Crystal Structure and Inhibitor Identifications Reveal Targeting Opportunity for the Atypical MAPK Kinase ERK3

 , and
, and 
Abstract
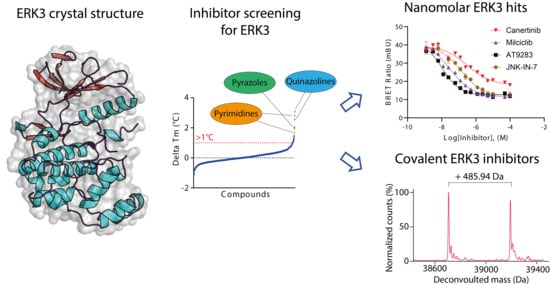
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
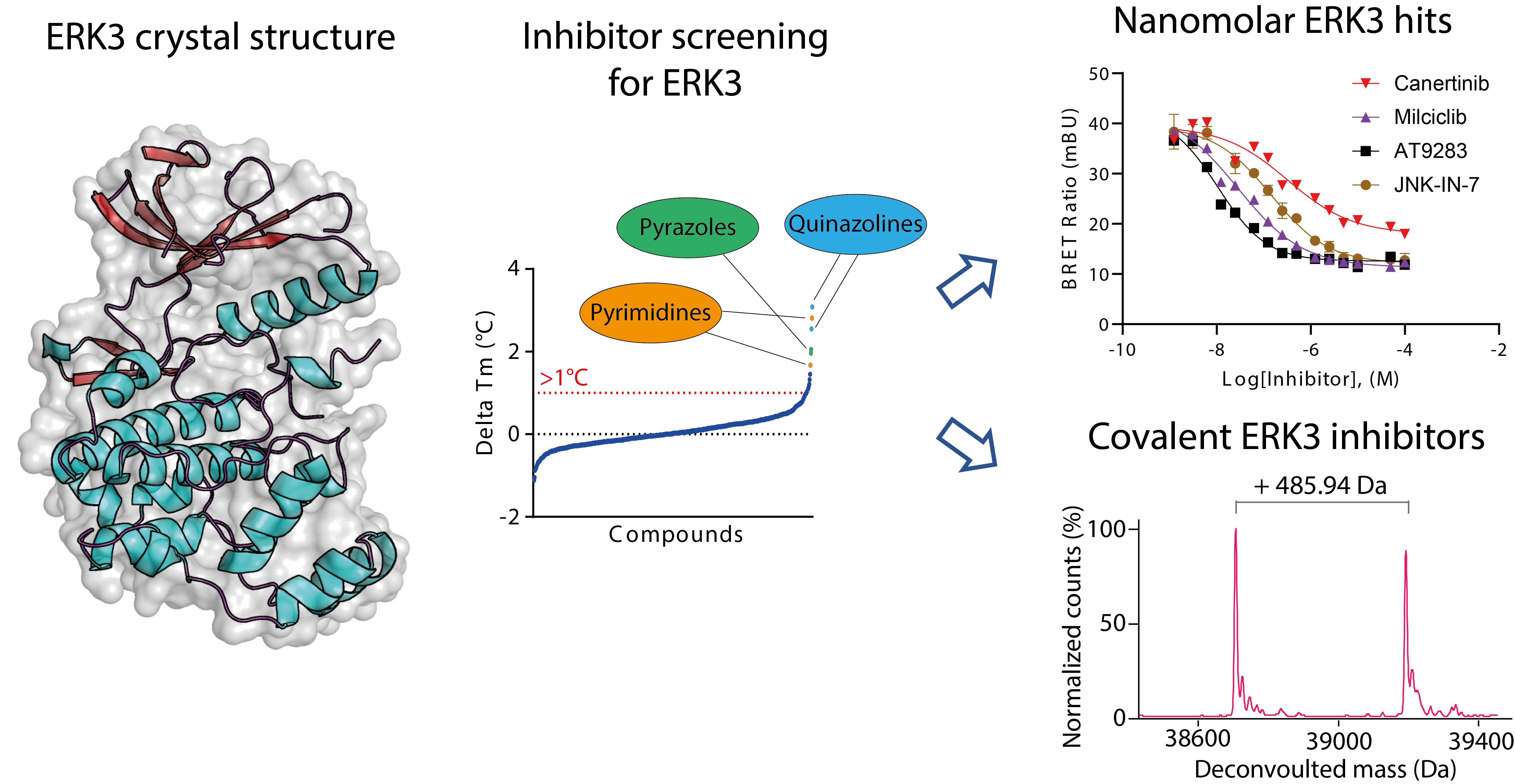
2.1. Crystal Structure of ERK3 Revealed Differences to the Classical MAPK ERK2
2.2. Screening and Characterization of Inhibitors for ERK3
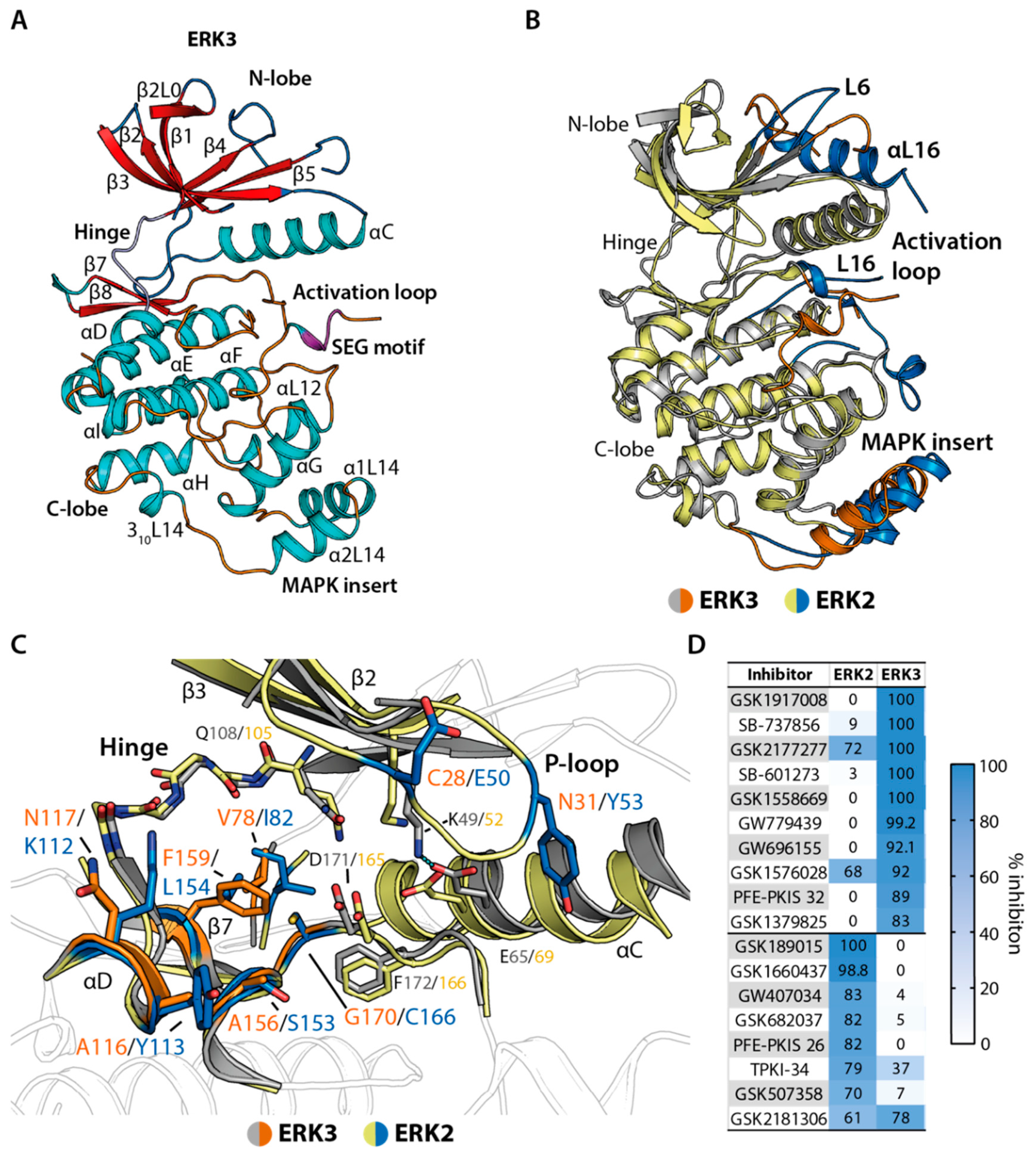
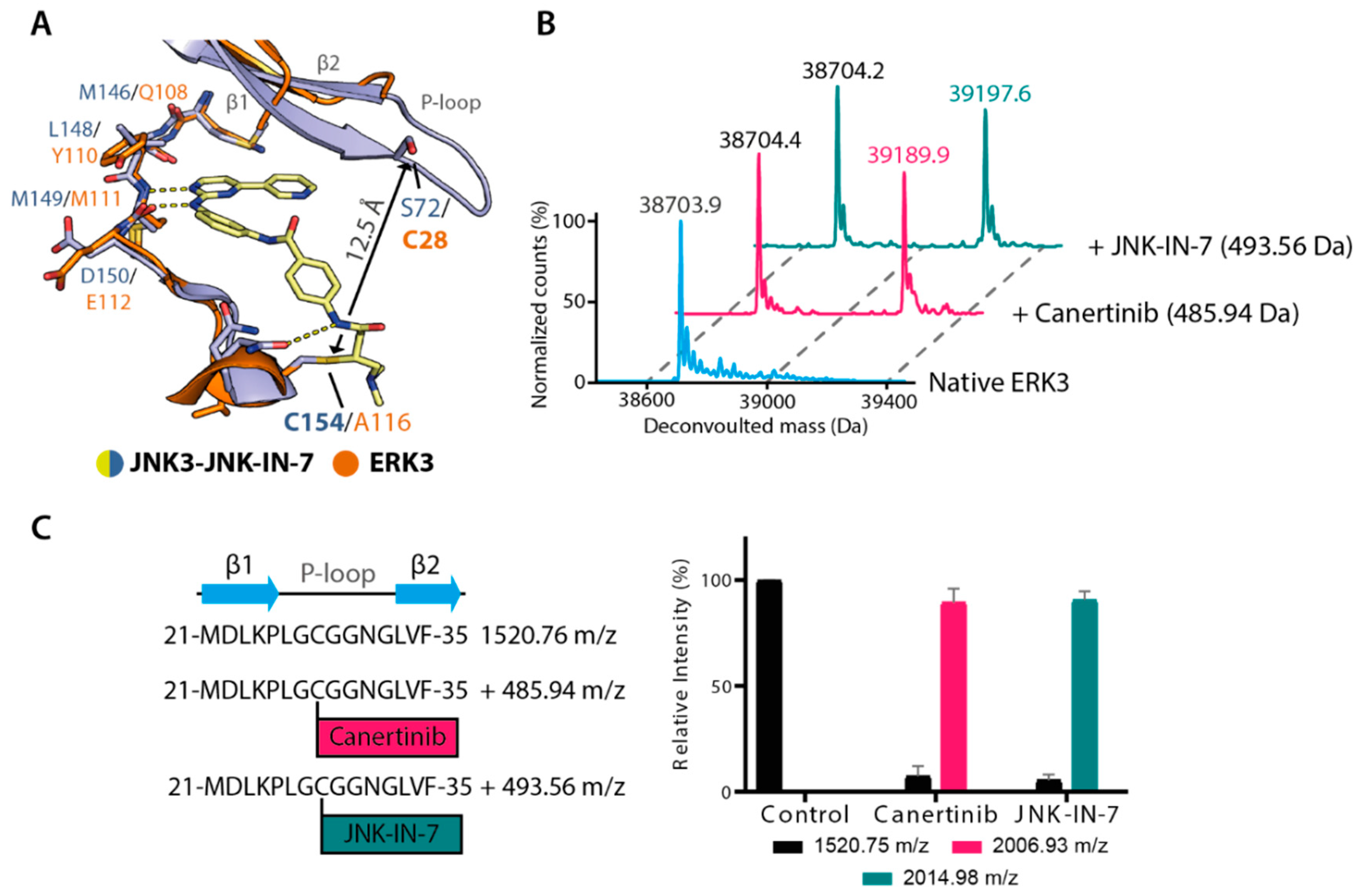
2.3. Irreversible Inhibitors Interacted with ERK3 via Covalent Targeting of the β1 Cysteine
2.4. Structural Comparison Provided Insights into Potential CAF052 Binding Mode in ERK3
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Structure Determination
4.3. Melting Temperature Assay (Differential Scanning Flourimetry (DSF))
4.4. NanoBRET
4.5. Isothermal Titration Calorimetry
4.6. Mass Spectrometry Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ERK3 | Extracellular signal-regulated kinase 3 |
| MAPK6 | Mitogen-activated protein kinase 6 |
| ERK2 | Extracellular signal-regulated kinase 2 |
| TxY | Threonine–x–tyrosine |
| SEG | Serine–glutamate–glycine |
| SRP | Serine–arginine–proline |
| APE | Alanine–proline–glutamate |
| MAP2K | Mitogen-activated protein kinase kinase |
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.M.; Isserlin, R.; Bader, G.D.; Frye, S.V.; Willson, T.M.; Yu, F.H. Too many roads not taken. Nat. Cell Biol. 2011, 470, 163–165. [Google Scholar] [CrossRef] [Green Version]
- Oprea, T.I.; Bologa, C.G.; Brunak, S.; Campbell, A.; Gan, G.N.; Gaulton, A.; Gomez, S.M.; Guha, R.; Hersey, A.; Holmes, J.; et al. Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov. 2018, 17, 317–332. [Google Scholar] [CrossRef]
- Fedorov, O.; Müller, S.; Knapp, S. The (un)targeted cancer kinome. Nat. Chem. Biol. 2010, 6, 166–169. [Google Scholar] [CrossRef]
- Drewry, D.H.; Wells, C.I.; Andrews, D.M.; Angell, R.; Al-Ali, H.; Axtman, A.D.; Capuzzi, S.J.; Elkins, J.M.; Ettmayer, P.; Frederiksen, M.; et al. Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS ONE 2017, 12, e0181585. [Google Scholar] [CrossRef]
- Elkins, J.M.; Fedele, V.; Szklarz, M.; Azeez, K.R.A.; Salah, E.; Mikolajczyk, J.; Romanov, S.; Sepetov, N.; Huang, X.-P.; Roth, B.L.; et al. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol. 2016, 34, 95–103. [Google Scholar] [CrossRef]
- Drewry, D.H.; Willson, T.M.; Zuercher, W.J. Seeding Collaborations to Advance Kinase Science with the GSK Published Kinase Inhibitor Set (PKIS). Curr. Top Med. Chem. 2014, 14, 340–342. [Google Scholar] [CrossRef]
- Wells, C.I.; Al-Ali, H.; Andrews, D.M.; Asquith, C.R.M.; Axtman, A.D.; Chung, M.; Dikic, I.; Ebner, D.V.; Elkins, J.M.; Ettmayer, P.; et al. The Kinase Chemogenomic Set (KCGS): An open science resource for kinase vulnerability identification. bioRxiv 2019, 12, 886523. [Google Scholar] [CrossRef] [Green Version]
- Knapp, S.; Arruda, P.; Blagg, J.; Burley, S.; Drewry, D.H.; Edwards, A.; Fabbro, D.; Gillespie, P.; Gray, N.S.; Kuster, B.; et al. A public-private partnership to unlock the untargeted kinome. Nat. Chem. Biol. 2012, 9, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, P.; Meloche, S. Atypical mitogen-activated protein kinases: Structure, regulation and functions. Biochim. Biophys. Acta 2007, 1773, 1376–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canagarajah, B.J.; Khokhlatchev, A.; Cobb, M.; Goldsmith, E.J. Activation Mechanism of the MAP Kinase ERK2 by Dual Phosphorylation. Cell 1997, 90, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Cobb, M.; Goldsmith, E.J. Dimerization in MAP-kinase signaling. Trends Biochem. Sci. 2000, 25, 7–9. [Google Scholar] [CrossRef]
- Mathien, S.; Soulez, M.; Klinger, S.; Meloche, S. Erk3 and Erk4. In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 1632–1638. [Google Scholar]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Mota-Peynado, A.; Chernoff, J.; Beeser, A. Identification of the Atypical MAPK Erk3 as a Novel Substrate for p21-activated Kinase (Pak) Activity. J. Biol. Chem. 2011, 286, 13603–13611. [Google Scholar] [CrossRef] [Green Version]
- Déléris, P.; Trost, M.; Topisirovic, I.; Tanguay, P.L.; Borden, K.L.; Thibault, P.; Meloche, S. Activation loop phosphorylation of ERK3/ERK4 by group I p21-activated kinases (PAKs) defines a novel PAK-ERK3/4-MAPK-activated protein kinase 5 signaling pathway. J. Biol. Chem. 2011, 286, 6470–6478. [Google Scholar] [CrossRef] [Green Version]
- Déléris, P.; Rousseau, J.; Coulombe, P.; Rodier, G.; Tanguay, P.-L.; Meloche, S. (Sylvain) Activation loop phosphorylation of the atypical MAP kinases ERK3 and ERK4 is required for binding, activation and cytoplasmic relocalization of MK5. J. Cell. Physiol. 2008, 217, 778–788. [Google Scholar] [CrossRef]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Seternes, O.M.; Mikalsen, T.; Johansen, B.; Michaelsen, E.; Armstrong, C.G.; Morrice, N.A.; Turgeon, B.; Meloche, S.; Moens, U.; Keyse, S.M. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 2004, 23, 4780–4791. [Google Scholar] [CrossRef]
- Åberg, E.; Torgersen, K.M.; Johansen, B.; Keyse, S.M.; Perander, M.; Seternes, O.-M. Docking of PRAK/MK5 to the Atypical MAPKs ERK3 and ERK4 Defines a Novel MAPK Interaction Motif. J. Biol. Chem. 2009, 284, 19392–19401. [Google Scholar] [CrossRef] [Green Version]
- El-Merahbi, R.; Viera, J.T.; Valdes, A.L.; Kolczynska, K.; Reuter, S.; Löffler, M.C.; Erk, M.; Ade, C.P.; Karwen, T.; Mayer, A.E.; et al. The adrenergic-induced ERK3 pathway drives lipolysis and suppresses energy dissipation. Genes Dev. 2020, 34, 495–510. [Google Scholar] [CrossRef] [Green Version]
- Long, W.; Foulds, C.E.; Qin, J.; Liu, J.; Ding, C.; Lonard, D.M.; Solis, L.M.; Wistuba, I.I.; Tsai, S.Y.; Tsai, M.-J.; et al. ERK3 signals through SRC-3 coactivator to promote human lung cancer cell invasion. J. Clin. Investig. 2012, 122, 1869–1880. [Google Scholar] [CrossRef]
- Bian, K.; Muppani, N.R.; Elkhadragy, L.; Wang, W.; Zhang, C.; Chen, T.; Jung, S.; Seternes, O.M.; Long, W. ERK3 regulates TDP2-mediated DNA damage response and chemoresistance in lung cancer cells. Oncotarget 2015, 7, 6665–6675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, F.; Schumacher, S.; Kant, S.; Menon, M.B.; Simon, R.; Turgeon, B.; Britsch, S.; Meloche, S.; Egaestel, M.; Kotlyarov, A. The Extracellular Signal-Regulated Kinase 3 (Mitogen-Activated Protein Kinase 6 [MAPK6])-MAPK-Activated Protein Kinase 5 Signaling Complex Regulates Septin Function and Dendrite Morphology. Mol. Cell. Biol. 2012, 32, 2467–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinger, S.; Turgeon, B.; Lévesque, K.; Wood, G.A.; Aagaard-Tillery, K.M.; Meloche, S. Loss of Erk3 function in mice leads to intrauterine growth restriction, pulmonary immaturity, and neonatal lethality. Proc. Natl. Acad. Sci. USA 2009, 106, 16710–16715. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Bian, K.; Vallabhaneni, S.; Zhang, B.; Wu, R.C.; O’Malley, B.W.; Long, W. ERK3 promotes endothelial cell functions by upregulating SRC-3/SP1-mediated VEGFR2 expression. J. Cell. Physiol. 2014, 229, 1529–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkhadragy, L.; Chen, M.; Miller, K.; Yang, M.H.; Long, W. A regulatory BMI1/let-7i/ERK3 pathway controls the motility of head and neck cancer cells. Mol. Oncol. 2017, 11, 194–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mahdi, R.; Babteen, N.; Thillai, K.; Holt, M.; Johansen, B.; Wetting, H.L.; Seternes, O.-M.; Wells, C.M. A novel role for atypical MAPK kinase ERK3 in regulating breast cancer cell morphology and migration. Cell Adhes. Migr. 2015, 9, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Inesta-Vaquera, F.; Niepel, M.; Zhang, J.; Ficarro, S.B.; Machleidt, T.; Xie, T.; Marto, J.A.; Kim, N.; Sim, T.; et al. Discovery of Potent and Selective Covalent Inhibitors of JNK. Chem. Biol. 2012, 19, 140–154. [Google Scholar] [CrossRef] [Green Version]
- Kooistra, A.J.; Kanev, G.K.; Van Linden, O.P.; Leurs, R.; De Esch, I.J.; De Graaf, C. KLIFS: A structural kinase-ligand interaction database. Nucleic Acids Res. 2015, 44, D365–D371. [Google Scholar] [CrossRef] [Green Version]
- Schröder, M.; Bullock, A.N.; Fedorov, O.; Bracher, F.; Chaikuad, A.; Knapp, S. DFG-1 residue controls inhibitor binding mode and affinity providing a basis for rational design of kinase inhibitor selectivity. J. Med. Chem. 2020, 63, 10224–10234. [Google Scholar] [CrossRef]
- Fedorov, O.; Niesen, F.H.; Knapp, S. Kinase Inhibitor Selectivity Profiling Using Differential Scanning Fluorimetry. Methods Mol. Biol. 2012, 795, 109–118. [Google Scholar] [CrossRef]
- Robers, M.; Friedman-Ohana, R.; Huber, K.; Kilpatrick, L.; Vasta, J.; Berger, B.-T.; Chaudhry, C.; Hill, S.; Müller, S.; Knapp, S.; et al. Quantifying Target Occupancy of Small Molecules Within Living Cells. Annu. Rev. Biochem. 2020, 89, 557–581. [Google Scholar] [CrossRef] [PubMed]
- Vasta, J.D.; Corona, C.R.; Wilkinson, J.; Zimprich, C.A.; Hartnett, J.R.; Ingold, M.R.; Zimmerman, K.; Machleidt, T.; Kirkland, T.A.; Huwiler, K.G.; et al. Quantitative, Wide-Spectrum Kinase Profiling in Live Cells for Assessing the Effect of Cellular ATP on Target Engagement. Cell Chem. Biol. 2018, 25, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.A.; Bethel, P.; Cook, C.; Davies, E.; Debreczeni, J.E.; Fairley, G.; Feron, L.; Flemington, V.; Graham, M.A.; Greenwood, R.; et al. Structure-Guided Discovery of Potent and Selective Inhibitors of ERK1/2 from a Modestly Active and Promiscuous Chemical Start Point. J. Med. Chem. 2017, 60, 3438–3450. [Google Scholar] [CrossRef]
- Stevens, K.L.; Reno, M.J.; Alberti, J.B.; Price, D.J.; Kane-Carson, L.S.; Knick, V.B.; Shewchuk, L.M.; Hassell, A.M.; Veal, J.M.; Davis, S.T.; et al. Synthesis and evaluation of pyrazolo[1,5-b]pyridazines as selective cyclin dependent kinase inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 5758–5762. [Google Scholar] [CrossRef]
- Schaenzer, A.J.; Wlodarchak, N.; Drewry, D.H.; Zuercher, W.J.; Rose, W.E.; Ferrer, C.A.; Sauer, J.-D.; Striker, R. GW779439X and Its Pyrazolopyridazine Derivatives Inhibit the Serine/Threonine Kinase Stk1 and Act As Antibiotic Adjuvants against β-Lactam-Resistant Staphylococcus aureus. ACS Infect. Dis. 2018, 4, 1508–1518. [Google Scholar] [CrossRef]
- Smalley, T.L.; Peat, A.J.; Boucheron, J.A.; Dickerson, S.; Garrido, D.; Preugschat, F.; Schweiker, S.L.; Thomson, S.A.; Wang, T.Y. Synthesis and evaluation of novel heterocyclic inhibitors of GSK-3. Bioorganic Med. Chem. Lett. 2006, 16, 2091–2094. [Google Scholar] [CrossRef]
- Smaill, J.B.; Rewcastle, G.W.; Loo, J.A.; Greis, K.D.; Chan, O.H.; Reyner, E.L.; Lipka, E.; Showalter, H.D.; Vincent, P.W.; Elliott, W.L.; et al. Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino)quinazoline- and 4-(phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides bearing additional solubilizing functions. J. Med. Chem. 2000, 43, 1380–1397. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Koch, P.; Laufer, S.A.; Knapp, S. The Cysteinome of Protein Kinases as a Target in Drug Development. Angew. Chem. Int. Ed. 2018, 57, 4372–4385. [Google Scholar] [CrossRef] [PubMed]
- Tavares, F.X.; Boucheron, J.A.; Dickerson, S.H.; Griffin, R.J.; Preugschat, F.; Thomson, S.A.; Wang, T.Y.; Zhou, H.-Q. N-Phenyl-4-pyrazolo[1,5-b]pyridazin-3-ylpyrimidin-2-amines as Potent and Selective Inhibitors of Glycogen Synthase Kinase 3 with Good Cellular Efficacy. J. Med. Chem. 2004, 47, 4716–4730. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; E Audia, J.; Austin, C.; Baell, J.B.; Bennett, J.; Blagg, J.; Bountra, C.; Brennan, P.E.; Brown, P.J.; E Bunnage, M.; et al. Corrigendum: The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 887. [Google Scholar] [CrossRef] [Green Version]
- Drewes, G.; Knapp, S. Chemoproteomics and Chemical Probes for Target Discovery. Trends Biotechnol. 2018, 36, 1275–1286. [Google Scholar] [CrossRef]
- Carter, A.; Kraemer, O.; Zwick, M.; Mueller-Fahrnow, A.; Arrowsmith, C.H.; Edwards, A.M. Target 2035: Probing the human proteome. Drug Discov. Today 2019, 24, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Heroven, C.; Georgi, V.; Ganotra, G.K.; Brennan, P.; Wolfreys, F.; Wade, R.C.; Fernandez-Montalvan, A.E.; Chaikuad, A.; Knapp, S. Halogen-Aromatic pi Interactions Modulate Inhibitor Residence Times. Angew Chem. Int. Ed. Engl. 2018, 57, 7220–7224. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Strand, A.; Robbins, D.; Cobb, M.; Goldsmith, E.J. Atomic structure of the MAP kinase ERK2 at 2.3 Å resolution. Nat. Cell Biol. 1994, 367, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Das, S.; Debreczeni, J.É.; Rellos, P.; Fedorov, O.; Niesen, F.H.; Guo, K.; Papagrigoriou, E.; Amos, A.L.; Cho, S.; et al. Kinase Domain Insertions Define Distinct Roles of CLK Kinases in SR Protein Phosphorylation. Structure 2009, 17, 352–362. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development ofCoot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Robers, M.B.; Vasta, J.D.; Corona, C.R.; Ohana, R.F.; Hurst, R.; Jhala, M.A.; Comess, K.M.; Wood, K.V. Quantitative, Real-Time Measurements of Intracellular Target Engagement Using Energy Transfer. Methods Mol. Biol. 2019, 1888, 45–71. [Google Scholar] [CrossRef] [Green Version]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schröder, M.; Filippakopoulos, P.; Schwalm, M.P.; Ferrer, C.A.; Drewry, D.H.; Knapp, S.; Chaikuad, A. Crystal Structure and Inhibitor Identifications Reveal Targeting Opportunity for the Atypical MAPK Kinase ERK3. Int. J. Mol. Sci. 2020, 21, 7953. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217953
Schröder M, Filippakopoulos P, Schwalm MP, Ferrer CA, Drewry DH, Knapp S, Chaikuad A. Crystal Structure and Inhibitor Identifications Reveal Targeting Opportunity for the Atypical MAPK Kinase ERK3. International Journal of Molecular Sciences. 2020; 21(21):7953. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217953
Chicago/Turabian StyleSchröder, Martin, Panagis Filippakopoulos, Martin P. Schwalm, Carla A. Ferrer, David H. Drewry, Stefan Knapp, and Apirat Chaikuad. 2020. "Crystal Structure and Inhibitor Identifications Reveal Targeting Opportunity for the Atypical MAPK Kinase ERK3" International Journal of Molecular Sciences 21, no. 21: 7953. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217953