SARS-CoV-2/Renin–Angiotensin System: Deciphering the Clues for a Couple with Potentially Harmful Effects on Skeletal Muscle

 ,
,
Abstract
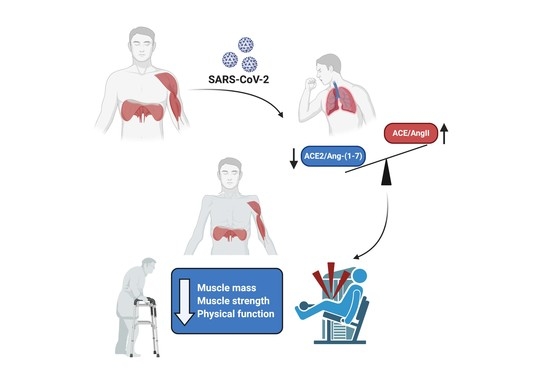
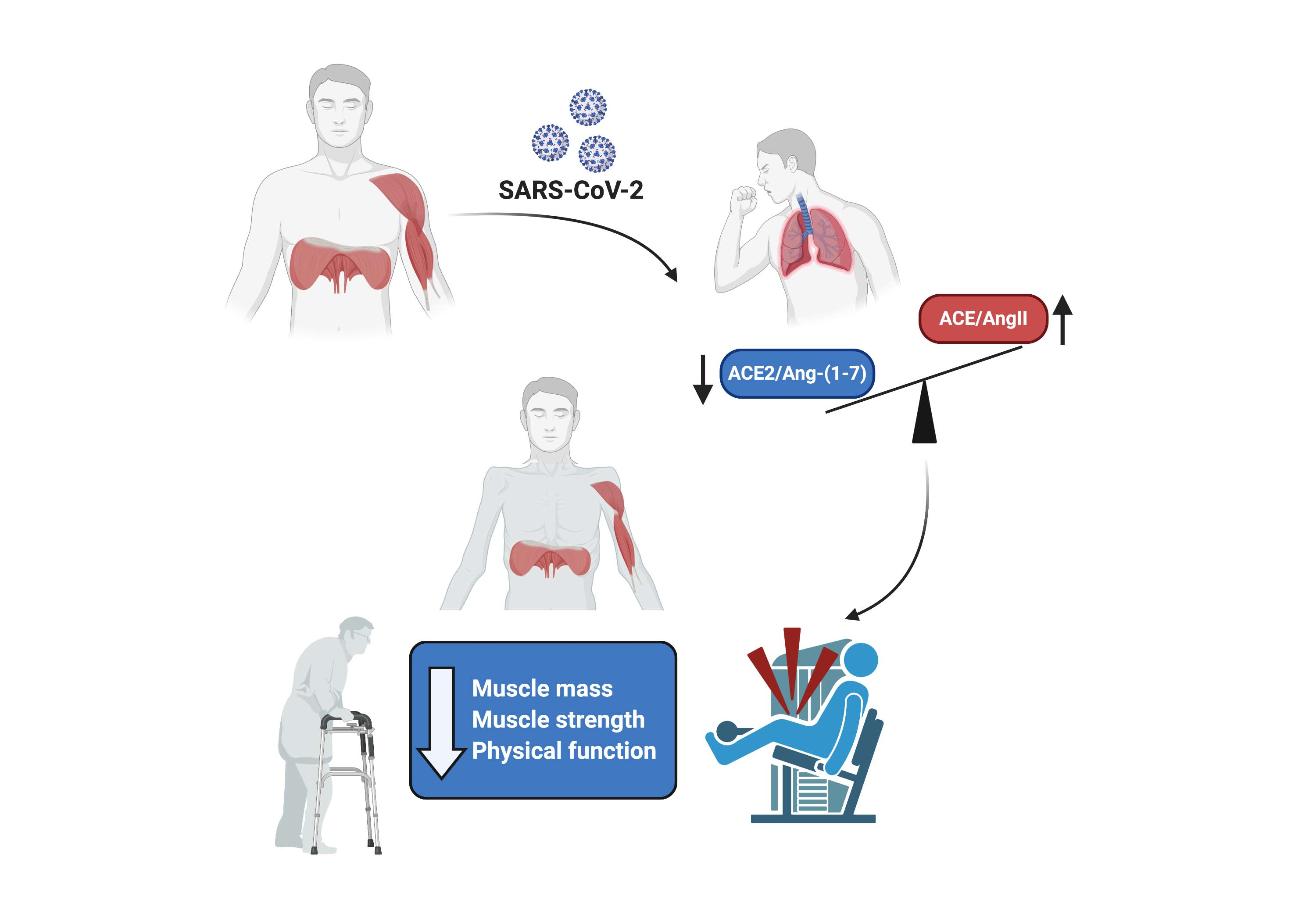
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
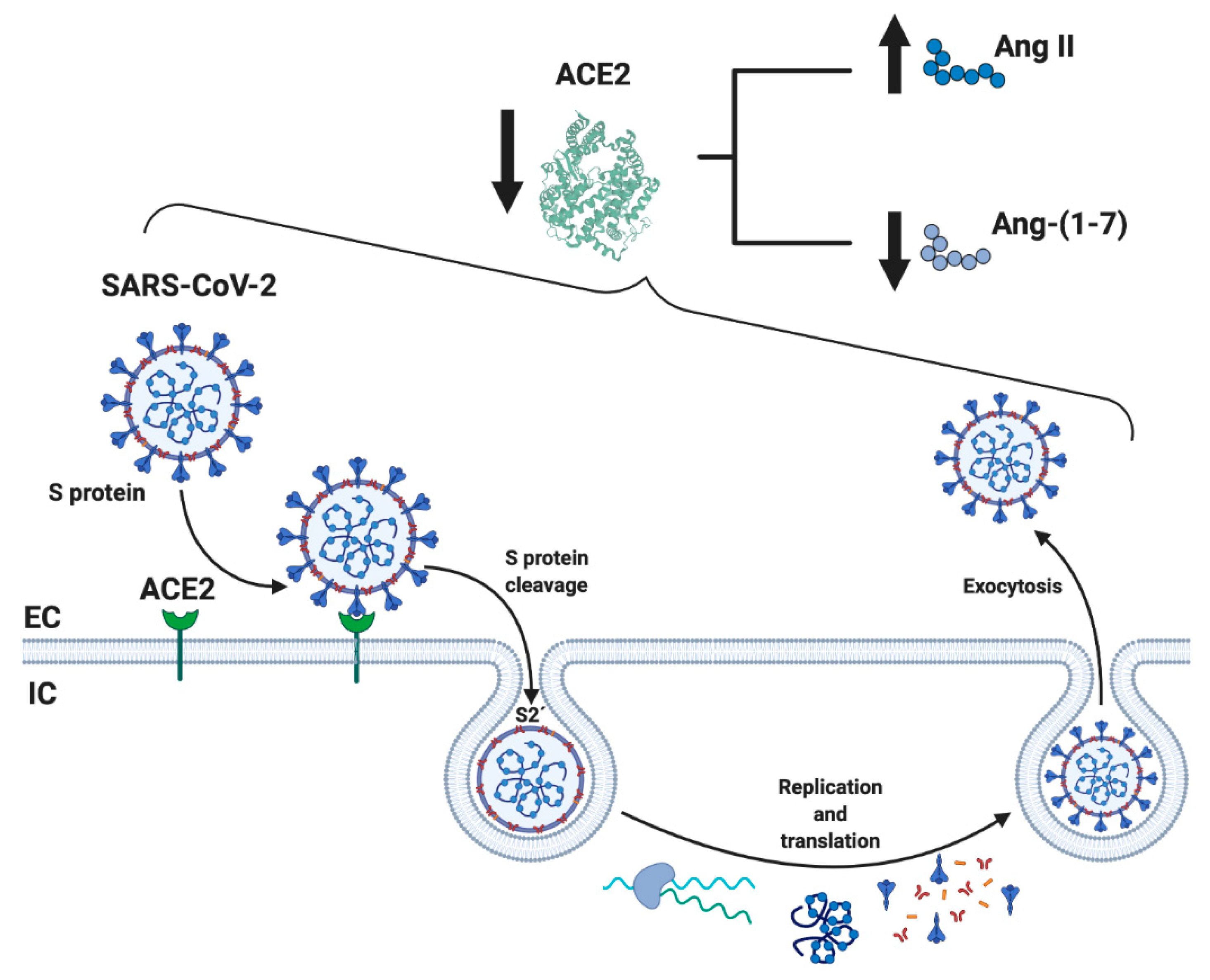
2. RAS Dysregulation and Its Relationship with COVID-19
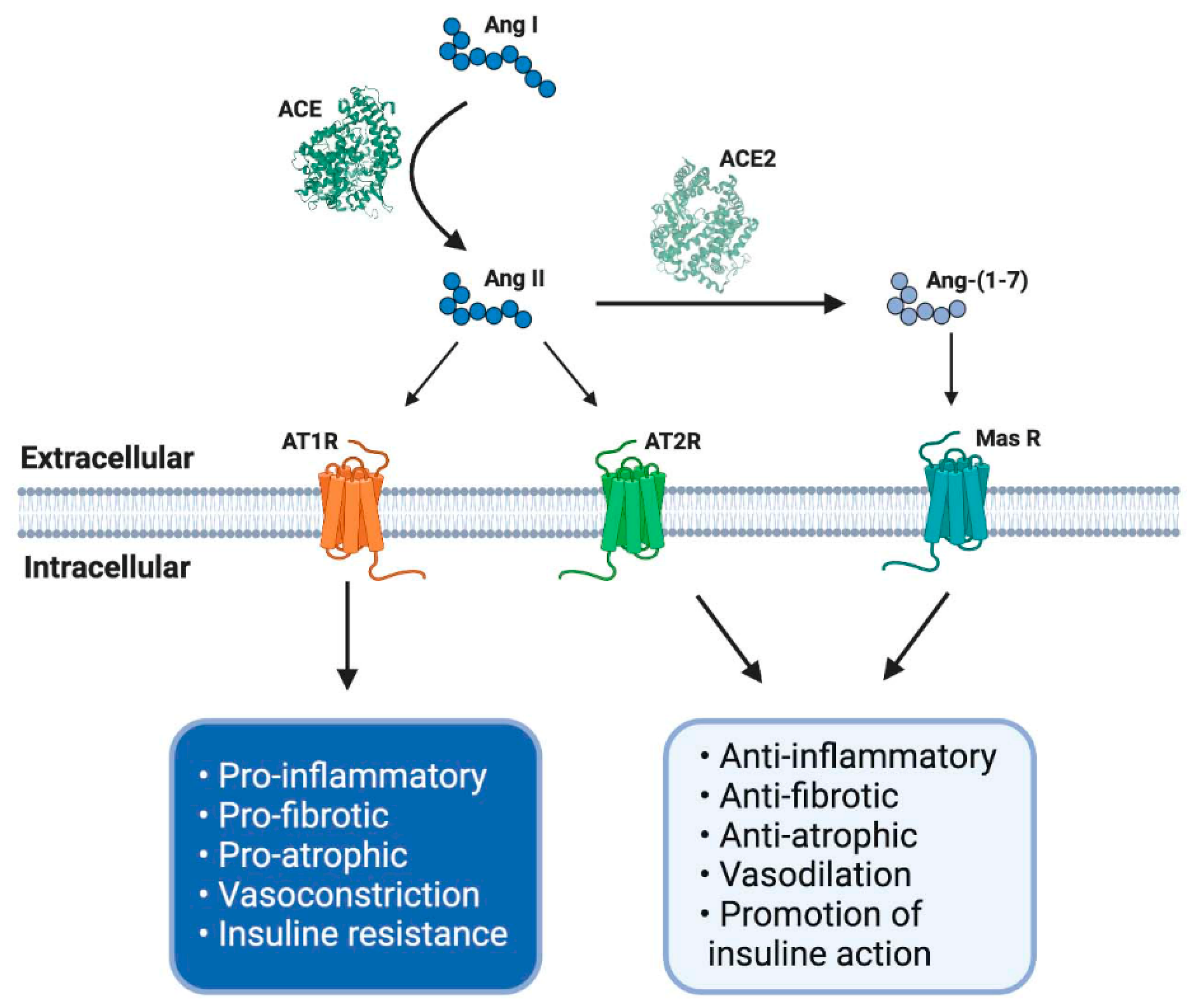
2.1. Classical Axis
2.2. Non-Classical Axis
3. RAS and Its Role in the Loss of Muscle Mass
4. Dysregulation of RAS by COVID-19 and Its Possible Harmful Effects on Skeletal Muscle
5. Diaphragmatic Dysfunction for Invasive Mechanical Ventilation in COVID-19 Patients
6. ICU-Acquired Weakness in COVID-19
7. Possible Therapeutic Interventions
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | angiotensin-converting enzyme |
| ACE2 | angiotensin-converting enzyme 2 |
| Akt | protein kinase B |
| ALI | acute lung injury |
| Ang I | angiotensin I |
| Ang II | angiotensin II |
| Ang-(1-7) | angiotensin (1-7) |
| Ang-(1-9) | angiotensin (1-9) |
| ARB | angiotensin II type 1 receptor blocker |
| ARDS | acute respiratory distress syndrome |
| AT1R | angiotensin II type 1 receptor |
| AT2R | angiotensin II type 2 receptor |
| CIM | critical illness myopathy |
| CIP | Critical illness polyneuropathy |
| CMV | controlled mechanical ventilation |
| COVID-19 | coronavirus disease 2019 |
| CVD | cardiovascular disease |
| DW | diaphragm weakness |
| ICU | intensive care unit |
| ICUAW | ICU-acquired weakness |
| IGF1 | insulin-like growth factor type 1 |
| IL-6 | Interleukin 6 |
| IMV | invasive mechanical ventilation |
| MasR | Mas receptor |
| mTOR | mammalian target of rapamycin |
| MuRF-1 | muscle RING-finger protein-1 |
| NF-κβ | Nuclear factor-kappa beta |
| PD | peptidase domain |
| RAS | renin–angiotensin system |
| ROS | reactive oxygen species |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| T2DM | Type 2 diabetes mellitus |
| TGFβ | transforming growth factor β |
| UPS | ubiquitin–proteasome system |
| VIDD | ventilator-induced diaphragmatic dysfunction |
References
- WHO. Coronavirus Disease (COVID-19) Pandemic [Situation Reports]. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 17 July 2020).
- Kai, H.; Kai, M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors-lessons from available evidence and insights into COVID-19. Hypertens Res. 2020, 43, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Lin, Q.; Jin, S.; You, L. Coronavirus 2019-nCoV: A brief perspective from the front line. J. Infect. 2020, 80, 373–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Lanza, K.; Perez, L.G.; Costa, L.B.; Cordeiro, T.M.; Palmeira, V.A.; Ribeiro, V.T.; Simoes, E.S.A.C. Covid-19: The renin-angiotensin system imbalance hypothesis. Clin. Sci. 2020, 134, 1259–1264. [Google Scholar] [CrossRef]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Kandeel, M.; Ibrahim, A.; Fayez, M.; Al-Nazawi, M. From SARS and MERS CoVs to SARS-CoV-2: Moving toward more biased codon usage in viral structural and nonstructural genes. J. Med. Virol. 2020, 92, 660–666. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Huang, H.-J.; Zhuang, D.-L.; Nasser, M.I.; Yang, M.-H.; Zhu, P.; Zhao, M.-Y. Biological, clinical and epidemiological features of COVID-19, SARS and MERS and AutoDock simulation of ACE2. Infect. Dis. Poverty 2020, 9, 99. [Google Scholar] [CrossRef]
- Malik, Y.S.; Sircar, S.; Bhat, S.; Sharun, K.; Dhama, K.; Dadar, M.; Tiwari, R.; Chaicumpa, W. Emerging novel coronavirus (2019-nCoV)-current scenario, evolutionary perspective based on genome analysis and recent developments. Vet. Q. 2020, 40, 68–76. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.A.-O.; Zhang, Y.A.-O.; Li, Y.; Xia, L.A.-O.; Guo, Y.; Zhou, Q.A.-O. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li W Fau-Farzan, M.; Farzan M Fau-Harrison, S.C.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.A.-O.; Wang, N.A.-O.X.; Corbett, K.A.-O.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.A.-O.; Graham, B.A.-O.; McLellan, J.A.-O.X. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1–7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [Green Version]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Cabello-Verrugio, C.; Rivera, J.C.; Garcia, D. Skeletal muscle wasting: New role of nonclassical renin-angiotensin system. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 158–163. [Google Scholar] [CrossRef]
- Ingraham, N.E.; Barakat, A.G.; Reilkoff, R.; Bezdicek, T.; Schacker, T.; Chipman, J.G.; Tignanelli, C.J.; Puskarich, M.A. Understanding the renin-angiotensin-aldosterone-SARS-CoV axis: A comprehensive review. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- Ghazi, L.; Drawz, P. Advances in understanding the renin-angiotensin-aldosterone system (RAAS) in blood pressure control and recent pivotal trials of RAAS blockade in heart failure and diabetic nephropathy. F1000Research 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, E.D.C.; Prodel, E.; Braz, I.D.; Giori, I.G.; Bargut, T.C.L.; Magliano, D.C.; Nobrega, A.C.L. Modulation of the renin-angiotensin system in white adipose tissue and skeletal muscle: Focus on exercise training. Clin. Sci. 2018, 132, 1487–1507. [Google Scholar] [CrossRef]
- Cabello-Verrugio, C.; Cordova, G.; Salas, J.D. Angiotensin II: Role in skeletal muscle atrophy. Curr. Protein Pept. Sci. 2012, 13, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Strawn, W.B.; Chappell, M.C.; Dean, R.H.; Kivlighn, S.; Ferrario, C.M. Inhibition of early atherogenesis by losartan in monkeys with diet-induced hypercholesterolemia. Circulation 2000, 101, 1586–1593. [Google Scholar] [CrossRef] [Green Version]
- Dandona, P.; Chaudhuri, A.; Ghanim, H.; Mohanty, P. Proinflammatory effects of glucose and anti-inflammatory effect of insulin: Relevance to cardiovascular disease. Am. J. Cardiol. 2007, 99, 15B–26B. [Google Scholar] [CrossRef]
- Cisternas, F.; Morales, M.G.; Meneses, C.; Simon, F.; Brandan, E.; Abrigo, J.; Vazquez, Y.; Cabello-Verrugio, C. Angiotensin-(1-7) decreases skeletal muscle atrophy induced by angiotensin II through a Mas receptor-dependent mechanism. Clin. Sci. 2015, 128, 307–319. [Google Scholar] [CrossRef]
- Winslow, M.A.; Hall, S.E. Muscle wasting: A review of exercise, classical and non-classical RAS axes. J. Cell. Mol. Med. 2019, 23, 5836–5845. [Google Scholar] [CrossRef]
- Warner, F.J.; Lew, R.A.; Smith, A.I.; Lambert, D.W.; Hooper, N.M.; Turner, A.J. Angiotensin-converting enzyme 2 (ACE2), but not ACE, is preferentially localized to the apical surface of polarized kidney cells. J. Biol. Chem. 2005, 280, 39353–39362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guo, F.; Liu, K.; Wang, H.; Rao, S.; Yang, P.; Jiang, C. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 2008, 136, 8–15. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef] [Green Version]
- D’Ardes, D.; Boccatonda, A.; Rossi, I.; Guagnano, M.T.; Santilli, F.; Cipollone, F.; Bucci, M. COVID-19 and RAS: Unravelling an Unclear Relationship. Int. J. Mol. Sci. 2020, 21, 3003. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes, E.S.A.C. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Wosten-van Asperen, R.M.; Lutter, R.; Specht, P.A.; Moll, G.N.; van Woensel, J.B.; van der Loos, C.M.; van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J. Pathol. 2011, 225, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Deshotels, M.R.; Xia, H.; Sriramula, S.; Lazartigues, E.; Filipeanu, C.M. Angiotensin II mediates angiotensin converting enzyme type 2 internalization and degradation through an angiotensin II type I receptor-dependent mechanism. Hypertension 2014, 64, 1368–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messerli, F.H.; Bangalore, S.; Bavishi, C.; Rimoldi, S.F. Angiotensin-Converting Enzyme Inhibitors in Hypertension: To Use or Not to Use? J. Am. Coll. Cardiol. 2018, 71, 1474–1482. [Google Scholar] [CrossRef]
- Gurwitz, D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev. Res. 2020, 81, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, K. Sarcopenia. Wien. Med. Wochenschr. 2019, 169, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyere, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, B.C.; Laakkonen, E.K.; Lowe, D.A. Aging of the musculoskeletal system: How the loss of estrogen impacts muscle strength. Bone 2019, 123, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Abrigo, J.; Marin, T.; Aguirre, F.; Tacchi, F.; Vilos, C.; Simon, F.; Arrese, M.; Cabrera, D.; Cabello-Verrugio, C. N-Acetyl Cysteine attenuates the sarcopenia and muscle apoptosis induced by chronic liver disease. Curr. Mol. Med. 2019, 20, 60–71. [Google Scholar] [CrossRef]
- Delafontaine, P.; Yoshida, T. The Renin-Angiotensin System and the Biology of Skeletal Muscle: Mechanisms of Muscle Wasting in Chronic Disease States. Trans. Am. Clin. Clim. Assoc. 2016, 127, 245–258. [Google Scholar]
- Abrigo, J.; Simon, F.; Cabrera, D.; Vilos, C.; Cabello-Verrugio, C. Mitochondrial Dysfunction in Skeletal Muscle Pathologies. Curr. Protein Pept. Sci. 2019, 20, 536–546. [Google Scholar] [CrossRef]
- Yoshida, T.; Tabony, A.M.; Galvez, S.; Mitch, W.E.; Higashi, Y.; Sukhanov, S.; Delafontaine, P. Molecular mechanisms and signaling pathways of angiotensin II-induced muscle wasting: Potential therapeutic targets for cardiac cachexia. Int. J. Biochem. Cell Biol. 2013, 45, 2322–2332. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Sowers, J.R.; Nistala, R.; Gong, H.; Uptergrove, G.M.; Clark, S.E.; Morris, E.M.; Szary, N.; Manrique, C.; Stump, C.S. Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells. J. Biol. Chem. 2006, 281, 35137–35146. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.H.; Li, Y.; Du, J.; Mitch, W.E.; Rosenthal, N.; Delafontaine, P. Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J. Clin. Investig. 2005, 115, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Semprun-Prieto, L.; Sukhanov, S.; Delafontaine, P. IGF-1 prevents ANG II-induced skeletal muscle atrophy via Akt- and Foxo-dependent inhibition of the ubiquitin ligase atrogin-1 expression. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1565–H1570. [Google Scholar] [CrossRef] [Green Version]
- Echeverría-Rodríguez, O.; Del Valle-Mondragón, L.; Hong, E. Angiotensin 1-7 improves insulin sensitivity by increasing skeletal muscle glucose uptake in vivo. Peptides 2014, 51, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.S.; Liu, C.; Tian, R.; Nishiyama, A.; Raij, L. Skeletal muscle insulin resistance in salt-sensitive hypertension: Role of angiotensin II activation of NFkappaB. Cardiovasc. Diabetol. 2015, 14, 45. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Du, J.; Hu, Z.; Han, G.; Delafontaine, P.; Garcia, G.; Mitch, W.E. IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J. Am. Soc. Nephrol. 2009, 20, 604–612. [Google Scholar] [CrossRef]
- Shen, C.; Zhou, J.; Wang, X.; Yu, X.Y.; Liang, C.; Liu, B.; Pan, X.; Zhao, Q.; Song, J.L.; Wang, J.; et al. Angiotensin-II-induced Muscle Wasting is Mediated by 25-Hydroxycholesterol via GSK3beta Signaling Pathway. EBioMedicine 2017, 16, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Costamagna, D.; Costelli, P.; Sampaolesi, M.; Penna, F. Role of Inflammation in Muscle Homeostasis and Myogenesis. Mediat. Inflamm 2015, 2015, 805172. [Google Scholar] [CrossRef] [Green Version]
- Morales, M.G.; Vazquez, Y.; Acuna, M.J.; Rivera, J.C.; Simon, F.; Salas, J.D.; Alvarez Ruf, J.; Brandan, E.; Cabello-Verrugio, C. Angiotensin II-induced pro-fibrotic effects require p38MAPK activity and transforming growth factor beta 1 expression in skeletal muscle cells. Int. J. Biochem. Cell Biol. 2012, 44, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.G.; Olguin, H.; Di Capua, G.; Brandan, E.; Simon, F.; Cabello-Verrugio, C. Endotoxin-induced skeletal muscle wasting is prevented by angiotensin-(1-7) through a p38 MAPK-dependent mechanism. Clin. Sci. 2015, 129, 461–476. [Google Scholar] [CrossRef]
- Morales, M.G.; Abrigo, J.; Acuna, M.J.; Santos, R.A.; Bader, M.; Brandan, E.; Simon, F.; Olguin, H.; Cabrera, D.; Cabello-Verrugio, C. Angiotensin-(1-7) attenuates disuse skeletal muscle atrophy in mice via its receptor, Mas. Dis. Models Mech. 2016, 9, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrigo, J.; Simon, F.; Cabrera, D.; Cabello-Verrugio, C. Angiotensin-(1-7) Prevents Skeletal Muscle Atrophy Induced by Transforming Growth Factor Type Beta (TGF-beta) via Mas Receptor Activation. Cell Physiol. Biochem. 2016, 40, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, F.; Abrigo, J.; Gonzalez, F.; Gonzalez, A.; Simon, F.; Cabello-Verrugio, C. Protective Effect of Angiotensin 1-7 on Sarcopenia Induced by Chronic Liver Disease in Mice. Int. J. Mol. Sci. 2020, 21, 3891. [Google Scholar] [CrossRef]
- Meneses, C.; Morales, M.G.; Abrigo, J.; Simon, F.; Brandan, E.; Cabello-Verrugio, C. The angiotensin-(1-7)/Mas axis reduces myonuclear apoptosis during recovery from angiotensin II-induced skeletal muscle atrophy in mice. Pflug. Arch. 2015, 467, 1975–1984. [Google Scholar] [CrossRef]
- Zambelli, V.; Sigurta, A.; Rizzi, L.; Zucca, L.; Delvecchio, P.; Bresciani, E.; Torsello, A.; Bellani, G. Angiotensin-(1-7) exerts a protective action in a rat model of ventilator-induced diaphragmatic dysfunction. Intensive Care Med. Exp. 2019, 7, 8. [Google Scholar] [CrossRef]
- Zangrillo, A.; Beretta, L.; Scandroglio, A.M.; Monti, G.; Fominskiy, E.; Colombo, S.; Morselli, F.; Belletti, A.; Silvani, P.; Crivellari, M.; et al. Characteristics, treatment, outcomes and cause of death of invasively ventilated patients with COVID-19 ARDS in Milan, Italy. Crit. Care Resusc. 2020, 22, 200–211. [Google Scholar]
- Guarracino, F.; Vetrugno, L.; Forfori, F.; Corradi, F.; Orso, D.; Bertini, P.; Ortalda, A.; Federici, N.; Copetti, R.; Bove, T. Lung, Heart, Vascular, and Diaphragm Ultrasound Examination of COVID-19 Patients: A Comprehensive Approach. J. Cardiothorac. Vasc. Anesth. 2020. [Google Scholar] [CrossRef]
- Arentz, M.; Yim, E.; Klaff, L.; Lokhandwala, S.; Riedo, F.X.; Chong, M.; Lee, M. Characteristics and Outcomes of 21 Critically Ill Patients With COVID-19 in Washington State. JAMA 2020, 323, 1612–1614. [Google Scholar] [CrossRef] [Green Version]
- Herridge, M.S.; Moss, M.; Hough, C.L.; Hopkins, R.O.; Rice, T.W.; Bienvenu, O.J.; Azoulay, E. Recovery and outcomes after the acute respiratory distress syndrome (ARDS) in patients and their family caregivers. Intensive Care Med. 2016, 42, 725–738. [Google Scholar] [CrossRef]
- Vetrugno, L.; Guadagnin, G.M.; Barbariol, F.; Langiano, N.; Zangrillo, A.; Bove, T. Ultrasound Imaging for Diaphragm Dysfunction: A Narrative Literature Review. J. Cardiothorac. Vasc. Anesth. 2019, 33, 2525–2536. [Google Scholar] [CrossRef]
- Inciardi, R.M.; Lupi, L.; Zaccone, G.; Italia, L.; Raffo, M.; Tomasoni, D.; Cani, D.S.; Cerini, M.; Farina, D.; Gavazzi, E.; et al. Cardiac Involvement in a Patient With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 819–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dres, M.; Demoule, A. Diaphragm dysfunction during weaning from mechanical ventilation: An underestimated phenomenon with clinical implications. Crit Care 2018, 22, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Thoracic Society; European Respiratory Society. ATS/ERS Statement on respiratory muscle testing. Am. J. Respir. Crit. Care Med. 2002, 166, 518–624. [Google Scholar] [CrossRef]
- Supinski, G.S.; Morris, P.E.; Dhar, S.; Callahan, L.A. Diaphragm Dysfunction in Critical Illness. Chest 2018, 153, 1040–1051. [Google Scholar] [CrossRef]
- Petrof, B.J.; Jaber, S.; Matecki, S. Ventilator-induced diaphragmatic dysfunction. Curr. Opin. Crit. Care 2010, 16, 19–25. [Google Scholar] [CrossRef]
- Jung, B.; Nougaret, S.; Conseil, M.; Coisel, Y.; Futier, E.; Chanques, G.; Molinari, N.; Lacampagne, A.; Matecki, S.; Jaber, S. Sepsis is associated with a preferential diaphragmatic atrophy: A critically ill patient study using tridimensional computed tomography. Anesthesiology 2014, 120, 1182–1191. [Google Scholar] [CrossRef]
- Jaber, S.; Jung, B.; Sebbane, M.; Ramonatxo, M.; Capdevila, X.; Mercier, J.; Eledjam, J.J.; Matecki, S. Alteration of the piglet diaphragm contractility in vivo and its recovery after acute hypercapnia. Anesthesiology 2008, 108, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-Y.; Li, L.-F. Ventilator-induced diaphragm dysfunction in critical illness. Exp. Biol. Med. 2018, 243, 1329–1337. [Google Scholar] [CrossRef]
- Jaber, S.; Jung, B.; Matecki, S.; Petrof, B.J. Clinical review: Ventilator-induced diaphragmatic dysfunction—Human studies confirm animal model findings! Crit Care 2011, 15, 206. [Google Scholar] [CrossRef] [Green Version]
- Levine, S.; Nguyen, T.; Taylor, N.; Friscia, M.E.; Budak, M.T.; Rothenberg, P.; Zhu, J.; Sachdeva, R.; Sonnad, S.; Kaiser, L.R.; et al. Rapid Disuse Atrophy of Diaphragm Fibers in Mechanically Ventilated Humans. N. Engl. J. Med. 2008, 358, 1327–1335. [Google Scholar] [CrossRef]
- Powers, S.K.; Wiggs, M.P.; Sollanek, K.J.; Smuder, A.J. Ventilator-induced diaphragm dysfunction: Cause and effect. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R464–R477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, S.K.; Kavazis, A.N.; Levine, S. Prolonged mechanical ventilation alters diaphragmatic structure and function. Crit. Care Med. 2009, 37, S347–S353. [Google Scholar] [CrossRef] [PubMed]
- Shanely, R.A.; Zergeroglu, M.A.; Lennon, S.L.; Sugiura, T.; Yimlamai, T.; Enns, D.; Belcastro, A.; Powers, S.K. Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am. J. Respir. Crit. Care Med. 2002, 166, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Luo, J.; Bourdon, J.; Lin, M.-C.; Gottfried, S.B.; Petrof, B.J. Controlled mechanical ventilation leads to remodeling of the rat diaphragm. Am. J. Respir. Crit. Care Med. 2002, 166, 1135–1140. [Google Scholar] [CrossRef]
- Shanely, R.A.; Van Gammeren, D.; Deruisseau, K.C.; Zergeroglu, A.M.; McKenzie, M.J.; Yarasheski, K.E.; Powers, S.K. Mechanical ventilation depresses protein synthesis in the rat diaphragm. Am. J. Respir. Crit. Care Med. 2004, 170, 994–999. [Google Scholar] [CrossRef]
- McClung, J.M.; Kavazis, A.N.; DeRuisseau, K.C.; Falk, D.J.; Deering, M.A.; Lee, Y.; Sugiura, T.; Powers, S.K. Caspase-3 regulation of diaphragm myonuclear domain during mechanical ventilation-induced atrophy. Am. J. Respir Crit Care Med. 2007, 175, 150–159. [Google Scholar] [CrossRef] [Green Version]
- McClung, J.M.; Whidden, M.A.; Kavazis, A.N.; Falk, D.J.; Deruisseau, K.C.; Powers, S.K. Redox regulation of diaphragm proteolysis during mechanical ventilation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, 1608–1617. [Google Scholar] [CrossRef] [Green Version]
- Sassoon, C.S.; Zhu, E.; Caiozzo, V.J.; Caiozzo, V.J. Assist-control mechanical ventilation attenuates ventilator-induced diaphragmatic dysfunction. Am. J. Respir. Crit. Care Med. 2003, 170, 626–632. [Google Scholar] [CrossRef]
- Falk, D.J.; Deruisseau, K.C.; Van Gammeren, D.L.; Deering, M.A.; Kavazis, A.N.; Powers, S.K. Mechanical ventilation promotes redox status alterations in the diaphragm. J. Appl. Physiol. 2006, 101, 1017–1024. [Google Scholar] [CrossRef] [Green Version]
- Zergeroglu, M.A.; McKenzie, M.J.; Shanely, R.A.; Van Gammeren, D.; DeRuisseau, K.C.; Powers, S.K. Mechanical ventilation-induced oxidative stress in the diaphragm. J. Appl. Physiol. 2003, 95, 1116–1124. [Google Scholar] [CrossRef] [Green Version]
- DeRuisseau, K.C.; Shanely, R.A.; Akunuri, N.; Hamilton, M.T.; Van Gammeren, D.; Zergeroglu, A.M.; McKenzie, M.; Powers, S.K. Diaphragm unloading via controlled mechanical ventilation alters the gene expression profile. Am. J. Respir. Crit. Care Med. 2005, 172, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.S.; Smuder, A.J.; Wiggs, M.P.; Hall, S.E.; Sollanek, K.J.; Morton, A.B.; Talbert, E.E.; Toklu, H.Z.; Tumer, N.; Powers, S.K. AT1 receptor blocker losartan protects against mechanical ventilation-induced diaphragmatic dysfunction. J. Appl. Physiol. 2015, 119, 1033–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermans, G.; Van den Berghe, G. Clinical review: Intensive care unit acquired weakness. Crit. Care 2015, 19, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolley, S.E.; Bunnell, A.E.; Hough, C.L. ICU-Acquired Weakness. Chest 2016, 150, 1129–1140. [Google Scholar] [CrossRef] [Green Version]
- Kramer, C.L. Intensive Care Unit-Acquired Weakness. Neurol. Clin. 2017, 35, 723–736. [Google Scholar] [CrossRef]
- De Jonghe, B.; Sharshar, T.; Lefaucheur, J.-P.; Authier, F.-J.; Durand-Zaleski, I.; Boussarsar, M.; Cerf, C.; Renaud, E.; Mesrati, F.; Carlet, J.; et al. Paresis acquired in the intensive care unit: A prospective multicenter study. JAMA 2002, 288, 2859–2867. [Google Scholar] [CrossRef] [Green Version]
- Batt, J.; dos Santos, C.C.; Cameron, J.I.; Herridge, M.S. Intensive care unit-acquired weakness: Clinical phenotypes and molecular mechanisms. Am. J. Respir. Crit. Care Med. 2013, 187, 238–246. [Google Scholar] [CrossRef]
- Fan, E.; Dowdy, D.W.; Colantuoni, E.; Mendez-Tellez, P.A.; Sevransky, J.E.; Shanholtz, C.; Himmelfarb, C.R.D.; Desai, S.V.; Ciesla, N.; Herridge, M.S.; et al. Physical complications in acute lung injury survivors: A two-year longitudinal prospective study. Crit. Care Med. 2014, 42, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yang, X.; Yang, L.; Zou, X.; Wang, Y.; Wu, Y.; Zhou, T.; Yuan, Y.; Qi, H.; Fu, S.; et al. Clinical course and predictors of 60-day mortality in 239 critically ill patients with COVID-19: A multicenter retrospective study from Wuhan, China. Crit. Care 2020, 24, 394. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.a.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Carfì, A.; Bernabei, R.; Landi, F.; for the Gemelli Against, C.-P.-A.C.S.G. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Polito, R.; Alfieri, A.; Mancini, A.; Imperlini, E.; Elce, A.; Krustrup, P.; Orru, S.; Buono, P.; Daniele, A. Molecular mechanisms involved in the positive effects of physical activity on coping with COVID-19. Eur. J. Appl. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Barker-Davies, R.M.; O’Sullivan, O.; Senaratne, K.P.P.; Baker, P.; Cranley, M.; Dharm-Datta, S.; Ellis, H.; Goodall, D.; Gough, M.; Lewis, S.; et al. The Stanford Hall consensus statement for post-COVID-19 rehabilitation. Br. J. Sports Med. 2020, 54, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Esler, M.; Esler, D. Can angiotensin receptor-blocking drugs perhaps be harmful in the COVID-19 pandemic? J. Hypertens 2020, 38, 781–782. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir’ Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Jarcho, J.A.; Ingelfinger, J.R.; Hamel, M.B.; D’Agostino, R.B., Sr.; Harrington, D.P. Inhibitors of the Renin-Angiotensin-Aldosterone System and Covid-19. N. Engl. J. Med. 2020, 382, 2462–2464. [Google Scholar] [CrossRef]
- Chung, M.K.; Karnik, S.; Saef, J.; Bergmann, C.; Barnard, J.; Lederman, M.M.; Tilton, J.; Cheng, F.; Harding, C.V.; Young, J.B.; et al. SARS-CoV-2 and ACE2: The biology and clinical data settling the ARB and ACEI controversy. EBioMedicine 2020, 58, 102907. [Google Scholar] [CrossRef]
- Mancia, G.; Rea, F.; Ludergnani, M.; Apolone, G.; Corrao, G. Renin-Angiotensin-Aldosterone System Blockers and the Risk of Covid-19. N. Engl. J. Med. 2020, 382, 2431–2440. [Google Scholar] [CrossRef]
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef]
- Reynolds, H.R.; Adhikari, S.; Pulgarin, C.; Troxel, A.B.; Iturrate, E.; Johnson, S.B.; Hausvater, A.; Newman, J.D.; Berger, J.S.; Bangalore, S.; et al. Renin-Angiotensin-Aldosterone System Inhibitors and Risk of Covid-19. N. Engl. J. Med. 2020, 382, 2441–2448. [Google Scholar] [CrossRef]
- Disser, N.P.; De Micheli, A.J.; Schonk, M.M.; Konnaris, M.A.; Piacentini, A.N.; Edon, D.L.; Toresdahl, B.G.; Rodeo, S.A.; Casey, E.K.; Mendias, C.L. Musculoskeletal Consequences of COVID-19. J. Bone Jt. Surg. Am. 2020, 102, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 323, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, A.; Misuraca, C.; Bianchi, A.; Borsa, N.; Limonta, S.; Maggiolini, S.; Bonardi, D.R.; Corsonello, A.; Di Rosa, M.; Soraci, L.; et al. Pulmonary function in patients surviving to COVID-19 pneumonia. Infection 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhang, W.; Yang, Y.; Zhang, J.; Li, Y.; Chen, Y. Respiratory rehabilitation in elderly patients with COVID-19: A randomized controlled study. Complement. Clin. Pr. 2020, 39, 101166. [Google Scholar] [CrossRef]
- Hawley, J.A.; Hargreaves, M.; Joyner, M.J.; Zierath, J.R. Integrative biology of exercise. Cell 2014, 159, 738–749. [Google Scholar] [CrossRef] [Green Version]
- Lau, H.M.; Ng, G.Y.; Jones, A.Y.; Lee, E.W.; Siu, E.H.; Hui, D.S. A randomised controlled trial of the effectiveness of an exercise training program in patients recovering from severe acute respiratory syndrome. Aust. J. Physiother. 2005, 51, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Li, P.; Lu, Y.; Wang, Z.; Li, J.; Liu, X.; Wu, W. Effects of resistance training on exercise capacity in elderly patients with chronic obstructive pulmonary disease: A meta-analysis and systematic review. Aging Clin. Exp. Res. 2020, 32, 1911–1922. [Google Scholar] [CrossRef]
- Nolan, C.M.; Rochester, C.L. Exercise Training Modalities for People with Chronic Obstructive Pulmonary Disease. COPD 2019, 16, 378–389. [Google Scholar] [CrossRef]
- Gomes-Santos, I.L.; Fernandes, T.; Couto, G.K.; Ferreira-Filho, J.C.; Salemi, V.M.; Fernandes, F.B.; Casarini, D.E.; Brum, P.C.; Rossoni, L.V.; de Oliveira, E.M.; et al. Effects of exercise training on circulating and skeletal muscle renin-angiotensin system in chronic heart failure rats. PLoS ONE 2014, 9, e98012. [Google Scholar] [CrossRef] [Green Version]
- Nunes-Silva, A.; Rocha, G.C.; Magalhaes, D.M.; Vaz, L.N.; Salviano de Faria, M.H.; Simoes, E.S.A.C. Physical Exercise and ACE2-Angiotensin-(1-7)-Mas Receptor Axis of the Renin Angiotensin System. Protein Pept. Lett. 2017, 24, 809–816. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez, A.; Orozco-Aguilar, J.; Achiardi, O.; Simon, F.; Cabello-Verrugio, C. SARS-CoV-2/Renin–Angiotensin System: Deciphering the Clues for a Couple with Potentially Harmful Effects on Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 7904. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217904
Gonzalez A, Orozco-Aguilar J, Achiardi O, Simon F, Cabello-Verrugio C. SARS-CoV-2/Renin–Angiotensin System: Deciphering the Clues for a Couple with Potentially Harmful Effects on Skeletal Muscle. International Journal of Molecular Sciences. 2020; 21(21):7904. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217904
Chicago/Turabian StyleGonzalez, Andrea, Josué Orozco-Aguilar, Oscar Achiardi, Felipe Simon, and Claudio Cabello-Verrugio. 2020. "SARS-CoV-2/Renin–Angiotensin System: Deciphering the Clues for a Couple with Potentially Harmful Effects on Skeletal Muscle" International Journal of Molecular Sciences 21, no. 21: 7904. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217904