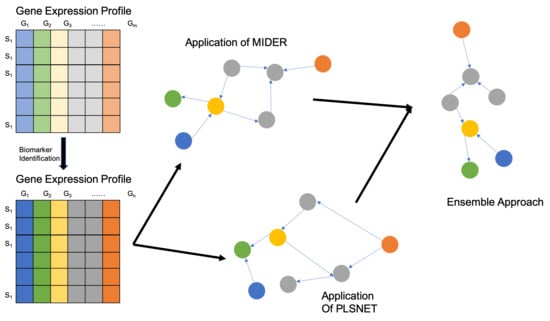
Biomarker Prioritisation and Power Estimation Using Ensemble Gene Regulatory Network Inference

 ,
,  ,
,  ,
,
Abstract
:
1. Introduction
2. Results
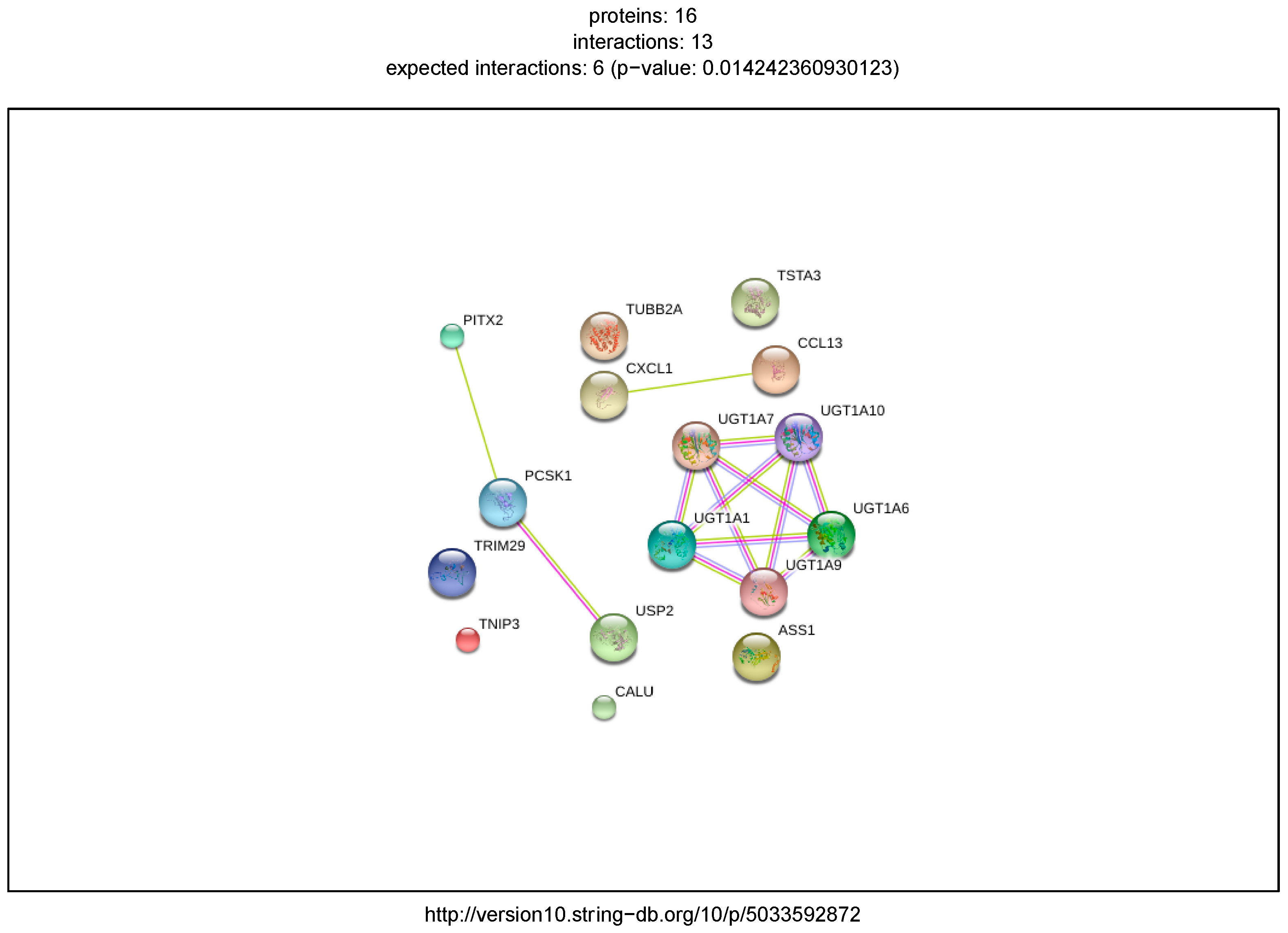
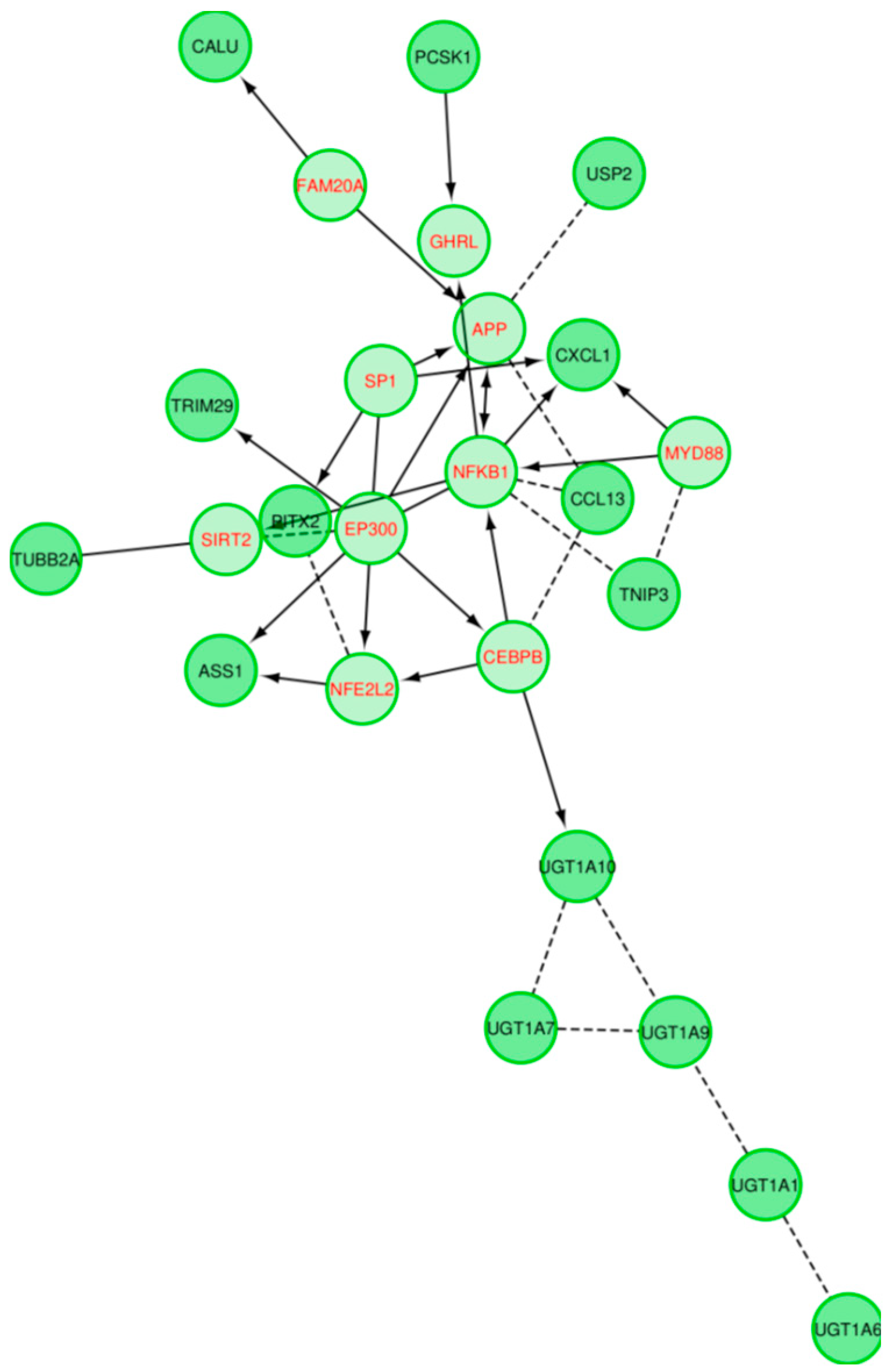
2.1. Inflammatory Bowel Disease
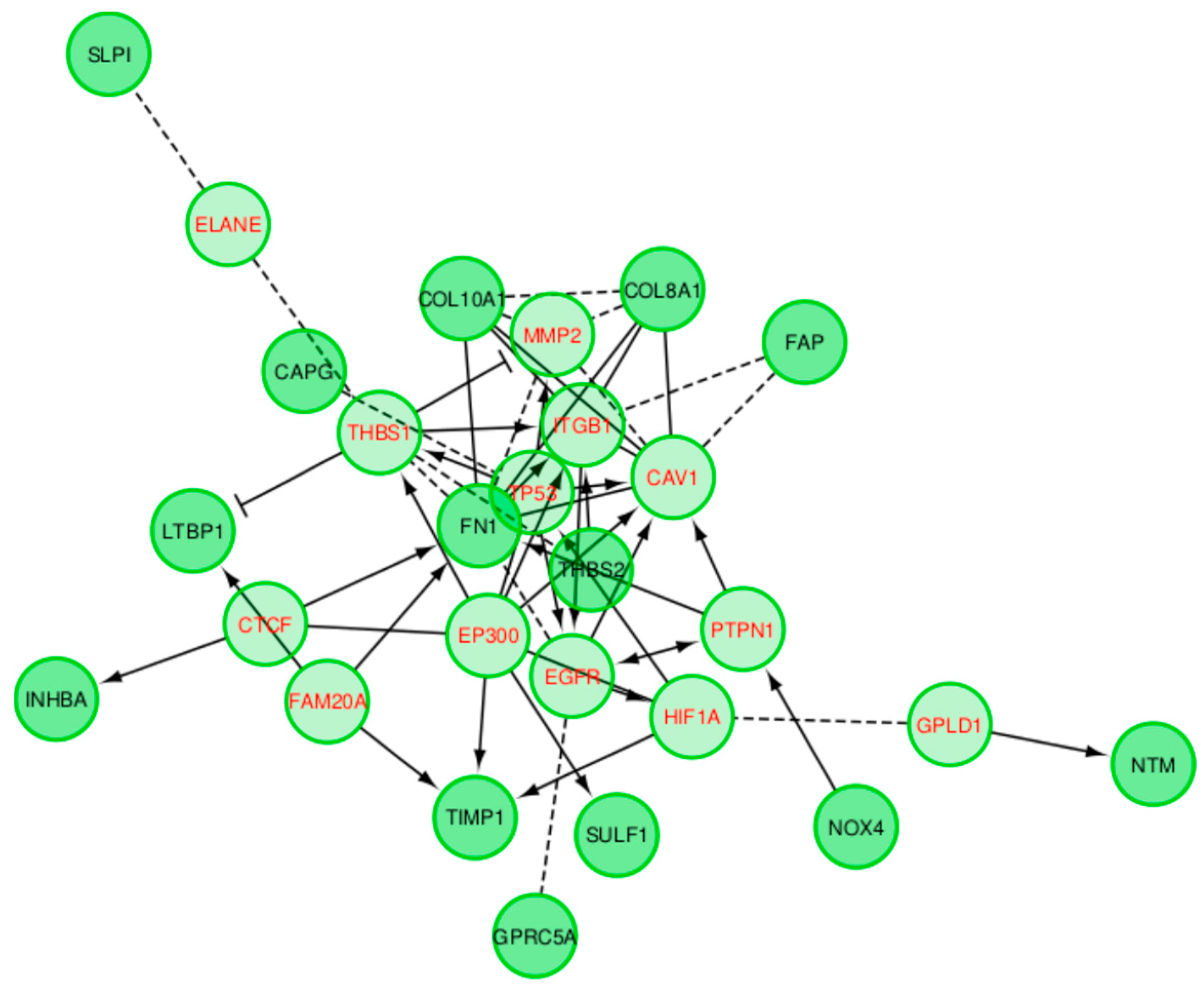
2.2. Pancreatic Ductal Adenocarcinoma
2.3. Acute Myeloid Leukaemia
2.4. DREAM
2.5. External Biological Validation
3. Discussion
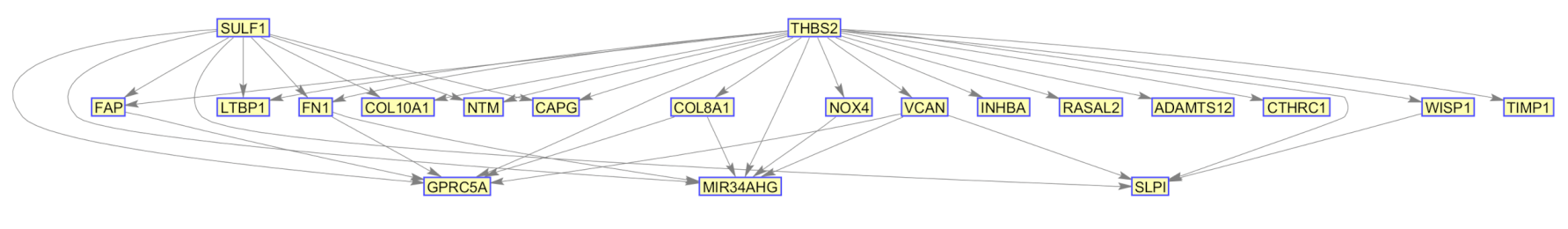
Biological Relevance of IBD and DPAC Networks
4. Materials and Methods
4.1. Datasets and Gene Selection
4.2. PLSNET
4.3. MIDER
- The algorithm estimates a number of statistical properties, including conditional entropies, transfer entropies, and mutual information from the data. These estimates are then employed at different stages of the network construction. Let be a discrete random variable with alphabet and probability mass function . Then the entropy is defined using Equation (4) as:For a continuous variable is replaced by . The joint entropy of two random variables and is defined using Equation (5) as:The conditional probability of a random variable conditioning upon another random variable is defined using Equation (6) as:Finally, the mutual information between two random variables is defined using Equation (7) as:
- Based on the mutual information estimates, a distance matrix between all the genes variables is constructed. The distance between two variables and is computed as . This distance matrix is used as a first approximation of the connections between variables. Since is symmetric, the distance matrix is also symmetric, i.e., .
- An entropy reduction, based on conditional entropies, is then applied to further refine the map. This allows for the discriminating between direct and indirect connections. One of the limitations of entropy reduction is that it requires a large amount of data to get a reliable estimate [38]. Instead of considering all the reactants, MIDER performs a limited reconstruction by considering only first important ones. In the MATLAB implementation of MIDER [2] algorithm, the authors have estimated joint entropies of up to 4-tuples of variables. In our case, since we have a limited number of samples, we have used only 3-tuples of variables (i.e., a value of ). This is also the default value used in the implementation of MIDER for computation reasons.
- Finally, the directions of the inferred links are assigned using transfer entropy, , which is a non-symmetric measure of causality [39]. Here, for every predicted link, MIDER calculates two transfer entropies (i.e., and ) and assigns the causality in the direction corresponding to the maximum of the two.
4.4. System Wide Analysis of GRN
- The first step is to convert the Bayesian gene network into an equivalent factor graph. A factor graphs is a bipartite graph with two types of nodes, i.e., a variable node that denotes each random variable and a factor node that denotes a local function.
- The next step is to discretise the data. For a GRN, this is considered an integral part of the model and is usually performed for computational efficiency. Without discretisation, a large amount of data is required to accurately learn the regulatory relations [40]. Furthermore, discretisation helps reduce noise in the continuous variables [40]. In the framework developed by Kotiang and Eslami [19], this step is done by using a Gaussian mixture model with different (at least two) quantisation levels.
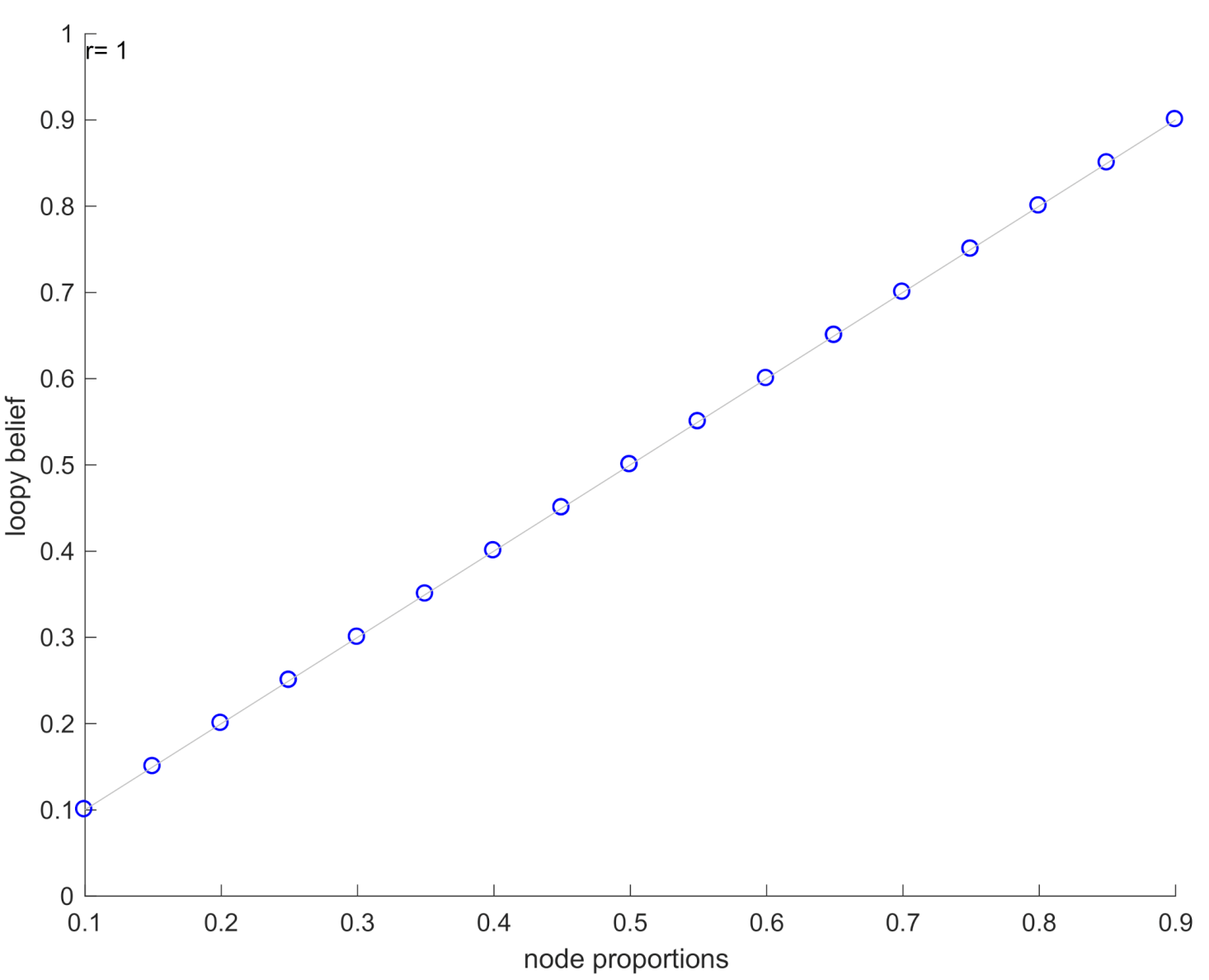
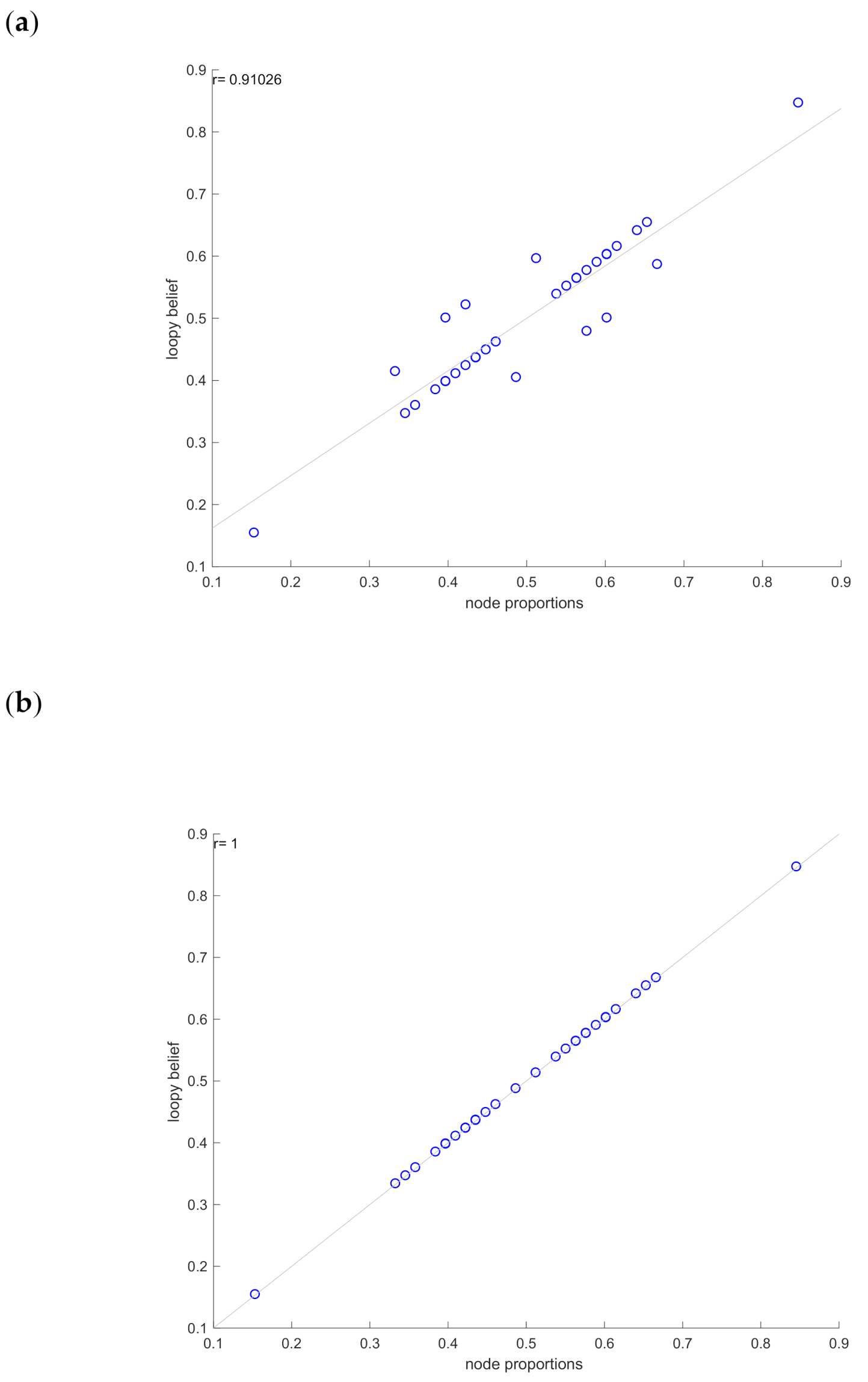
- To approximate the marginal posterior distributions across all genes, the loopy-belief propagation (LBP) algorithm is applied. LBP is a popular message passing algorithm that can be used to infer probabilities in a loopy graph. It is an iterative procedure that minimises the Bethe free energy [18] and achieves a good approximation if the solution converges in fixed number of iterations [41].
- Finally, the predicted marginals are compared with node proportions to estimate the performance of the inferred GRN.
4.5. Ensemble Approach
4.6. Biological Investigation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AML | Acute Myeloid Leukemia |
| GRN | Gene Regulatory Network |
| IBD | Inflammatory Bowel Disease |
| LBP | Loopy Belief Propagation |
| MIDER | Mutual Information Distance and Entropy Reduction |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| PLSNET | Partial Least Square based Network |
References
- De Smet, R.; Marchal, K. Advantages and limitations of current network inference methods. Nat. Rev. Microbiol. 2010, 8, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Villaverde, A.F.; Ross, J.; Morán, F.; Banga, J.R. MIDER: Network Inference with Mutual Information Distance and Entropy Reduction. PLoS ONE 2014, 9, e96732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faith, J.J.; Hayete, B.; Thaden, J.T.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.J.; Gardner, T.S. Large-Scale Mapping and Validation of Escherichia coli Transcriptional Regulation from a Compendium of Expression Profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef] [PubMed]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [Green Version]
- Butte, A.J.; Kohane, I.S. Mutual information relevance networks: Functional genomic clustering using pairwise entropy measurements. Pac. Symp. Biocomput. 2000, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Dalla Favera, R.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006, 7, S7. [Google Scholar] [CrossRef] [Green Version]
- Meyer, P.E.; Kontos, K.; Lafitte, F.; Bontempi, G. Information-theoretic inference of large transcriptional regulatory networks. EURASIP J. Bioinform. Syst. Biol. 2007, 2007, 79879. [Google Scholar] [CrossRef] [Green Version]
- Tourassi, G.D.; Frederick, E.D.; Markey, M.K.; Floyd, C.E.J. Application of the mutual information criterion for feature selection in computer-aided diagnosis. Med. Phys. 2001, 28, 2394–2402. [Google Scholar] [CrossRef]
- Ding, C.; Peng, H. Minimum redundancy feature selection from microarray gene expression data. J. Bioinform. Comput. Biol. 2005, 3, 185–205. [Google Scholar] [CrossRef]
- Haury, A.-C.; Mordelet, F.; Vera-Licona, P.; Vert, J.-P. TIGRESS: Trustful Inference of Gene REgulation using Stability Selection. BMC Syst. Biol. 2012, 6, 145. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R.; Johnstone, I.; Hastie, T.; Efron, B. Least angle regression. Ann. Stat. 2004, 32, 407–499. [Google Scholar] [CrossRef] [Green Version]
- Ruyssinck, J.; Huynh-Thu, V.A.; Geurts, P.; Dhaene, T.; Demeester, P.; Saeys, Y. NIMEFI: Gene Regulatory Network Inference using Multiple Ensemble Feature Importance Algorithms. PLoS ONE 2014, 9, e92709. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Jiang, Q.; Chen, L.; Guo, D. Gene regulatory network inference using PLS-based methods. BMC Bioinform. 2016, 17, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quraishi, M.N.; Acharjee, A.; Beggs, A.D.; Horniblow, R.; Tselepis, C.; Gkoutus, G.; Ghosh, S.; Rossiter, A.; Loman, N.; van Schaik, W.; et al. A Pilot Integrative Analysis of Colonic Gene Expression, Gut Microbiota, and Immune Infiltration in Primary Sclerosing Cholangitis-Inflammatory Bowel Disease: Association of Disease With Bile Acid Pathways. J. Crohn’s Colitis 2020, 14, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajamani, D.; Bhasin, M.K. Identification of key regulators of pancreatic cancer progression through multidimensional systems-level analysis. Genome Med. 2016, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Mills, K.I.; Kohlmann, A.; Williams, P.M.; Wieczorek, L.; Liu, W.M.; Li, R.; Wei, W.; Bowen, D.T.; Loeffler, H.; Hernandez, J.M.; et al. Microarray-based classifiers and prognosis models identify subgroups with distinct clinical outcomes and high risk of AML transformation of myelodysplastic syndrome. Blood 2009, 114, 1063–1072. [Google Scholar] [CrossRef] [Green Version]
- Yedidia, J.S.; Freeman, W.T.; Weiss, Y. Constructing free-energy approximations and generalized belief propagation algorithms. IEEE Trans. Inf. Theory 2005, 51, 2282–2312. [Google Scholar] [CrossRef]
- Kotiang, S.; Eslami, A. A probabilistic graphical model for system-wide analysis of gene regulatory networks. Bioinformatics 2020, 36, 3192–3199. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- De Vries, H.S.; Morsche, R.H.M.T.; Jenniskens, K.; Peters, W.H.M.; de Jong, D.J. A functional polymorphism in UGT1A1 related to hyperbilirubinemia is associated with a decreased risk for Crohn’s disease. J. Crohn’s Colitis 2012, 6, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdmann, P.; Bruckmueller, H.; Martin, P.; Busch, D.; Haenisch, S.; Müller, J.; Wiechowska-Kozlowska, A.; Partecke, L.I.; Heidecke, C.D.; Cascorbi, I.; et al. Dysregulation of Mucosal Membrane Transporters and Drug-Metabolizing Enzymes in Ulcerative Colitis. J. Pharm. Sci. 2019, 108, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Leníček, M.; Ďuricová, D.; Hradsky, O.; Dušátková, P.; Jirásková, A.; Lukáš, M.; Nachtigal, P.; Vítek, L. The Relationship Between Serum Bilirubin and Crohn’s Disease. Inflamm. Bowel Dis. 2014, 20, 481–487. [Google Scholar] [CrossRef] [PubMed]
- TWeismüller, J.; Zhou, T.; Kalthoff, S.; Lenzen, H.; Manns, M.P.; Strassburg, C.P. Genetic variants of UDP-glucuronosyltransferase 1A genes are associated with disease presentation and outcome in primary sclerosing cholangitis. Liver Int. 2020, 40, 1645–1654. [Google Scholar] [CrossRef]
- Gabitova-Cornell, L.; Surumbayeva, A.; Peri, S.; Franco-Barraza, J.; Restifo, D.; Weitz, N.; Ogier, C.; Goldman, A.R.; Hartman, T.R.; Francescone, R.; et al. Cholesterol Pathway Inhibition Induces TGF-β Signaling to Promote Basal Differentiation in Pancreatic Cancer. Cancer Cell 2020. [Google Scholar] [CrossRef]
- Tan, M.; de Muckadell, O.B.S.; Joergensen, M.T. Gene Expression Network Analysis of Precursor Lesions in Familial Pancreatic Cancer. J. Pancreat. Cancer 2020, 6, 73–84. [Google Scholar] [CrossRef]
- Simpson, R.E.; Yip-Schneider, M.T.; Wu, H.; Fan, H.; Liu, Z.; Korc, M.; Zhang, J.; Schmidt, C.M. Circulating Thrombospondin-2 enhances prediction of malignant intraductal papillary mucinous neoplasm. Am. J. Surg. 2019, 217, 425–428. [Google Scholar] [CrossRef]
- Le Large, T.Y.; Meijer, L.L.; Paleckyte, R.; Boyd, L.N.; Kok, B.; Wurdinger, T.; Schelfhorst, T.; Piersma, S.R.; Pham, T.V.; van Grieken, N.C.; et al. Combined Expression of Plasma Thrombospondin-2 and CA19-9 for Diagnosis of Pancreatic Cancer and Distal Cholangiocarcinoma: A Proteome Approach. Oncologist 2020, 25, e634–e643. [Google Scholar] [CrossRef] [Green Version]
- Bausch, D.; Fritz, S.; Bolm, L.; Wellner, U.F.; Fernandez-del-Castillo, C.; Warshaw, A.L.; Thayer, S.P.; Liss, A.S. Hedgehog signaling promotes angiogenesis directly and indirectly in pancreatic cancer. Angiogenesis 2020, 23, 479–492. [Google Scholar] [CrossRef]
- Wang, W.; Xing, H.; Huang, C.; Pan, H.; Li, D. Identification of pancreatic cancer type related factors by Weighted Gene Co-Expression Network Analysis. Med. Oncol. 2020, 37, 33. [Google Scholar] [CrossRef]
- Pospisil, P.; Korideck, H.; Wang, K.; Yang, Y.; Iyer, L.K.; Kassis, A.I. Computational and Biological Evaluation of Quinazolinone Prodrug for Targeting Pancreatic Cancer. Chem. Biol. Drug Des. 2012, 79, 926–934. [Google Scholar] [CrossRef]
- Lyu, Y.; Cheng, Y.; Wang, B.; Chen, L.; Zhao, S. Sulfatase 1 expression in pancreatic cancer and its correlation with clinicopathological features and postoperative prognosis. Cancer Biomark. 2018, 22, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.M.S.; Michael, A.; Westley, L.; Kocher, H.M.; Murphy, J.I.; Dhoot, G.K. SULF1/SULF2 splice variants differentially regulate pancreatic tumour growth progression. Exp. Cell Res. 2014, 324, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Merodio, L.; Williams, J.A.; Gkoutos, G.V.; Acharjee, A. Omics biomarker identification pipeline for translational medicine. J. Transl. Med. 2019, 17, 155. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and Variable Selection via the Elastic Net. J. R. Stat. Soc. Ser. B 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Marbach, D.; Schaffter, T.; Mattiussi, C.; Floreano, D. Generating Realistic In Silico Gene Networks for Performance Assessment of Reverse Engineering Methods. J. Comput. Biol. 2009, 16, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Samoilov, M.; Arkin, A.; Ross, J. On the deduction of chemical reaction pathways from measurements of time series of concentrations. Chaos An Interdiscip. J. Nonlinear Sci. 2001, 11, 108–114. [Google Scholar] [CrossRef]
- Schreiber, T. Measuring Information Transfer. Phys. Rev. Lett. 2000, 85, 461–464. [Google Scholar] [CrossRef] [Green Version]
- Gallo, C.A.; Cecchini, R.L.; Carballido, J.A.; Micheletto, S.; Ponzoni, I. Discretization of gene expression data revised. Brief. Bioinform. 2016, 17, 758–770. [Google Scholar] [CrossRef]
- Murphy, K.P.; Weiss, Y.; Jordan, M.I. Loopy Belief Propagation for Approximate Inference: An Empirical Study. arXiv, 2013; arXiv:abs/1301.6725. [Google Scholar]
- Chlebus, B.S.; Nguyen, S.H. On Finding Optimal Discretizations for Two Attributes. In Rough Sets and Current Trends in Computing; Springer: Berlin/Heidelberg, Germany, 1998; pp. 537–544. [Google Scholar]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]













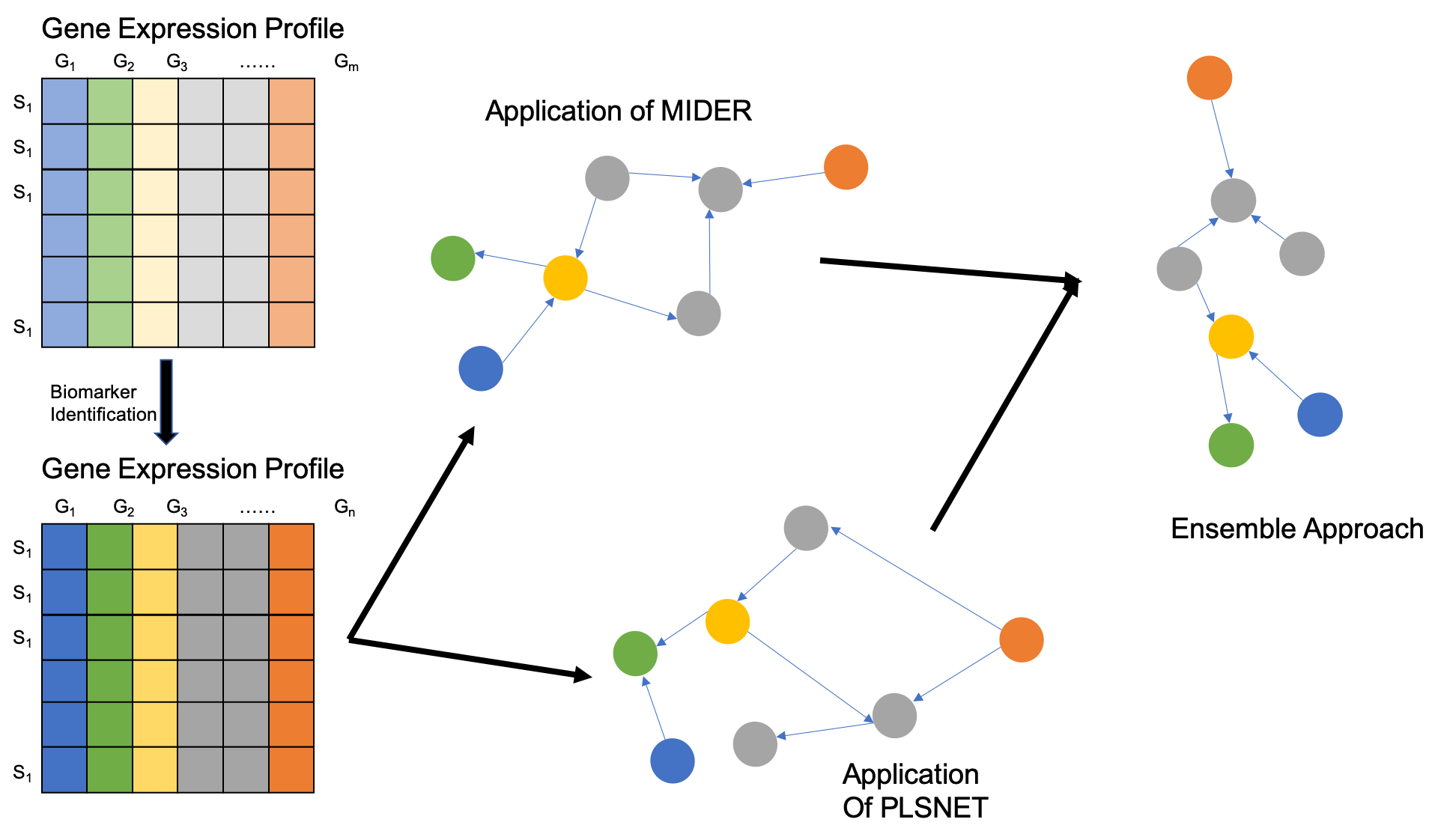{kind=link}
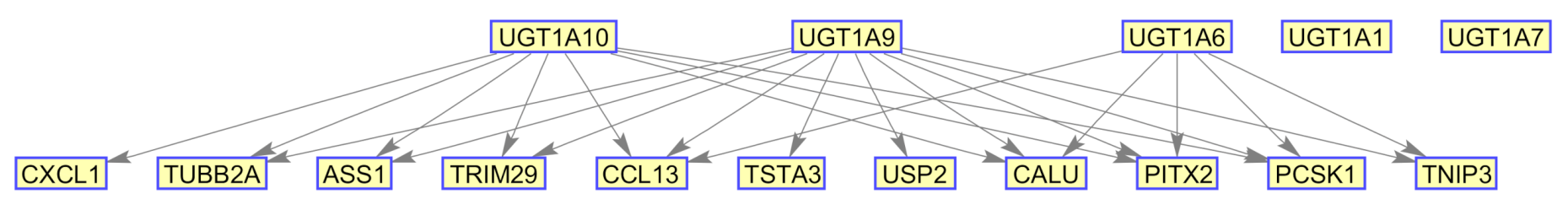{kind=link}
{kind=link}
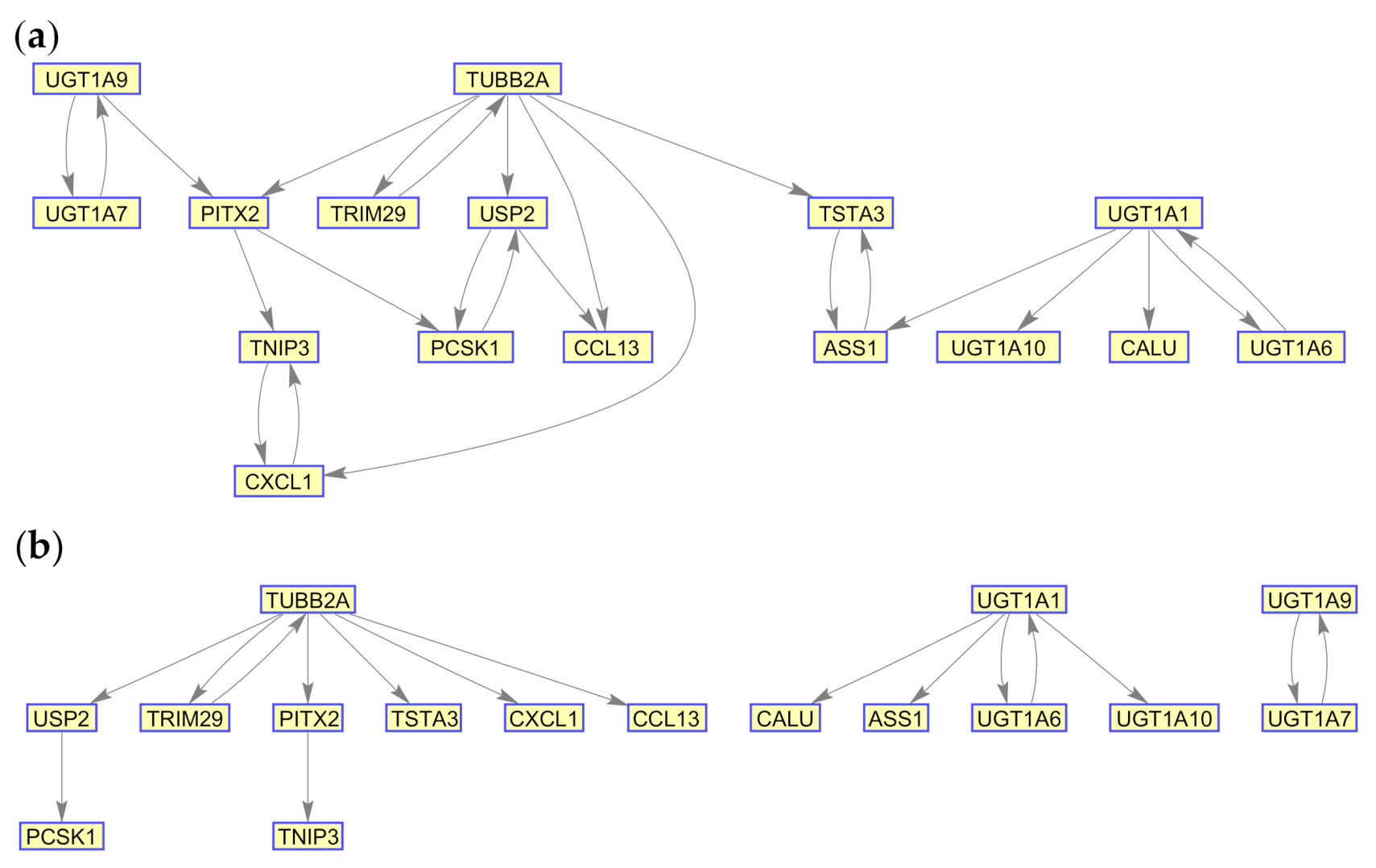{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Top 2% | Top 5% | Top 10% | Top 15% | Top 20% | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R | T | I | R | T | I | R | T | I | R | T | I | R | T | I | |
| TUBB2A | 0 | 66 | 0 | 0 | 98 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| CALU | 0 | 36 | 0 | 0 | 97 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| USP2 | 0 | 1 | 0 | 0 | 23 | 0 | 0 | 98 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| UGT1A1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 14 | 0 | 0 | 84 | 2 | 0 | 90 | 0 |
| ASS1 | 0 | 49 | 0 | 0 | 94 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| UGT1A6 | 7 | 0 | 0 | 26 | 1 | 0 | 86 | 1 | 2 | 87 | 0 | 12 | 23 | 0 | 77 |
| UGT1A10 | 81 | 1 | 0 | 99 | 1 | 0 | 99 | 0 | 1 | 92 | 0 | 8 | 33 | 0 | 67 |
| UGT1A9 | 82 | 0 | 0 | 95 | 0 | 0 | 97 | 0 | 2 | 95 | 0 | 5 | 59 | 0 | 41 |
| UGT1A7 | 5 | 0 | 0 | 12 | 0 | 0 | 40 | 3 | 0 | 46 | 12 | 37 | 0 | 1 | 99 |
| TRIM29 | 0 | 31 | 0 | 0 | 93 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| PITX2 | 0 | 99 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| TSTA3 | 0 | 3 | 0 | 0 | 45 | 0 | 0 | 96 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| PCSK1 | 0 | 20 | 1 | 0 | 75 | 1 | 0 | 99 | 1 | 0 | 99 | 1 | 0 | 99 | 1 |
| CXCL1 | 0 | 2 | 0 | 1 | 28 | 0 | 0 | 88 | 3 | 0 | 96 | 4 | 0 | 94 | 6 |
| CCL13 | 0 | 27 | 0 | 0 | 95 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| TNIP3 | 1 | 2 | 0 | 2 | 26 | 0 | 1 | 85 | 1 | 0 | 98 | 2 | 0 | 97 | 3 |
| Genes | Predicted Marginals | Observed States | ||
|---|---|---|---|---|
| 0 | 1 | 0 | 1 | |
| TUBB2A | 0.55 | 0.45 | 0.55 | 0.45 |
| CALU | 0.9 | 0.1 | 0.9 | 0.1 |
| USP2 | 0.2 | 0.8 | 0.2 | 0.8 |
| UGT1A1 | 0.5498 | 0.4502 | 0.55 | 0.45 |
| ASS1 | 0.9 | 0.1 | 0.9 | 0.1 |
| UGT1A6 | 0.5999 | 0.4001 | 0.6 | 0.4 |
| UGT1A10 | 0.5999 | 0.4001 | 0.6 | 0.4 |
| UGT1A9 | 0.5998 | 0.4002 | 0.6 | 0.4 |
| UGT1A7 | 0.3501 | 0.6499 | 0.35 | 0.65 |
| TRIM29 | 0.45 | 0.55 | 0.45 | 0.55 |
| PITX2 | 0.5 | 0.5 | 0.5 | 0.5 |
| TSTA3 | 0.75 | 0.25 | 0.75 | 0.25 |
| PCSK1 | 0.85 | 0.15 | 0.85 | 0.15 |
| CXCL1 | 0.35 | 0.65 | 0.35 | 0.65 |
| CCL13 | 0.25 | 0.75 | 0.25 | 0.75 |
| TNIP3 | 0.7 | 0.3 | 0.7 | 0.3 |
| Genes | Top 2% | Top 5% | Top 10% | Top 15% | Top 20% | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R | T | I | R | T | I | R | T | I | R | T | I | R | T | I | |
| SULF1 | 100 | 0 | 0 | 100 | 0 | 0 | 65 | 0 | 35 | 3 | 0 | 97 | 0 | 0 | 100 |
| COL8A1 | 63 | 1 | 0 | 6 | 5 | 89 | 0 | 1 | 99 | 0 | 0 | 100 | 0 | 0 | 100 |
| INHBA | 0 | 0 | 0 | 2 | 5 | 0 | 3 | 69 | 24 | 0 | 29 | 71 | 0 | 4 | 96 |
| FN1 | 7 | 0 | 0 | 29 | 27 | 30 | 0 | 4 | 96 | 0 | 0 | 100 | 0 | 0 | 100 |
| COL10A1 | 0 | 0 | 0 | 0 | 98 | 0 | 0 | 97 | 3 | 0 | 69 | 31 | 0 | 18 | 82 |
| THBS2 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 76 | 0 | 24 | 14 | 0 | 86 |
| NTM | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 100 | 0 | 0 | 94 | 6 | 0 | 80 | 20 |
| NOX4 | 0 | 0 | 0 | 21 | 31 | 15 | 0 | 13 | 87 | 0 | 1 | 99 | 0 | 0 | 100 |
| RASAL2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 75 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| ADAMTS12 | 0 | 0 | 0 | 0 | 12 | 0 | 0 | 98 | 2 | 0 | 49 | 51 | 0 | 17 | 83 |
| CAPG | 0 | 96 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 95 | 5 |
| LTBP1 | 0 | 9 | 0 | 0 | 98 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| CTHRC1 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 78 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| FAP | 0 | 13 | 0 | 0 | 78 | 21 | 0 | 24 | 76 | 0 | 2 | 98 | 0 | 0 | 100 |
| WISP1 | 26 | 0 | 0 | 61 | 8 | 27 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 |
| VCAN | 2 | 0 | 0 | 15 | 0 | 0 | 36 | 11 | 41 | 0 | 4 | 96 | 0 | 0 | 100 |
| TIMP1 | 0 | 0 | 0 | 0 | 66 | 0 | 0 | 100 | 0 | 0 | 88 | 12 | 0 | 39 | 61 |
| MIR34AHG | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| SLPI | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| GPRC5A | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 0 |
| Author Name | Disease Type | N | Reference |
|---|---|---|---|
| Quraishi et al. | Inflammatory bowel disease | 16 | [15] |
| Rajamani et al. | Pancreatic ductal adenocarcinoma | 20 | [16] |
| Mills et al. | Acute myeloid leukemia | 60 | [17] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aziz, F.; Acharjee, A.; Williams, J.A.; Russ, D.; Bravo-Merodio, L.; Gkoutos, G.V. Biomarker Prioritisation and Power Estimation Using Ensemble Gene Regulatory Network Inference. Int. J. Mol. Sci. 2020, 21, 7886. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217886
Aziz F, Acharjee A, Williams JA, Russ D, Bravo-Merodio L, Gkoutos GV. Biomarker Prioritisation and Power Estimation Using Ensemble Gene Regulatory Network Inference. International Journal of Molecular Sciences. 2020; 21(21):7886. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217886
Chicago/Turabian StyleAziz, Furqan, Animesh Acharjee, John A. Williams, Dominic Russ, Laura Bravo-Merodio, and Georgios V. Gkoutos. 2020. "Biomarker Prioritisation and Power Estimation Using Ensemble Gene Regulatory Network Inference" International Journal of Molecular Sciences 21, no. 21: 7886. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217886