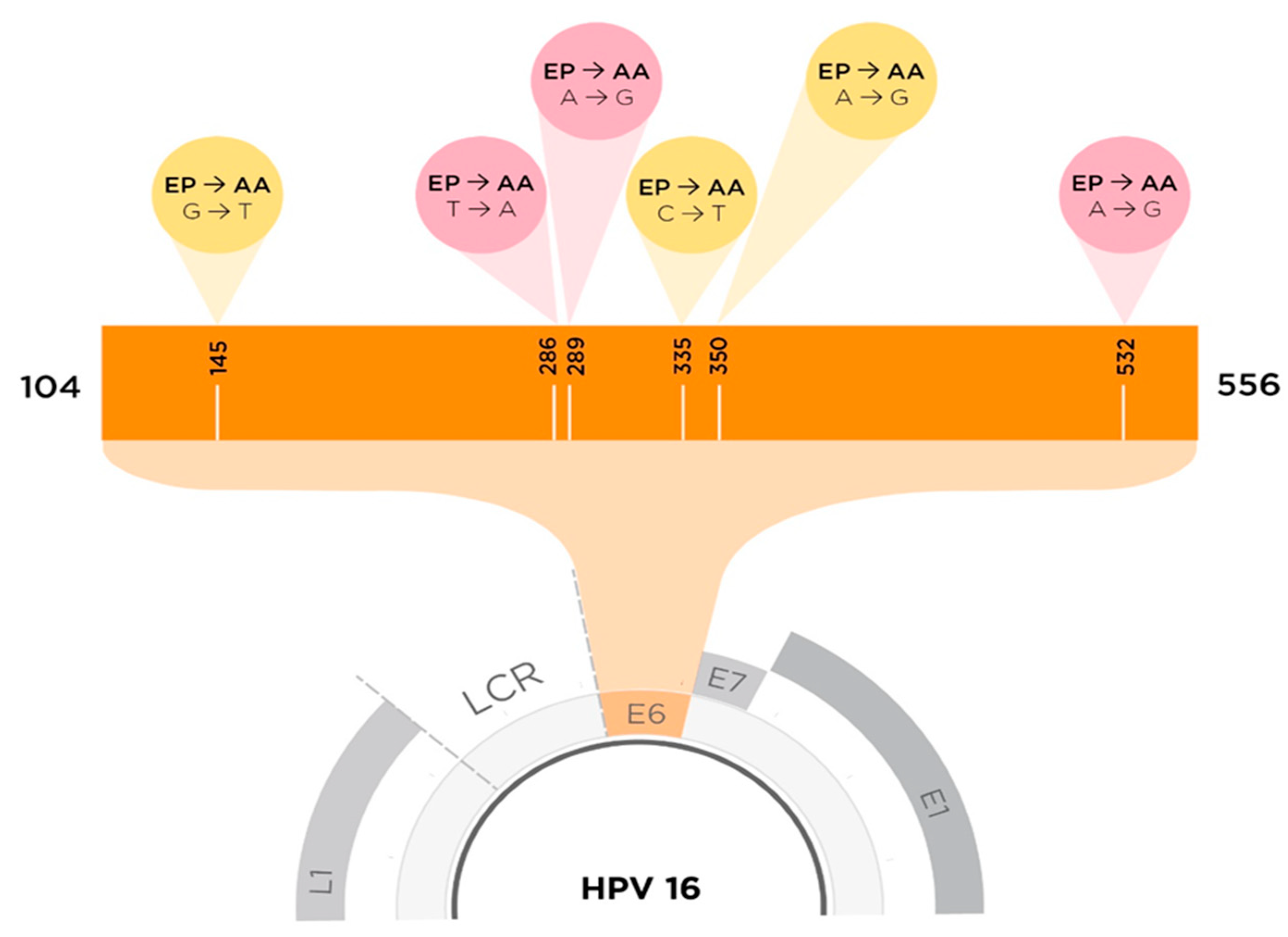
Virus–Host Protein–Protein Interactions between Human Papillomavirus 16 E6 A1 and D2/D3 Sub-Lineages: Variances and Similarities


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
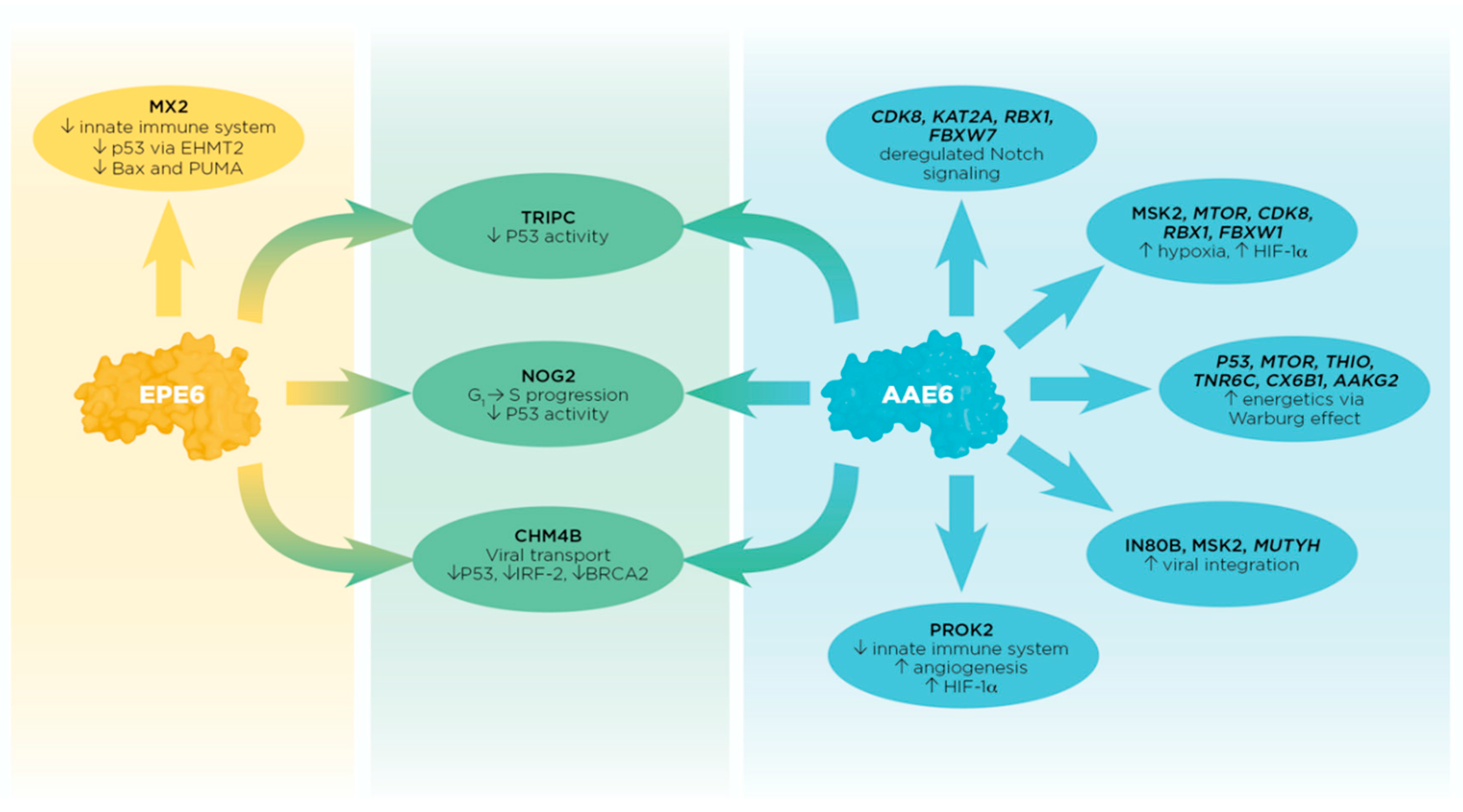
2.1. Peptide Method: E6 Activities Relate to P53 Tumour Suppression, Hypoxia, Energetics, Chromosome Remodeling, and Innate Immunity
2.1.1. AAE6 Affects Chromosome Remodeling, Hypoxia, and Innate Immunity
2.1.2. EPE6 Interferes with Host-Cellular Immune Surveillance
2.1.3. AAE6 and EPE6 Both Disrupt P53 Activities
2.2. Protein-Pathway Method: AAE6 Is More “Successful” than EPE6 in the Malignant Transformation Process
2.2.1. AAE6 Affects Notch Signaling, Hypoxia, Metabolism, and DNA Base Excision Repair
2.2.2. EPE6 Is Associated with DNA Damage Repair and Cancer Pathways
2.2.3. AAE6 and EPE6 Both Prevent P53 Activity
3. Discussion
4. Materials and Methods
4.1. Wet Lab Methods
4.2. Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
4.3. Databases Used for Contaminant Removal and Identification of Cellular Targets
4.4. Post LC-MS/MS Data Contaminant Removal
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.F.; Peto, J.; Meijer, C.J.L.M.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Crow, J.M. HPV: The global burden. Nat. Cell Biol. 2012, 488, S2–S3. [Google Scholar] [CrossRef] [PubMed]
- Garbuglia, A.R. Human Papillomavirus in Head and Neck Cancer. Cancers 2014, 6, 1705–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghittoni, R.; Accardi, R.; Hasan, U.; Gheit, T.; Sylla, B.; Tommasino, M. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes 2009, 40, 1–13. [Google Scholar] [CrossRef]
- Klingelhutz, A.J.; Roman, A. Cellular transformation by human papillomaviruses: Lessons learned by comparing high-and low-risk viruses. Virology 2012, 424, 77–98. [Google Scholar] [CrossRef] [Green Version]
- Pol, S.B.V.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [Green Version]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [Green Version]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus Type 16 Oncogenes Downregulate Expression of Interferon-Responsive Genes and Upregulate Proliferation-Associated and NF-κB-Responsive Genes in Cervical Keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef] [Green Version]
- Rincon-Orozco, B.; Halec, G.; Rosenberger, S.; Muschik, D.; Nindl, I.; Bachmann, A.; Ritter, T.M.; Dondog, B.; Ly, R.; Bosch, F.X.; et al. Epigenetic Silencing of Interferon- in Human Papillomavirus Type 16-Positive Cells. Cancer Res. 2009, 69, 8718–8725. [Google Scholar] [CrossRef] [Green Version]
- DeCarlo, C.A.; Severini, A.; Edler, L.; Escott, N.G.; Lambert, P.F.; Ulanova, M.; Zehbe, I. IFN-κ, a novel type I IFN, is undetectable in HPV-positive human cervical keratinocytes. Lab. Investig. 2010, 90, 1482–1491. [Google Scholar] [CrossRef] [Green Version]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Münch, P.; Iftner, T.; Stubenrauch, F. High-Risk Human Papillomaviruses Repress Constitutive Kappa Interferon Transcription via E6 To Prevent Pathogen Recognition Receptor and Antiviral-Gene Expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gruijl, T.D.; Bontkes, H.J.; Walboomers, J.M.; Coursaget, P.; Stukart, M.J.; Dupuy, C.; Kueter, E.; Verheijen, R.H.; Helmerhorst, T.J.; Duggan-Keen, M.F.; et al. Immune responses against human papillomavirus (HPV) type 16 virus-like particles in a cohort study of women with cervical intraepithelial neoplasia. I. Differential T-helper and IgG responses in relation to HPV infection and disease outcome. J. Gen. Virol. 1999, 80, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, K.; Leong, C.M.; Baxter, L.; Inglis, E.; Yun, K.; Bäckström, B.T.; Doorbar, J.; Hibma, M. Depletion of Langerhans Cells in Human Papillomavirus Type 16-Infected Skin Is Associated with E6-Mediated Down Regulation of E-Cadherin. J. Virol. 2003, 77, 8378–8385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingelhutz, A.J.; Foster, S.A.; McDougall, J.K. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nat. Cell Biol. 1996, 380, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus E6 and Myc proteins associatein vivoand bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216. [Google Scholar] [CrossRef] [Green Version]
- Gewin, L.; Myers, H.; Kiyono, T.; Galloway, D.A. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 2004, 18, 2269–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzenellenbogen, R.A.; Egelkrout, E.M.; Vliet-Gregg, P.; Gewin, L.C.; Gafken, P.R.; Galloway, D.A. NFX1-123 and Poly(A) Binding Proteins Synergistically Augment Activation of Telomerase in Human Papillomavirus Type 16 E6-Expressing Cells. J. Virol. 2007, 81, 3786–3796. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Zanier, K.; Sidi, A.O.M.O.; Boulade-Ladame, C.; Rybin, V.; Chappelle, A.; Atkinson, A.; Kieffer, B.; Travé, G. Solution Structure Analysis of the HPV16 E6 Oncoprotein Reveals a Self-Association Mechanism Required for E6-Mediated Degradation of p53. Structure 2012, 20, 604–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranjec, C.; Banks, L. A Systematic Analysis of Human Papillomavirus (HPV) E6 PDZ Substrates Identifies MAGI-1 as a Major Target of HPV Type 16 (HPV-16) and HPV-18 Whose Loss Accompanies Disruption of Tight Junctions. J. Virol. 2010, 85, 1757–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Laura, R.; Hepner, K.; Guccione, E.; Sawyers, C.; Lasky, L.; Banks, L. Oncogenic human papillomavirus E6 proteins target the MAGI-2 and MAGI-3 proteins for degradation. Oncogene 2002, 21, 5088–5096. [Google Scholar] [CrossRef]
- Nakagawa, S.; Huibregtse, J.M. Human Scribble (Vartul) Is Targeted for Ubiquitin-Mediated Degradation by the High-Risk Papillomavirus E6 Proteins and the E6AP Ubiquitin-Protein Ligase. Mol. Cell. Biol. 2000, 20, 8244–8253. [Google Scholar] [CrossRef]
- Kiyono, T.; Hiraiwa, A.; Fujita, M.; Hayashi, Y.; Akiyama, T.; Ishibashi, M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 11612–11616. [Google Scholar] [CrossRef] [Green Version]
- Gardiol, D.; Kühne, C.; Glaunsinger, B.; Lee, S.S.; Javier, R.; Banks, L. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene 1999, 18, 5487–5496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Terai, M.; Fu, L.; Herrero, R.; DeSalle, R.; Burk, R.D. Diversifying Selection in Human Papillomavirus Type 16 Lineages Based on Complete Genome Analyses. J. Virol. 2005, 79, 7014–7023. [Google Scholar] [CrossRef] [Green Version]
- Burk, R.D.; Harari, A.; Chen, Z. Human papillomavirus genome variants. Virology 2013, 445, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Zehbe, I.; Wilander, E.; Delius, H.; Tommasino, M. Human papillomavirus 16 E6 variants are more prevalent in invasive cervical carcinoma than the prototype. Cancer Res. 1998, 58, 58. [Google Scholar]
- Zehbe, I.; Richard, C.; Decarlo, C.A.; Shai, A.; Lambert, P.F.; Lichtig, H.; Tommasino, M.; Sherman, L. Human papillomavirus 16 E6 variants differ in their dysregulation of human keratinocyte differentiation and apoptosis. Virology 2009, 383, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacapala-Gómez, A.E.; Del Moral-Hernández, O.; Villegas-Sepúlveda, N.; Hidalgo-Miranda, A.; Romero-Cordoba, S.L.; Beltrán-Anaya, F.O.; Leyva-Vázquez, M.A.; Alarcón-Romero, L.D.C.; Illades-Aguiar, B. Changes in global gene expression profiles induced by HPV 16 E6 oncoprotein variants in cervical carcinoma C33-A cells. Virology 2016, 488, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Hochmann, J.; Ferreira, S.; Sobrinho, J.S.; Sichero, L. Cooperative role of c-MYC and E6/E7 from two molecular variants of human papillomavirus type 16 upon proliferation and in vitro transformation of primary human keratinocytes/Papel cooperativo de c-MYC e E6/E7 de duas variantes moleculares do Papilomavirus Humano tipo 16 na proliferacao e transformacao in vitro de queratinocitos humanos primarios. Rev. Med. 2019, 98, 52–59. [Google Scholar]
- Xi, L.F.; Koutsky, L.A.; Galloway, D.A.; Kiviat, N.B.; Kuypers, J.; Hughes, J.P.; Wheeler, C.M.; Holmes, K.K. Genomic Variation of Human Papillomavirus Type 16 and Risk for High Grade Cervical Intraepithelial Neoplasia. J. Natl. Cancer Inst. 1997, 89, 796–802. [Google Scholar] [CrossRef] [Green Version]
- Villa, L.L.; Sichero, L.; Rahal, P.; Caballero, O.; Ferenczy, A.; Rohan, T.; Franco, E.L. Molecular variants of human papillomavirus types 16 and 18 preferentially associated with cervical neoplasia. J. Gen. Virol. 2000, 81, 2959–2968. [Google Scholar] [CrossRef]
- Berumen, J.; Ordonez, R.M.; Lazcano, E.; Salmeron, J.; Galvan, S.C.; Estrada, R.A.; Yunes, E.; Garcia-Carranca, A.; Gonzalez-Lira, G.; La Campa, A.M.-D. Asian-American Variants of Human Papillomavirus 16 and Risk for Cervical Cancer: A Case-Control Study. J. Natl. Cancer Inst. 2001, 93, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.F.; Koutsky, L.A.; Hildesheim, A.; Galloway, D.A.; Wheeler, C.M.; Winer, R.L.; Ho, J.; Kiviat, N.B. Risk for High-Grade Cervical Intraepithelial Neoplasia Associated with Variants of Human Papillomavirus Types 16 and 18. Cancer Epidemiol. Biomark. Prev. 2007, 16, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togtema, M.; Jackson, R.; Richard, C.; Niccoli, S.; Zehbe, I. The human papillomavirus 16 European-T350G E6 variant can immortalize but not transform keratinocytes in the absence of E7. Virology 2015, 485, 274–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yang, B.; Zhang, A.; Zhou, A.; Yuan, J.; Wang, Y.; Sun, L.; Cao, H.; Wang, J.; Zheng, W. Association between human papillomavirus type 16 E6 and E7 variants with subsequent persistent infection and recurrence of cervical high-grade squamous intraepithelial lesion after conization. J. Med. Virol. 2016, 88, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, S.; Abraham, S.; Richard, C.; Zehbe, I. The Asian-American E6 Variant Protein of Human Papillomavirus 16 Alone Is Sufficient To Promote Immortalization, Transformation, and Migration of Primary Human Foreskin Keratinocytes. J. Virol. 2012, 86, 12384–12396. [Google Scholar] [CrossRef] [Green Version]
- Richard, C.; Lanner, C.; Naryzhny, S.N.; Sherman, L.; Lee, H.; Lambert, P.F.; Zehbe, I. The immortalizing and transforming ability of two common human papillomavirus 16 E6 variants with different prevalences in cervical cancer. Oncogene 2010, 29, 3435–3445. [Google Scholar] [CrossRef] [Green Version]
- Jackson, R.; Togtema, M.; Lambert, P.F.; Zehbe, I. Tumourigenesis Driven by the Human Papillomavirus Type 16 Asian-American E6 Variant in a Three-Dimensional Keratinocyte Model. PLoS ONE 2014, 9, e101540. [Google Scholar] [CrossRef]
- Cuninghame, S.; Jackson, R.; Lees, S.J.; Zehbe, I. Two common variants of human papillomavirus type 16 E6 differentially deregulate sugar metabolism and hypoxia signalling in permissive human keratinocytes. J. Gen. Virol. 2017, 98, 2310–2319. [Google Scholar] [CrossRef]
- Chakrabarti, O.; Veeraraghavalu, K.; Liu, Y.; Androphy, E.J.; Margaret, A.; Krishna, S.; Tergaonkar, V.; Stanley, M.A. Human Papillomavirus Type 16 E6 Amino Acid 83 Variants Enhance E6-Mediated MAPK Signaling and Differentially Regulate Tumorigenesis by Notch Signaling and Oncogenic Ras Human Papillomavirus Type 16 E6 Amino Acid 83 Variants Enhance E6-Mediated MAPK Signal. J. Virol. 2004, 78, 5934–5945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sichero, L.; Sobrinho, J.S.; Villa, L.L. Oncogenic potential diverge among human papillomavirus type 16 natural variants. Virology 2012, 432, 127–132. [Google Scholar] [CrossRef]
- Hochmann, J.; Sobrinho, J.S.; Villa, L.L.; Sichero, L. The Asian-American variant of human papillomavirus type 16 exhibits higher activation of MAPK and PI3K/AKT signaling pathways, transformation, migration and invasion of primary human keratinocytes. Virology 2016, 492, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Lichtig, H.; Algrisi, M.; Botzer, L.E.; Abadi, T.; Verbitzky, Y.; Jackman, A.; Tommasino, M.; Zehbe, I.; Sherman, L. HPV16 E6 natural variants exhibit different activities in functional assays relevant to the carcinogenic potential of E6. Virology 2006, 350, 216–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifford, G.M.; Tenet, V.; Georges, D.; Alemany, L.; Pavón, M.A.; Chen, Z.; Yeager, M.; Cullen, M.; Boland, J.F.; Bass, S.; et al. Human papillomavirus 16 sub-lineage dispersal and cervical cancer risk worldwide: Whole viral genome sequences from 7116 HPV16-positive women. Papillomavirus Res. 2019, 7, 67–74. [Google Scholar] [CrossRef]
- Togtema, M.; Hussack, G.; Dayer, G.; Teghtmeyer, M.R.; Raphael, S.; Tanha, J.; Zehbe, I. Single-Domain Antibodies Represent Novel Alternatives to Monoclonal Antibodies as Targeting Agents against the Human Papillomavirus 16 E6 Protein. Int. J. Mol. Sci. 2019, 20, 2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.A.; Kramer, R.E.; Tan, M.J.A.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive Analysis of Host Cellular Interactions with Human Papillomavirus E6 Proteins Identifies New E6 Binding Partners and Reflects Viral Diversity. J. Virol. 2012, 86, 13174–13186. [Google Scholar] [CrossRef] [Green Version]
- Min, J.-N.; Tian, Y.; Xiao, Y.; Wu, L.; Li, L.; Chang, S. The mINO80 chromatin remodeling complex is required for efficient telomere replication and maintenance of genome stability. Cell Res. 2013, 23, 1396–1413. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, J.; Chen, A.; Lyu, J.; Ai, G.; Zeng, Q.; Sun, Y.; Chen, C.; Wang, J.; Qiu, J.; et al. Ino80 promotes cervical cancer tumorigenesis by activating Nanog expression. Oncotarget 2016, 7, 72250–72262. [Google Scholar] [CrossRef] [Green Version]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M.; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nat. Cell Biol. 2012, 487, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Gross-Mesilaty, S.; Reinstein, E.; Bercovich, B.; Tobias, K.E.; Schwartz, A.L.; Kahana, C.; Ciechanover, A. Basal and human papillomavirus E6 oncoprotein-induced degradation of Myc proteins by the ubiquitin pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 8058–8063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalkat, M.; Resetca, D.; Lourenco, C.; Chan, P.-K.; Wei, Y.; Shiah, Y.-J.; Vitkin, N.; Tong, Y.; Sunnerhagen, M.; Done, S.J.; et al. MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol. Cell 2018, 72, 836–848.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deak, M.; Clifton, A.D.; Lucocq, J.M.; Alessi, D.R. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activationofCREB. EMBO J. 1998, 17, 4426–4441. [Google Scholar] [CrossRef] [Green Version]
- Wiggin, G.R.; Soloaga, A.; Foster, J.M.; Murray-Tait, V.; Cohen, P.; Arthur, J.S.C. MSK1 and MSK2 Are Required for the Mitogen-and Stress-Induced Phosphorylation of CREB and ATF1 in Fibroblasts. Mol. Cell. Biol. 2002, 22, 2871–2881. [Google Scholar] [CrossRef] [Green Version]
- Ananieva, O.; Darragh, J.; Johansen, C.; Carr, J.M.; McIlrath, J.; Park, J.M.; Wingate, A.D.; Monk, C.E.; Toth, R.; Santos, S.G.; et al. The kinases MSK1 and MSK2 act as negative regulators of Toll-like receptor signaling. Nat. Immunol. 2008, 9, 1028–1036. [Google Scholar] [CrossRef] [Green Version]
- Soloaga, A.; Thomson, S.; Wiggin, G.R.; Rampersaud, N.; Dyson, M.H.; Hazzalin, C.A.; Mahadevan, L.C.; Arthur, J.S.C. MSK2 and MSK1 mediate the mitogen-and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003, 22, 2788–2797. [Google Scholar] [CrossRef] [Green Version]
- Längst, G.; Manelyte, L. Chromatin Remodelers: From Function to Dysfunction. Genes 2015, 6, 299–324. [Google Scholar] [CrossRef]
- Llanos, S.; Cuadrado, A.; Serrano, M. MSK2 Inhibits p53 Activity in the Absence of Stress. Sci. Signal. 2009, 2, ra57. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Li, H.; Wang, H.; Zhang, F.; Cao, H.; Xu, S. MSK2 promotes proliferation and tumor formation in squamous cervical cancer via PAX8/RB-E2F1/cyclin A2 axis. J. Cell. Biochem. 2019, 120, 11432–11440. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, L.; Berghe, W.V.; Beck, I.M.; De Bosscher, K.; Haegeman, G. The versatile role of MSKs in transcriptional regulation. Trends Biochem. Sci. 2009, 34, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Hashimura, M.; Mastumoto, T.; Tazo, Y.; Inoue, H.; Kuwata, T.; Saegusa, M. Transcriptional upregulation of HIF-1α by NF-κB/p65 and its associations with β-catenin/p300 complexes in endometrial carcinoma cells. Lab. Investig. 2013, 93, 1184–1193. [Google Scholar] [CrossRef] [Green Version]
- Lauttia, S.; Sihto, H.; Kavola, H.; Koljonen, V.; Bohling, T.; Joensuu, H. Prokineticins and Merkel cell polyomavirus infection in Merkel cell carcinoma. Br. J. Cancer 2014, 110, 1446–1455. [Google Scholar] [CrossRef]
- Zhou, Q.-Y. The prokineticins: A novel pair of regulatory peptides. Mol. Interv. 2006, 6, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurebayashi, H.; Goi, T.; Shimada, M.; Tagai, N.; Naruse, T.; Nakazawa, T.; Kimura, Y.; Hirono, Y.; Yamaguchi, A. Prokineticin 2 (PROK2) is an important factor for angiogenesis in colorectal cancer. Oncotarget 2015, 6, 26242–26251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeCouter, J.; Kowalski, J.; Foster, J.; Hass, P.; Zhang, Z.; Dillard-Telm, L.; Frantz, G.; Rangell, L.; DeGuzman, L.; Keller, G.-A.; et al. Identification of an angiogenic mitogen selective for endocrine gland endothelium. Nat. Cell Biol. 2001, 412, 877–884. [Google Scholar] [CrossRef]
- LeCouter, J.; Lin, R.; Tejada, M.; Frantz, G.; Peale, F.; Hillan, K.J.; Ferrara, N. The endocrine-gland-derived VEGF homologue Bv8 promotes angiogenesis in the testis: Localization of Bv8 receptors to endothelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 2685–2690. [Google Scholar] [CrossRef] [Green Version]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef]
- Pett, M.R.; Herdman, M.T.; Palmer, R.D.; Yeo, G.S.; Shivji, M.K.; Stanley, M.A.; Coleman, N. Selection of cervical keratinocytes containing integrated HPV16 associates with episome loss and an endogenous antiviral response. Proc. Natl. Acad. Sci. USA 2006, 103, 3822–3827. [Google Scholar] [CrossRef] [Green Version]
- Rolland, T.; Taşan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A Proteome-Scale Map of the Human Interactome Network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Rada, M.; Vasileva, E.; Lezina, L.; Marouco, D.; Antonov, A.V.; Macip, S.; Melino, G.; Barlev, N.A. Human EHMT2/G9a activates p53 through methylation-independent mechanism. Oncogene 2016, 36, 922–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, D.; Anbarasu, K.; Rajabather, S.; Priya, R.S.; Desai, P.; Mahalingam, S. Nucleolar GTP-binding Protein-1 (NGP-1) Promotes G1to S Phase Transition by Activating Cyclin-dependent Kinase Inhibitor p21Cip1/Waf1. J. Biol. Chem. 2015, 290, 21536–21552. [Google Scholar] [CrossRef] [Green Version]
- Essers, P.B.; Pereboom, T.C.; Goos, Y.J.; Paridaen, J.T.; MacInnes, A.W. A comparative study of nucleostemin family members in zebrafish reveals specific roles in ribosome biogenesis. Dev. Biol. 2014, 385, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racevskis, J.; Dill, A.; Stockert, R.; Fineberg, S.A. Cloning of a Novel Nucleolar Guanosine 5’-Triphosphate Binding Protein Autoantigen from a Breast Tumor. Cell Growth Differ. 1996, 7, 271–280. [Google Scholar]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef] [Green Version]
- Cloutier, P.; Poitras, C.; Durand, M.; Hekmat, O.; Fiola-Masson, É.; Bouchard, A.; Faubert, D.; Chabot, B.; Coulombe, B. R2TP/Prefoldin-like component RUVBL1/RUVBL2 directly interacts with ZNHIT2 to regulate assembly of U5 small nuclear ribonucleoprotein. Nat. Commun. 2017, 8, 15615. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nat. Cell Biol. 2017, 545, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Zanier, K.; Charbonnier, S.; Sidi, A.O.M.O.; McEwen, A.G.; Ferrario, M.G.; Poussin-Courmontagne, P.; Cura, V.; Brimer, N.; Babah, K.O.; Ansari, T.; et al. Structural Basis for Hijacking of Cellular LxxLL Motifs by Papillomavirus E6 Oncoproteins. Science 2013, 339, 694–698. [Google Scholar] [CrossRef] [Green Version]
- Larrieu, D.; Brunet, M.; Vargas, C.; Hanoun, N.; Ligat, L.; Dagnon, L.; Lulka, H.; Pommier, R.M.; Selves, J.; Jády, B.E.; et al. The E3 ubiquitin ligase TRIP12 participates in cell cycle progression and chromosome stability. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. The TRIP from ULF to ARF. Cancer Cell 2010, 17, 317–318. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Shan, J.; Zhu, W.-G.; Qin, J.; Gu, W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nat. Cell Biol. 2010, 464, 624–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chio, I.I.C.; Sasaki, M.; Ghazarian, D.; Moreno, J.; Done, S.J.; Ueda, T.; Inoue, S.; Chang, Y.-L.; Chen, N.-J.; Mak, T.W. TRADD contributes to tumour suppression by regulating ULF-dependent p19Arf ubiquitylation. Nat. Cell Biol. 2012, 14, 625–633. [Google Scholar] [CrossRef]
- Keppler, B.K.; Archer, T.K. Ubiquitin-dependent and Ubiquitin-independent Control of Subunit Stoichiometry in the SWI/SNF Complex. J. Biol. Chem. 2010, 285, 35665–35674. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Archer, T.K. Regulating SWI/SNF Subunit Levels via Protein-Protein Interactions and Proteasomal Degradation: BAF155 and BAF170 Limit Expression of BAF57. Mol. Cell. Biol. 2005, 25, 9016–9027. [Google Scholar] [CrossRef] [Green Version]
- Cristofaro, M.F.D.; Betz, B.L.; Rorie, C.J.; Reisman, D.N.; Wang, W.; Weissman, B.E. Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J. Cell. Physiol. 2001, 186, 136–145. [Google Scholar] [CrossRef]
- Lee, K.; Lee, A.-Y.; Kwon, Y.K.; Kwon, H. Suppression of HPV E6 and E7 expression by BAF53 depletion in cervical cancer cells. Biochem. Biophys. Res. Commun. 2011, 412, 328–333. [Google Scholar] [CrossRef]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Göttlinger, H.G. AIP1/ALIX Is a Binding Partner for HIV-1 p6 and EIAV p9 Functioning in Virus Budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- Hu, B.; Jiang, D.; Chen, Y.; Wei, L.; Zhang, S.; Zhao, F.; Ni, R.-Z.; Lu, C.; Wan, C. High CHMP4B expression is associated with accelerated cell proliferation and resistance to doxorubicin in hepatocellular carcinoma. Tumor Biol. 2015, 36, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Gollin, S.M. Cytogenetic alterations and their molecular genetic correlates in head and neck squamous cell carcinoma: A next generation window to the biology of disease. Genes Chromosom. Cancer 2014, 53, 972–990. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, I.; Fujita, K.; Jenkins, L.M.M.; Hiyoshi, Y.; Mondal, A.M.; Vojtesek, B.; Lane, D.P.; Appella, E.; Harris, C.C. Autophagic degradation of the inhibitory p53 isoform Δ133p53α as a regulatory mechanism for p53-mediated senescence. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubel, P.; Urban, C.; Bergant, V.; Schneider, W.M.; Knauer, B.; Stukalov, A.; Scaturro, P.; Mann, A.; Brunotte, L.; Hoffmann, H.H.; et al. A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat. Immunol. 2019, 20, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Saito, H.; Takaoka, M.; Miki, Y.; Nakanishi, A. BRCA2 mediates centrosome cohesion via an interaction with cytoplasmic dynein. Cell Cycle 2016, 15, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Neilson, L.J.; Zhong, H.; Murray, P.S.; Zanivan, S.; Zaidel-Bar, R.; Rao, M.V. E-cadherin interactome complexity and robustness resolved by quantitative proteomics. Sci. Signal. 2014, 7, rs7. [Google Scholar] [CrossRef] [Green Version]
- Popko-Scibor, A.E.; Lindberg, M.J.; Hansson, M.L.; Holmlund, T.; Wallberg, A.E. Ubiquitination of Notch1 is regulated by MAML1-mediated p300 acetylation of Notch1. Biochem. Biophys. Res. Commun. 2011, 416, 300–306. [Google Scholar] [CrossRef]
- Loganathan, S.K.; Schleicher, K.; Malik, A.; Quevedo, R.; Langille, E.; Teng, K.; Oh, R.H.; Rathod, B.; Tsai, R.; Samavarchi-Tehrani, P.; et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 2020, 367, 1264–1269. [Google Scholar] [CrossRef]
- Verheyen, E.M.; Gottardi, C.J. Regulation of Wnt/β-catenin signaling by protein kinases. Dev. Dyn. 2009, 239, 34–44. [Google Scholar] [CrossRef] [Green Version]
- Mazzei, F.; Viel, A.; Bignami, M. Role of MUTYH in human cancer. Mutat. Res. Mol. Mech. Mutagen. 2013, 33–43. [Google Scholar] [CrossRef]
- Jackson, R.; Rosa, B.A.; Lameiras, S.; Cuninghame, S.; Bernard, J.; Floriano, W.B.; Lambert, P.F.; Nicolas, A.; Zehbe, I. Erratum to: Functional variants of human papillomavirus type 16 demonstrate host genome integration and transcriptional alterations corresponding to their unique cancer epidemiology. BMC Genom. 2016, 17, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoder, K.E.; Espeseth, A.; Wang, X.-H.; Fang, Q.; Russo, M.T.; Lloyd, R.S.; Hazuda, D.; Sobol, R.W.; Fishel, R. The Base Excision Repair Pathway Is Required for Efficient Lentivirus Integration. PLoS ONE 2011, 6, e17862. [Google Scholar] [CrossRef] [Green Version]
- Zapatka, M.; Pathogens, P.; Borozan, I.; Brewer, D.S.; Iskar, M.; Grundhoff, A.; Alawi, M.; Desai, N.; Sültmann, H.; Moch, H.; et al. The landscape of viral associations in human cancers. Nat. Genet. 2020, 52, 320–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, J.M.; Czaja, W. Facilitation of base excision repair by chromatin remodeling. DNA Repair 2015, 36, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A Employs CDK8-Mediator to Stimulate RNAPII Elongation in Response to Hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heir, P.; Sufan, R.I.; Greer, S.N.; Poon, B.P.; Lee, J.E.; Ohh, M. DCNL1 Functions as a Substrate Sensor and Activator of Cullin 2-RING Ligase. Mol. Cell. Biol. 2013, 33, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Perez-Perri, J.I.; Dengler, V.L.; Audetat, K.A.; Pandey, A.; Bonner, E.A.; Urh, M.; Mendez, J.; Daniels, D.L.; Wappner, P.; Galbraith, M.D.; et al. The TIP60 Complex Is a Conserved Coactivator of HIF1A. Cell Rep. 2016, 16, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Knaup, K.X.; Jozefowski, K.; Schmidt, R.; Bernhardt, W.M.; Weidemann, A.; Juergensen, J.S.; Warnecke, C.; Eckardt, K.-U.; Wiesener, M.S. Mutual Regulation of Hypoxia-Inducible Factor and Mammalian Target of Rapamycin as a Function of Oxygen Availability. Mol. Cancer Res. 2009, 7, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Onnis, B.; Fer, N.; Rapisarda, A.; Perez, V.S.; Melillo, G. Autocrine production of IL-11 mediates tumorigenicity in hypoxic cancer cells. J. Clin. Investig. 2013, 123, 1615–1629. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Gullberg, U.; Olsson, I.; Ajore, R. Myeloid Translocation Gene-16 Co-Repressor Promotes Degradation of Hypoxia-Inducible Factor 1. PLoS ONE 2015, 10, e0123725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minchenko, A.; Leshchinsky, I.; Opentanova, I.; Sang, N.; Srinivas, V.; Armstead, V.; Caro, J. Hypoxia-inducible Factor-1-mediated Expression of the 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) Gene. J. Biol. Chem. 2001, 277, 6183–6187. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Gui, L.; Zhao, X.; Zhu, L.; Li, Q. FOXO1 is a tumor suppressor in cervical cancer. Genet. Mol. Res. 2015, 14, 6605–6616. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M.T. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Gu, Y.; Mahoney, W.; Lee, S.-H.; Singh, K.K.; Lu, A.-L. Human Homolog of the MutY Repair Protein (hMYH) Physically Interacts with Proteins Involved in Long Patch DNA Base Excision Repair. J. Biol. Chem. 2000, 276, 5547–5555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, L.; Gilkes, D.M.; Hu, H.; Luo, W.; Bullen, J.W.; Liang, H.; Semenza, G.L. HIF-1α and TAZ serve as reciprocal co-activators in human breast cancer cells. Oncotarget 2015, 6, 11768–11778. [Google Scholar] [CrossRef] [Green Version]
- Xiang, L.; Gilkes, D.M.; Hu, H.; Takano, N.; Luo, W.; Lu, H.; Bullen, J.W.; Samanta, D.; Liang, H.; Semenza, G.L. Hypoxia-inducible factor 1 mediates TAZ expression and nuclear localization to induce the breast cancer stem cell phenotype. Oncotarget 2014, 5, 12509–12527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia Requires Notch Signaling to Maintain the Undifferentiated Cell State. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Grace, M.; Munger, K. Proteomic analysis of the gamma human papillomavirus type 197 E6 and E7 associated cellular proteins. Virology 2017, 500, 71–81. [Google Scholar] [CrossRef]
- The UniProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [CrossRef] [Green Version]
- Oughtred, R.; Stark, C.; Breitkreutz, B.-J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2018, 47, D529–D541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2019, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner-Martinez, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dayer, G.; Masoom, M.L.; Togtema, M.; Zehbe, I. Virus–Host Protein–Protein Interactions between Human Papillomavirus 16 E6 A1 and D2/D3 Sub-Lineages: Variances and Similarities. Int. J. Mol. Sci. 2020, 21, 7980. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217980
Dayer G, Masoom ML, Togtema M, Zehbe I. Virus–Host Protein–Protein Interactions between Human Papillomavirus 16 E6 A1 and D2/D3 Sub-Lineages: Variances and Similarities. International Journal of Molecular Sciences. 2020; 21(21):7980. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217980
Chicago/Turabian StyleDayer, Guillem, Mehran L. Masoom, Melissa Togtema, and Ingeborg Zehbe. 2020. "Virus–Host Protein–Protein Interactions between Human Papillomavirus 16 E6 A1 and D2/D3 Sub-Lineages: Variances and Similarities" International Journal of Molecular Sciences 21, no. 21: 7980. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217980