Downregulation of miR-140-3p Contributes to Upregulation of CD38 Protein in Bronchial Smooth Muscle Cells


Abstract
:1. Introduction
2. Results
2.1. Differentially Expressed miRNAs in BSMs of the Antigen-Induced AHR Mice
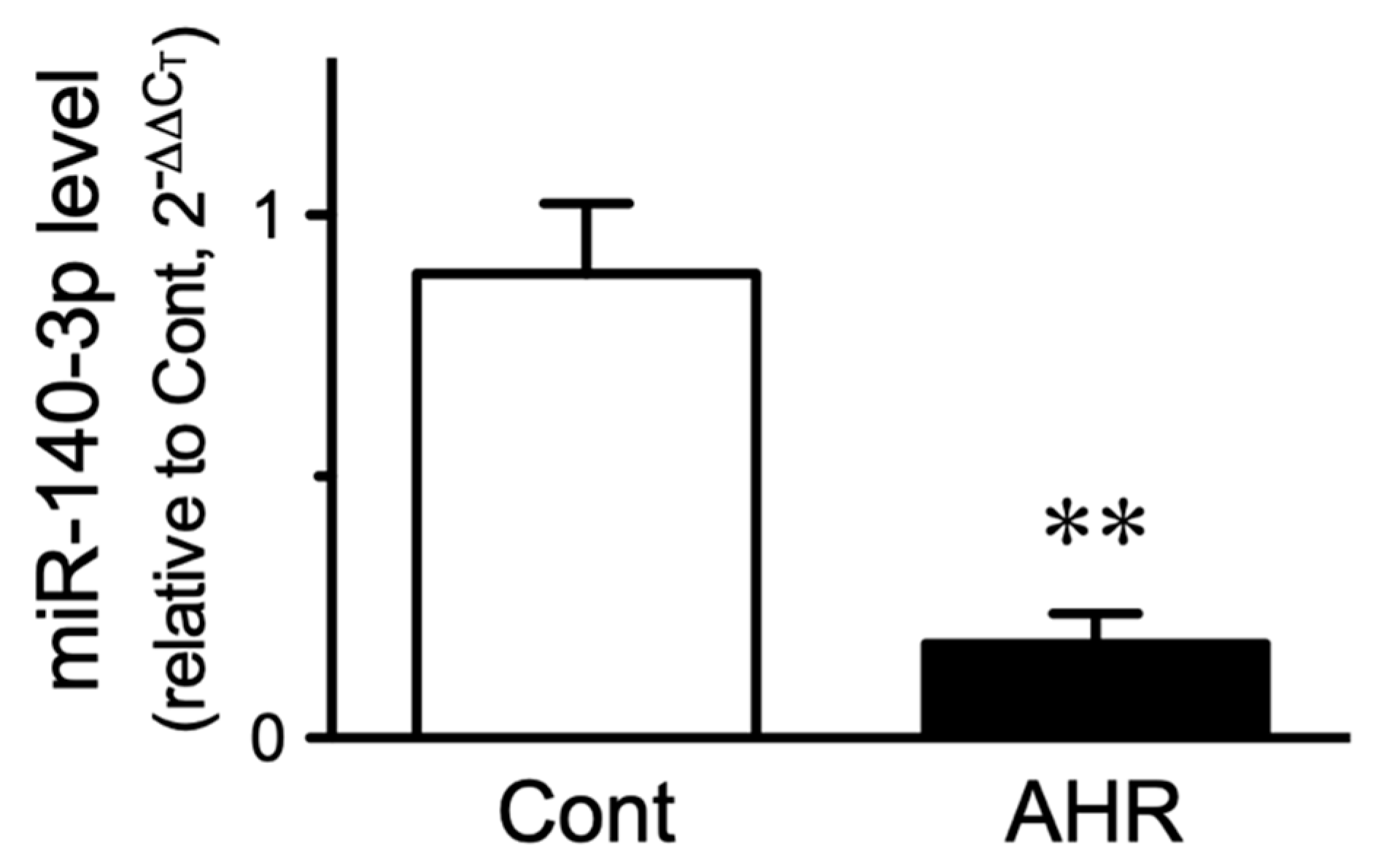
2.2. Change in miR-140-3p Level in BSMs of the Antigen-Induced AHR Mice
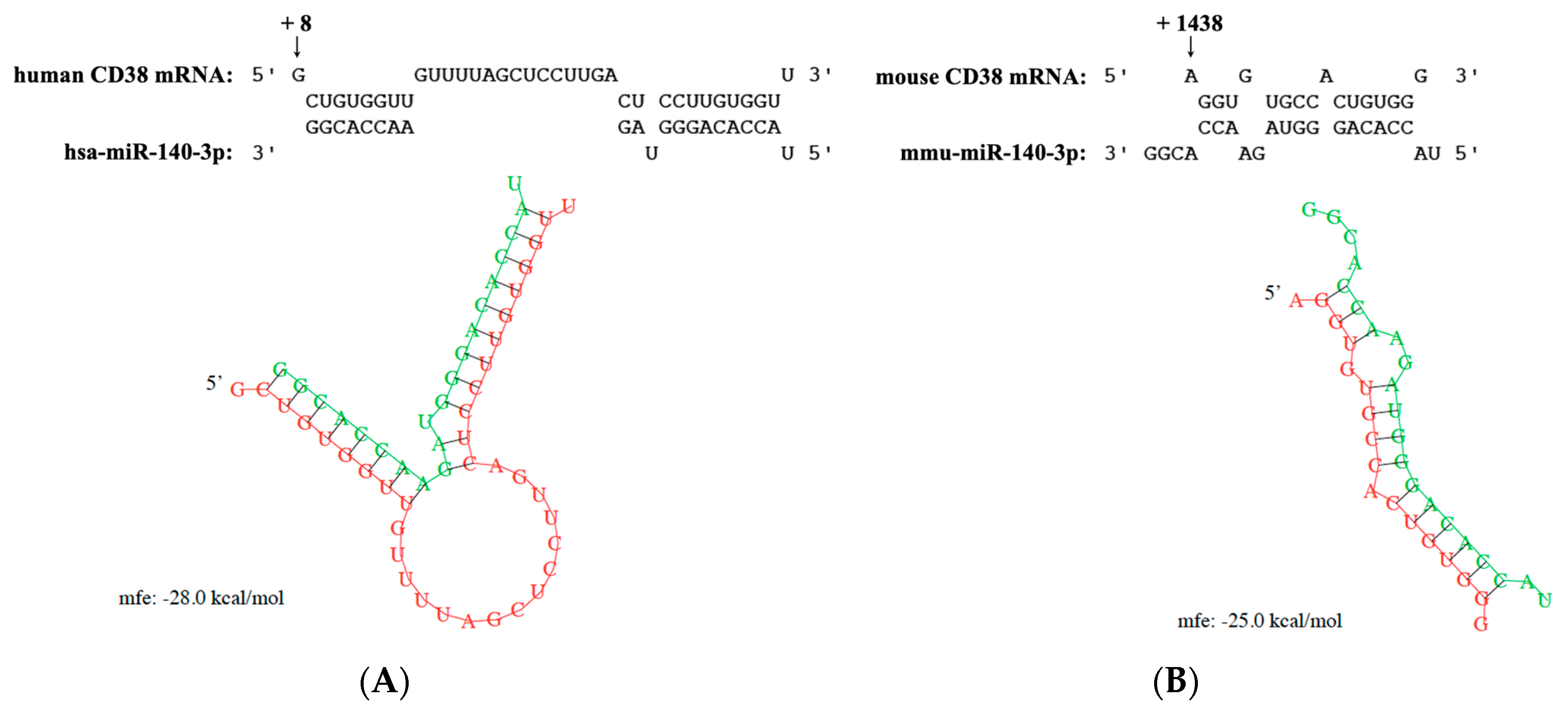
2.3. CD38 mRNA as a Predicted Target of miR-140-3p
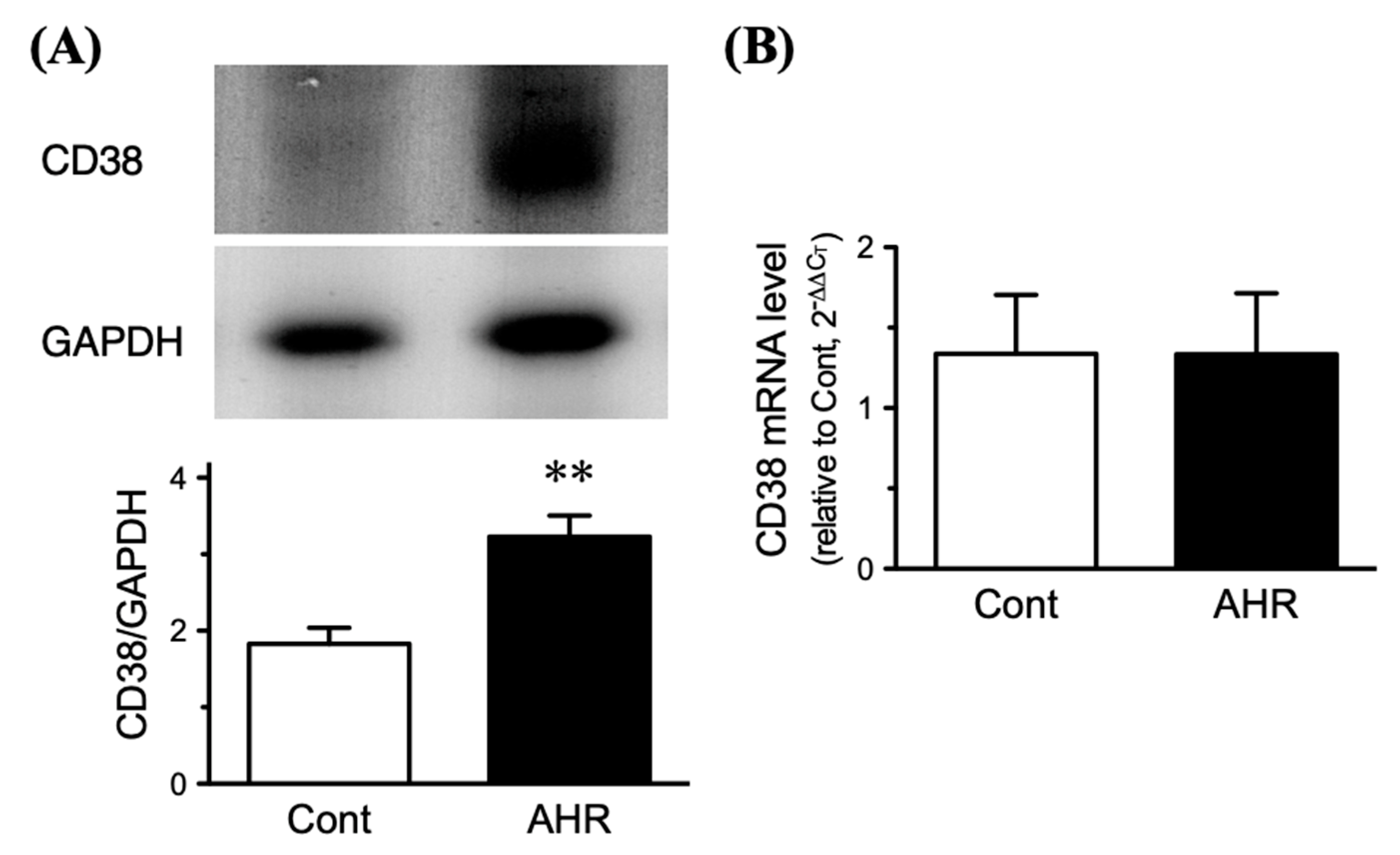
2.4. Upregulation of CD38 Protein in BSMs of the Antigen-Induced AHR Mice
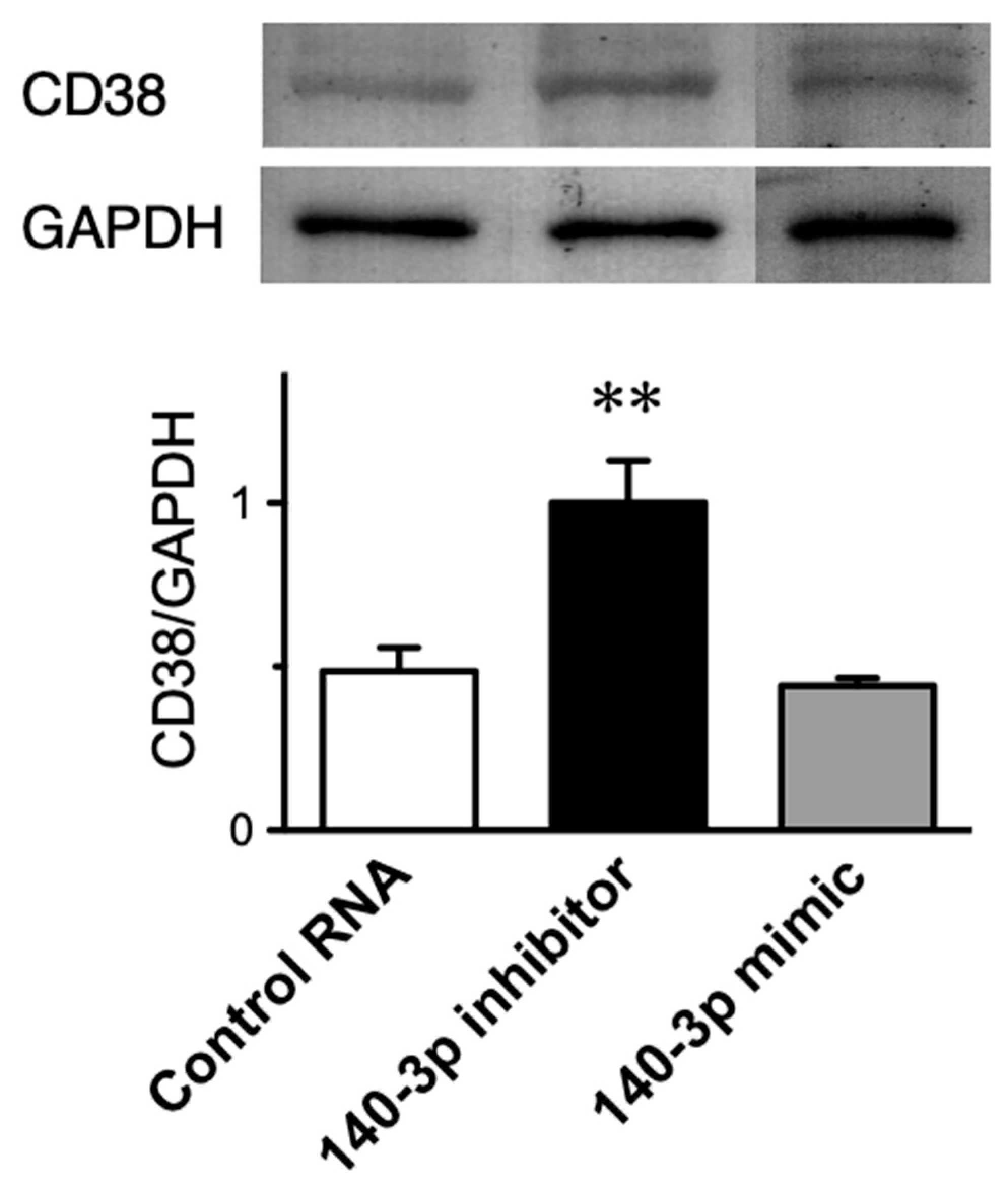
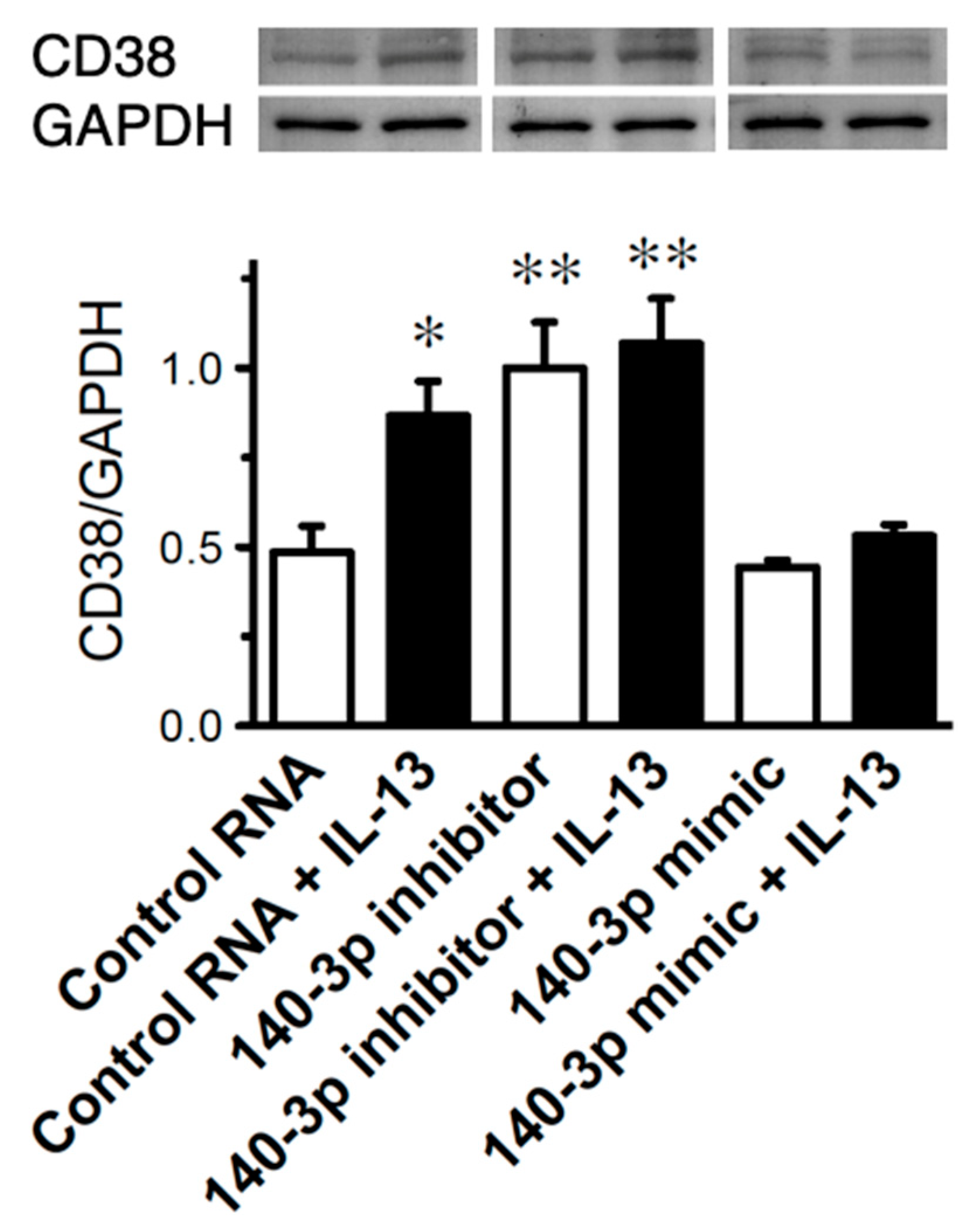
2.5. Effect of Inhibition of miR-140-3p on CD38 Protein Expression in Cultured Human BSM Cells (hBSMCs)
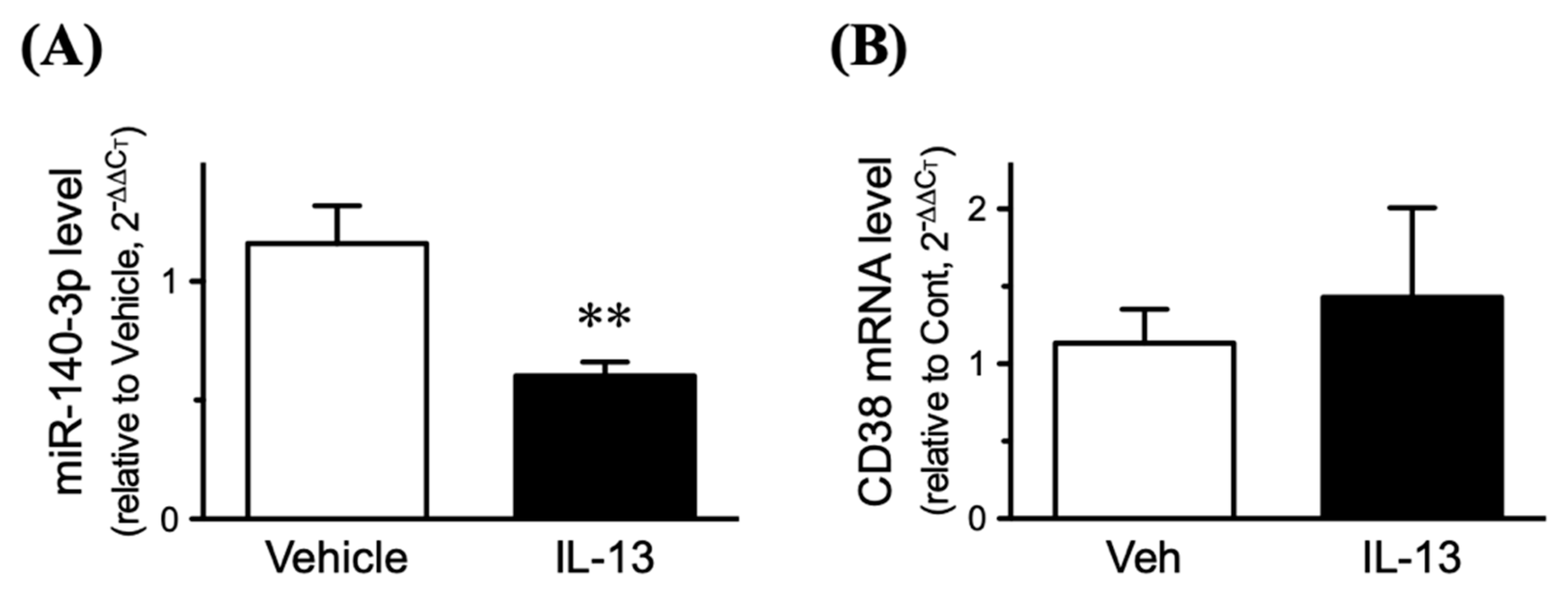
2.6. Effect of Inhibition of miR-140-3p on CD38 Protein Exp 2.6. Effects of IL-13 on Expression Levels of CD38 and miR-140-3p in Cultured Human BSM Cells (hBSMCs)
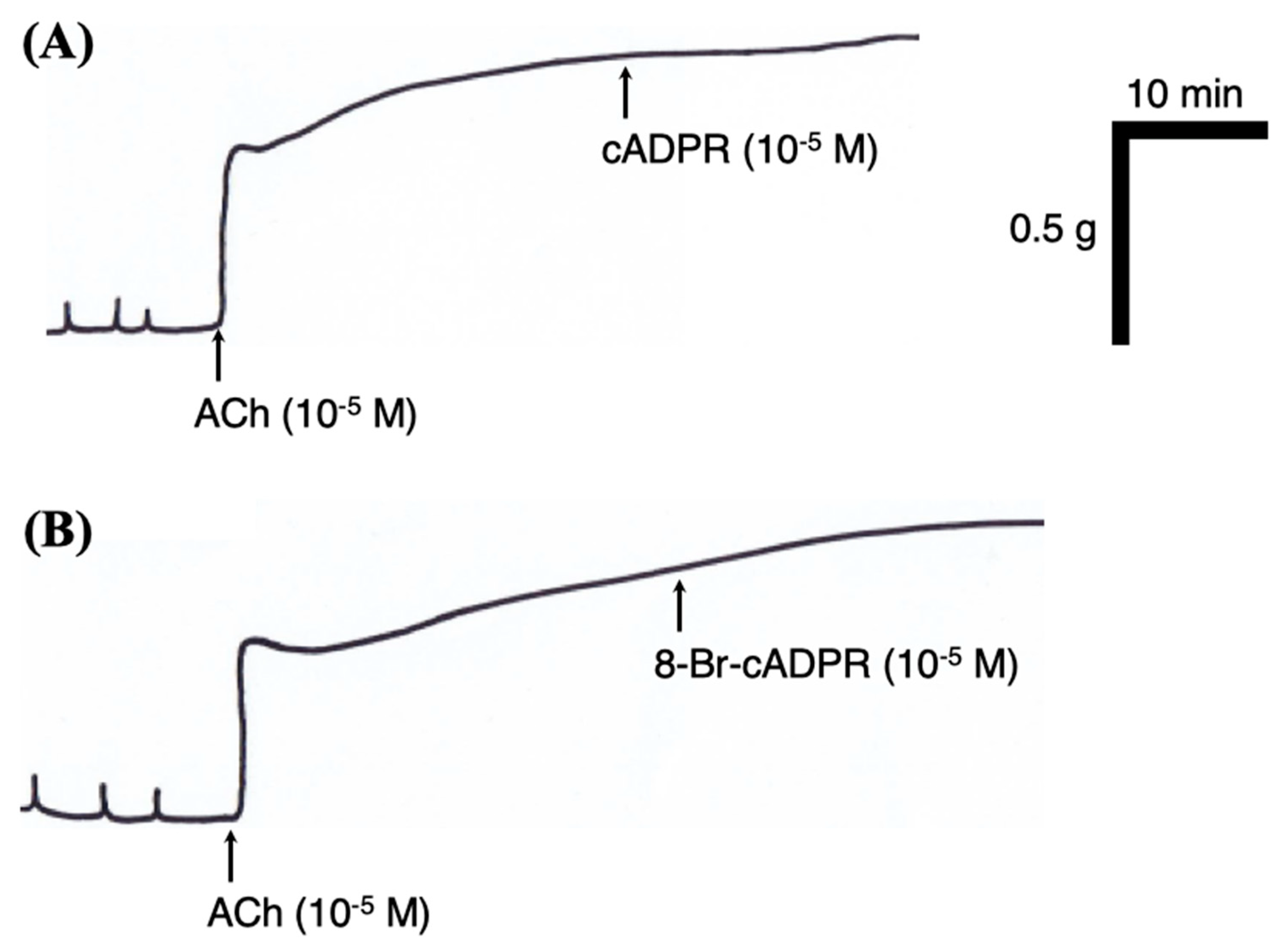
2.7. Effects of Cyclic ADP Ribose (cADPR) and Its Antagonist 8-Bromo-cADPR (8-Br-cADPR) on BSM Function
3. Discussion
4. Materials and Methods
4.1. Mouse Model of Allergic Bronchial Asthma
4.2. miRNA Microarray Analyses
4.3. Cell Culture and Sample Collection
4.4. Transfection of miR-140-3p Inhibitor and Mimic
4.5. Quantitative RT-PCR Analyses
4.6. Western Blot Analyses
4.7. Data and Statistical Analyses
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACh | acetylcholine |
| AHR | airway hyperresponsiveness |
| ANOVA | analysis of variance |
| ASM | airway smooth muscle |
| BSM | bronchial smooth muscle |
| cADPR | cyclic ADP-ribose |
| COPD | chronic obstructive pulmonary disease |
| CT | threshold cycle |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| hBSMC | human bronchial smooth muscle cell |
| hEGF | human epidermal growth factor |
| hFGF-b | human fibroblast growth factor-basic |
| IL-13 | interleukin-13 |
| LNA | locked nucleic acid |
| miRNA | microRNA |
| mRNA | messenger RNA |
| NAD | nicotinamide adenine dinucleotide |
| OA | ovalbumin |
| pre-miRNA | precursor miRNA |
| PCR | polymerase chain reaction |
| pri-miRNA | primary miRNA |
| PVDF | polyvinylidene difluoride |
| RT | reverse transcription |
| RT-qPCR | quantitative real-time RT-PCR |
| snRNA | small nuclear RNA |
| UTR | untranslated region |
References
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.E.; Moschos, S.A.; Perry, M.M.; Barnes, P.J.; Lindsay, M.A. Maternally imprinted microRNAs are differentially expressed during mouse and human lung development. Dev. Dyn. 2007, 236, 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schembri, F.; Sridhar, S.; Perdomo, C.; Gustafson, A.M.; Zhang, X.; Ergun, A.; Lu, J.; Liu, G.; Zhang, X.; Bowers, J.; et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 2319–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pottelberge, G.R.; Mestdagh, P.; Bracke, K.R.; Thas, O.; Durme, Y.M.; Joos, G.F.; Vandesompele, J.; Brusselle, G.G. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 898–906. [Google Scholar] [CrossRef]
- Faiz, A.; Steiling, K.; Roffel, M.P.; Postma, D.S.; Spira, A.; Lenburg, M.E.; Borggrewe, M.; Eijgenraam, T.R.; Jonker, M.R.; Koppelman, G.H.; et al. Effect of long-term corticosteroid treatment on microRNA and gene-expression profiles in COPD. Eur. Respir. J. 2019, 53, 1801202. [Google Scholar] [CrossRef]
- Mei, D.; Tan, W.S.D.; Tay, Y.; Mukhopadhyay, A.; Wong, W.S.F. Therapeutic RNA Strategies for Chronic Obstructive Pulmonary Disease. Trends Pharmacol. Sci. 2020, 41, 475–486. [Google Scholar] [CrossRef]
- Chiba, Y.; Tanabe, M.; Goto, K.; Sakai, H.; Misawa, M. Down-regulation of miR-133a contributes to up-regulation of Rhoa in bronchial smooth muscle cells. Am. J. Respir. Crit. Care Med. 2009, 180, 713–719. [Google Scholar] [CrossRef]
- Clifford, R.L.; Singer, C.A.; John, A.E. Epigenetics and miRNA emerge as key regulators of smooth muscle cell phenotype and function. Pulm. Pharmacol. Ther. 2013, 26, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Tost, J. A translational perspective on epigenetics in allergic diseases. J. Allergy Clin. Immunol. 2018, 142, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Boudewijn, I.M.; Roffel, M.P.; Vermeulen, C.J.; Nawijn, M.C.; Kok, K.; Terpstra, M.M.; Koppelman, G.H.; Guryev, V.; van den Berge, M. INDURAIN-investigators. A Novel Role of Bronchial MicroRNAs and Long Non-coding RNAs in Asthma Remission. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Ando, Y.; Fujii, S.; Miyakawa, Y.; Suto, W.; Kamei, J.; Sakai, H.; Hanazaki, M. Down-regulation of miR-140-3p is a cause of up-regulation of RhoA protein in bronchial smooth muscle of murine experimental asthma. Am. J. Respir. Cell Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Boushey, H.A.; Holtzman, M.J.; Sheller, J.R.; Nadel, J.A. Bronchial hyperreactivity. Am. Rev. Respir. Dis. 1980, 121, 389–413. [Google Scholar]
- Jeffery, P.K. Microscopic structure of airway secretory cells: Variation in hypersecretory disease and effects of drugs. In Airway Secretion: Physiological Basis for the Control of Mucus Hypersecretion; Takishima, T., Shimura, S., Eds.; Marcel Dekker: New York, NY, USA, 1993; pp. 149–215. [Google Scholar]
- Wiggs, B.R.; Moreno, R.; Hogg, J.C.; Hilliam, C.; Pare, P.D. A model of the mechanics of airway narrowing. J. Appl. Physiol. 1990, 69, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Seow, C.Y.; Schellenberg, R.R.; Pare, P.D. Structural and functional changes in the airway smooth muscle of asthmatic subjects. Am. J. Respir. Crit. Care Med. 1998, 158, S179–S186. [Google Scholar] [CrossRef]
- Martin, J.G.; Duguet, A.; Eidelman, D.H. The contribution of airway smooth muscle to airway narrowing and airway hyperresponsiveness in disease. Eur. Respir. J. 2000, 16, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Santos, M.D.; Álvarez-González, M.; Estrada-Soto, S.; Bazán-Perkins, B. Regulation of Myosin Light-Chain Phosphatase Activity to Generate Airway Smooth Muscle Hypercontractility. Front. Physiol. 2020, 11, 701. [Google Scholar] [CrossRef] [PubMed]
- Guedes, A.G.; Dileepan, M.; Jude, J.A.; Deshpande, D.A.; Walseth, T.F.; Kannan, M.S. Role of CD38/cADPR signaling in obstructive pulmonary diseases. Curr. Opin. Pharmacol. 2020, 51, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.E.; Larner-Svensson, H.; Perry, M.M.; Campbell, G.A.; Herrick, S.E.; Adcock, I.M.; Erjefalt, J.S.; Chung, K.F.; Lindsay, M.A. MicroRNA expression profiling in mild asthmatic human airways and effect of corticosteroid therapy. PLoS ONE 2009, 4, e5889. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.R.; Schlauch, K.; Lao, R.; Halayko, A.J.; Gerthoffer, W.T.; Singer, C.A. MicroRNA expression in human airway smooth muscle cells: Role of miR-25 in regulation of airway smooth muscle phenotype. Am. J. Respir. Cell Mol. Biol. 2010, 42, 506–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, D.A.; Dogan, S.; Walseth, T.F.; Miller, S.M.; Amrani, Y.; Panettieri, R.A.; Kannan, M.S. Modulation of calcium signaling by interleukin-13 in human airway smooth muscle: Role of CD38/cyclic adenosine diphosphate ribose pathway. Am. J. Respir. Cell Mol. Biol. 2004, 31, 36–42. [Google Scholar] [CrossRef]
- Sieck, G.C.; White, T.A.; Thompson, M.A.; Pabelick, C.M.; Wylam, M.E.; Prakash, Y.S. Regulation of store-operated Ca2+ entry by CD38 in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 29, L378–L385. [Google Scholar] [CrossRef]
- Chiba, Y.; Suto, W.; Sakai, H. Augmented Pla2g4c/Ptgs2/Hpgds axis in bronchial smooth muscle tissues of experimental asthma. PLoS ONE 2018, 13, e0202623. [Google Scholar] [CrossRef] [Green Version]
- Chiba, Y.; Nakazawa, S.; Todoroki, M.; Shinozaki, K.; Sakai, H.; Misawa, M. Interleukin-13 augments bronchial smooth muscle contractility with an up-regulation of RhoA protein. Am. J. Respir. Cell Mol. Biol. 2009, 40, 159–167. [Google Scholar] [CrossRef]
- Chiba, Y.; Todoroki, M.; Nishida, Y.; Tanabe, M.; Misawa, M. A novel STAT6 inhibitor AS1517499 ameliorates antigen-induced bronchial hypercontractility in mice. Am. J. Respir. Cell Mol. Biol. 2009, 41, 516–524. [Google Scholar] [CrossRef]
- Amrani, Y.; Panettieri, R.A., Jr. Modulation of calcium homeostasis as a mechanism for altering smooth muscle responsiveness in asthma. Curr. Opin. Allergy Clin. Immunol. 2002, 2, 39–45. [Google Scholar] [CrossRef]
- Kato, Y.; Manabe, T.; Tanaka, Y.; Mochizuki, H. Effect of an orally active Th1/Th2 balance modulator, M50367, on IgE production, eosinophilia, and airway hyperresponsiveness in mice. J. Immunol. 1999, 162, 7470–7479. [Google Scholar]
- Chiba, Y.; Ueno, A.; Shinozaki, K.; Takeyama, H.; Nakazawa, S.; Sakai, H.; Misawa, M. Involvement of RhoA-mediated Ca2+ sensitization in antigen-induced bronchial smooth muscle hyperresponsiveness in mice. Respir. Res. 2005, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, Y.; Ueno, A.; Sakai, H.; Misawa, M. Hyperresponsiveness of bronchial but not tracheal smooth muscle in a murine model of allergic bronchial asthma. Inflamm. Res. 2004, 53, 636–642. [Google Scholar] [CrossRef]
- Berry, M.A.; Parker, D.; Neale, N.; Woodman, L.; Morgan, A.; Monk, P.; Bradding, P.; Wardlaw, A.J.; Pavord, I.D.; Brightling, C.E. Sputum and bronchial submucosal IL-13 expression in asthma and eosinophilic bronchitis. J. Allergy Clin. Immunol. 2004, 114, 1106–1109. [Google Scholar] [CrossRef]
- Saha, S.K.; Berry, M.A.; Parker, D.; Siddiqui, S.; Morgan, A.; May, R.; Monk, P.; Bradding, P.; Wardlaw, A.J.; Pavord, I.D.; et al. Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J. Allergy Clin. Immunol. 2008, 121, 685–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavi, S.R.; Ahmadi, A.; Jamalkandi, S.A.; Salimian, J. Involvement of microRNAs in physiological and pathological processes in asthma. J. Cell Physiol. 2019, 234, 21547–21559. [Google Scholar] [CrossRef] [PubMed]
- Taka, S.; Tzani-Tzanopoulou, P.; Wanstall, H.; Papadopoulos, N.G. MicroRNAs in Asthma and Respiratory Infections: Identifying Common Pathways. Allergy Asthma Immunol. Res. 2020, 12, 4–23. [Google Scholar] [CrossRef]
- Jude, J.A.; Dileepan, M.; Subramanian, S.; Solway, J.; Panettieri, R.A., Jr.; Walseth, T.F.; Kannan, M.S. miR-140-3p regulation of TNF-α-induced CD38 expression in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L460–L480. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, D.A.; Guedes, A.G.P.; Graeff, R.; Dogan, S.; Subramanian, S.; Walseth, T.F.; Kannan, M.S. CD38/cADPR Signaling Pathway in Airway Disease: Regulatory Mechanisms. Mediat. Inflamm. 2018, 2018, 8942042. [Google Scholar] [CrossRef]
- Specjalski, K.; Jassem, E. MicroRNAs: Potential Biomarkers and Targets of Therapy in Allergic Diseases? Arch. Immunol. Ther. Exp. 2019, 67, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Chiba, Y.; Matsusue, K.; Misawa, M. RhoA, a possible target for treatment of airway hyperresponsiveness in bronchial asthma. J. Pharmacol. Sci. 2010, 114, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Akiyama, Y.; Yuasa, Y. Multiple-to-multiple relationships between microRNAs and target genes in gastric cancer. PLoS ONE 2013, 8, e62589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Song, Y.; Yu, H.; Luo, X. Identification of lncRNA TRPM2-AS/miR-140-3p/PYCR1 axis’s proliferates and anti-apoptotic effect on breast cancer using co-expression network analysis. Cancer Biol. Ther. 2019, 20, 760–773. [Google Scholar] [CrossRef]
- Liang, S.; Ren, K.; Li, B.; Li, F.; Liang, Z.; Hu, J.; Xu, B.; Zhang, A. LncRNA SNHG1 alleviates hypoxia-reoxygenation-induced vascular endothelial cell injury as a competing endogenous RNA through the HIF-1α/VEGF signal pathway. Mol. Cell Biochem. 2020, 465, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Lian, L.; Ding, H.; Hu, Y.; Xiao, Z.; Xiong, K.; Yang, Q. LncRNA ANCR promotes hepatocellular carcinoma metastasis through upregulating HNRNPA1 expression. RNA Biol. 2020, 17, 381–394. [Google Scholar] [CrossRef]
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santos-Argumedo, L.; Parkhouse, R.M.; Walseth, T.F.; Lee, H.C. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059. [Google Scholar] [CrossRef]
- White, T.A.; Johnson, S.; Walseth, T.F.; Lee, H.C.; Graeff, R.M.; Munshi, C.B.; Prakash, Y.S.; Sieck, G.C.; Kannan, M.S. Subcellular localization of cyclic ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities in porcine airway smooth muscle. Biochim. Biophys. Acta 2000, 1498, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Franco, L.; Bruzzone, S.; Song, P.; Guida, L.; Zocchi, E.; Walseth, T.F.; Crimi, E.; Usai, C.; De Flora, A.; Brusasco, V. Extracellular cyclic ADP-ribose potentiates ACh-induced contraction in bovine tracheal smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L98–L106. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, D.A.; White, T.A.; Dogan, S.; Walseth, T.F.; Panettieri, R.A.; Kannan, M.S. CD38/cyclic ADP-ribose signaling: Role in the regulation of calcium homeostasis in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L773–L788. [Google Scholar] [CrossRef]
- Deshpande, D.A.; White, T.A.; Guedes, A.G.; Milla, C.; Walseth, T.F.; Lund, F.E.; Kannan, M.S. Altered airway responsiveness in CD38-deficient mice. Am. J. Respir. Cell Mol. Biol. 2005, 32, 149–156. [Google Scholar] [CrossRef]
- Guedes, A.G.; Paulin, J.; Rivero-Nava, L.; Kita, H.; Lund, F.E.; Kannan, M.S. CD38-deficient mice have reduced airway hyperresponsiveness following IL-13 challenge. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1286–L1293. [Google Scholar] [CrossRef] [Green Version]
- Guedes, A.G.; Jude, J.A.; Paulin, J.; Kita, H.; Lund, F.E.; Kannan, M.S. Role of CD38 in TNF-alpha-induced airway hyperresponsiveness. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L290–L299. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.F.; Mignery, G.A.; Somlyo, A.V. Immunogold localization of inositol 1,4,5-trisphosphate receptors and characterization of ultrastructural features of the sarcoplasmic reticulum in phasic and tonic smooth muscle. J. Muscle Res. Cell Motil. 1994, 15, 682–700. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Yoshii, A.; Dobashi, K.; Horie, T.; Mori, M.; Nakazawa, T. InsP3, but not novel Ca2+ releasers, contributes to agonist-initiated contraction in rabbit airway smooth muscle. J. Physiol. 1998, 511, 915–933. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Sakai, H.; Suenaga, H.; Kamata, K.; Misawa, M. Enhanced Ca2+ sensitization of the bronchial smooth muscle contraction in antigen-induced airway hyperresponsive rats. Res. Commun. Mol. Pathol. Pharmacol. 1999, 106, 77–85. [Google Scholar]
- Guedes, A.G.; Deshpande, D.A.; Dileepan, M.; Walseth, T.F.; Panettieri, R.A., Jr.; Subramanian, S.; Kannan, M.S. CD38 and airway hyper-responsiveness: Studies on human airway smooth muscle cells and mouse models. Can. J. Physiol. Pharmacol. 2015, 93, 145–153. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA (mmu) | Accession | Mature Sequence | Human Expression | AHR/Cont |
|---|---|---|---|---|
| miR-140-3p | MIMAT0000152 | 5′-UACCACAGGGUAGAACCACGG-3′ | Yes | 0.448 |
| miR-140-5p | MIMAT0000151 | 5′-CAGUGGUUUUACCCUAUGGUAG-3′ | Yes | 0.449 |
| miR-133a-3p | MIMAT0000145 | 5′-UUUGGUCCCCUUCAACCAGCUG-3′ | Yes | 0.465 |
| miR-1971 | MIMAT0009446 | 5′-GUAAAGGCUGGGCUGAGA-3′ | No | 2.004 |
| miR-142-3p | MIMAT0000155 | 5′-UGUAGUGUUUCCUACUUUAUGGA-3′ | Yes | 2.101 |
| miR-669c-5p | MIMAT0003479 | 5′-AUAGUUGUGUGUGGAUGUGUGU-3′ | No | 2.123 |
| miR-1897-5p | MIMAT0007864 | 5′-CUUUGGAUGGAGAAAGAGGGGG-3′ | No | 2.257 |
| miR-300-5p | MIMAT0004578 | 5′-UUGAAGAGAGGUUAUCCUUUGU-3′ | Yes | 2.262 |
| miR-1196-5p | MIMAT0005857 | 5′-AAAUCUACCUGCCUCUGCCU-3′ | No | 2.488 |
| miR-302a-3p | MIMAT0000380 | 5′-UAAGUGCUUCCAUGUUUUGGUGA-3′ | Yes | 2.525 |
| miR-133b-3p | MIMAT0000769 | 5′-UUUGGUCCCCUUCAACCAGCUA-3′ | Yes | 3.134 |
| miR-1947-3p | MIMAT0017343 | 5′-GCACUGAGCUAGCUCUCCCUCC-3′ | No | 6.173 |
| miR-3100-3p | MIMAT0014920 | 5′-CUGUGACACACCCGCUCCCAG-3′ | No | 6.410 |
| miR-3474 | MIMAT0015646 | 5′-CCCUGGGAGGAGACGUGGAUUC-3′ | No | 12.195 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiba, Y.; Matsumoto, M.; Hanazaki, M.; Sakai, H. Downregulation of miR-140-3p Contributes to Upregulation of CD38 Protein in Bronchial Smooth Muscle Cells. Int. J. Mol. Sci. 2020, 21, 7982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217982
Chiba Y, Matsumoto M, Hanazaki M, Sakai H. Downregulation of miR-140-3p Contributes to Upregulation of CD38 Protein in Bronchial Smooth Muscle Cells. International Journal of Molecular Sciences. 2020; 21(21):7982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217982
Chicago/Turabian StyleChiba, Yoshihiko, Mayumi Matsumoto, Motohiko Hanazaki, and Hiroyasu Sakai. 2020. "Downregulation of miR-140-3p Contributes to Upregulation of CD38 Protein in Bronchial Smooth Muscle Cells" International Journal of Molecular Sciences 21, no. 21: 7982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217982