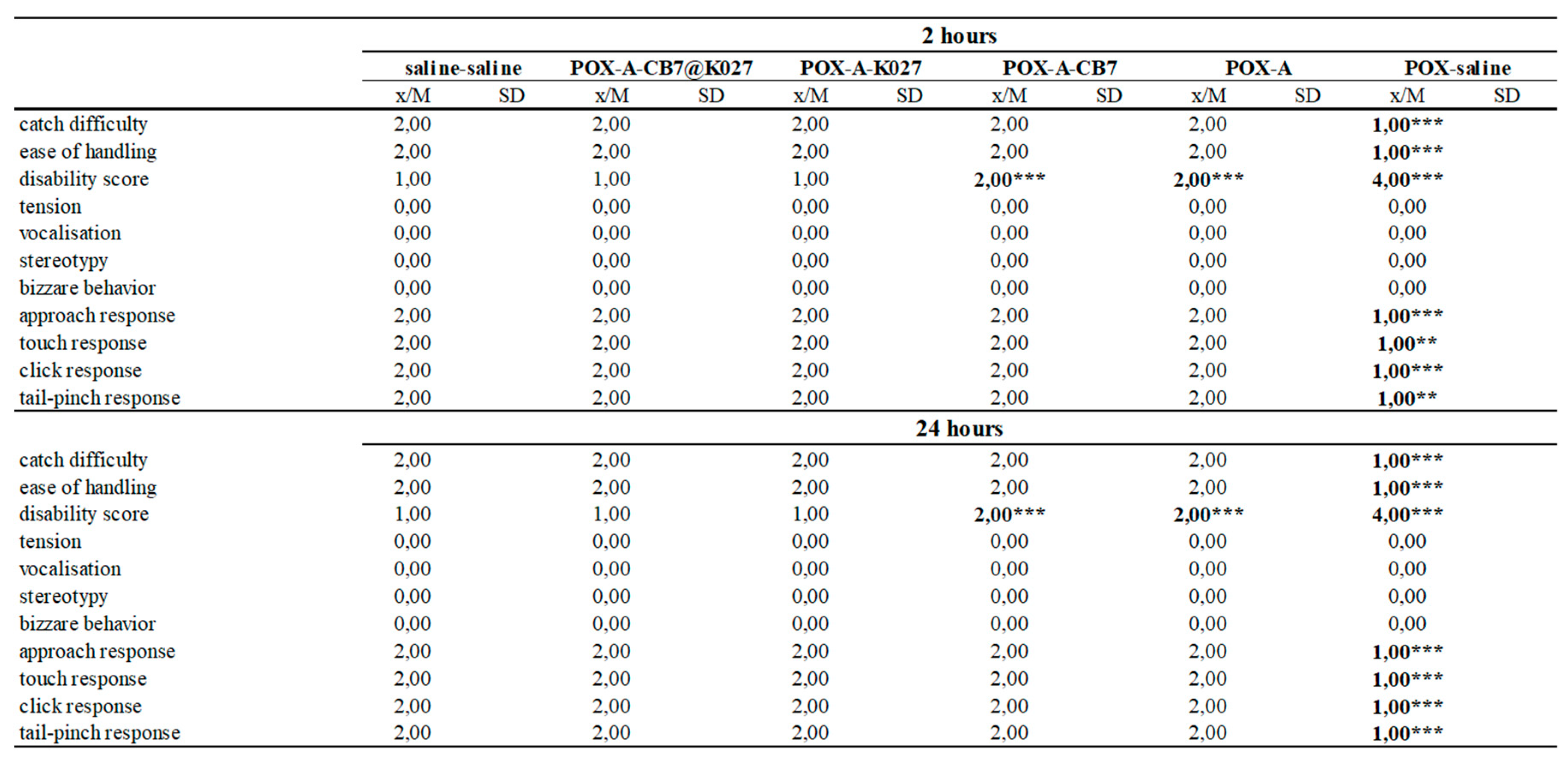
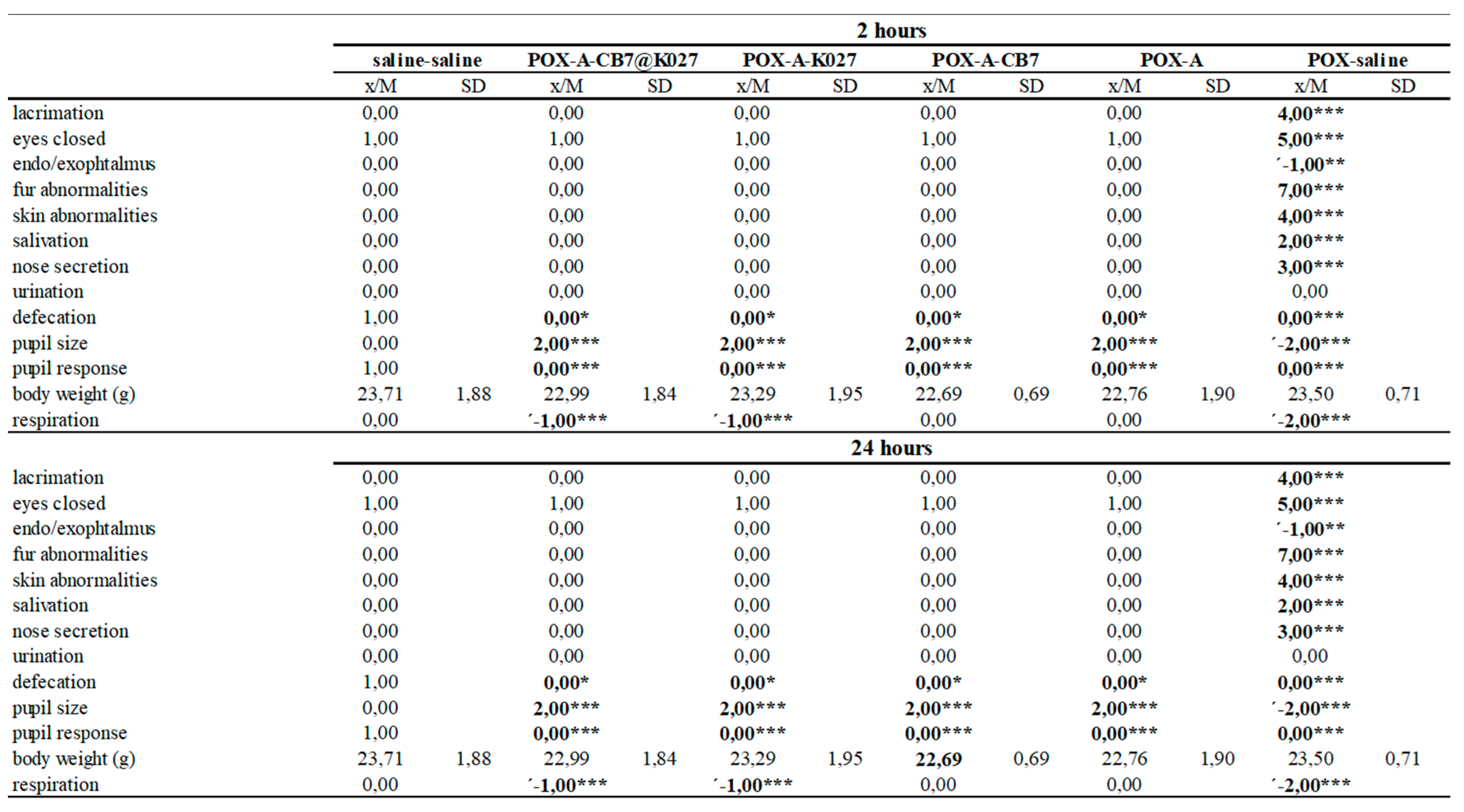
2.6. Functional Observatory Battery
3. Discussion
4. Materials and Methods
4.1. In Silico Prediction
4.2. Animals
4.3. Chemicals
4.4. LC-HRMS Analysis
4.4.1. LC-HRMS Parameters
4.4.2. Sample Preparation and Method Validation
4.5. Design of In Vivo Experiments
4.5.1. Toxicokinetics of Paraoxon Versus Acetylcholinesterase Inhibition in the Blood and Brain—Pharmacokinetic Study of Atropine
4.5.2. Dosing in Reactivation Study and Functional Observatory Battery
4.5.3. Acetylcholinesterase Activity Assessment
4.5.4. Functional Observatory Battery
4.6. Data Evaluation
4.6.1. Toxicokinetics of Paraoxon, Pharmacokinetics of Atropine
4.6.2. Acetylcholinesterase Reactivations
4.6.3. Functional Observatory Battery
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A | Atropine |
| AChE | Acetylcholinesterase |
| AUCtotal | The area under the concentration–time curve from zero to infinity |
| Cmax | Peak plasma/brain concentration |
| CB[7] | Cucurbit[7]uril |
| xx@CB[7] | Compounds (xx—atropine, paraoxon, or oxime K027) encapsulated into cucurbit[7]uril |
| FOB | Functional observatory battery |
| OPs | Organophosphates—nerve agents and pesticides |
| PP | Protein precipitation |
| POX | Paraoxon |
| QC | Quality control |
| Tmax | Time to Cmax |
References
- Eyer, P. The role of oximes in the management of organophosphorus pesticide poisoning. Toxicol. Rev. 2003, 22, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Musilek, K.; Pohanka, M.; Zdarova Karasova, J.; Soukup, O. Prophylaxis and post-exposure treatment of intoxications caused by nerve agents and organophosphorus pesticides. Mini Rev. Med. Chem. 2013, 13, 2102–2115. [Google Scholar] [CrossRef] [PubMed]
- Jokanovic, M. Medical treatment of acute poisoning with organophosphorus and carbamate pesticides. Tox. Lett. 2009, 190, 107–115. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.; Aaron, C.K. Organophosphate and carbamate poisoning. Emerg. Med. Clin. N. Am. 2015, 33, 133–151. [Google Scholar] [CrossRef]
- Karasova, J.Z.; Zemek, F.; Musilek, K.; Kuca, K. Time-dependent changes of oxime K027 concentrations in different parts of rat central nervous system. Neurotox. Res. 2013, 23, 63–68. [Google Scholar] [CrossRef]
- Karasova, J.Z.; Zemek, F.; Kassa, J.; Kuca, K. Entry of oxime K027 into the different parts of rat brain: Comparison with obidoxime and oxime HI-6. J. Appl. Biomed. 2014, 12, 25–29. [Google Scholar] [CrossRef]
- Karasova, J.Z.; Kvetina, J.; Tacheci, I.; Radochova, V.; Musilek, K.; Kuca, K.; Bures, J. Pharmacokinetic profile of promising acetylcholinesterase reactivators K027 and K203 in experimental pigs. Tox. Lett. 2017, 273, 20–25. [Google Scholar] [CrossRef]
- Lagona, J.; Fettinger, J.C.; Isaacs, L. Cucurbi[n]urils: Synthetic and Mechanistic studies. J. Org. Chem 2005, 70, 10381–10392. [Google Scholar] [CrossRef]
- Liu, S.M.; Wu, C.T. Recent progress in studies of cucurbituril. Prog. Chem. 2005, 17, 143–150. [Google Scholar]
- Wang, R.; Bardelang, D.; Waite, M.; Udachin, K.A.; Leek, D.M.; Yu, K.; Ratcliffe, C.I.; Ripmeester, J.A. Inclusion complexes of coumarin in cucurbiturils. Org. Biomol. Chem. 2009, 7, 2435–2439. [Google Scholar] [CrossRef]
- Wang, R.; Macartney, D.H. Cucurbit[7]uril host-guest complexes of the histamine H2-receptor antagonist ranitidine. Org. Biomol. Chem. 2008, 6, 1955–1960. [Google Scholar] [CrossRef] [PubMed]
- McInnes, F.J.; Anthony, N.G.; Kennedy, A.R.; Wheate, N.J. Solid state stabilisation of the orally delivered drugs atenolol, glibenclamide, memantine and paracetamol through their complexation with cucurbit[7]uril. Org. Biomol. Chem. 2010, 8, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Wyman, I.W.; Macartney, D.H. Host-guest complexations of local anaesthetics by cucurbit[7]uril in aqueous solution. Org. Biomol. Chem. 2010, 8, 247–252. [Google Scholar] [CrossRef]
- Wheate, N.J.; Buck, D.P.; Day, A.I.; Collins, J.G. Cucurbit[n]uril binding of platinum anti-cancer complexes. Dalton Trans. 2006, 3, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrýs, R.; Klusoňová, A.; Lísa, M.; Karasová, J.Ž. Encapsulation of oxime acetylcholinesterase reactivators: Influence of physiological conditions on the stability of oxime-cucurbit[7]uril complexes. New J. Chem. 2020. [Google Scholar] [CrossRef]
- Zdarova Karasova, J.; Hepnarova, V.; Andrys, R.; Lisa, M.; Jost, P.; Muckova, L.; Pejchal, J.; Herman, D.; Jun, D.; Kassa, J.; et al. Encapsulation of oxime K027 into cucurbit[7]uril: In vivo evaluation of safety, absorption, brain distribution and reactivation effectiveness. Toxicol. Lett. 2020, 320, 64–72. [Google Scholar] [CrossRef]
- Plumb, J.A.; Venugopal, B.; Oun, R.; Gomez-Roman, N.; Kawazoe, Y.; Venkataramanan, N.S.; Wheate, N.J. Cucurbit[7]uril encapsulated cisplatin overcomes cisplatin resistance via a pharmacokinetic effect. Metallomics 2012, 4, 561–567. [Google Scholar] [CrossRef] [Green Version]
- EMA. Guideline on Bioanalytical Method Validation. 21 July 2011. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf (accessed on 27 July 2020).
- Center for Drug Evaluation and Research (CDER); Center for Veterinary Medicine (CVM); FDA. Guidance for Industry, Bioanalytical Method Validation. May 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 27 July 2018).
- Li, B.; Sedlacek, M.; Manoharan, I.; Boopathy, M.; Duysen, E.G.; Masson, P.; Lockride, O. Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem. Pharmacol. 2005, 70, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Ortigoza-Ferado, J.; Richter, R.J.; Hornung, S.K.; Motulsky, A.G.; Furlong, C.E. Paraoxon hydrolysis in human serum mediated by a genetically variable arylesterase and albumin. Am. J. Hum. Genet. 1984, 36, 295–305. [Google Scholar]
- Moser, V.C.; Tilson, H.A.; MacPhail, R.C.; Becking, G.C.; Cuomo, V.; Frantík, E.; Kulig, B.M.; Winneke, G. The IPCS Collaborative Study on Neurobehavioral Screening Methods: II. Protocol design and testing procedures. Neurotoxicology 1997, 18, 929–938. [Google Scholar]
- Assaf, K.I.; Nau, W.M. Cucurbiturils: From synthesis to high-affinity binding and catalysis. Chem. Soc. Rev. 2015, 44, 394–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biedermann, F.; Uzunova, V.D.; Scherman, O.A.; Nau, W.M.; De Simone, A. Release of high-energy water as an essential driving force for the high-affinity binding of cucurbit[n]urils. J. Am. Chem. Soc. 2012, 134, 15318–15323. [Google Scholar] [CrossRef] [PubMed]
- Proakis, A.G.; Harris, G.B. Comparative penetration of glycopyrrolate and atropine across the blood—brain and placental barriers in anesthetized dogs. Anesthesiology 1978, 48, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.D.; Bosin, T.R.; Maickel, R.P. Physiological disposition of atropine in the rat. Pharmacol. Biochem. Behav. 1974, 2, 843–845. [Google Scholar] [CrossRef]
- Chowdhary, S.; Bhattacharya, R.; Banerjee, D. Acute organophosphorus poisoning. Clin. Chim. Acta 2014, 431, 66–76. [Google Scholar] [CrossRef]
- Kanto, J.; Virtanen, R.; Iisalo, E.; Mäenpää, K.; Liukko, P. Placental transfer and pharmacokinetics of atropine after a single maternal intravenous and intramuscular administration. Acta Anaesthesiol. Scand. 1981, 25, 85–88. [Google Scholar] [CrossRef]
- Kassa, J. Importance of cholinolytic drug selection for the efficacy of HI-6 against soman in rats. Toxicology 1997, 116, 147–152. [Google Scholar] [CrossRef]
- Karasova, J.; Bajgar, J.; Jun, D.; Pavlikova, R.; Kuca, K. Time-course changes of acetylcholinesterase activity in blood and some tissues in rats after intoxication by Russian VX. Neurotox. Res. 2009, 16, 356–360. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, X.; Li, S.; Li, L.; Zhang, J.; Wang, R. A Synthetic Receptor as a Specific Antidote for Paraquat Poisoning. Theranostics 2019, 9, 633–645. [Google Scholar] [CrossRef]
- Kassa, J.; Musilek, K.; Karasova, J.Z.; Kuca, K.; Bajgar, J. Two possibilities how to increase the efficacy of antidotal treatment of nerve agent poisonings. Mini Med. Chem. Rev. 2012, 12, 24–34. [Google Scholar] [CrossRef]
- Katalinic, M.; Hrvat, N.M.; Karasova, J.Z.; Misik, J.; Kovarik, Z. Translation of in vitro to in vivo pyridinium oxime potential in tabun poisoning. Arh. Hig. Rada Toksikol. 2015, 66, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collombet, J.M.; Four, E.; Fauquette, W.; Burckhart, M.F.; Masqueliez, C.; Bernabe, D.; Baubichon, D.; Lallement, G. Soman poisoning induces delayed astrogliotic scar and angiogenesis in damaged mouse brain areas. Neurotoxicology 2007, 28, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Collombet, J.M. Nerve agent intoxication: Recent neuropathophysiological findings and subsequent impact on medical management prospects. Toxicol. Appl. Pharmacol. 2011, 255, 229–241. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hub, J.S.; de Groot, B.L.; van der Spoel, D. G_wham—A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem Theory Comput. 2010, 6, 3713–3720. [Google Scholar] [CrossRef] [Green Version]
- Aldrige, W.N. Serum esterases. II. An enzyme hydrolysing diethyl p-nitrophenyl phosphate (E600) and its identity with the A-esterase of mammalian sera. Biochem. J. 1953, 53, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassa, J.; Korabecny, J.; Nepovimova, E.; Jun, D. The influence of modulators of acetylcholinesterase on the resistance of mice against soman and on the effectiveness of antidotal treatment of soman poisoning in mice. J. Appl. Biomed. 2018, 16, 10–14. [Google Scholar] [CrossRef]
- Karasova, J.Z.; Maderycova, Z.; Tumova, M.; Jun, D.; Rehacek, V.; Kuca, K.; Misik, J. Activity of cholinesterases in a young and healthy middle-European population: Relevance for toxicology, pharmacology and clinical praxis. Toxicol. Lett. 2017, 277, 24–31. [Google Scholar] [CrossRef]
- Clement, J.G. Central activity of acetylcholinesterase oxime reactivators. Toxicol. Appl. Pharmacol. 1992, 112, 104–109. [Google Scholar] [CrossRef]
- Kassa, J.; Misik, J.; Hatlapatkova, J.; Zdarova Karasova, J.; Sepsova, V.; Caisberber, F.; Pejchal, J. The evaluation of the reactivating and neuroprotective efficacy of two newly prepared bispyridinium oximes (K305, K307) in tabun-poisoned rats—A comparison with trimedoxime and the oxime K203. Molecules 2017, 22, 1152. [Google Scholar] [CrossRef] [Green Version]
- Kassa, J.; Zdarova Karasova, J.; Tesarova, S. A comparison of the neuroprotective efficacy of individual oxime (HI-6) and combinations of oximes (HI-6+trimedoxime, HI-6+K203) in soman-poisoned rats. Drug Chem. Toxicol. 2011, 34, 233–239. [Google Scholar] [CrossRef]
- Shih, T.-M.; McDonough, J.H. Neurochemical mechanisms in soman-induced seizures. J. Appl. Toxicol. 1997, 17, 255–264. [Google Scholar] [CrossRef]
- Lushchekina, S.V.; Schopfer, L.M.; Grigorenko, B.L.; Nemukhin, A.V.; Varfolomeev, S.D.; Lockridge, O.; Masson, P. Optimization of cholinesterase-based catalytic bioscavengers against organophosphorus agents. Front. Pharmacol 2018, 9, 211. [Google Scholar] [CrossRef]
- Joosen, M.J.A.; van der Schansa, M.J.; van Dijka, C.h.G.M.; Kuijpersa, W.C.; Wortelboerb, H.M.; van Helden, H.P.M. Increasing oxime efficacy by blood-brain barrier modulation. Toxicol. Lett. 2011, 206, 67–71. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLASMA | Atropine | Atropine-CB[7] |
|---|---|---|
| Cmax (ng/mL) | 860.48 ± 48.74 | 849.17 ± 56.96 |
| Tmax (min) | 6.67 ± 1.36 | 5.00 ± 0.00 |
| AUCtotal (min × ng/mL) | 31,028 ± 2956 | 27,643 ± 2903 |
| λz (L/min) | 0.024 ± 0.01 | 0.027 ± 0.00 |
| Half-life (min) | 28.67 ± 0.93 | 25.96 ± 0.03 |
| MRT (min) | 31.25 ± 0.90 | 33.10 ± 2.08 |
| CL (L/min/kg) | 0.33 ± 0.03 | 0.37 ± 0.04 |
| Vz (L/kg) | 13.84 ± 1.80 | 13.97 ± 1.42 |
| Vss (L/kg) | 10.32 ± 0.96 | 12.12 ± 0.54 |
| BRAIN | ||
| Cmax (ng/mL) | 99.58 ± 8.43 | 107.28 ± 4.89 |
| Tmax (min) | 33.33 ± 2.72 | 31.67 ± 13.00 |
| AUCtotal (min × ng/mL) | 17,361 ± 2092 | 19,582 ± 4343 |
| λz (L/min) | 0.007 ± 0.001 | 0.006 ± 0.002 |
| Half-life (min) | 105.87 ± 8.71 | 153.66 ± 52.82 |
| MRT (min) | 164.53 ± 12.01 | 231.54 ± 72.24 |
| PLASMA | POX | POX-CB[7] |
|---|---|---|
| Cmax (ng/mL) | 137.41 ± 16.25 | 126.90 ± 15.11 |
| Tmax (min) | 5.00 ± 0.00 | 5.00 ± 0.00 |
| AUCtotal (min × ng/mL) | 1588 ± 144 | 1524 ±122 |
| λz (L/min) | 0.070 ± 0.002 | 0.063 ± 0.015 |
| Half-life (min) | 9.91 ± 0.26 | 14.75 ± 5.11 |
| MRT (min) | 12.47 ± 0.40 | 13.66 ± 0.71 |
| CL (L/min/kg) | 0.33 ± 0.03 | 0.34 ± 0.03 |
| Vz (L/kg) | 4.75 ± 0.39 | 7.39 ± 2.62 |
| Vss (L/kg) | 4.12 ± 0.26 | 4.72 ± 0.44 |
| BRAIN | ||
| Cmax (ng/mL) | 71.44 ± 3.23 | 63.57 ± 4.10 |
| Tmax (min) | 5.00 ± 0.00 | 5.00 ± 0.00 |
| AUCtotal (min × ng/mL) | 899 ± 90 | 723 ± 86 |
| λz (L/min) | 0.114 ± 0.029 | 0.103 ± 0.003 |
| Half-life (min) | 8.57 ± 3.20 | 6.76 ± 0.19 |
| MRT (min) | 11.35 ± 0.85 | 11.62 ± 0.77 |
| Name | Molecular Formula | Theoretical Mass [M+H]+ | Retention Time (min) |
|---|---|---|---|
| Paraoxon-ethyl | C10H14NO6P | 276.06315 | 2.86 |
| Paraoxon-ethyl-d10 | C10H4D10NO6P | 286.12592 | 2.85 |
| Atropine | C17H23NO3 | 290.17507 | 2.01 |
| Amisulpride | C17H27N3O4S | 370.1795 | 2.03 |
| Name | Molecular Formula | Theoretical Mass [M+H+]− | Retention Time (min) |
| 4-Nitrophenol | C6H5NO3 | 138.01967 | 2.24 |
| 4-Nitrophenol-d4 | C6HD4NO3 | 142.04477 | 2.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zdarova Karasova, J.; Mzik, M.; Kucera, T.; Vecera, Z.; Kassa, J.; Sestak, V. Interaction of Cucurbit[7]uril with Oxime K027, Atropine, and Paraoxon: Risky or Advantageous Delivery System? Int. J. Mol. Sci. 2020, 21, 7883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217883
Zdarova Karasova J, Mzik M, Kucera T, Vecera Z, Kassa J, Sestak V. Interaction of Cucurbit[7]uril with Oxime K027, Atropine, and Paraoxon: Risky or Advantageous Delivery System? International Journal of Molecular Sciences. 2020; 21(21):7883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217883
Chicago/Turabian StyleZdarova Karasova, Jana, Martin Mzik, Tomas Kucera, Zbynek Vecera, Jiri Kassa, and Vit Sestak. 2020. "Interaction of Cucurbit[7]uril with Oxime K027, Atropine, and Paraoxon: Risky or Advantageous Delivery System?" International Journal of Molecular Sciences 21, no. 21: 7883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217883