Protection against Doxorubicin-Induced Cardiac Dysfunction Is Not Maintained Following Prolonged Autophagy Inhibition

 ,
,  , , , , ,
, , , , , 
Abstract
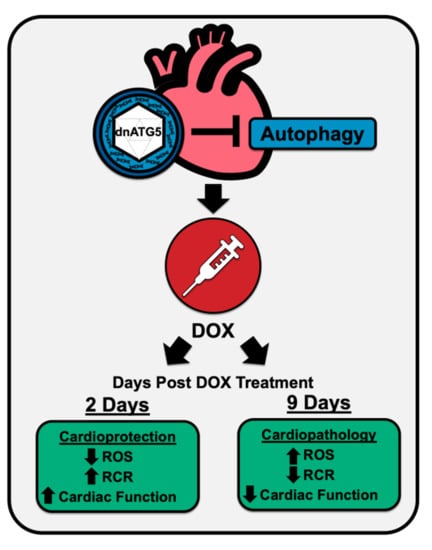
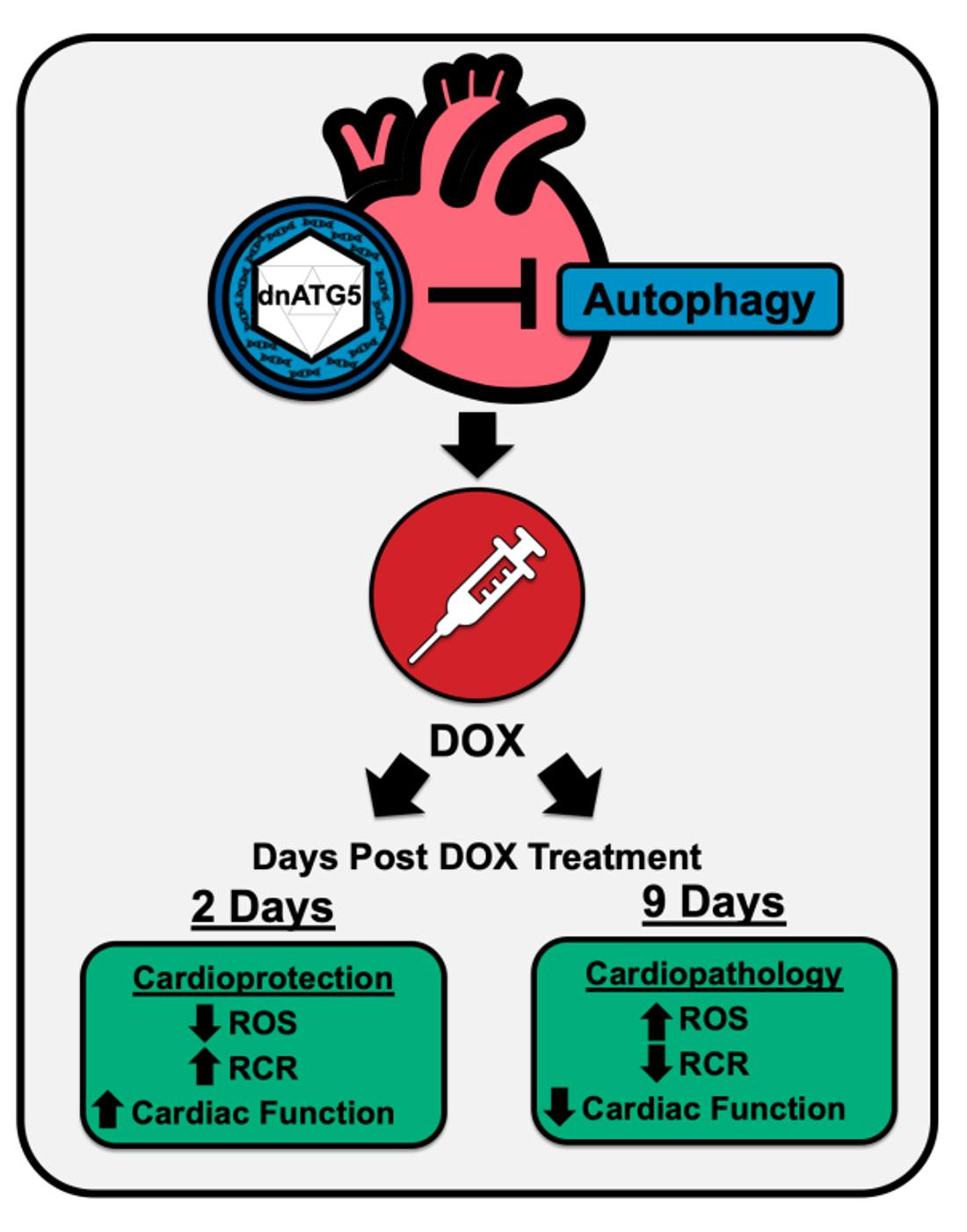
:
1. Introduction
2. Results
2.1. Biological Response to dnATG5 and Doxorubicin Exposure
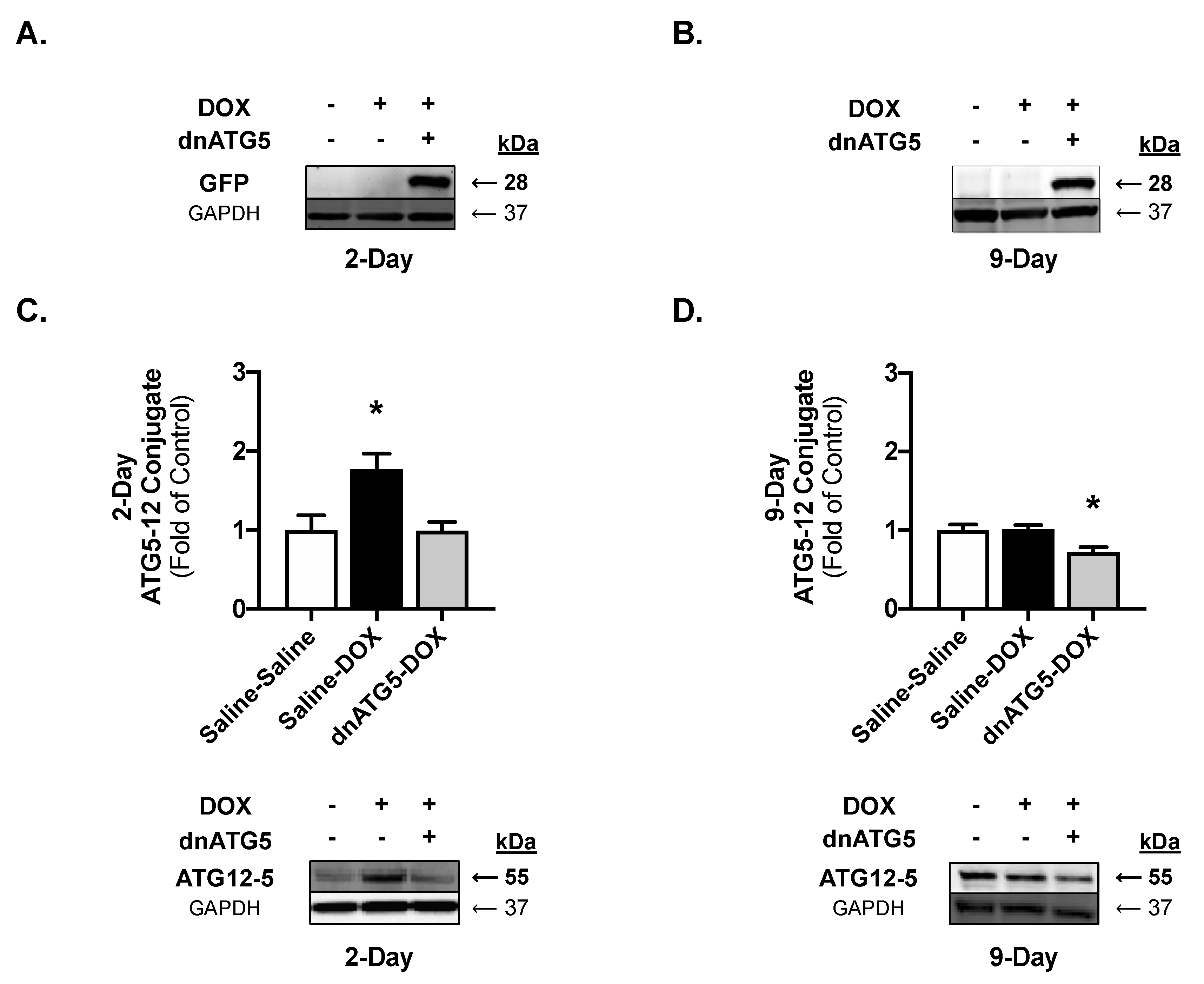
2.2. Validation of the Experimental Treatment
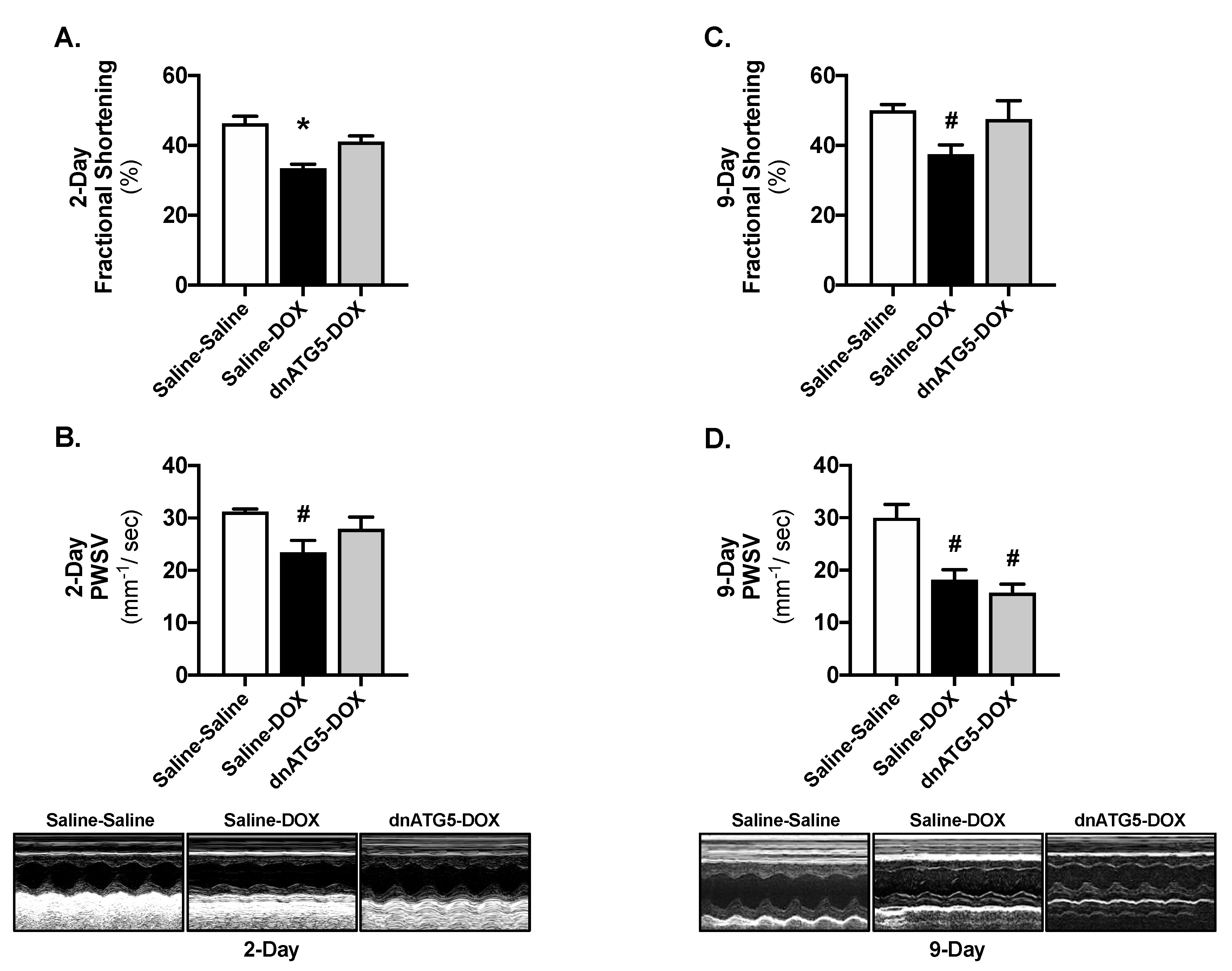
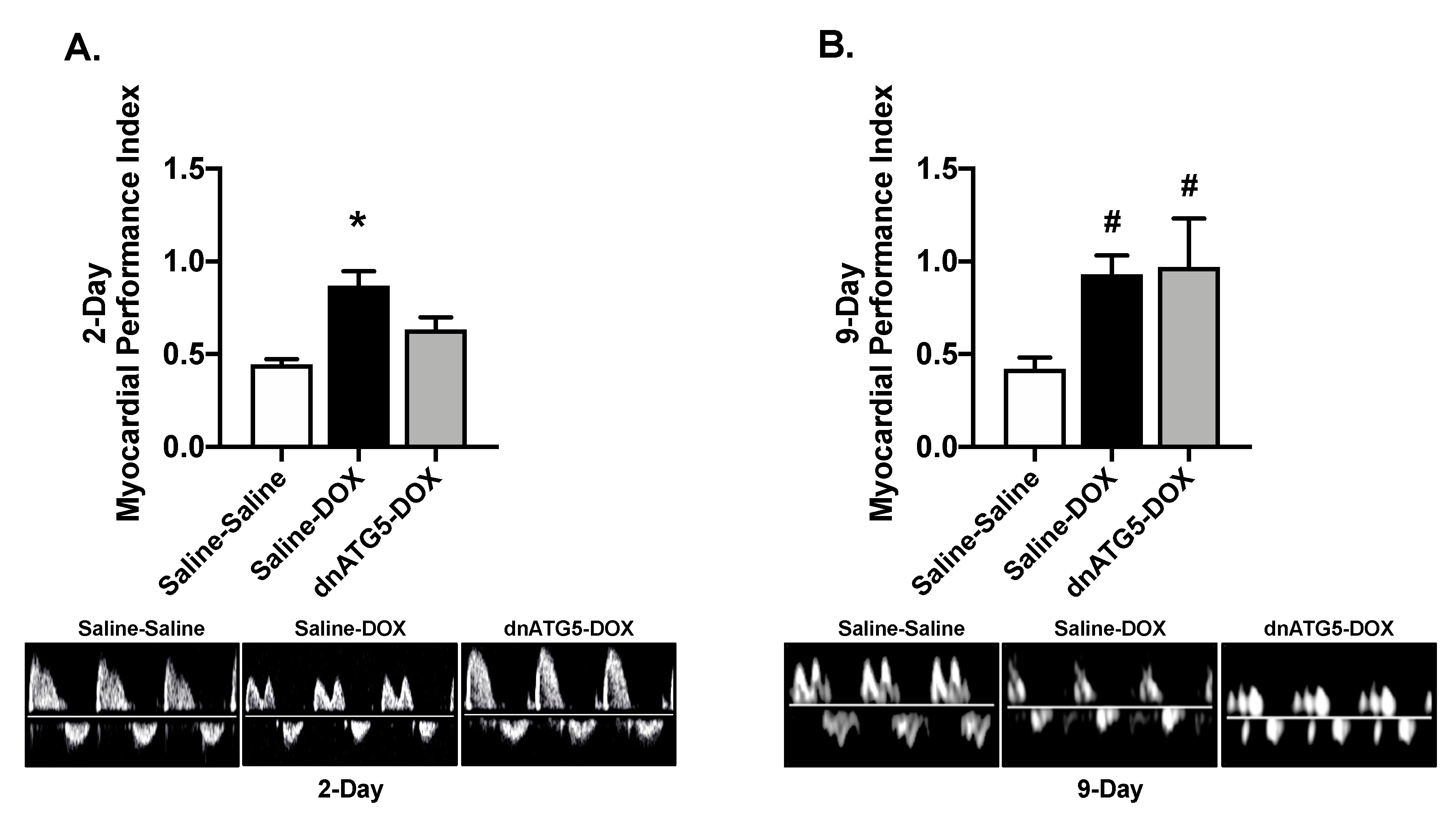
2.3. Inhibition of Autophagosome Formation Protects against Acute DOX-Induced Cardiomyopathy
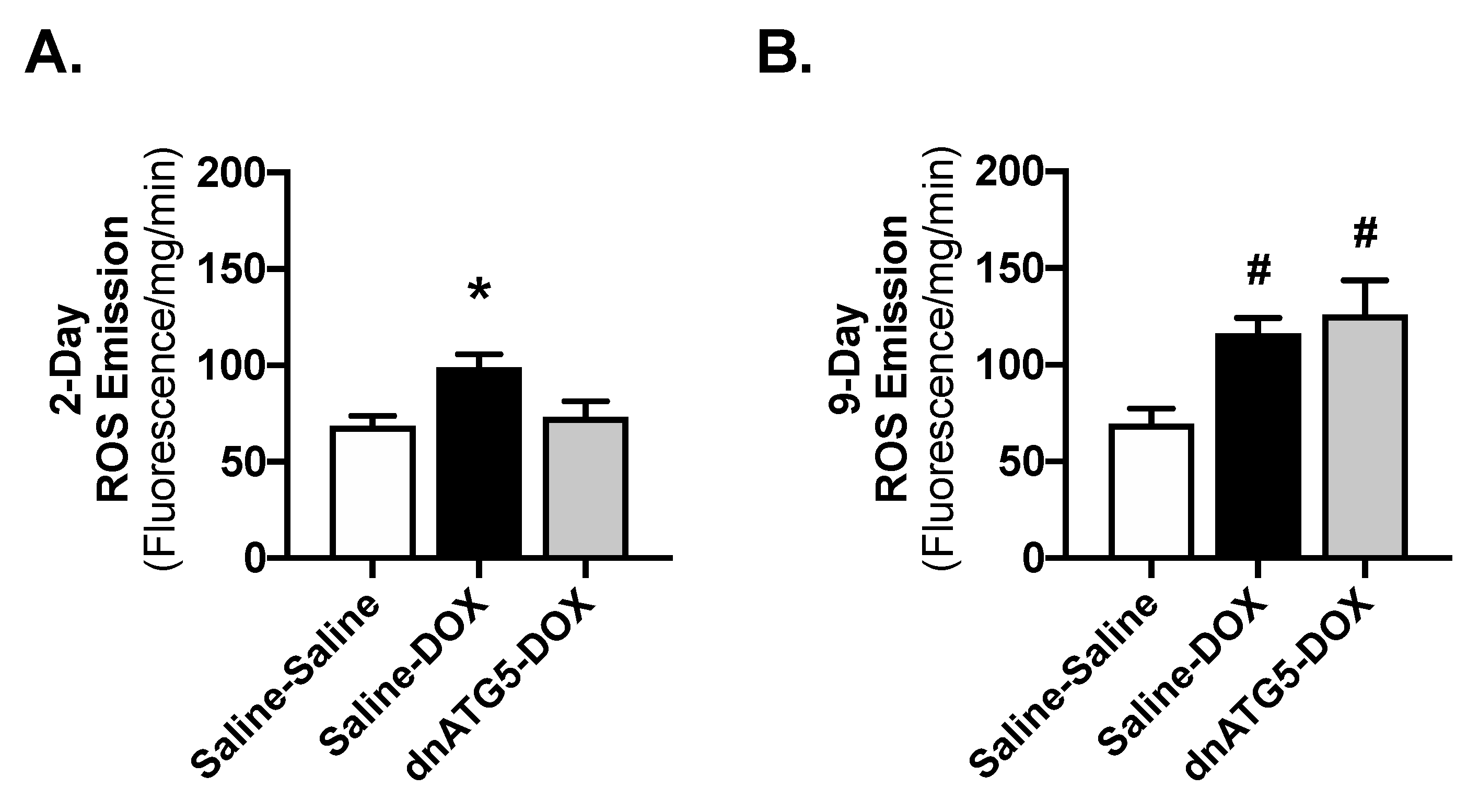
2.4. Inhibition of Autophagosome Formation Protects against Acute DOX-Induced Mitochondrial Dysfunction and ROS Production
3. Discussion
3.1. DOX-Induced Autophagy Signaling
3.2. Cardiac Function and DOX-Induced Autophagy
3.3. Relationship between Autophagy and Oxidative Stress
4. Methods
4.1. Experimental Animals
4.2. Echocardiography
4.3. Cardiac Muscle Permeabilization
4.4. Mitochondrial Respiration
4.5. Mitochondrial ROS Emission
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonadonna, G.; Monfardini, S.; De Lena, M.; Fossati-Bellani, F. Clinical evaluation of adriamycin, a new antitumour antibiotic. Br. Med. J. 1969, 3, 503–506. [Google Scholar] [CrossRef] [Green Version]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.A.; Cho, H.; Lee, N.; Jung, S.Y.; Sim, S.H.; Park, I.H.; Lee, S.; Lee, E.S.; Kim, H.J. Doxorubicin-induced heart failure in cancer patients: A cohort study based on the Korean National Health Insurance Database. Cancer Med. 2018, 7, 6084–6092. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, P.A. Anthracycline cardiotoxicity: An update on mechanisms, monitoring and prevention. Heart 2018, 104, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Cardinale, D.; Colombo, A.; Lamantia, G.; Colombo, N.; Civelli, M.; De Giacomi, G.; Rubino, M.; Veglia, F.; Fiorentini, C.; Cipolla, C.M. Anthracycline-induced cardiomyopathy: Clinical relevance and response to pharmacologic therapy. J. Am. Coll. Cardiol. 2010, 55, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Dirks-Naylor, A.J. The role of autophagy in doxorubicin-induced cardiotoxicity. Life Sci. 2013, 93, 913–916. [Google Scholar] [CrossRef]
- Xiao, B.; Hong, L.; Cai, X.; Mei, S.; Zhang, P.; Shao, L. The true colors of autophagy in doxorubicin-induced cardiotoxicity. Oncol. Lett. 2019, 18, 2165–2172. [Google Scholar] [CrossRef] [Green Version]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavandero, S.; Troncoso, R.; Rothermel, B.A.; Martinet, W.; Sadoshima, J.; Hill, J.A. Cardiovascular autophagy: Concepts, controversies, and perspectives. Autophagy 2013, 9, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, D.; Wiersma, M.; Brundel, B. Role of Autophagy in Proteostasis: Friend and Foe in Cardiac Diseases. Cells 2018, 7, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, K.; Kwon, O.S.; Smuder, A.J.; Wiggs, M.P.; Sollanek, K.J.; Christou, D.D.; Yoo, J.K.; Hwang, M.H.; Szeto, H.H.; Kavazis, A.N.; et al. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J. Physiol. 2015, 593, 2017–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, A.B.; Mor Huertas, A.; Hinkley, J.M.; Ichinoseki-Sekine, N.; Christou, D.D.; Smuder, A.J. Mitochondrial accumulation of doxorubicin in cardiac and diaphragm muscle following exercise preconditioning. Mitochondrion 2018, 45, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Prathumsap, N.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. Effects of doxorubicin on the heart: From molecular mechanisms to intervention strategies. Eur. J. Pharmacol. 2020, 866, 172818. [Google Scholar] [CrossRef]
- Tanaka, K.; Kawano, M.; Iwasaki, T.; Itonaga, I.; Tsumura, H. A meta-analysis of randomized controlled trials that compare standard doxorubicin with other first-line chemotherapies for advanced/metastatic soft tissue sarcomas. PLoS ONE 2019, 14, e0210671. [Google Scholar] [CrossRef]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Wojtacki, J.; Lewicka-Nowak, E.; Lesniewski-Kmak, K. Anthracycline-induced cardiotoxicity: Clinical course, risk factors, pathogenesis, detection and prevention--review of the literature. Med. Sci. Monit. 2000, 6, 411–420. [Google Scholar]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc. Res. 2005, 68, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lullmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Sohal, D.S.; Nghiem, M.; Crackower, M.A.; Witt, S.A.; Kimball, T.R.; Tymitz, K.M.; Penninger, J.M.; Molkentin, J.D. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 2001, 89, 20–25. [Google Scholar] [CrossRef]
- Nishida, K.; Kyoi, S.; Yamaguchi, O.; Sadoshima, J.; Otsu, K. The role of autophagy in the heart. Cell Death Differ. 2009, 16, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, C.S.; Alam, S.; Aishwarya, R.; Miriyala, S.; Bhuiyan, M.A.N.; Panchatcharam, M.; Pattillo, C.B.; Orr, A.W.; Sadoshima, J.; Hill, J.A.; et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 2019, 9, 2002. [Google Scholar] [CrossRef]
- Campbell, T.L.; Quadrilatero, J. Data on skeletal muscle apoptosis, autophagy, and morphology in mice treated with doxorubicin. Data Brief. 2016, 7, 786–793. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Wu, W.; Yan, J.; Li, X.; Yu, H.; Yu, X. Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int. J. Cardiol. 2009, 134, 82–90. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.L.; Chen, H.L.; Wu, D.; Chen, J.X.; Wang, X.X.; Li, R.L.; He, J.H.; Mo, L.; Cen, X.; et al. Ghrelin inhibits doxorubicin cardiotoxicity by inhibiting excessive autophagy through AMPK and p38-MAPK. Biochem. Pharmacol. 2014, 88, 334–350. [Google Scholar] [CrossRef]
- Sun, A.; Cheng, Y.; Zhang, Y.; Zhang, Q.; Wang, S.; Tian, S.; Zou, Y.; Hu, K.; Ren, J.; Ge, J. Aldehyde dehydrogenase 2 ameliorates doxorubicin-induced myocardial dysfunction through detoxification of 4-HNE and suppression of autophagy. J. Mol. Cell. Cardiol. 2014, 71, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, K.; Kobayashi, S.; Timm, D.; Liang, Q. Resveratrol attenuates doxorubicin-induced cardiomyocyte death via inhibition of p70 S6 kinase 1-mediated autophagy. J. Pharm. Exp. Ther 2012, 341, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Xu, X.; Kobayashi, S.; Timm, D.; Jepperson, T.; Liang, Q. Caloric restriction mimetic 2-deoxyglucose antagonizes doxorubicin-induced cardiomyocyte death by multiple mechanisms. J. Biol. Chem. 2011, 286, 21993–22006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Volden, P.; Timm, D.; Mao, K.; Xu, X.; Liang, Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 2010, 285, 793–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, D.; Kim, J.; Shaw, R.J.; Guan, K.L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef] [Green Version]
- Montalvo, R.N.; Doerr, V.; Min, K.; Szeto, H.H.; Smuder, A.J. Doxorubicin-induced oxidative stress differentially regulates proteolytic signaling in cardiac and skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R227–R233. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Xu, J.; Kim, J.S.; Dunn, W.A., Jr.; Leeuwenburgh, C. Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity. Autophagy 2013, 9, 328–344. [Google Scholar] [CrossRef] [Green Version]
- Roca-Agujetas, V.; de Dios, C.; Leston, L.; Mari, M.; Morales, A.; Colell, A. Recent Insights into the Mitochondrial Role in Autophagy and Its Regulation by Oxidative Stress. Oxid. Med. Cell. Longev. 2019, 2019, 3809308. [Google Scholar] [CrossRef] [Green Version]
- Doerr, V.; Montalvo, R.N.; Kwon, O.S.; Talbert, E.E.; Hain, B.A.; Houston, F.E.; Smuder, A.J. Prevention of Doxorubicin-Induced Autophagy Attenuates Oxidative Stress and Skeletal Muscle Dysfunction. Antioxidants (Basel) 2020, 9, 263. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, H.; Wu, Z. Microglia-aging: Roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging. Behav. Brain Res. 2009, 201, 1–7. [Google Scholar] [CrossRef]
- Kubota, C.; Torii, S.; Hou, N.; Saito, N.; Yoshimoto, Y.; Imai, H.; Takeuchi, T. Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J. Biol. Chem. 2010, 285, 667–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smuder, A.J.; Sollanek, K.J.; Nelson, W.B.; Min, K.; Talbert, E.E.; Kavazis, A.N.; Hudson, M.B.; Sandri, M.; Szeto, H.H.; Powers, S.K. Crosstalk between autophagy and oxidative stress regulates proteolysis in the diaphragm during mechanical ventilation. Free Radic. Biol. Med. 2018, 115, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar] [CrossRef] [Green Version]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NRC. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2011; Volume 8, pp. 1–220. [Google Scholar]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.; Griendling, K.K.; Harrison, D.G. Measurement of reactive oxygen species in cardiovascular studies. Hypertension 2007, 49, 717–727. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Saline-Saline | Saline-DOX | dnATG5-DOX | |
|---|---|---|---|
| 2-Day | |||
| Initial weight (g) | 278.6 ± 3.3 | 283.1 ± 5.5 | 280.1 ± 3.4 |
| Treatment weight (g) | 296.5 ± 4.4 | 302.8 ± 5.1 | 299.9 ± 4.5 |
| Final weight (g) | 295.9 ± 4.5 | 287.1 ± 6.6 | 290.5 ± 6.3 |
| Heart weight (mg) | 870.5 ± 18.8 | 792.4 ± 27.3 | 822.0 ± 28.1 |
| 9-Day | |||
| Initial weight (g) | 268.8 ± 3.6 | 273.0 ± 5.8 | 278.3 ± 3.3 |
| Treatment weight (g) | 286.5 ± 6.8 | 287.4 ± 6.0 | 293.3 ± 4.8 |
| Final weight (g) | 282.6 ± 5.6 | 229.4 ± 13.8 # | 229.0 ± 5.9 # |
| Heart weight (mg) | 796.0 ± 30.6 | 597.5 ± 43.1 # | 581.1 ± 13.5 # |
| Saline-Saline | Saline-DOX | dnATG5-DOX | |
|---|---|---|---|
| 2-Day | |||
| SWTd (mm) | 1.41 ± 0.03 | 1.24 ± 0.05 # | 1.24 ± 0.04 # |
| SWTs (mm) | 2.54 ± 0.07 | 2.13 ± 0.08 # | 2.25 ± 0.09 # |
| PWTd (mm) | 1.43 ± 0.04 | 1.36 ± 0.09 | 1.36 ± 0.05 |
| PWTs (mm) | 2.54 ± 0.06 | 1.90 ± 0.11 # | 2.16 ± 0.09 # |
| 9-Day | |||
| SWTd (mm) | 1.74 ± 0.07 | 2.08 ± 0.23 | 1.95 ± 0.17 |
| SWTs (mm) | 2.80 ± 0.10 | 2.85 ± 0.20 | 2.63 ± 0.18 |
| PWTd (mm) | 1.81 ± 0.07 | 2.04 ± 0.15 | 2.30 ± 0.27 |
| PWTs (mm) | 2.86 ± 0.12 | 2.91 ± 0.20 | 3.21 ± 0.36 |
| Saline-Saline | Saline-DOX | dnATG5-DOX | |
|---|---|---|---|
| 2-Day | |||
| State 3 (nmoles O2/mg/min) | 9.81 ± 0.83 | 9.75 ± 0.67 | 10.60 ± 0.66 |
| State 4 (nmoles O2/mg/min) | 2.13 ± 0.23 | 3.79 ± 0.24 * | 2.67 ± 0.13 |
| RCR (State 3/State 4) | 4.93 ± 0.37 | 2.67 ± 0.26 * | 4.06 ± 0.23 |
| 9-Day | |||
| State 3 (nmoles O2/mg/min) | 9.50 ± 1.57 | 8.40 ± 1.12 | 11.29 ± 1.14 |
| State 4 (nmoles O2/mg/min) | 2.16 ± 0.41 | 2.37 ± 0.20 | 3.21 ± 0.30 |
| RCR (State 3/State 4) | 4.53 ± 0.18 | 3.52 ± 0.27 # | 3.54 ± 0.20 # |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montalvo, R.N.; Doerr, V.; Kwon, O.S.; Talbert, E.E.; Yoo, J.-K.; Hwang, M.-H.; Nguyen, B.L.; Christou, D.D.; Kavazis, A.N.; Smuder, A.J. Protection against Doxorubicin-Induced Cardiac Dysfunction Is Not Maintained Following Prolonged Autophagy Inhibition. Int. J. Mol. Sci. 2020, 21, 8105. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218105
Montalvo RN, Doerr V, Kwon OS, Talbert EE, Yoo J-K, Hwang M-H, Nguyen BL, Christou DD, Kavazis AN, Smuder AJ. Protection against Doxorubicin-Induced Cardiac Dysfunction Is Not Maintained Following Prolonged Autophagy Inhibition. International Journal of Molecular Sciences. 2020; 21(21):8105. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218105
Chicago/Turabian StyleMontalvo, Ryan N., Vivian Doerr, Oh Sung Kwon, Erin E. Talbert, Jeung-Ki Yoo, Moon-Hyon Hwang, Branden L. Nguyen, Demetra D. Christou, Andreas N. Kavazis, and Ashley J. Smuder. 2020. "Protection against Doxorubicin-Induced Cardiac Dysfunction Is Not Maintained Following Prolonged Autophagy Inhibition" International Journal of Molecular Sciences 21, no. 21: 8105. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218105