Heterogeneity of Neuroinflammatory Responses in Amyotrophic Lateral Sclerosis: A Challenge or an Opportunity?



Abstract
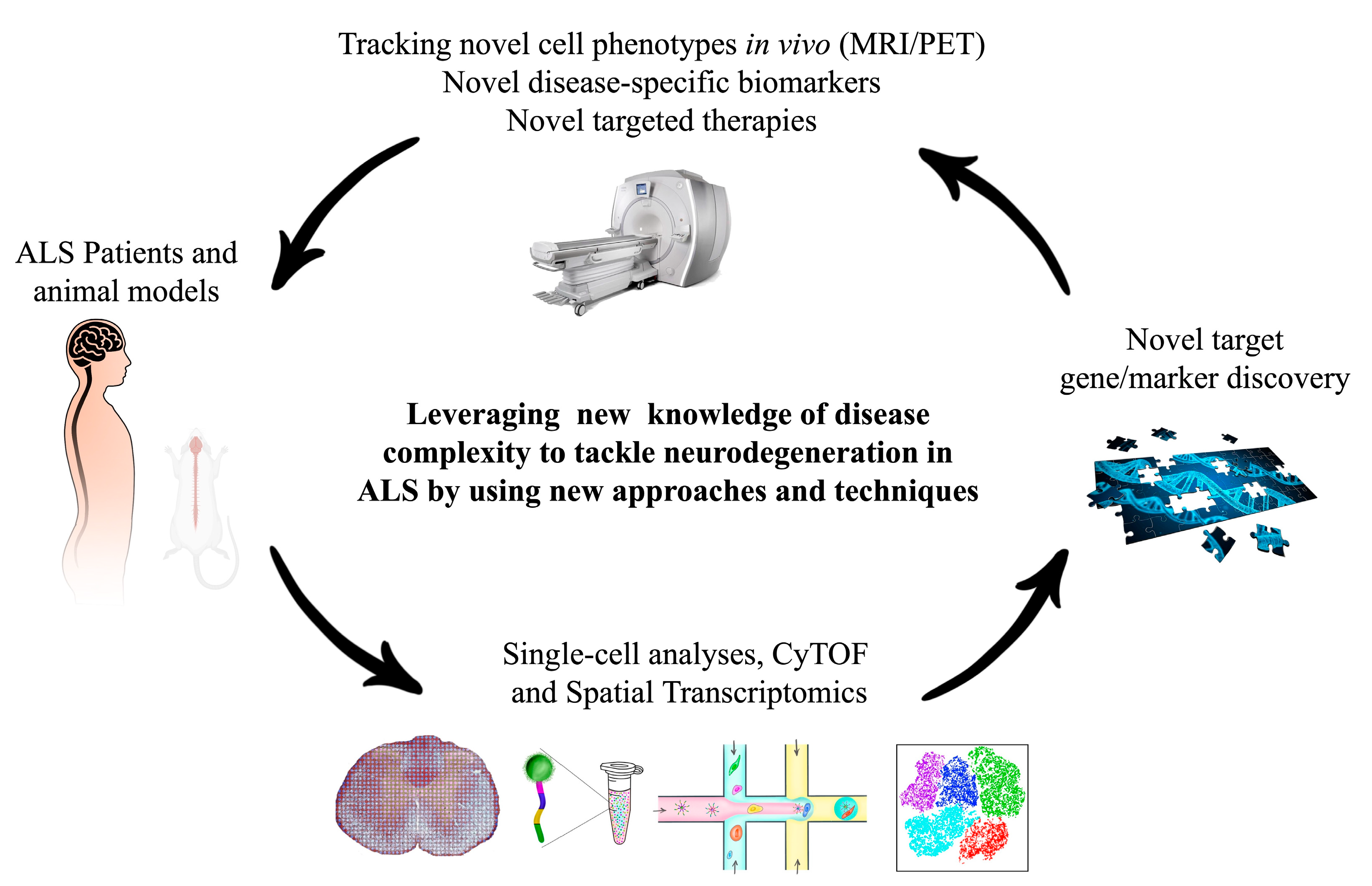
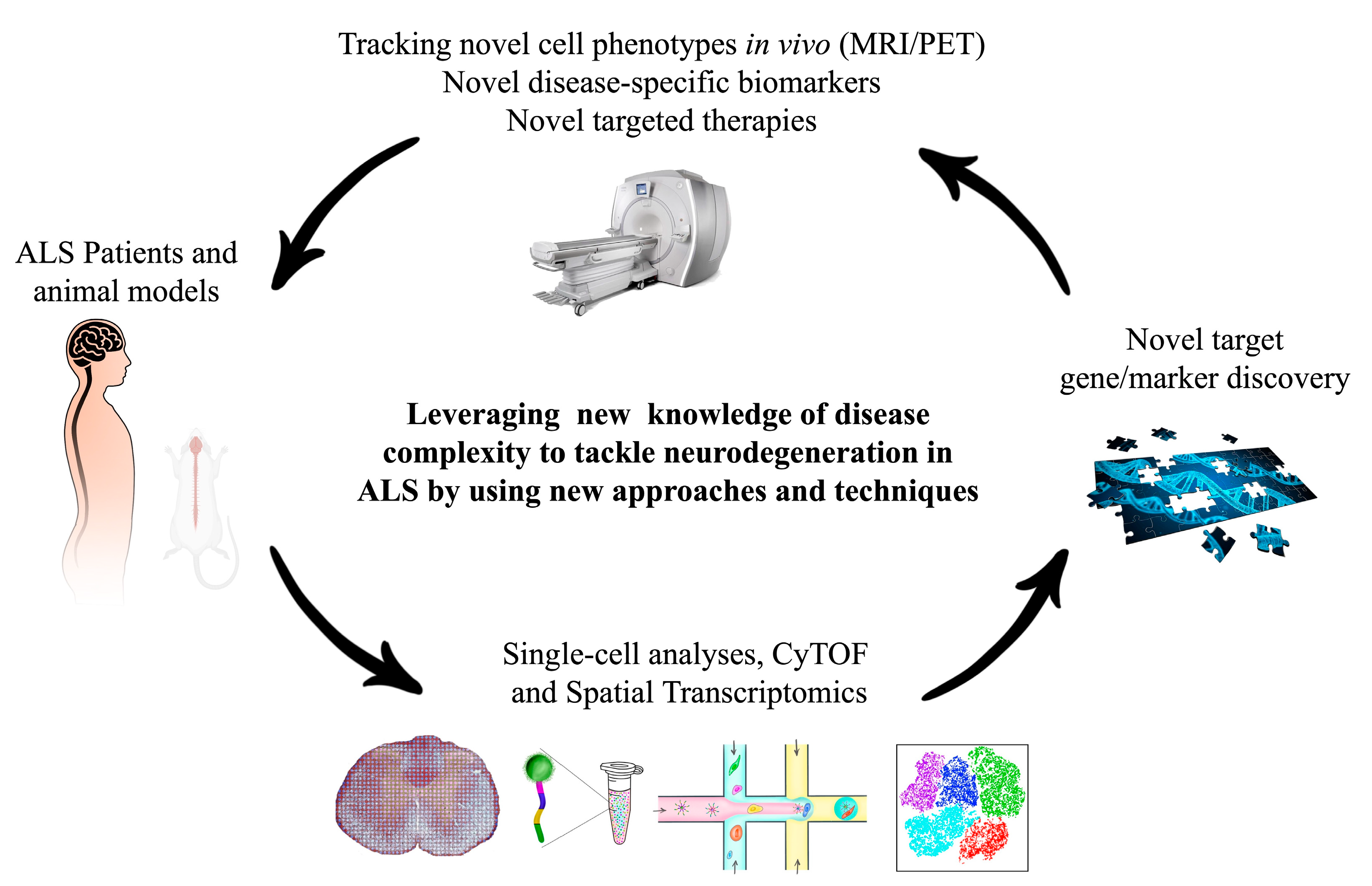
:1. Introduction
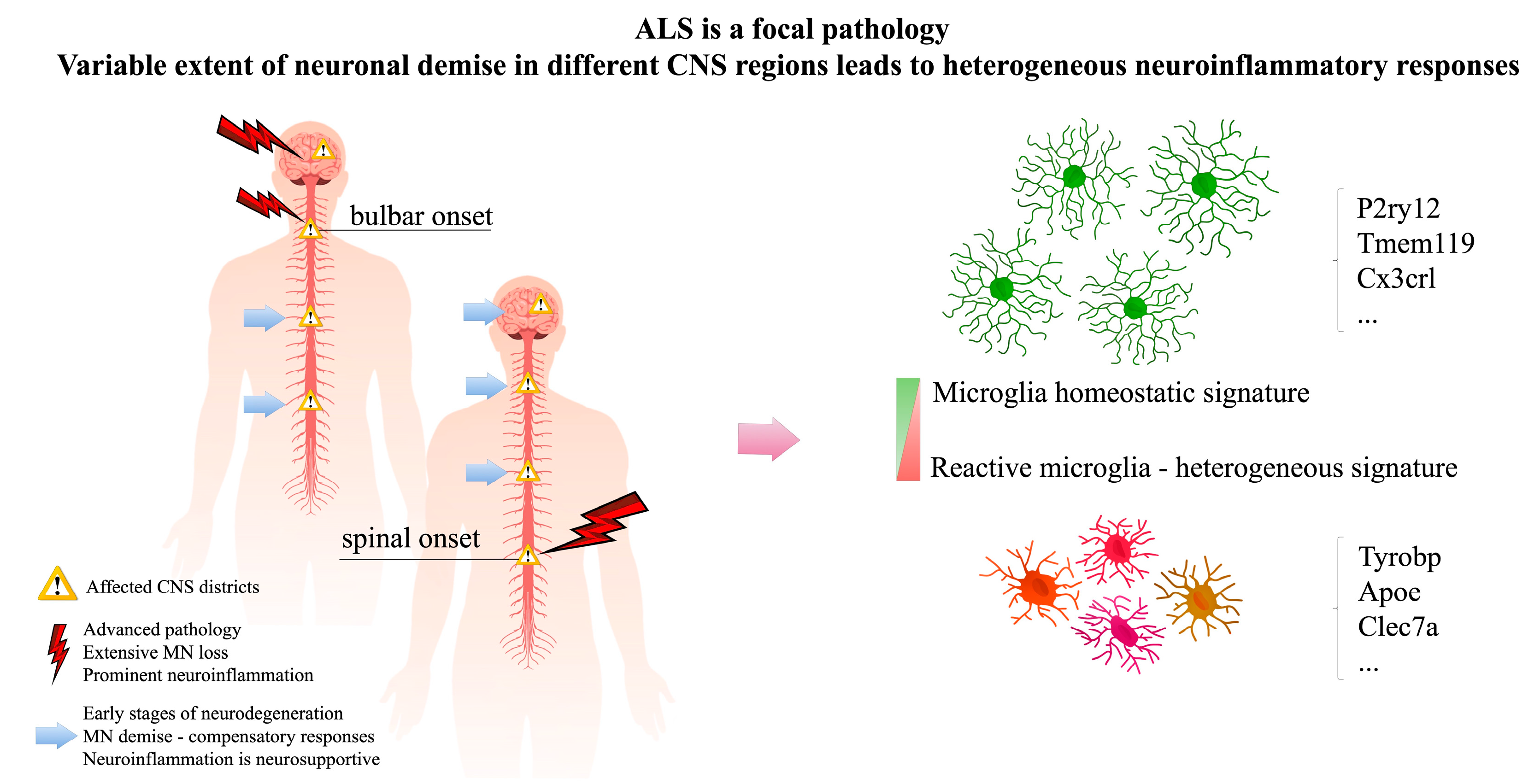
2. ALS Is a Focal Pathology, Neuroinflammatory Responses Are Intrinsically Heterogeneous
3. The Signature of Activated Microglia in ALS Is Disease Specific
4. Single-Cell and Spatial Transcriptomics Approaches: Powerful Tools to Unveil the Complexity of Neuroinflammatory Responses
5. Neuroinflammation as a Diagnostic/Prognostic Marker
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Silani, V. Therapy in Amyotrophic Lateral Sclerosis (ALS): An unexpected evolving scenario. Arch. Ital. Biol. 2017, 155, 118–130. [Google Scholar] [PubMed]
- Molteni, M.; Rossetti, C. Neurodegenerative diseases: The immunological perspective. J. Neuroimmunol. 2017, 313, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Navarro, V.; Sanchez-Mejias, E.; Jimenez, S.; Muñoz-Castro, C.; Sanchez-Varo, R.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Microglia in Alzheimer’s Disease: Activated, Dysfunctional or Degenerative. Front. Aging Neurosci. 2018, 10, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brites, D.; Vaz, A.R. Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front. Cell. Neurosci. 2014, 8, 117. [Google Scholar] [CrossRef]
- Li, Q.; Haney, M.S. The role of glia in protein aggregation. Neurobiol. Dis. 2020, 143. [Google Scholar] [CrossRef]
- Grad, L.I.; Pokrishevsky, E.; Silverman, J.M.; Cashman, N.R. Exosome-dependent and independent mechanisms are involved in prion-like transmission of propagated Cu/Zn superoxide dismutase misfolding. Prion 2014, 8, 331–335. [Google Scholar] [CrossRef]
- Pöyhönen, S.; Er, S.; Domanskyi, A.; Airavaara, M. Effects of neurotrophic factors in glial cells in the central nervous system: Expression and properties in neurodegeneration and injury. Front. Physiol. 2019, 10, 486. [Google Scholar] [CrossRef]
- Frühbeis, C.; Fröhlich, D.; Krämer-Albers, E.M. Emerging roles of exosomes in neuron-glia communication. Front. Physiol. 2012, 3 APR. [Google Scholar] [CrossRef] [Green Version]
- Van Den Bosch, L.; Tilkin, P.; Lemmens, G.; Robberecht, W. Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport 2002, 13, 1067–1070. [Google Scholar] [CrossRef]
- Keller, A.F.; Gravel, M.; Kriz, J. Treatment with minocycline after disease onset alters astrocyte reactivity and increases microgliosis in SOD1 mutant mice. Exp. Neurol. 2011, 228, 69–79. [Google Scholar] [CrossRef]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The dual role of microglia in ALS: Mechanisms and therapeutic approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, J.S.; Beers, D.R.; Zhao, W.; Appel, S.H. Microglia in ALS: The good, the bad, and the resting. J. Neuroimmune Pharmacol. 2009, 4, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Côté, F.; Collard, J.F.; Julien, J.P. Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: A mouse model of amyotrophic lateral sclerosis. Cell 1993, 73, 35–46. [Google Scholar] [CrossRef]
- Kennel, P.F.; Finiels, F.; Revah, F.; Mallet, J. Neuromuscular functicon impairment is not caused by motor neurone loss in FALS mice: An electromyographic study. Neuroreport 1996, 7, 1427–1431. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Spinelli, E.G.; Riva, N.; Rancoita, P.M.V.; Schito, P.; Doretti, A.; Poletti, B.; Di Serio, C.; Silani, V.; Filippi, M.; Agosta, F. Structural MRI outcomes and predictors of disease progression in amyotrophic lateral sclerosis. NeuroImage Clin. 2020, 27. [Google Scholar] [CrossRef]
- Rajagopalan, V.; Pioro, E.P. Unbiased MRI analyses identify micropathologic differences between upper motor neuron-predominant ALS phenotypes. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Rizzo, G.; Marliani, A.; Battaglia, S.; Albini Riccioli, L.; De Pasqua, S.; Vacchiano, V.; Infante, R.; Avoni, P.; Donadio, V.; Passaretti, M.; et al. Diagnostic and Prognostic Value of Conventional Brain MRI in the Clinical Work-Up of Patients with Amyotrophic Lateral Sclerosis. J. Clin. Med. 2020, 9, 2538. [Google Scholar] [CrossRef]
- Oeckl, P.; Weydt, P.; Thal, D.R.; Weishaupt, J.H.; Ludolph, A.C.; Otto, M. Proteomics in cerebrospinal fluid and spinal cord suggests UCHL1, MAP2 and GPNMB as biomarkers and underpins importance of transcriptional pathways in amyotrophic lateral sclerosis. Acta Neuropathol. 2019, 139, 119–134. [Google Scholar] [CrossRef]
- Raoul, C.; Estévez, A.G.; Nishimune, H.; Cleveland, D.W.; DeLapeyrière, O.; Henderson, C.E.; Haase, G.; Pettmann, B. Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS-linked SOD1 mutations. Neuron 2002, 35, 1067–1083. [Google Scholar] [CrossRef] [Green Version]
- Appel, S.H.; Zhao, W.; Beers, D.R.; Henkel, J.S. The microglial-motoneuron dialogue in ALS. Acta Myol. 2011, 30, 4–8. [Google Scholar] [PubMed]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillée, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, F.; Komine, O.; Fujimori-Tonou, N.; Katsuno, M.; Jin, S.; Watanabe, S.; Sobue, G.; Dezawa, M.; Wyss-Coray, T.; Yamanaka, K. Astrocyte-Derived TGF-β1 Accelerates Disease Progression in ALS Mice by Interfering with the Neuroprotective Functions of Microglia and T Cells. Cell Rep. 2015. [Google Scholar] [CrossRef] [Green Version]
- Costagli, M.; Donatelli, G.; Biagi, L.; Caldarazzo Ienco, E.; Siciliano, G.; Tosetti, M.; Cosottini, M. Magnetic susceptibility in the deep layers of the primary motor cortex in Amyotrophic Lateral Sclerosis. NeuroImage Clin. 2016, 12, 965–969. [Google Scholar] [CrossRef]
- Jara, J.H.; Genç, B.; Stanford, M.J.; Pytel, P.; Roos, R.P.; Weintraub, S.; Mesulam, M.M.; Bigio, E.H.; Miller, R.J.; Özdinler, P.H. Evidence for an early innate immune response in the motor cortex of ALS. J. Neuroinflamm. 2017, 14. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.W.; Sidman, R.L.; Taksir, T.V.; Treleaven, C.M.; Fidler, J.A.; Cheng, S.H.; Dodge, J.C.; Shihabuddin, L.S. Relationship between neuropathology and disease progression in the SOD1G93A ALS mouse. Exp. Neurol. 2011, 227, 287–295. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Kevin, D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haidet-phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Matthew, J.; Foust, K.D.; et al. Astrocytes from Familial and Sporadic ALS Patients are Toxic to Motor Neurons. Nat Biotechnol 2012, 29, 824–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swarup, V.; Phaneuf, D.; Dupré, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.-P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB–mediated pathogenic pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef] [PubMed]
- Ouali Alami, N.; Schurr, C.; Olde Heuvel, F.; Tang, L.; Li, Q.; Tasdogan, A.; Kimbara, A.; Nettekoven, M.; Ottaviani, G.; Raposo, C.; et al. NF-κB activation in astrocytes drives a stage-specific beneficial neuroimmunological response in ALS. EMBO J. 2018, e98697. [Google Scholar] [CrossRef] [PubMed]
- Crosio, C.; Valle, C.; Casciati, A.; Iaccarino, C.; Carrì, M.T. Astroglial inhibition of NF-κb does not ameliorate disease onset and progression in a mouse model for amyotrophic lateral sclerosis (ALS). PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, H.; Hale, V.A.; Dolcet, X.; Davies, A. NF- B signalling regulates the growth of neural processes in the developing PNS and CNS. Development 2005, 132, 1713–1726. [Google Scholar] [CrossRef] [Green Version]
- Kaltschmidt, B.; Ndiaye, D.; Korte, M.; Pothion, S.; Arbibe, L.; Prullage, M.; Pfeiffer, J.; Lindecke, A.; Staiger, V.; Israel, A.; et al. NF- B Regulates Spatial Memory Formation and Synaptic Plasticity through Protein Kinase A/CREB Signaling. Mol. Cell. Biol. 2006, 26, 2936–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taoufik, E.; Tseveleki, V.; Chu, S.Y.; Tselios, T.; Karin, M.; Lassmann, H.; Szymkowski, D.E.; Probert, L. Transmembrane tumour necrosis factor is neuroprotective and regulates experimental autoimmune encephalomyelitis via neuronal nuclear factor-κB. Brain 2011, 134, 2722–2735. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Oliveira, C.R. Inhibition of NF-kB renders cells more vulnerable to apoptosis induced by amyloid beta peptides. Free Radic. Res. 2003, 37, 967–973. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Boillee, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [Green Version]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef] [Green Version]
- Chiu, I.M.; Morimoto, E.T.A.; Goodarzi, H.; Liao, J.T.; O’Keeffe, S.; Phatnani, H.P.; Muratet, M.; Carroll, M.C.; Levy, S.; Tavazoie, S.; et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013, 4, 385–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.-C.; Ré, D.B.; Nagai, M.; Ischiropoulos, H.; Przedborski, S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12132–12137. [Google Scholar] [CrossRef] [Green Version]
- Potolicchio, I.; Carven, G.J.; Xu, X.; Stipp, C.; Riese, R.J.; Stern, L.J.; Santambrogio, L. Proteomic analysis of microglia-derived exosomes: Metabolic role of the aminopeptidase CD13 in neuropeptide catabolism. J. Immunol. 2005, 175, 2237–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, C.; Sainz-Fuertes, R.; Lynham, S.; Hye, A.; Killick, R.; Warley, A.; Bolondi, C.; Pocock, J.; Lovestone, S. Wnt3a induces exosome secretion from primary cultured rat microglia. BMC Neurosci. 2012, 13, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turola, E.; Furlan, R.; Bianco, F.; Matteoli, M.; Verderio, C. Microglial microvesicle secretion and intercellular signaling. Front. Physiol. 2012, 3, 149. [Google Scholar] [CrossRef] [Green Version]
- Parisi, C.; Arisi, I.; D’Ambrosi, N.; Storti, A.E.; Brandi, R.; D’Onofrio, M.; Volonté, C. Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis. 2013, 4, e959. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef] [PubMed]
- Koval, E.D.; Shaner, C.; Zhang, P.; du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 2013, 22, 4127–4135. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef] [Green Version]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e10. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [Green Version]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Butovsky, O.; Siddiqui, S.; Gabriely, G.; Lanser, A.J.; Dake, B.; Murugaiyan, G.; Doykan, C.E.; Wu, P.M.; Gali, R.R.; Iyer, L.K.; et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J. Clin. Investig. 2012, 122, 3063–3087. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, S.; Äijö, T.; Vickovic, S.; Braine, C.; Kang, K.; Mollbrink, A.; Fagegaltier, D.; Andrusivová, Ž.; Saarenpää, S.; Saiz-Castro, G.; et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 2019, 364, 89–93. [Google Scholar] [CrossRef]
- Chen, Z.; Jalabi, W.; Shpargel, K.B.; Farabaugh, K.T.; Dutta, R.; Yin, X.; Kiddds, G.J.; Bergmann, C.C.; Stohlman, S.A.; Trapp, B.D. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J. Neurosci. 2012, 32, 11706–11715. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, C.; Pagé, J.; Bédard, A.; Tremblay, P.; Vallières, L. G protein-coupled receptor 84, a microglia-associated protein expressed in neuroinflammatory conditions. Glia 2007, 55, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Recio, C.; Lucy, D.; Purvis, G.S.D.; Iveson, P.; Zeboudj, L.; Iqbal, A.J.; Lin, D.; O’Callaghan, C.; Davison, L.; Griesbach, E.; et al. Activation of the immune-metabolic receptor GPR84 enhances inflammation and phagocytosis in macrophages. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Nicolas, A.; Kenna, K.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e6. [Google Scholar] [CrossRef] [Green Version]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dols-Icardo, O.; Montal, V.; Sirisi, S.; López-Pernas, G.; Cervera-Carles, L.; Querol-Vilaseca, M.; Muñoz, L.; Belbin, O.; Alcolea, D.; Molina-Porcel, L.; et al. Motor cortex transcriptome reveals microglial key events in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.L.; Tan, C.C.; Hou, X.H.; Cao, X.P.; Tan, L.; Yu, J.T. TREM2 Variants and Neurodegenerative Diseases: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2019, 68, 1171–1184. [Google Scholar] [CrossRef]
- Svahn, A.J.; Don, E.K.; Badrock, A.P.; Cole, N.J.; Graeber, M.B.; Yerbury, J.J.; Chung, R.; Morsch, M. Nucleo-cytoplasmic transport of TDP-43 studied in real time: Impaired microglia function leads to axonal spreading of TDP-43 in degenerating motor neurons. Acta Neuropathol. 2018, 136, 445–459. [Google Scholar] [CrossRef] [Green Version]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef]
- Böttcher, C.; Schlickeiser, S.; Sneeboer, M.A.M.; Kunkel, D.; Knop, A.; Paza, E.; Fidzinski, P.; Kraus, L.; Snijders, G.J.L.; Kahn, R.S.; et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat. Neurosci. 2019, 22, 78–90. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Stickels, R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.; Di Bella, D.; Arlotta, P.; Macosko, E.; Chen, F. Sensitive spatial genome wide expression profiling at cellular resolution. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ajami, B.; Samusik, N.; Wieghofer, P.; Ho, P.P.; Crotti, A.; Bjornson, Z.; Prinz, M.; Fantl, W.J.; Nolan, G.P.; Steinman, L. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.I.; Appel, S.H. IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch. Neurol. 1990, 47, 1210–1216. [Google Scholar] [CrossRef]
- Banati, R.B.; Gehrmann, J.; Kellner, M.; Holsboer, F. Antibodies against microglia/brain macrophages in the cerebrospinal fluid of a patient with acute amyotrophic lateral sclerosis and presenile dementia. Clin. Neuropathol. 1995, 14, 197–200. [Google Scholar]
- Henkel, J.S.; Engelhardt, J.I.; Siklós, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 2004, 55, 221–235. [Google Scholar] [CrossRef]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Fan, J.; Lindemann, P.; Feuilloley, M.G.J.; Papadopoulos, V. Structural and functional evolution of the translocator protein (18 kDa). Curr. Mol. Med. 2012, 12, 369–386. [Google Scholar] [CrossRef]
- Gatliff, J.; Campanella, M. TSPO: Kaleidoscopic 18-kDa amid biochemical pharmacology, control and targeting of mitochondria. Biochem. J. 2016, 473, 107–121. [Google Scholar] [CrossRef] [Green Version]
- Veenman, L.; Gavish, M. The role of 18 kDa mitochondrial translocator protein (TSPO) in programmed cell death, and effects of steroids on TSPO expression. Curr. Mol. Med. 2012, 12, 398–412. [Google Scholar]
- Hardwick, M.; Fertikh, D.; Culty, M.; Li, H.; Vidic, B.; Papadopoulos, V. Peripheral-type benzodiazepine receptor (PBR) in human breast cancer: Correlation of breast cancer cell aggressive phenotype with PBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport of cholesterol. Cancer Res. 1999, 59, 831–842. [Google Scholar] [PubMed]
- Olson, J.M.; Ciliax, B.J.; Mancini, W.R.; Young, A.B. Presence of peripheral-type benzodiazepine binding sites on human erythrocyte membranes. Eur. J. Pharmacol. 1988, 152, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Prinz, W.A. Bridging the gap: Membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 2014, 205, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 2016, 131, 505–523. [Google Scholar] [CrossRef] [Green Version]
- Boutin, H.; Murray, K.; Pradillo, J.; Maroy, R.; Smigova, A.; Gerhard, A.; Jones, P.A.; Trigg, W. 18F-GE-180: A novel TSPO radiotracer compared to 11C-R-PK11195 in a preclinical model of stroke. Eur. J. Nucl. Med. Mol. Imaging 2014, 42, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Le, K.X.; Park, M.; Wang, S.; Belanger, A.P.; Dubey, X.; Frost, J.L.; Holton, P.; Reiser, V.; Jones, X.P.A.; et al. In Vivo Detection of Age- and Disease-Related Increases in Neuroinflammation by 18 F-GE180 TSPO MicroPET Imaging in Wild-Type and Alzheimer’s Transgenic Mice. J. Neurosci. 2015, 35, 15716–15730. [Google Scholar] [CrossRef] [Green Version]
- Gargiulo, S.; Anzilotti, S.; Coda, A.R.D.; Gramanzini, M.; Greco, A.; Panico, M.; Vinciguerra, A.; Zannetti, A.; Vicidomini, C.; Dollé, F.; et al. Imaging of brain TSPO expression in a mouse model of amyotrophic lateral sclerosis with 18F-DPA-714 and micro-PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Tauber, C.; Vercoullie, J.; Arlicot, N.; Prunier, C.; Praline, J.; Nicolas, G.; Venel, Y.; Hommet, C.; Baulieu, J.-L.; et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52941. [Google Scholar] [CrossRef]
- Visigalli, I.; Moresco, R.M.; Belloli, S.; Politi, L.S.; Gritti, A.; Ungaro, D.; Matarrese, M.; Turolla, E.; Falini, A.; Scotti, G.; et al. Monitoring disease evolution and treatment response in lysosomal disorders by the peripheral benzodiazepine receptor ligand PK11195. Neurobiol. Dis. 2009, 34, 51–62. [Google Scholar] [CrossRef]
- Salsano, E.; Marotta, G.; Manfredi, V.; Giovagnoli, A.R.; Farina, L.; Savoiardo, M.; Pareyson, D.; Benti, R.; Uziel, G. Brain fluorodeoxyglucose PET in adrenoleukodystrophy. Neurology 2014, 83, 981–989. [Google Scholar] [CrossRef]
- Lavisse, S.; Guillermier, M.; Hérard, A.-S.; Petit, F.; Delahaye, M.; Van Camp, N.; Ben Haim, L.; Lebon, V.; Remy, P.; Dollé, F.; et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J. Neurosci. 2012, 32, 10809–10818. [Google Scholar] [CrossRef] [Green Version]
- Venneti, S.; Wang, G.; Nguyen, J.; Wiley, C.A. The positron emission tomography ligand DAA1106 binds with high affinity to activated microglia in human neurological disorders. J. Neuropathol. Exp. Neurol. 2008, 67, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-K.; Guilarte, T.R. Translocator protein 18 kDa (TSPO): Molecular sensor of brain injury and repair. Pharmacol. Ther. 2008, 118, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Lopresti, B.J.; Wang, G.; Hamilton, R.L.; Mathis, C.A.; Klunk, W.E.; Apte, U.M.; Wiley, C.A. PK11195 labels activated microglia in Alzheimer’s disease and in vivo in a mouse model using PET. Neurobiol. Aging 2009, 30, 1217–1226. [Google Scholar] [CrossRef] [Green Version]
- Van Weehaeghe, D.; Babu, S.; De Vocht, J.; Zurcher, N.R.; Chew, S.; Tseng, C.-E.J.; Loggia, M.; Koole, M.; Rezaei, A.; Schramm, G.; et al. Moving towards multicenter therapeutic trials in ALS: Feasibility of data pooling using different TSPO positron emission tomography (PET) radioligands. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Petit-Taboué, M.C.; Baron, J.C.; Barré, L.; Travère, J.M.; Speckel, D.; Camsonne, R.; MacKenzie, E.T. Brain kinetics and specific binding of [11C]PK 11195 to ω3 sites in baboons: Positron emission tomography study. Eur. J. Pharmacol. 1991, 200, 347–351. [Google Scholar] [CrossRef]
- Shah, F.; Hume, S.P.; Pike, V.W.; Ashworth, S.; McDermott, J. Synthesis of the enantiomers of [N-methyl-11C]PK 11195 and comparison of their behaviours as radioligands for PK binding sites in rats. Nucl. Med. Biol. 1994, 21, 573–581. [Google Scholar] [CrossRef]
- James, M.L.; Fulton, R.R.; Vercoullie, J.; Henderson, D.J.; Garreau, L.; Chalon, S.; Dolle, F.; Selleri, S.; Guilloteau, D.; Kassiou, M. DPA-714, a new translocator protein-specific ligand: Synthesis, radiofluorination, and pharmacologic characterization. J. Nucl. Med. 2008, 49, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, S.; Lepelletier, F.-X.; Trigg, W.; Banister, S.; Reekie, T.; Kassiou, M.; Gerhard, A.; Hinz, R.; Boutin, H. Comparative Evaluation of Three TSPO PET Radiotracers in a LPS-Induced Model of Mild Neuroinflammation in Rats. Mol. Imaging Biol. 2017, 19, 77–89. [Google Scholar] [CrossRef]
- Paganoni, S.; Alshikho, M.J.; Zürcher, N.R.; Cernasov, P.; Babu, S.; Loggia, M.L.; Chan, J.; Chonde, D.B.; Garcia, D.I.; Catana, C.; et al. Imaging of glia activation in people with primary lateral sclerosis. NeuroImage Clin. 2018, 17, 347–353. [Google Scholar] [CrossRef]
- Zürcher, N.R.; Loggia, M.L.; Lawson, R.; Chonde, D.B.; Izquierdo-Garcia, D.; Yasek, J.E.; Akeju, O.; Catana, C.; Rosen, B.R.; Cudkowicz, M.E.; et al. Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: Assessed with [11C]-PBR28. NeuroImage Clin. 2015, 7, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, D.R.; Yeo, A.J.; Gunn, R.N.; Song, K.; Wadsworth, G.; Lewis, A.; Rhodes, C.; Pulford, D.J.; Bennacef, I.; Parker, C.A.; et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 2012, 32, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tondo, G.; Iaccarino, L.; Cerami, C.; Vanoli, G.E.; Presotto, L.; Masiello, V.; Coliva, A.; Salvi, F.; Bartolomei, I.; Mosca, L.; et al. C-PK11195 PET – based molecular study of microglia activation in SOD1 amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2020. [Google Scholar] [CrossRef]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldovan, R.P.; Teodoro, R.; Gao, Y.; Deuther-Conrad, W.; Kranz, M.; Wang, Y.; Kuwabara, H.; Nakano, M.; Valentine, H.; Fischer, S.; et al. Development of a High-Affinity PET Radioligand for Imaging Cannabinoid Subtype 2 Receptor. J. Med. Chem. 2016, 59, 7840–7855. [Google Scholar] [CrossRef]
- Slavik, R.; Grether, U.; Müller Herde, A.; Gobbi, L.; Fingerle, J.; Ullmer, C.; Krämer, S.D.; Schibli, R.; Mu, L.; Ametamey, S.M. Discovery of a high affinity and selective pyridine analog as a potential positron emission tomography imaging agent for cannabinoid type 2 receptor. J. Med. Chem. 2015, 58, 4266–4277. [Google Scholar] [CrossRef]
- Espejo-Porras, F.; Piscitelli, F.; Verde, R.; Ramos, J.A.; Di Marzo, V.; de Lago, E.; Fernández-Ruiz, J. Changes in the endocannabinoid signaling system in CNS structures of TDP-43 transgenic mice: Relevance for a neuroprotective therapy in TDP-43-related disorders. J. Neuroimmune Pharmacol. 2015, 10, 233–244. [Google Scholar] [CrossRef]
- Shoemaker, J.L.; Seely, K.A.; Reed, R.L.; Crow, J.P.; Prather, P.L. The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J. Neurochem. 2007, 101, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Kim, J. CB2 Cannabinoid Receptor Knockout in Mice Impairs Contextual Long-Term Memory and Enhances Spatial Working Memory. Neural Plast. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Palazuelos, J.; Ortega, Z.; Díaz-Alonso, J.; Guzmán, M.; Galve-Roperh, I. CB 2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J. Biol. Chem. 2012, 287, 1198–1209. [Google Scholar] [CrossRef] [Green Version]
- Duff, G.; Argaw, A.; Cecyre, B.; Cherif, H.; Tea, N.; Zabouri, N.; Casanova, C.; Ptito, M.; Bouchard, J.-F. Cannabinoid Receptor CB2 Modulates Axon Guidance. PLoS ONE 2013, 8, e70849. [Google Scholar] [CrossRef] [Green Version]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid Signaling and Synaptic Function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Li, Y. Chronic activation of CB2 cannabinoid receptors in the hippocampus increases excitatory synaptic transmission. J. Physiol. 2015, 593, 871–886. [Google Scholar] [CrossRef] [Green Version]
- Calignano, A.; La Rana, G.; Giuffrida, A.; Piomelli, D. Control of pain initiation by endogenous cannabinoids. Nature 1998, 394, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, G.; Lee, G.; Valenzano, K. The Role of the Cannabinoid CB2 Receptor in Pain Transmission and Therapeutic Potential of Small Molecule CB2 Receptor Agonists. Curr. Med. Chem. 2007, 14, 917–936. [Google Scholar] [CrossRef]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic Cannabinoid Receptors Regulate Microglial Cell Migration. Microglial Cell Migration. 2003, 23, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Guida, F.; Luongo, L.; Boccella, S.; Giordano, M.E.; Romano, R.; Bellini, G.; Manzo, I.; Furiano, A.; Rizzo, A.; Imperatore, R.; et al. Palmitoylethanolamide induces microglia changes associated with increased migration and phagocytic activity: Involvement of the CB2 receptor. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jia, J.; Liu, X.; Bai, F.; Wang, Q.; Xiong, L. Activation of murine microglial N9 cells is attenuated through cannabinoid receptor CB2 signaling. Biochem. Biophys. Res. Commun. 2015, 458, 92–97. [Google Scholar] [CrossRef]
- Tanaka, M.; Sackett, S.; Zhang, Y. Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef]
- Malek, N.; Popiolek-Barczyk, K.; Mika, J.; Przewlocka, B.; Starowicz, K. Anandamide, acting via CB2 receptors, alleviates LPS-induced neuroinflammation in rat primary microglial cultures. Neural Plast. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Zoppi, S.; Madrigal, J.L.; Caso, J.R.; García-Gutiérrez, M.S.; Manzanares, J.; Leza, J.C.; García-Bueno, B. Regulatory role of the cannabinoid CB2 receptor in stress-induced neuroinflammation in mice. Br. J. Pharmacol. 2014, 171, 2814–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apolloni, S.; Amadio, S.; Parisi, C.; Matteucci, A.; Potenza, R.L.; Armida, M.; Popoli, P.; D’Ambrosi, N.; Volonté, C. Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis. Model. Mech. 2014, 7, 1101–1109. [Google Scholar] [CrossRef] [Green Version]
- Horti, A.G.; Gao, Y.; Ravert, H.T.; Finley, P.; Valentine, H.; Wong, D.F.; Endres, C.J.; Savonenko, A.V.; Dannals, R.F. Synthesis and biodistribution of [11C]A-836339, a new potential radioligand for PET imaging of cannabinoid type 2 receptors (CB2). Bioorganic Med. Chem. 2010, 18, 5202–5207. [Google Scholar] [CrossRef] [Green Version]
- Savonenko, A.V.; Melnikova, T.; Wang, Y.; Ravert, H.; Gao, Y.; Koppel, J.; Lee, D.; Pletnikova, O.; Cho, E.; Sayyida, N.; et al. Cannabinoid CB2 Receptors in a Mouse Model of Aβ Amyloidosis: Immunohistochemical Analysis and Suitability as a PET Biomarker of Neuroinflammation. PLoS ONE 2015, 10, e0129618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evens, N.; Vandeputte, C.; Coolen, C.; Janssen, P.; Sciot, R.; Baekelandt, V.; Verbruggen, A.M.; Debyser, Z.; Van Laere, K.; Bormans, G.M. Preclinical evaluation of [ 11C]NE40, a type 2 cannabinoid receptor PET tracer. Nucl. Med. Biol. 2012, 39, 389–399. [Google Scholar] [CrossRef]
- Ahmad, R.; Koole, M.; Evens, N.; Serdons, K.; Verbruggen, A.; Bormans, G.; Van Laere, K. Whole-body biodistribution and radiation dosimetry of the cannabinoid type 2 receptor ligand [11C]-NE40 in healthy subjects. Mol. Imaging Biol. 2013, 15, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoya, T.; Fukumoto, D.; Kakiuchi, T.; Nishiyama, S.; Yamamoto, S.; Ohba, H.; Tsukada, H.; Ueki, T.; Sato, K.; Ouchi, Y. In vivo TSPO and cannabinoid receptor type 2 availability early in post-stroke neuroinflammation in rats: A positron emission tomography study. J. Neuroinflamm. 2017, 14, 69. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.; Iga, Y.; Nakamura, M.; Takizawa, C.; Fukumoto, D.; Kakiuchi, T.; Nishiyama, S.; Ohba, H.; Tsukada, H.; Sato, K.; et al. Upregulation of cannabinoid receptor type 2, but not TSPO, in senescence-accelerated neuroinflammation in mice: A positron emission tomography study. J. Neuroinflamm. 2019, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavik, R.; Bieri, D.; Ermak, S.Č.; Müller, A.; Krämer, S.D.; Weber, M.; Schibli, R.; Ametamey, S.M.; Mu, L. Development and evaluation of novel pet tracers for imaging cannabinoid receptor type 2 in brain. CHIMIA Int. J. Chem. 2014, 68, 208–210. [Google Scholar]
- Haider, A.; Kretz, J.; Gobbi, L.; Ahmed, H.; Atz, K.; Bürkler, M.; Bartelmus, C.; Fingerle, J.; Guba, W.; Ullmer, C.; et al. Structure-Activity Relationship Studies of Pyridine-Based Ligands and Identification of a Fluorinated Derivative for Positron Emission Tomography Imaging of Cannabinoid Type 2 Receptors. J. Med. Chem. 2019, 62, 11165–11181. [Google Scholar] [CrossRef]
- Boutin, H.; Prenant, C.; Maroy, R.; Galea, J.; Greenhalgh, A.D.; Smigova, A.; Cawthorne, C.; Julyan, P.; Wilkinson, S.M.; Banister, S.D.; et al. [18F]DPA-714: Direct Comparison with [11C]PK11195 in a Model of Cerebral Ischemia in Rats. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Ratai, E.M.; Alshikho, M.J.; Zürcher, N.R.; Loggia, M.L.; Cebulla, C.L.; Cernasov, P.; Reynolds, B.; Fish, J.; Seth, R.; Babu, S.; et al. Integrated imaging of [11C]-PBR28 PET, MR diffusion and magnetic resonance spectroscopy 1H-MRS in amyotrophic lateral sclerosis. NeuroImage Clin. 2018, 20, 357–364. [Google Scholar] [CrossRef]
- Postnov, A.; Ahmad, R.; Evens, N.; Versijpt, J.; Vandenbulcke, M.; Yaqub, M.; Verbruggen, A.; Bormans, G.; Vandenberghe, W.; Laere, K. Quantification of 11C-NE40, a novel PET radioligand for CB2 receptor imaging. J. Nucl. Med. 2013, 54, 188. [Google Scholar]
- Slavik, R.; Herde, A.M.; Bieri, D.; Weber, M.; Schibli, R.; Krämer, S.D.; Ametamey, S.M.; Mu, L. Synthesis, radiolabeling and evaluation of novel 4-oxo-quinoline derivatives as PET tracers for imaging cannabinoid type 2 receptor. Eur. J. Med. Chem. 2015, 92, 554–564. [Google Scholar] [CrossRef]
- Territo, P.R.; Meyer, J.A.; Peters, J.S.; Riley, A.A.; McCarthy, B.P.; Gao, M.; Min, W.; Green, M.A.; Zheng, Q.H.; Hutchins, G.D. Characterization of 11C-GSK1482160 for Targeting the P2X7 receptor as a biomarker for neuroinflammation. J. Nucl. Med. 2017, 58, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Farmer, W.T.; Murai, K. Resolving astrocyte heterogeneity in the CNS. Front. Cell. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Marques, S.; Zeisel, A.; Codeluppi, S.; Van Bruggen, D.; Falcão, A.M.; Xiao, L.; Li, H.; Häring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 2016, 352, 1326–1329. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; Manno, G.L.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| (a) Homeostatic microglia | ||||
|---|---|---|---|---|
| Gene | Species | CNS Regions Analyzed | Validation Methods | References |
| Fcrls | Mus musculus (C57BL6) | Brain and SC | RNA-seq, RT-qPCR, IHC, FACS | [47,48,49,50] |
| P2ry12 | M. musculus (C57BL6) | Brain and SC | RNA-seq, RT-qPCR, IHC, FACS | [47,49,51,52,53] |
| Tmem119 | M. musculus (C57BL6) | Brain and SC | RNA-seq, RT-qPCR | [39,47,49,50,51,53] |
| Tgfbr1 | M. musculus (C57BL6) | Brain and SC | RNA-seq | [47,54] |
| Csfr1 | M. musculus (C57BL6) | Brain and SC | RNA-seq | [47,54] |
| Sparc | M. musculus (C57BL6, DBA/2J & C57/SJL) | Brain and SC | RNA-seq, RT-qPCR | [47,55] |
| Cx3cr1 | M. musculus (C57BL6, DBA/2J & C57/SJL) | Brain and SC | RNA-seq, RT-qPCR | [47,50,52,53,55] |
| Hexb | M. musculus (C57BL6) | Brain and SC | RNA-seq, RT-qPCR | [47,52] |
| Olfml3 | M. musculus (C57BL6) | Brain and SC | RNA-seq, RT-qPCR, IHC, FACS | [39,47,49,50] |
| Ltc4s | M. musculus (C57BL6) | Brain and SC | RNA-seq | [47,50] |
| SiglecH | M. musculus (C57BL6) | Brain and SC | RNA-seq, IHC, FACS | [39,47,49,51] |
| Gpr34 | M. musculus (C57BL6) | Brain and SC | RT-qPCR, RNA-seq | [47,49,50] |
| (b) ALS microglia | ||||
| Gene | Species | CNS Regions Analyzed | Validation Methods | References |
| Axl * | M. musculus (B6/SJL-SOD1G93A, C57BL6-SOD1G93A) | SC | Microarray, RNA-seq, scRNA-seq | [39,45,56] |
| Apoe * | M. musculus (B6/SJL-SOD1G93A, C57BL6-SOD1G93A) | SC | Microarray, RNA-seq, scRNA-seq | [39,45,56] |
| Homo sapiens (ALS patients) | Lumbar SC | RNA-seq, scRNA-seq, RT-qPCR, IHC, FACS | [45] | |
| Spp1 * | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, scRNA-seq, RT-qPCR | [39,56] |
| Csf1 * | M. musculus (B6/SJL-SOD1G93A, C57BL6-SOD1G93A) | Brain and SC | Microarray, RNA-seq, scRNA-seq | [45,54] |
| Cybb (Nox2) | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, RT-qPCR, IHC | [39,40] |
| Igf-1 | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, RT-qPCR | [39] |
| Grn | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, RT-qPCR | [39] |
| Optn | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, RT-qPCR | [39] |
| Mmp-12 | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, Microarray | [39,57] |
| Tyrobp * (Dap-12) | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, scRNA-seq, RT-qPCR, ST | [39,56,58] |
| H. sapiens (ALS patient) | Cervical and lumbar SC | ST | [58] | |
| Trem2 * | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, scRNA-seq, ST | [56,58] |
| H. sapiens (ALS patient) | SC | ST | [58] | |
| Lpl * | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, scRNA-seq, ST | [56,58] |
| H. sapiens (ALS patient) | Cervical and lumbar SC | ST | [58] | |
| B2m * | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, scRNA-seq, ST | [56,58] |
| H. sapiens (ALS patient) | Cervical and lumbar SC | ST | [58] | |
| Ctsl * | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, scRNA-seq | [39,56] |
| Itgax * | M. musculus (C57BL6-SOD1G93A) | Brain & SC | RNA-seq, scRNA-seq | [54,56] |
| Clec7a * | M. musculus (C57BL6-SOD1G93A) | SC | RNA-seq, scRNA-seq | [39,54,56] |
| (c) LPS-stimulated microglia | ||||
| Gene | Species | CNS Regions Analyzed | Validation Methods | References |
| Stat3 | M. musculus (C57BL6, DBA/2J and C57/SJL) | Brain and SC | RNA-seq | [39,55] |
| Socs3 | M. musculus (DBA/2J and C57/SJL) | Brain | RNA-seq | [49,55,59] |
| Map3k8 | M. musculus (DBA/2 J and C57/SJL) | Brain | RNA-seq | [55] |
| Ccl2 | M. musculus (C57BL6) | Brain | RNA-seq | [49] |
| Gpr84 | M. musculus (C57BL6) | Brain | RT-qPCR | [49,60,61] |
| Tracer and Radioisotope | Target | Tested in | Notes | References |
|---|---|---|---|---|
| [11C]-(R)-PK11195 | TSPO | human ALS patients and healthy controls | signals are not influenced by patient’s TSPO genotype | [103,131] |
| rat model of cerebral ischemia (Wistar rats) | poor specificity | |||
| [18F]DPA-714 | TSPO | ALS model (SOD1G93A mouse) | signals correlate to increased TSPO expression and compromised brain regions | [87,131] |
| rat model of cerebral ischemia (Wistar rats) | higher affinity and better signal-to-noise ratio than PK11195 | |||
| [11C]-PBR28 | TSPO | human ALS, PLS patients and healthy controls | signals correlate to glial activation and inflammation | [102,132] |
| human healthy subjects | binding is affected by TSPO polymorphism | |||
| [11C]A-836339 | CB2 | neuroinflammation-induced/AD models (CD-1 and APPswe/PS1ΔE9 mouse) | first CB2 radiotracer tested | [123,124] |
| AD model (APPswe/PS1ΔE9 mouse) | detection of neuroinflammation very early in the pathology | |||
| [11C]NE40 | CB2 | AD, PD patients and healthy controls | no differences between disease and control cases | [127,128,133] |
| ischemic stroke model (Sprague–Dawley rat) | signal in peri-infarct area, concomitant to CB2 up-regulation | |||
| senescence-accelerated model (SAMP10 mouse) | detection of early signs of neuroinflammation in cortex | |||
| [11C]KD2 | CB2 | ALS patients | selective binding in post-mortem ALS spinal cord specimens | [129,134] |
| neuroinflammation-induced model (CD-1 mouse) | limited target specificity and excessive lipophilicity | |||
| [11C]RS-016 | CB2 | neuroinflammation-induced model (CD-1 mouse) | high blood stability and CB2 specificity | [134] |
| ALS patients | selective binding in post-mortem ALS spinal cord specimens | |||
| [18F]29 | CB2 | neuroinflammation-induced model (CD-1 mouse) | CB2 specific tracing but very rapid metabolism | [105] |
| [18F]3 | CB2 | Wistar rats | rapid washout from brain tissue | [130] |
| ALS patients and healthy controls | selective binding in post-mortem ALS spinal cord specimens | |||
| [11C]GSK1482160 | P2X7 | neuroinflammation-induced model (C57BL6) | increased signals in CNS sites with prominent neuroinflammation | [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cipollina, G.; Davari Serej, A.; Di Nolfi, G.; Gazzano, A.; Marsala, A.; Spatafora, M.G.; Peviani, M. Heterogeneity of Neuroinflammatory Responses in Amyotrophic Lateral Sclerosis: A Challenge or an Opportunity? Int. J. Mol. Sci. 2020, 21, 7923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217923
Cipollina G, Davari Serej A, Di Nolfi G, Gazzano A, Marsala A, Spatafora MG, Peviani M. Heterogeneity of Neuroinflammatory Responses in Amyotrophic Lateral Sclerosis: A Challenge or an Opportunity? International Journal of Molecular Sciences. 2020; 21(21):7923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217923
Chicago/Turabian StyleCipollina, Giada, Arash Davari Serej, Gianluca Di Nolfi, Andrea Gazzano, Andrea Marsala, Mauro G. Spatafora, and Marco Peviani. 2020. "Heterogeneity of Neuroinflammatory Responses in Amyotrophic Lateral Sclerosis: A Challenge or an Opportunity?" International Journal of Molecular Sciences 21, no. 21: 7923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217923