Delicate Role of PD-L1/PD-1 Axis in Blood Vessel Inflammatory Diseases: Current Insight and Future Significance


Abstract
:1. Introduction
2. Overview of General Biological Activities of PD-L1 and PD-1 Molecules
3. Relevance of PD-L1/PD-1 Axis on Vascular Endothelium and Barrier Functions
4. Regulation of Blood Vessel Inflammation by PD-L1/PD-1 Axis
4.1. PD-1/PD-L1 Axis in Experimental Atherosclerosis
4.2. PD-1/PD-L1 Axis in Human Atherosclerosis
5. Delicate Restoration of Protective Immunity to Curtail Infections by Targeting PD-1 Pathway in Underlying Blood Vessel Inflammation
5.1. PD-1/PD-L1 Axis during Acute Infections in Blood Vessel Inflammation
5.2. PD-1/PD-L1 Axis during Chronic Infections in Blood Vessel Inflammation
5.2.1. Role of PD-1/PD-L1 Axis in Cytomegalovirus-Associated Atherosclerosis
5.2.2. Role of PD-1/PD-L1 Axis in Chlamydia-Associated Atherosclerosis
5.2.3. Role of PD-1/PD-L1 Axis in Helicobacter-Associated Atherosclerosis
6. Implication of PD-1/PD-L1 Axis in the Altered Metabolism during Blood Vessel Inflammation
7. Therapeutic Relevance of PD-L1/PD-1 Axis in Blood Vessel Inflammation
7.1. Current Understanding
7.2. Future Perspectives
8. Concluding Remarks
Funding
Conflicts of Interest
References
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vasc. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Bondareva, O.; Sheikh, B.N. Vascular Homeostasis and Inflammation in Health and Disease-Lessons from Single Cell Technologies. Int. J. Mol. Sci. 2020, 21, 4688. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Corbera-Bellalta, M.; Audia, S.; Planas-Rigol, E.; Martin, L.; Cid, M.C.; Bonnotte, B. Recent advances in our understanding of giant cell arteritis pathogenesis. Autoimmun. Rev. 2017, 16, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Shavit, E.; Alavi, A.; Sibbald, R.G. Vasculitis-What Do We Have to Know? A Review of Literature. Int. J. Low. Extrem. Wounds 2018, 17, 218–226. [Google Scholar] [CrossRef]
- Seyahi, E. Takayasu arteritis: An update. Curr. Opin. Rheumatol. 2017, 29, 51–56. [Google Scholar] [CrossRef]
- Dejaco, C.; Brouwer, E.; Mason, J.C.; Buttgereit, F.; Matteson, E.L.; Dasgupta, B. Giant cell arteritis and polymyalgia rheumatica: Current challenges and opportunities. Nat. Reviews. Rheumatol. 2017, 13, 578–592. [Google Scholar] [CrossRef]
- Harky, A.; Fok, M.; Balmforth, D.; Bashir, M. Pathogenesis of large vessel vasculitis: Implications for disease classification and future therapies. Vasc. Med. 2019, 24, 79–88. [Google Scholar] [CrossRef]
- Sun, X.L.; Law, B.Y.; de Seabra Rodrigues Dias, I.R.; Mok, S.W.F.; He, Y.Z.; Wong, V.K. Pathogenesis of thromboangiitis obliterans: Gene polymorphism and immunoregulation of human vascular endothelial cells. Atherosclerosis 2017, 265, 258–265. [Google Scholar] [CrossRef]
- Agarwal, S.; Agrawal, D.K. Kawasaki disease: Etiopathogenesis and novel treatment strategies. Expert Rev. Clin. Immunol. 2017, 13, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demir, S.; Sag, E.; Dedeoglu, F.; Ozen, S. Vasculitis in Systemic Autoinflammatory Diseases. Front. Pediatrics 2018, 6, 377. [Google Scholar] [CrossRef] [PubMed]
- López-Mejías, R.; Castañeda, S.; Genre, F.; Remuzgo-Martínez, S.; Carmona, F.D.; Llorca, J.; Blanco, R.; Martín, J.; González-Gay, M.A. Genetics of immunoglobulin-A vasculitis (Henoch-Schönlein purpura): An updated review. Autoimmun. Rev. 2018, 17, 301–315. [Google Scholar] [CrossRef]
- Greco, A.; Marinelli, C.; Fusconi, M.; Macri, G.F.; Gallo, A.; De Virgilio, A.; Zambetti, G.; de Vincentiis, M. Clinic manifestations in granulomatosis with polyangiitis. Int. J. Immunopathol. Pharmacol. 2016, 29, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopalco, G.; Rigante, D.; Venerito, V.; Emmi, G.; Anelli, M.G.; Lapadula, G.; Iannone, F.; Cantarini, L. Management of Small Vessel Vasculitides. Curr. Rheumatol. Rep. 2016, 18, 36. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. New Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, R.; Jhaveri, V.M.; Kay, S.S.; Greer, A.; Sutherland, K.J.; McMurry, H.S.; Lin, N.; Mittal, J.; Malhotra, A.K.; Patel, A.P. Recent Advances in Understanding the Pathogenesis of Cardiovascular Diseases and Development of Treatment Modalities. Cardiovasc. Hematol. Disord. Drug Targets 2019, 19, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Sayols-Baixeras, S.; Lluís-Ganella, C.; Lucas, G.; Elosua, R. Pathogenesis of coronary artery disease: Focus on genetic risk factors and identification of genetic variants. Appl. Clin. Genet. 2014, 7, 15–32. [Google Scholar] [PubMed] [Green Version]
- Campia, U.; Gerhard-Herman, M.; Piazza, G.; Goldhaber, S.Z. Peripheral Artery Disease: Past, Present, and Future. Am. J. Med. 2019, 132, 1133–1141. [Google Scholar] [CrossRef]
- Meschia, J.F.; Klaas, J.P.; Brown, R.D., Jr.; Brott, T.G. Evaluation and Management of Atherosclerotic Carotid Stenosis. Mayo Clin. Proc. 2017, 92, 1144–1157. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Reviews. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes: The Task Force for the diagnosis and management of chronic coronary syndromes of the European Society of Cardiology (ESC). Eur. Heart J. 2019, 41, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Lacey, B.; Herrington, W.G.; Preiss, D.; Lewington, S.; Armitage, J. The Role of Emerging Risk Factors in Cardiovascular Outcomes. Curr. Atheroscler. Rep. 2017, 19, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, S.J.; Krist, A.H.; Owens, D.K.; Barry, M.J.; Caughey, A.B.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W., Jr.; Kemper, A.R.; Kubik, M.; et al. Risk Assessment for Cardiovascular Disease With Nontraditional Risk Factors: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 320, 272–280. [Google Scholar]
- Lafferty, K.J.; Cunningham, A.J. A new analysis of allogeneic interactions. Aust. J. Exp. Biol. Med Sci. 1975, 53, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Bottomly, K. Signals and signs for lymphocyte responses. Cell 1994, 76, 275–285. [Google Scholar] [CrossRef]
- Allison, J.P. CD28-B7 interactions in T-cell activation. Curr. Opin. Immunol. 1994, 6, 414–419. [Google Scholar] [CrossRef]
- Zhu, Y.; Yao, S.; Chen, L. Cell surface signaling molecules in the control of immune responses: A tide model. Immunity 2011, 34, 466–478. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The interaction properties of costimulatory molecules revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Watanabe, R.; Berry, G.J.; Vaglio, A.; Liao, Y.J.; Warrington, K.J.; Goronzy, J.J.; Weyand, C.M. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc. Natl. Acad. Sci. USA 2017, 114, E970–E979. [Google Scholar] [CrossRef] [Green Version]
- Hid Cadena, R.; Reitsema, R.D.; Huitema, M.G.; van Sleen, Y.; van der Geest, K.S.M.; Heeringa, P.; Boots, A.M.H.; Abdulahad, W.H.; Brouwer, E. Decreased Expression of Negative Immune Checkpoint VISTA by CD4+ T Cells Facilitates T Helper 1, T Helper 17, and T Follicular Helper Lineage Differentiation in GCA. Front. Immunol. 2019, 10, 1638. [Google Scholar] [CrossRef]
- Chun, J.K.; Kang, D.W.; Yoo, B.W.; Shin, J.S.; Kim, D.S. Programmed death-1 (PD-1) gene polymorphisms lodged in the genetic predispositions of Kawasaki Disease. Eur. J. Pediatrics 2010, 169, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.H.; Park, M.J.; Park, S.; Lee, E.S. Altered expression of costimulatory molecules in Behçet’s disease according to clinical activity. Br. J. Dermatol. 2011, 164, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Kijlstra, A.; Yang, P. The genetics of Behçet’s disease in a Chinese population. Front. Med. 2012, 6, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Guo, H.; Hou, S.; Jiang, Z.; Kijlstra, A.; Yang, P. Lack of an association of PD-1 and its ligand genes with Behcet’s disease in a Chinese Han population. PLoS ONE 2011, 6, e25345. [Google Scholar] [CrossRef]
- Wilde, B.; Hua, F.; Dolff, S.; Jun, C.; Cai, X.; Specker, C.; Feldkamp, T.; Kribben, A.; Cohen Tervaert, J.W.; Witzke, O. Aberrant expression of the negative costimulator PD-1 on T cells in granulomatosis with polyangiitis. Rheumatology 2012, 51, 1188–1197. [Google Scholar] [CrossRef] [Green Version]
- Slot, M.C.; Sokolowska, M.G.; Savelkouls, K.G.; Janssen, R.G.; Damoiseaux, J.G.; Tervaert, J.W. Immunoregulatory gene polymorphisms are associated with ANCA-related vasculitis. Clin. Immunol. 2008, 128, 39–45. [Google Scholar] [CrossRef]
- Sakthivel, P.; Giscombe, R.; Ramanujam, R.; Lefvert, A.K. Polymorphisms in PDCD1 gene are not associated with Wegener’s granulomatosis. Rheumatol. Int. 2009, 29, 1247–1250. [Google Scholar] [CrossRef]
- Dermani, F.K.; Samadi, P.; Rahmani, G.; Kohlan, A.K.; Najafi, R. PD-1/PD-L1 immune checkpoint: Potential target for cancer therapy. J. Cell. Physiol. 2019, 234, 1313–1325. [Google Scholar] [CrossRef]
- Rotte, A.; D’Orazi, G.; Bhandaru, M. Nobel committee honors tumor immunologists. J. Exp. Clin. Cancer Res. 2018, 37, 262. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.W.; Chang, J.W. Immune checkpoint inhibitors win the 2018 Nobel Prize. Biomed. J. 2019, 42, 299–306. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Zasada, M.; Lenart, M.; Rutkowska-Zapała, M.; Stec, M.; Durlak, W.; Grudzień, A.; Krzeczkowska, A.; Mól, N.; Pilch, M.; Siedlar, M.; et al. Analysis of PD-1 expression in the monocyte subsets from non-septic and septic preterm neonates. PLoS ONE 2017, 12, e0186819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Ni, Z.; Liu, X.; Feng, S.; Dong, X.; Shi, X.; Zhai, J.; Mai, S.; Jiang, J.; Wang, Z.; et al. Beyond T Cells: Understanding the Role of PD-1/PD-L1 in Tumor-Associated Macrophages. J. Immunol. Res. 2019, 2019, 1919082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Greenwald, R.J.; Latchman, Y.E.; Sharpe, A.H. Negative co-receptors on lymphocytes. Curr. Opin. Immunol. 2002, 14, 391–396. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef]
- Patsoukis, N.; Duke-Cohan, J.S.; Chaudhri, A.; Aksoylar, H.I.; Wang, Q.; Council, A.; Berg, A.; Freeman, G.J.; Boussiotis, V.A. Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun. Biol. 2020, 3, 128. [Google Scholar] [CrossRef]
- Marasco, M.; Berteotti, A.; Weyershaeuser, J.; Thorausch, N.; Sikorska, J.; Krausze, J.; Brandt, H.J.; Kirkpatrick, J.; Rios, P.; Schamel, W.W.; et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci. Adv. 2020, 6, eaay4458. [Google Scholar] [CrossRef] [Green Version]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Riella, L.V.; Chock, S.; Liu, T.; Zhao, X.; Yuan, X.; Paterson, A.M.; Watanabe, T.; Vanguri, V.; Yagita, H.; et al. The novel costimulatory programmed death ligand 1/B7.1 pathway is functional in inhibiting alloimmune responses in vivo. J. Immunol. 2011, 187, 1113–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.M.G.; Wen, T.; Dong, H. Bidirectional signals of PD-L1 in T cells that fraternize with cancer cells. Nat. Immunol. 2020, 21, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Harrison, D.L.; Song, Y.; Ji, J.; Huang, J.; Hui, E. Antigen-Presenting Cell-Intrinsic PD-1 Neutralizes PD-L1 in cis to Attenuate PD-1 Signaling in T Cells. Cell Rep. 2018, 24, 379–390.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, B.; Du, X.; Wang, Q.; Chen, Y.; Zhang, X. Increased PD-1 on CD4(+)CD28(-) T cell and soluble PD-1 ligand-1 in patients with T2DM: Association with atherosclerotic macrovascular diseases. Metab. Clin. Exp. 2013, 62, 778–785. [Google Scholar] [CrossRef]
- Chang, B.; Huang, T.; Wei, H.; Shen, L.; Zhu, D.; He, W.; Chen, Q.; Zhang, H.; Li, Y.; Huang, R.; et al. The correlation and prognostic value of serum levels of soluble programmed death protein 1 (sPD-1) and soluble programmed death-ligand 1 (sPD-L1) in patients with hepatocellular carcinoma. Cancer Immunol. Immunother. 2019, 68, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Hebbar, M.; Jeannin, P.; Magistrelli, G.; Hatron, P.Y.; Hachulla, E.; Devulder, B.; Bonnefoy, J.Y.; Delneste, Y. Detection of circulating soluble CD28 in patients with systemic lupus erythematosus, primary Sjögren’s syndrome and systemic sclerosis. Clin. Exp. Immunol. 2004, 136, 388–392. [Google Scholar] [CrossRef]
- Kakoulidou, M.; Giscombe, R.; Zhao, X.; Lefvert, A.K.; Wang, X. Human Soluble CD80 is generated by alternative splicing, and recombinant soluble CD80 binds to CD28 and CD152 influencing T-cell activation. Scand. J. Immunol. 2007, 66, 529–537. [Google Scholar] [CrossRef]
- Wan, B.; Nie, H.; Liu, A.; Feng, G.; He, D.; Xu, R.; Zhang, Q.; Dong, C.; Zhang, J.Z. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J. Immunol. 2006, 177, 8844–8850. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Lang, J. Soluble PD-1 and PD-L1: Predictive and prognostic significance in cancer. Oncotarget 2017, 8, 97671–97682. [Google Scholar] [CrossRef] [Green Version]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A., Jr.; Topper, J.N.; Nagel, T.; Anderson, K.R.; Garcia-Cardeña, G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann. N. Y. Acad. Sci. 2000, 902, 230–239, discussion 239–240. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Shimokawa, H.; Tang, E.H.; Feletou, M. Endothelial dysfunction and vascular disease. Acta Physiol. 2009, 196, 193–222. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Qin, T.; Liu, Z.; Wang, J.; Jia, Y.; Feng, Y.; Gao, Y.; Li, K. anlotinib alters tumor immune microenvironment by downregulating PD-L1 expression on vascular endothelial cells. Cell Death Dis. 2020, 11, 309. [Google Scholar] [CrossRef]
- Bichsel, C.A.; Wang, L.; Froment, L.; Berezowska, S.; Müller, S.; Dorn, P.; Marti, T.M.; Peng, R.W.; Geiser, T.; Schmid, R.A.; et al. Increased PD-L1 expression and IL-6 secretion characterize human lung tumor-derived perivascular-like cells that promote vascular leakage in a perfusable microvasculature model. Sci. Rep. 2017, 7, 10636. [Google Scholar] [CrossRef]
- Dimou, P.; Wright, R.D.; Budge, K.L.; Midgley, A.; Satchell, S.C.; Peak, M.; Beresford, M.W. The human glomerular endothelial cells are potent pro-inflammatory contributors in an in vitro model of lupus nephritis. Sci. Rep. 2019, 9, 8348. [Google Scholar] [CrossRef] [Green Version]
- Eppihimer, M.J.; Gunn, J.; Freeman, G.J.; Greenfield, E.A.; Chernova, T.; Erickson, J.; Leonard, J.P. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation 2002, 9, 133–145. [Google Scholar] [CrossRef]
- Papaioannou, T.G.; Stefanadis, C. Vascular wall shear stress: Basic principles and methods. Hell. J. Cardiol. Hjc = Hell. Kardiol. Ep. 2005, 46, 9–15. [Google Scholar]
- Zheng, X.; Fernando, V.; Sharma, V.; Walia, Y.; Letson, J.; Furuta, S. Correction of arginine metabolism with sepiapterin-the precursor of nitric oxide synthase cofactor BH(4)-induces immunostimulatory-shift of breast cancer. Biochem. Pharmacol. 2020, 176, 113887. [Google Scholar] [CrossRef]
- Rodig, N.; Ryan, T.; Allen, J.A.; Pang, H.; Grabie, N.; Chernova, T.; Greenfield, E.A.; Liang, S.C.; Sharpe, A.H.; Lichtman, A.H.; et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur. J. Immunol. 2003, 33, 3117–3126. [Google Scholar] [CrossRef]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef]
- Taguchi, K.; Onoe, T.; Yoshida, T.; Yamashita, Y.; Tanaka, Y.; Ohdan, H. Tumor Endothelial Cell-Mediated Antigen-Specific T-cell Suppression via the PD-1/PD-L1 Pathway. Mol. Cancer Res. 2020, 18, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.M.; Tsai, C.L.; Kao, J.T.; Hsieh, C.T.; Shieh, D.C.; Lee, Y.J.; Tsay, G.J.; Cheng, K.S.; Wu, Y.Y. PD-1 and PD-L1 Up-regulation Promotes T-cell Apoptosis in Gastric Adenocarcinoma. Anticancer Res. 2018, 38, 2069–2078. [Google Scholar] [PubMed]
- Pober, J.S.; Merola, J.; Liu, R.; Manes, T.D. Antigen Presentation by Vascular Cells. Front. Immunol. 2017, 8, 1907. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Frebel, H.; Nindl, V.; Schuepbach, R.A.; Braunschweiler, T.; Richter, K.; Vogel, J.; Wagner, C.A.; Loffing-Cueni, D.; Kurrer, M.; Ludewig, B.; et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J. Exp. Med. 2012, 209, 2485–2499. [Google Scholar] [CrossRef]
- Aquila, G.; Pannella, M.; Morelli, M.B.; Caliceti, C.; Fortini, C.; Rizzo, P.; Ferrari, R. The role of Notch pathway in cardiovascular diseases. Glob. Cardiol. Sci. Pract. 2013, 2013, 364–371. [Google Scholar] [CrossRef]
- Pannella, M.; Caliceti, C.; Fortini, F.; Aquila, G.; Vieceli Dalla Sega, F.; Pannuti, A.; Fortini, C.; Morelli, M.B.; Fucili, A.; Francolini, G.; et al. Serum From Advanced Heart Failure Patients Promotes Angiogenic Sprouting and Affects the Notch Pathway in Human Endothelial Cells. J. Cell. Physiol. 2016, 231, 2700–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.; Liu, Z.; Yin, J.; Zhou, T.; Liu, J.; Qu, H. Notch Signaling Pathway Was Involved in Regulating Programmed Cell Death 1 Expression during Sepsis-Induced Immunosuppression. Mediat. Inflamm. 2015, 2015, 539841. [Google Scholar] [CrossRef] [PubMed]
- Mansour, F.A.; Al-Mazrou, A.; Al-Mohanna, F.; Al-Alwan, M.; Ghebeh, H. PD-L1 is overexpressed on breast cancer stem cells through notch3/mTOR axis. Oncoimmunology 2020, 9, 1729299. [Google Scholar] [CrossRef] [Green Version]
- Sugita, S.; Usui, Y.; Horie, S.; Futagami, Y.; Yamada, Y.; Ma, J.; Kezuka, T.; Hamada, H.; Usui, T.; Mochizuki, M.; et al. Human corneal endothelial cells expressing programmed death-ligand 1 (PD-L1) suppress PD-1+ T helper 1 cells by a contact-dependent mechanism. Investig. Ophthalmol. Vis. Sci. 2009, 50, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Jin, Y.; Freeman, G.J.; Sharpe, A.H.; Dana, M.R. The function of donor versus recipient programmed death-ligand 1 in corneal allograft survival. J. Immunol. 2007, 179, 3672–3679. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Chauhan, S.K.; El Annan, J.; Annan, J.E.; Sage, P.T.; Sharpe, A.H.; Dana, R. A novel function for programmed death ligand-1 regulation of angiogenesis. Am. J. Pathol. 2011, 178, 1922–1929. [Google Scholar] [CrossRef]
- Amaya, C.N.; Wians, F.H., Jr.; Bryan, B.A.; Torabi, A. Enhanced expression of Programmed cell death 1 (PD-1) protein in benign vascular anomalies. Pathology 2017, 49, 292–296. [Google Scholar] [CrossRef]
- Hutchins, N.A.; Wang, F.; Wang, Y.; Chung, C.S.; Ayala, A. Kupffer cells potentiate liver sinusoidal endothelial cell injury in sepsis by ligating programmed cell death ligand-1. J. Leukoc. Biol. 2013, 94, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Lomas-Neira, J.; Monaghan, S.F.; Huang, X.; Fallon, E.A.; Chung, C.S.; Ayala, A. Novel Role for PD-1:PD-L1 as Mediator of Pulmonary Vascular Endothelial Cell Functions in Pathogenesis of Indirect ARDS in Mice. Front. Immunol. 2018, 9, 3030. [Google Scholar] [CrossRef] [Green Version]
- Tewalt, E.F.; Cohen, J.N.; Rouhani, S.J.; Guidi, C.J.; Qiao, H.; Fahl, S.P.; Conaway, M.R.; Bender, T.P.; Tung, K.S.; Vella, A.T.; et al. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood 2012, 120, 4772–4782. [Google Scholar] [CrossRef]
- Grabie, N.; Gotsman, I.; DaCosta, R.; Pang, H.; Stavrakis, G.; Butte, M.J.; Keir, M.E.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation 2007, 116, 2062–2071. [Google Scholar] [CrossRef] [Green Version]
- LaGier, A.J.; Pober, J.S. Immune accessory functions of human endothelial cells are modulated by overexpression of B7-H1 (PDL1). Hum. Immunol. 2006, 67, 568–578. [Google Scholar] [CrossRef]
- Pittet, C.L.; Newcombe, J.; Prat, A.; Arbour, N. Human brain endothelial cells endeavor to immunoregulate CD8 T cells via PD-1 ligand expression in multiple sclerosis. J. Neuroinflammation 2011, 8, 155. [Google Scholar] [CrossRef] [Green Version]
- Mazanet, M.M.; Hughes, C.C. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J. Immunol. 2002, 169, 3581–3588. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.J.; Hu, X.F.; Yan, M.; Zhang, W.Y.; Mao, X.B.; Shu, Y.W. Human umbilical vein endothelial cells promote the inhibitory activation of CD4(+)CD25(+)Foxp3(+) regulatory T cells via PD-L1. Atherosclerosis 2016, 244, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Latchman, Y.E.; Buhlmann, J.E.; Tomczak, M.F.; Horwitz, B.H.; Freeman, G.J.; Sharpe, A.H. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur. J. Immunol. 2003, 33, 2706–2716. [Google Scholar] [CrossRef]
- Duong, C.N.; Vestweber, D. Mechanisms Ensuring Endothelial Junction Integrity Beyond VE-Cadherin. Front. Physiol. 2020, 11, 519. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Anderson, J.M. Two classes of tight junctions are revealed by ZO-1 isoforms. Am. J. Physiol. 1993, 264, C918–C924. [Google Scholar] [CrossRef]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akwii, R.G.; Sajib, M.S.; Zahra, F.T.; Mikelis, C.M. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells 2019, 8, 471. [Google Scholar] [CrossRef] [Green Version]
- Thurston, G.; Daly, C. The complex role of angiopoietin-2 in the angiopoietin-tie signaling pathway. Cold Spring Harb. Perspect. Med. 2012, 2, a006550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eklund, L.; Kangas, J.; Saharinen, P. Angiopoietin-Tie signalling in the cardiovascular and lymphatic systems. Clin. Sci. 2017, 131, 87–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meurs, M.; Kümpers, P.; Ligtenberg, J.J.; Meertens, J.H.; Molema, G.; Zijlstra, J.G. Bench-to-bedside review: Angiopoietin signalling in critical illness—A future target? Crit. Care 2009, 13, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemieux, C.; Maliba, R.; Favier, J.; Théorêt, J.F.; Merhi, Y.; Sirois, M.G. Angiopoietins can directly activate endothelial cells and neutrophils to promote proinflammatory responses. Blood 2005, 105, 1523–1530. [Google Scholar] [CrossRef]
- Tayeh, M.A.; Scicli, A.G. Angiotensin II and bradykinin regulate the expression of P-selectin on the surface of endothelial cells in culture. Proc. Assoc. Am. Physicians 1998, 110, 412–421. [Google Scholar]
- Alvarez, A.; Cerdá-Nicolás, M.; Naim Abu Nabah, Y.; Mata, M.; Issekutz, A.C.; Panés, J.; Lobb, R.R.; Sanz, M.J. Direct evidence of leukocyte adhesion in arterioles by angiotensin II. Blood 2004, 104, 402–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potente, M.; Urbich, C.; Sasaki, K.; Hofmann, W.K.; Heeschen, C.; Aicher, A.; Kollipara, R.; DePinho, R.A.; Zeiher, A.M.; Dimmeler, S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Investig. 2005, 115, 2382–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity 2014, 41, 802–814. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Giobbie-Hurder, A.; Liao, X.; Connelly, C.; Connolly, E.M.; Li, J.; Manos, M.P.; Lawrence, D.; McDermott, D.; Severgnini, M.; et al. Angiopoietin-2 as a Biomarker and Target for Immune Checkpoint Therapy. Cancer Immunol. Res. 2017, 5, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Pacurari, M.; Kafoury, R.; Tchounwou, P.B.; Ndebele, K. The Renin-Angiotensin-aldosterone system in vascular inflammation and remodeling. Int. J. Inflamm. 2014, 2014, 689360. [Google Scholar] [CrossRef]
- Otani, A.; Takagi, H.; Oh, H.; Koyama, S.; Honda, Y. Angiotensin II induces expression of the Tie2 receptor ligand, angiopoietin-2, in bovine retinal endothelial cells. Diabetes 2001, 50, 867–875. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Zhou, J.; Chen, Y.; Chen, Y.; Chen, L.; Zhang, P.; Ma, L.; Jiang, Z.; Bian, J.; Yin, W. Angiotensin II contributes to intratumoral immunosuppressionvia induction of PD-L1 expression in non-small cell lung carcinoma. Int. Immunopharmacol. 2020, 84, 106507. [Google Scholar] [CrossRef]
- Lichtman, A.H. T cell costimulatory and coinhibitory pathways in vascular inflammatory diseases. Front. Physiol. 2012, 3, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Li, L.; Wu, Y.; Yang, K. PD-1/PD-L1 in cardiovascular disease. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 505, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Gotsman, I.; Grabie, N.; Dacosta, R.; Sukhova, G.; Sharpe, A.; Lichtman, A.H. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J. Clin. Investig. 2007, 117, 2974–2982. [Google Scholar] [CrossRef]
- Koga, N.; Suzuki, J.; Kosuge, H.; Haraguchi, G.; Onai, Y.; Futamatsu, H.; Maejima, Y.; Gotoh, R.; Saiki, H.; Tsushima, F.; et al. Blockade of the interaction between PD-1 and PD-L1 accelerates graft arterial disease in cardiac allografts. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2057–2062. [Google Scholar] [CrossRef] [Green Version]
- Cochain, C.; Chaudhari, S.M.; Koch, M.; Wiendl, H.; Eckstein, H.H.; Zernecke, A. Programmed cell death-1 deficiency exacerbates T cell activation and atherogenesis despite expansion of regulatory T cells in atherosclerosis-prone mice. PLoS ONE 2014, 9, e93280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, D.X.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1100–1107. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Zhu, L.; Wang, J.Z.; Wang, X.J.; Chen, J.Z.; Song, L.; Wu, Y.J.; Sun, K.; Yuan, Z.Y.; Hui, R. Methylation of FOXP3 in regulatory T cells is related to the severity of coronary artery disease. Atherosclerosis 2013, 228, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhang, K.; Li, J.; Dong, M.; Yang, J.; An, G.; Qin, W.; Gao, F.; Zhang, C.; Zhang, Y. Statins induce the accumulation of regulatory T cells in atherosclerotic plaque. Mol. Med. 2012, 18, 598–605. [Google Scholar] [CrossRef]
- Nus, M.; Sage, A.P.; Lu, Y.; Masters, L.; Lam, B.Y.H.; Newland, S.; Weller, S.; Tsiantoulas, D.; Raffort, J.; Marcus, D.; et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat. Med. 2017, 23, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, R.; Gao, W.; He, Z.; Wu, F.; Chu, Y.; Wu, J.; Ma, L.; Liang, C. Circulating CD4(+)CXCR5(+) T cells contribute to proinflammatory responses in multiple ways in coronary artery disease. Int. Immunopharmacol. 2017, 52, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Shirai, T.; Namkoong, H.; Zhang, H.; Berry, G.J.; Wallis, B.B.; Schaefgen, B.; Harrison, D.G.; Tremmel, J.A.; Giacomini, J.C.; et al. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J. Clin. Investig. 2017, 127, 2725–2738. [Google Scholar] [CrossRef]
- Qiu, M.K.; Wang, S.C.; Dai, Y.X.; Wang, S.Q.; Ou, J.M.; Quan, Z.W. PD-1 and Tim-3 Pathways Regulate CD8+ T Cells Function in Atherosclerosis. PLoS ONE 2015, 10, e0128523. [Google Scholar] [CrossRef]
- Jones, D.; Como, C.N.; Jing, L.; Blackmon, A.; Neff, C.P.; Krueger, O.; Bubak, A.N.; Palmer, B.E.; Koelle, D.M.; Nagel, M.A. Varicella zoster virus productively infects human peripheral blood mononuclear cells to modulate expression of immunoinhibitory proteins and blocking PD-L1 enhances virus-specific CD8+ T cell effector function. Plos Pathog. 2019, 15, e1007650. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.H.; Chuang, Y.S.; Lin, Y.T. Does Herpes Zoster Increase the Risk of Stroke and Myocardial Infarction? A Comprehensive Review. J. Clin. Med. 2019, 8, 547. [Google Scholar] [CrossRef] [Green Version]
- Cronin, D.C., 2nd; Stack, R.; Fitch, F.W. IL-4-producing CD8+ T cell clones can provide B cell help. J. Immunol. 1995, 154, 3118–3127. [Google Scholar]
- Asciutto, G.; Wigren, M.; Fredrikson, G.N.; Mattisson, I.Y.; Grönberg, C.; Alm, R.; Björkbacka, H.; Dias, N.V.; Edsfeldt, A.; Gonçalves, I.; et al. Apolipoprotein B-100 Antibody Interaction With Atherosclerotic Plaque Inflammation and Repair Processes. Stroke 2016, 47, 1140–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitriu, I.E. The life (and death) of CD4+ CD28(null) T cells in inflammatory diseases. Immunology 2015, 146, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Behnamfar, N.; Zibaeenezhad, M.J.; Golmoghaddam, H.; Doroudchi, M. CD45RO+ memory T-cells produce IL-17 in patients with atherosclerosis. Cell. Mol. Biol. (Noisy-Le-Grandfrance) 2015, 61, 17–23. [Google Scholar]
- Lio, W.M.; Cercek, B.; Yano, J.; Yang, W.; Ghermezi, J.; Zhao, X.; Zhou, J.; Zhou, B.; Freeman, M.R.; Chyu, K.Y.; et al. Sex as a Determinant of Responses to a Coronary Artery Disease Self-Antigen Identified by Immune-Peptidomics. Front. Immunol. 2020, 11, 694. [Google Scholar] [CrossRef] [Green Version]
- Mihailovic, P.M.; Lio, W.M.; Herscovici, R.; Chyu, K.Y.; Yano, J.; Zhao, X.; Zhou, J.; Zhou, B.; Freeman, M.R.; Yang, W.; et al. Keratin 8 is a potential self-antigen in the coronary artery disease immunopeptidome: A translational approach. PLoS ONE 2019, 14, e0213025. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhuang, Y.; Wei, X.; Shang, F.; Wang, J.; Zhang, Y.; Liu, X.; Yang, Y.; Liu, L.; Zheng, Q. Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J. Mol. Cell. Cardiol. 2009, 46, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Ye, J.; Ma, Y.; Hua, P.; Huang, Y.; Fu, X.; Li, D.; Yuan, M.; Xia, Z. Function of T regulatory type 1 cells is down-regulated and is associated with the clinical presentation of coronary artery disease. Hum. Immunol. 2018, 79, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, P.; Karyampudi, L.; Shreeder, B.; Krempski, J.; Bahr, D.; Daum, J.; Kalli, K.R.; Goode, E.L.; Block, M.S.; Cannon, M.J.; et al. IL10 Release upon PD-1 Blockade Sustains Immunosuppression in Ovarian Cancer. Cancer Res. 2017, 77, 6667–6678. [Google Scholar] [CrossRef] [Green Version]
- Porichis, F.; Hart, M.G.; Zupkosky, J.; Barblu, L.; Kwon, D.S.; McMullen, A.; Brennan, T.; Ahmed, R.; Freeman, G.J.; Kavanagh, D.G.; et al. Differential impact of PD-1 and/or interleukin-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions. J. Virol. 2014, 88, 2508–2518. [Google Scholar] [CrossRef] [Green Version]
- Salem, J.E.; Manouchehri, A.; Moey, M.; Lebrun-Vignes, B.; Bastarache, L.; Pariente, A.; Gobert, A.; Spano, J.P.; Balko, J.M.; Bonaca, M.P.; et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: An observational, retrospective, pharmacovigilance study. Lancet. Oncol. 2018, 19, 1579–1589. [Google Scholar] [CrossRef]
- Young, M.R. Endothelial cells in the eyes of an immunologist. Cancer Immunol. Immunother. 2012, 61, 1609–1616. [Google Scholar] [CrossRef] [Green Version]
- Naghavi, M.; Wyde, P.; Litovsky, S.; Madjid, M.; Akhtar, A.; Naguib, S.; Siadaty, M.S.; Sanati, S.; Casscells, W. Influenza infection exerts prominent inflammatory and thrombotic effects on the atherosclerotic plaques of apolipoprotein E-deficient mice. Circulation 2003, 107, 762–768. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.C.; Ed Rainger, G.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial dysfunction in COVID-19: A position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef]
- Warren-Gash, C.; Smeeth, L.; Hayward, A.C. Influenza as a trigger for acute myocardial infarction or death from cardiovascular disease: A systematic review. Lancet. Infect. Dis. 2009, 9, 601–610. [Google Scholar] [CrossRef]
- Cappuccio, F.P.; Siani, A. Covid-19 and cardiovascular risk: Susceptibility to infection to SARS-CoV-2, severity and prognosis of Covid-19 and blockade of the renin-angiotensin-aldosterone system. An evidence-based viewpoint. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1227–1235. [Google Scholar] [CrossRef]
- Madjid, M.; Aboshady, I.; Awan, I.; Litovsky, S.; Casscells, S.W. Influenza and cardiovascular disease: Is there a causal relationship? Tex. Heart Inst. J. 2004, 31, 4–13. [Google Scholar]
- Rutigliano, J.A.; Sharma, S.; Morris, M.Y.; Oguin, T.H., 3rd; McClaren, J.L.; Doherty, P.C.; Thomas, P.G. Highly pathological influenza A virus infection is associated with augmented expression of PD-1 by functionally compromised virus-specific CD8+ T cells. J. Virol. 2014, 88, 1636–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valero-Pacheco, N.; Arriaga-Pizano, L.; Ferat-Osorio, E.; Mora-Velandia, L.M.; Pastelin-Palacios, R.; Villasís-Keever, M.; Alpuche-Aranda, C.; Sánchez-Torres, L.E.; Isibasi, A.; Bonifaz, L.; et al. PD-L1 expression induced by the 2009 pandemic influenza A(H1N1) virus impairs the human T cell response. Clin. Dev. Immunol. 2013, 2013, 989673. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.S.; Aslam, S.; Kontopantelis, E.; Myint, P.K.; Zaman, M.J.; Buchan, I.; Loke, Y.K.; Mamas, M.A. Influenza, influenza-like symptoms and their association with cardiovascular risks: A systematic review and meta-analysis of observational studies. Int. J. Clin. Pract. 2015, 69, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Bentebibel, S.E.; Khurana, S.; Schmitt, N.; Kurup, P.; Mueller, C.; Obermoser, G.; Palucka, A.K.; Albrecht, R.A.; Garcia-Sastre, A.; Golding, H.; et al. ICOS(+)PD-1(+)CXCR3(+) T follicular helper cells contribute to the generation of high-avidity antibodies following influenza vaccination. Sci. Rep. 2016, 6, 26494. [Google Scholar] [CrossRef]
- Elsner, R.A.; Ernst, D.N.; Baumgarth, N. Single and coexpression of CXCR4 and CXCR5 identifies CD4 T helper cells in distinct lymph node niches during influenza virus infection. J. Virol. 2012, 86, 7146–7157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.Y.; Ma, Y.T.; Zhang, J.Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Reviews. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G. Virology, Epidemiology, Pathogenesis, and Control of COVID-19. Viruses 2020, 12, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellesi, S.; Metafuni, E.; Hohaus, S.; Maiolo, E.; Marchionni, F.; D’Innocenzo, S.; La Sorda, M.; Ferraironi, M.; Ramundo, F.; Fantoni, M.; et al. Increased CD95 (Fas) and PD-1 expression in peripheral blood T lymphocytes in COVID-19 patients. Br. J. Haematol. 2020, 191, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.K.; Han, G.C.; Kim, M.; Kim, G.; Shin, H.M.; Song, K.H.; Choe, P.G.; Park, W.B.; Kim, E.S.; Kim, H.B.; et al. Aberrant hyperactivation of cytotoxic T-cell as a potential determinant of COVID-19 severity. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2020, 97, 313–321. [Google Scholar]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Reviews. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Mayr, M.; Kiechl, S.; Willeit, J.; Wick, G.; Xu, Q. Infections, immunity, and atherosclerosis: Associations of antibodies to Chlamydia pneumoniae, Helicobacter pylori, and cytomegalovirus with immune reactions to heat-shock protein 60 and carotid or femoral atherosclerosis. Circulation 2000, 102, 833–839. [Google Scholar] [CrossRef] [Green Version]
- Inozume, T.; Hanada, K.; Wang, Q.J.; Ahmadzadeh, M.; Wunderlich, J.R.; Rosenberg, S.A.; Yang, J.C. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J. Immunother. 2010, 33, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Adam, E.; Melnick, J.L.; Probtsfield, J.L.; Petrie, B.L.; Burek, J.; Bailey, K.R.; McCollum, C.H.; DeBakey, M.E. High levels of cytomegalovirus antibody in patients requiring vascular surgery for atherosclerosis. Lancet 1987, 2, 291–293. [Google Scholar] [CrossRef]
- Yang, F.J.; Shu, K.H.; Chen, H.Y.; Chen, I.Y.; Lay, F.Y.; Chuang, Y.F.; Wu, C.S.; Tsai, W.C.; Peng, Y.S.; Hsu, S.P.; et al. Anti-cytomegalovirus IgG antibody titer is positively associated with advanced T cell differentiation and coronary artery disease in end-stage renal disease. Immun. Ageing 2018, 15, 15. [Google Scholar] [CrossRef] [Green Version]
- Beyaz, M.O.; Ugurlucan, M.; Oztas, D.M.; Meric, M.; Conkbayir, C.; Agacfidan, A.; Onel, M.; Alpagut, U. Evaluation of the relationship between plaque formation leading to symptomatic carotid artery stenosis and cytomegalovirus by investigating the virus DNA. Arch. Med Sci. Atheroscler. Dis. 2019, 4, e19–e24. [Google Scholar] [CrossRef] [Green Version]
- Rahbar, A.; Söderberg-Nauclér, C. Human cytomegalovirus infection of endothelial cells triggers platelet adhesion and aggregation. J. Virol. 2005, 79, 2211–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvain, J.; Collet, J.P.; Nagaswami, C.; Beygui, F.; Edmondson, K.E.; Bellemain-Appaix, A.; Cayla, G.; Pena, A.; Brugier, D.; Barthelemy, O.; et al. Composition of coronary thrombus in acute myocardial infarction. J. Am. Coll. Cardiol. 2011, 57, 1359–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, J.; Shmeleva, E.V.; Boag, S.E.; Fiser, K.; Bagnall, A.; Murali, S.; Dimmick, I.; Pircher, H.; Martin-Ruiz, C.; Egred, M.; et al. Myocardial ischemia and reperfusion leads to transient CD8 immune deficiency and accelerated immunosenescence in CMV-seropositive patients. Circ. Res. 2015, 116, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Góis, J.; Higuchi, M.; Reis, M.; Diament, J.; Sousa, J.; Ramires, J.; Oliveira, S. Infectious agents, inflammation, and growth factors: How do they interact in the progression or stabilization of mild human atherosclerotic lesions? Ann. Vasc. Surg. 2006, 20, 638–645. [Google Scholar] [CrossRef]
- Cao, J.; Mao, Y.; Dong, B.; Guan, W.; Shi, J.; Wang, S. Detection of specific Chlamydia pneumoniae and cytomegalovirus antigens in human carotid atherosclerotic plaque in a Chinese population. Oncotarget 2017, 8, 55435–55442. [Google Scholar] [CrossRef] [Green Version]
- Jaff, M.R.; Dale, R.A.; Creager, M.A.; Lipicky, R.J.; Constant, J.; Campbell, L.A.; Hiatt, W.R. Anti-chlamydial antibiotic therapy for symptom improvement in peripheral artery disease: Prospective evaluation of rifalazil effect on vascular symptoms of intermittent claudication and other endpoints in Chlamydia pneumoniae seropositive patients (PROVIDENCE-1). Circulation 2009, 119, 452–458. [Google Scholar]
- Evani, S.J.; Ramasubramanian, A.K. Biophysical regulation of Chlamydia pneumoniae-infected monocyte recruitment to atherosclerotic foci. Sci. Rep. 2016, 6, 19058. [Google Scholar] [CrossRef] [Green Version]
- Tumurkhuu, G.; Dagvadorj, J.; Porritt, R.A.; Crother, T.R.; Shimada, K.; Tarling, E.J.; Erbay, E.; Arditi, M.; Chen, S. Chlamydia pneumoniae Hijacks a Host Autoregulatory IL-1β Loop to Drive Foam Cell Formation and Accelerate Atherosclerosis. Cell Metab. 2018, 28, 432–448.e4. [Google Scholar] [CrossRef] [Green Version]
- Porritt, R.A.; Crother, T.R. Chlamydia pneumoniae Infection and Inflammatory Diseases. Forum Immunopathol. Dis. Ther. 2016, 7, 237–254. [Google Scholar] [CrossRef]
- Starkey, M.R.; Nguyen, D.H.; Brown, A.C.; Essilfie, A.T.; Kim, R.Y.; Yagita, H.; Horvat, J.C.; Hansbro, P.M. Programmed Death Ligand 1 Promotes Early-Life Chlamydia Respiratory Infection-Induced Severe Allergic Airway Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 493–503. [Google Scholar] [CrossRef]
- Cardilo-Reis, L.; Gruber, S.; Schreier, S.M.; Drechsler, M.; Papac-Milicevic, N.; Weber, C.; Wagner, O.; Stangl, H.; Soehnlein, O.; Binder, C.J. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. Embo Mol. Med. 2012, 4, 1072–1086. [Google Scholar] [CrossRef] [PubMed]
- Berry, A.; Balard, P.; Coste, A.; Olagnier, D.; Lagane, C.; Authier, H.; Benoit-Vical, F.; Lepert, J.C.; Séguéla, J.P.; Magnaval, J.F.; et al. IL-13 induces expression of CD36 in human monocytes through PPARgamma activation. Eur. J. Immunol. 2007, 37, 1642–1652. [Google Scholar] [CrossRef]
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieniazek, P.; Karczewska, E.; Duda, A.; Tracz, W.; Pasowicz, M.; Konturek, S.J. Association of Helicobacter pylori infection with coronary heart disease. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 1999, 50, 743–751. [Google Scholar]
- Murray, L.J.; Bamford, K.B.; Kee, F.; McMaster, D.; Cambien, F.; Dallongeville, J.; Evans, A. Infection with virulent strains of Helicobacter pylori is not associated with ischaemic heart disease: Evidence from a population-based case-control study of myocardial infarction. Atherosclerosis 2000, 149, 379–385. [Google Scholar] [CrossRef]
- Sawayama, Y.; Hamada, M.; Otaguro, S.; Maeda, S.; Ohnishi, H.; Fujimoto, Y.; Taira, Y.; Hayashi, J. Chronic Helicobacter pylori infection is associated with peripheral arterial disease. J. Infect. Chemother. Off. J. Jpn. Soc. Chemother. 2008, 14, 250–254. [Google Scholar] [CrossRef]
- Pietroiusti, A.; Diomedi, M.; Silvestrini, M.; Cupini, L.M.; Luzzi, I.; Gomez-Miguel, M.J.; Bergamaschi, A.; Magrini, A.; Carrabs, T.; Vellini, M.; et al. Cytotoxin-associated gene-A--positive Helicobacter pylori strains are associated with atherosclerotic stroke. Circulation 2002, 106, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Elkind, M.S.; Luna, J.M.; Moon, Y.P.; Boden-Albala, B.; Liu, K.M.; Spitalnik, S.; Rundek, T.; Sacco, R.L.; Paik, M.C. Infectious burden and carotid plaque thickness: The northern Manhattan study. Stroke 2010, 41, e117–e122. [Google Scholar] [CrossRef] [Green Version]
- Adachi, K.; Arima, N.; Takashima, T.; Miyaoka, Y.; Yuki, M.; Ono, M.; Komazawa, Y.; Kawamura, A.; Fujishiro, H.; Ishihara, S.; et al. Pulse-wave velocity and cardiovascular risk factors in subjects with Helicobacter pylori infection. J. Gastroenterol. Hepatol. 2003, 18, 771–777. [Google Scholar] [CrossRef]
- Huang, B.; Chen, Y.; Xie, Q.; Lin, G.; Wu, Y.; Feng, Y.; Li, J.; Zhuo, Y.; Zhang, P. CagA-positive Helicobacter pylori strains enhanced coronary atherosclerosis by increasing serum OxLDL and HsCRP in patients with coronary heart disease. Dig. Dis. Sci. 2011, 56, 109–114. [Google Scholar] [CrossRef]
- Iwai, N.; Okuda, T.; Oka, K.; Hara, T.; Inada, Y.; Tsuji, T.; Komaki, T.; Inoue, K.; Dohi, O.; Konishi, H.; et al. Helicobacter pylori eradication increases the serum high density lipoprotein cholesterol level in the infected patients with chronic gastritis: A single-center observational study. PLoS ONE 2019, 14, e0221349. [Google Scholar] [CrossRef]
- Das, S.; Suarez, G.; Beswick, E.J.; Sierra, J.C.; Graham, D.Y.; Reyes, V.E. Expression of B7-H1 on gastric epithelial cells: Its potential role in regulating T cells during Helicobacter pylori infection. J. Immunol. 2006, 176, 3000–3009. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.Y.; Lin, C.W.; Cheng, K.S.; Lin, C.; Wang, Y.M.; Lin, I.T.; Chou, Y.H.; Hsu, P.N. Increased programmed death-ligand-1 expression in human gastric epithelial cells in Helicobacter pylori infection. Clin. Exp. Immunol. 2010, 161, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Beswick, E.J.; Pinchuk, I.V.; Das, S.; Powell, D.W.; Reyes, V.E. Expression of the programmed death ligand 1, B7-H1, on gastric epithelial cells after Helicobacter pylori exposure promotes development of CD4+ CD25+ FoxP3+ regulatory T cells. Infect. Immun. 2007, 75, 4334–4341. [Google Scholar] [CrossRef] [Green Version]
- Hueso, M.; Mallén, A.; Casas, Á.; Guiteras, J.; Sbraga, F.; Blasco-Lucas, A.; Lloberas, N.; Torras, J.; Cruzado, J.M.; Navarro, E. Integrated miRNA/mRNA Counter-Expression Analysis Highlights Oxidative Stress-Related Genes CCR7 and FOXO1 as Blood Markers of Coronary Arterial Disease. Int. J. Mol. Sci. 2020, 21, 1943. [Google Scholar] [CrossRef] [Green Version]
- Paredes, S.; Fonseca, L.; Ribeiro, L.; Ramos, H.; Oliveira, J.C.; Palma, I. Novel and traditional lipid profiles in Metabolic Syndrome reveal a high atherogenicity. Sci. Rep. 2019, 9, 11792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, R.J.; Peng, S.K. The role of cholesterol oxidation products in the pathogenesis of atherosclerosis. Ann. Clin. Lab. Sci. 1989, 19, 225–237. [Google Scholar]
- Kuo, A.; Lee, M.Y.; Sessa, W.C. Lipid Droplet Biogenesis and Function in the Endothelium. Circ. Res. 2017, 120, 1289–1297. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Singh, R. Autophagy and Lipid Droplets in the Liver. Annu. Rev. Nutr. 2015, 35, 215–237. [Google Scholar] [CrossRef]
- Saito, T.; Kuma, A.; Sugiura, Y.; Ichimura, Y.; Obata, M.; Kitamura, H.; Okuda, S.; Lee, H.C.; Ikeda, K.; Kanegae, Y.; et al. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 2019, 10, 1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [Green Version]
- Staiger, H.; Staiger, K.; Stefan, N.; Wahl, H.G.; Machicao, F.; Kellerer, M.; Häring, H.U. Palmitate-induced interleukin-6 expression in human coronary artery endothelial cells. Diabetes 2004, 53, 3209–3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Zhang, H.; Yuan, Y.; He, Q.; Zhou, J.; Li, S.; Sun, Y.; Li, D.Y.; Qiu, H.B.; Wang, W.; et al. Fatty Acid Oxidation Controls CD8(+) Tissue-Resident Memory T-cell Survival in Gastric Adenocarcinoma. Cancer Immunol. Res. 2020, 8, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Wei, G.; Huang, J.; Pang, S.; Liu, L.; Yan, B. Decreased gene expression of LC3 in peripheral leucocytes of patients with coronary artery disease. Eur. J. Clin. Investig. 2011, 41, 958–963. [Google Scholar] [CrossRef]
- Kang, J.; Ding, Z.; Luo, Y.; Yang, Y. Expression of autophagy related gene 5 and cyclin E in coronary heart disease and its clinical significance. Zhong Nan Da Xue Xue Bao. Yi Xue Ban J. Cent. South Univ. Med Sci. 2020, 45, 17–23. [Google Scholar]
- Khalil, H.; Abd El Maksoud, A.I.; Alian, A.; El-Hamady, W.A.; Daif, A.A.; Awad, A.M.; Guirgis, A.A. Interruption of Autophagosome Formation in Cardiovascular Disease, an Evidence for Protective Response of Autophagy. Immunol. Investig. 2020, 49, 249–263. [Google Scholar] [CrossRef]
- Wang, C.; Xu, W.; Liang, M.; Huang, D.; Huang, K. CTRP13 inhibits atherosclerosis via autophagy-lysosome-dependent degradation of CD36. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 2290–2300. [Google Scholar] [CrossRef] [Green Version]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef] [Green Version]
- Hubler, M.J.; Kennedy, A.J. Role of lipids in the metabolism and activation of immune cells. J. Nutr. Biochem. 2016, 34, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wu, W.K.K.; Gao, J.; Li, Z.; Dong, B.; Lin, X.; Li, Y.; Li, Y.; Gong, J.; Qi, C.; et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J. Exp. Clin. Cancer Res. 2019, 38, 140. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Zhang, J.; Ren, X. PD-L1 regulates tumorigenesis and autophagy of ovarian cancer by activating mTORC signaling. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus-Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, M.K.; Ghatge, M.; Flora, G.D.; Dhanesha, N.; Jain, M.; Markan, K.R.; Potthoff, M.J.; Lentz, S.R.; Chauhan, A.K. Metabolic enzyme pyruvate kinase M2 regulates platelet function and arterial thrombosis. Blood 2020. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Folco, E.J.; Sukhova, G.K.; Quillard, T.; Libby, P. Moderate hypoxia potentiates interleukin-1β production in activated human macrophages. Circ. Res. 2014, 115, 875–883. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. New Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Lutgens, E.; Seijkens, T.T.P. Cancer patients receiving immune checkpoint inhibitor therapy are at an increased risk for atherosclerotic cardiovascular disease. J. Immunother. Cancer 2020, 8, e000300. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, K.; Ieda, M. Cardiac Complications in Immune Checkpoint Inhibition Therapy. Front. Cardiovasc. Med. 2019, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, M.; Béjot, Y.; Rothwell, P.M.; Touzé, E. Long-Term Risk of Myocardial Infarction Compared to Recurrent Stroke After Transient Ischemic Attack and Ischemic Stroke: Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2018, 7, e007267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornhill, J.F.; Herderick, E.E.; Vince, D.G. The clinical morphology of human atherosclerotic lesions. Lessons from the PDAY Study. Pathobiological Determinants of Atherosclerosis in Youth. Wien. Klin. Wochenschr. 1995, 107, 540–543. [Google Scholar] [PubMed]
- Strong, J.P. Natural history and risk factors for early human atherogenesis. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Clin. Chem. 1995, 41, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.M.; Cheng, R.; Feldman, L.E. Hepatotoxicity and Recurrent NSTEMI While on Pembrolizumab for Metastatic Giant Cell Bone Tumor. Am. J. Med Sci. 2019, 357, 343–347. [Google Scholar] [CrossRef]
- Xia, W.; Zou, C.; Chen, H.; Xie, C.; Hou, M. Immune checkpoint inhibitor induces cardiac injury through polarizing macrophages via modulating microRNA-34a/Kruppel-like factor 4 signaling. Cell Death Dis. 2020, 11, 575. [Google Scholar] [CrossRef]
- Taleb, S.; Tedgui, A. IL-17 in atherosclerosis: The good and the bad. Cardiovasc. Res. 2018, 114, 7–9. [Google Scholar] [CrossRef]
- Weyand, C.M.; Berry, G.J.; Goronzy, J.J. The immunoinhibitory PD-1/PD-L1 pathway in inflammatory blood vessel disease. J. Leukoc. Biol. 2018, 103, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]



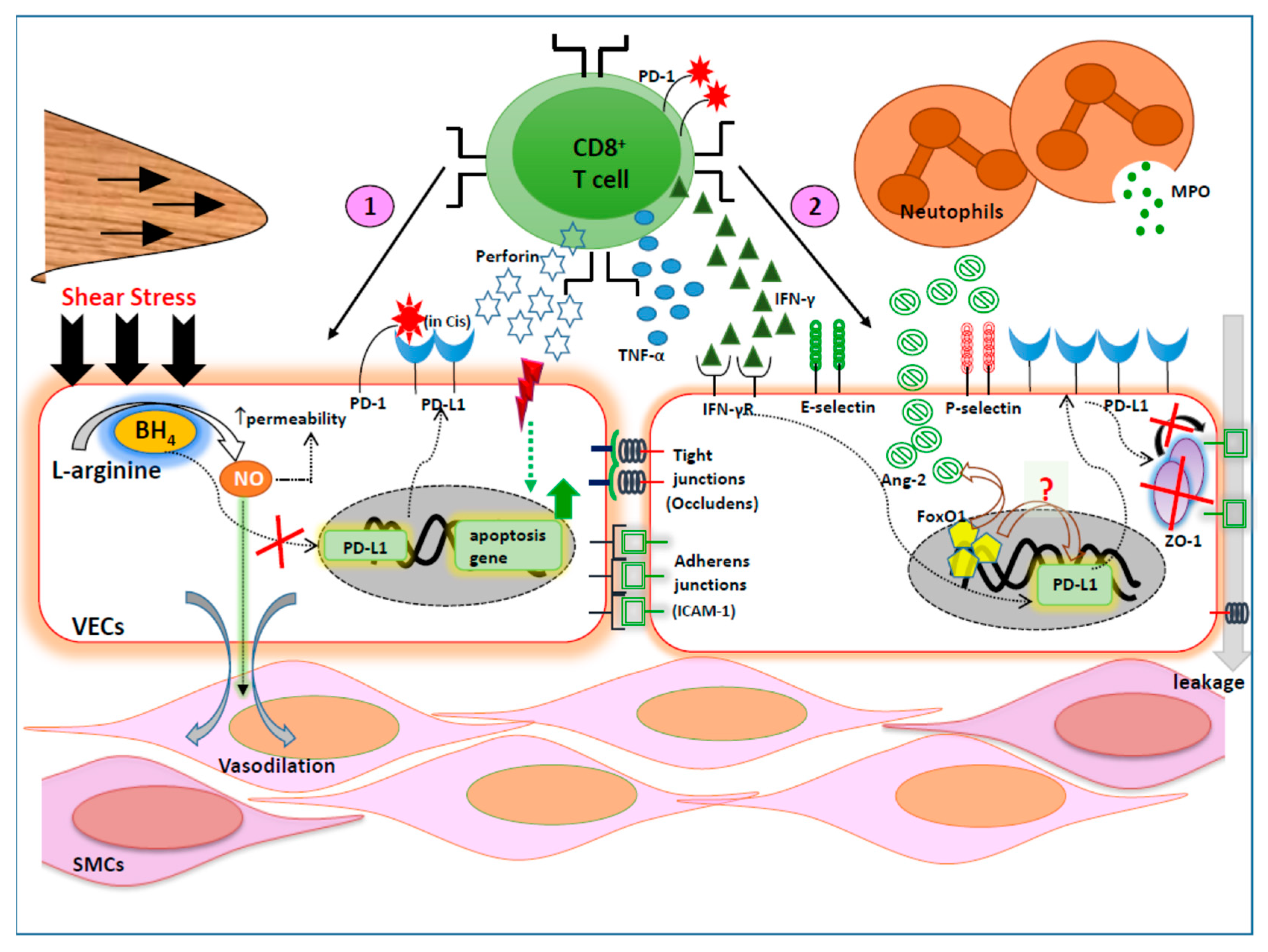{kind=link}
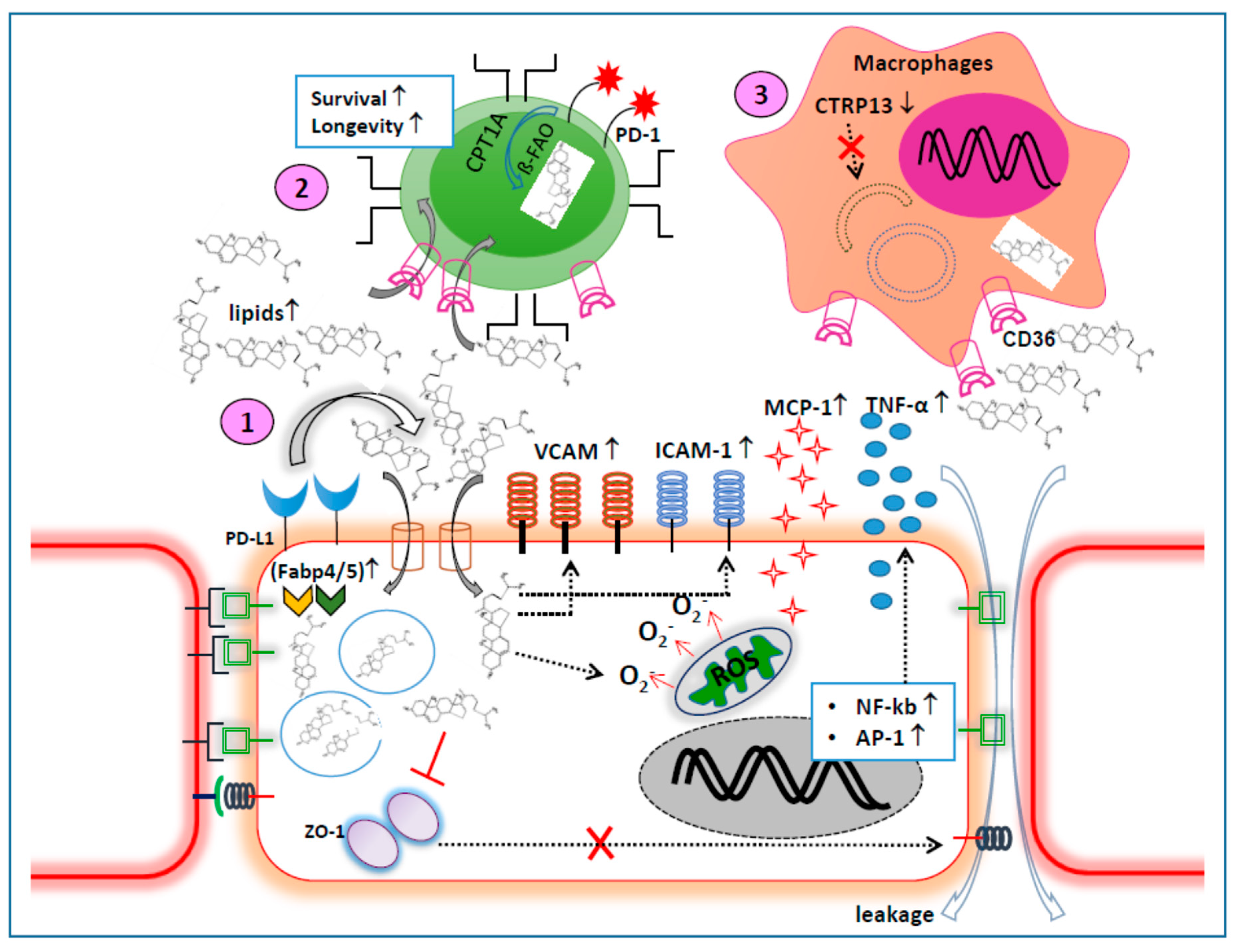{kind=link}
| Blood Vessel Diseases and References | Disease-Specific PD-1, PD-L1/PD-L2 Expression, Sample Origin (Cells/Tissues) and Sample Type (DNA/Transcript/Protein) | Inferences | Species Reported | Population Studied |
|---|---|---|---|---|
| GCA [29,30] | 1. ↑ PD-1 (t) and ↓ PD-L1 (t) on temporal arteries; ↑ PD-1 (p) and ↓ PD-L1 (p) on vascular T cells and DCs, respectively | 1. Inhibiting PD-1 pathway (i) increases vascular inflammation; (ii) aggravates maladaptive remodeling of arterial wall | human and mice (NOD. Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG)) | Caucasian /Hispanian/ African-American |
| 2. ↓ PD-1 (p) on blood CD4+T cells and no differences PD-L1/PD-L2 (p) on blood monocytes; ↑ PD-1 (p) and PD-L1 (p) on temporal arteries | 2. Minimizing immune activation and preventing further damage the vessel wall | Human | Dutch | |
| KD [31] | ↑ T allele frequency of PD-1 gene SNP (rs41386349) | PD-1 genetic predisposition in contribution to KD | Human | Korean |
| BD [32,33,34] | 1. ↓ PD-L1 (p) on APCs and cutaneous lesions and ↓ PD-L1 (t) in PBMCs | 1. Disrupted PD-L1 contribute to the development of BD | Human | Korean |
| 2. DNA from blood samples lack gene polymorphisms in PD-1, PD-L1 and PD-L2 (SNP’s: PD-1 rs2227981 and rs10204525, PD-L1 rs1970000 and PD-L2 rs7854303) | 2. Negligible role of PD-1 and its ligands in BD | Human | Chinese Han | |
| GPA (fWG) [35,36,37] | 1. ↑ PD-1 (p) on T cells; No PD-1 (p) on lesional T cells of renal biopsies with necrotizing & crescentic glomerulonephritis | 1. ↑ PD-1+ T cells were positively correlated with activation state including CD28null memory Th cells as well as T effector memory cells, IFN-γ+ T cells, induction of PD-1 by chronic CMV infection; PD-1+CD4+ CD25+ T cells were negatively correlated with the relapse rate | Human | German |
| 2. DNA isolated from blood lack PD-1.3G/A polymorphism (+ 7146G/A) and PD-1.5C/T polymorphism (+ 7785C/T) and also SNP’s in intron 4 and exon-5 in PDCD1 gene | 2. Co-occurance of PD-1.5 T allele with CTLA4 + 49 AA homozygosity was reduced among the patients. Apart, no obvious role of PD-1 in GPA and ANCA-associated GPA | Human | Swedish, Dutch and Caucasians | |
| CSV [36] | DNA isolated from blood lack PD-1.3G/A polymorphism (+ 7146G/A) and PD-1.5C/T polymorphism (+ 7785C/T) in PDCD1 gene | No obvious role of PD-1 in CSV and ANCA-associated CSV | Human | Dutch and Caucasians |
| Endothelial Cell Origin and References | Basal Expression of PD-L1/PD-1 | Inflamed Expression of PD-L1/PD-1 | Species Studied |
|---|---|---|---|
| Corneal EC [84] | Constitutive PD-L1 expression | Enhanced expression by IFN-γ | Human |
| Corneal EC [85] | No PD-L1 expression | Enhanced PD-L1 expression after electrocautery | Mouse |
| Lung and heart EC, microvascular EC line MS-1 [86] | Constitutive expression of PD-L1 | Not investigated | Mouse |
| Infantile haemangiomas and venous malformalies ECs [87] | No PD-L1 expression High PD-1 expression | Not investigated | Human |
| Skin tissue EC [87] | No PD-L1 and PD-1 expression | Not investigated | Human |
| Microvascular pancreatic ECs [70] | Constitutive expression | Enhanced PD-L1 expression by IFN-α, -β and -γ | Mouse |
| Lung, heart, pancreas and stomach ECs [70] | Constitutive expression | Not investigated | Mouse |
| Brain tissue EC [70] | Not investigated | Enhanced PD-L1 expression by IL-12 | Mouse |
| Liver sinuid EC [88] | Basal PD-L1 expression | Enhanced PD-L1 expression in sepsis model of induced peritonitis | Mouse |
| Lung EC [89] | Basal PD-L1 expression | Enhanced PD-L1 expression in hemorrhagic shock model | Mouse |
| Lymphatic EC [90] | Basal expression of PD-L1 | Not investigated | Mouse |
| HUVEC [73] | No basal PD-L1 expression | Enhanced by IFN-γ and TNF-α | Human |
| Heart EC [73] | No basal PD-L1 expression | Enhanced by IFN-γ and TNF-α | Mouse |
| Heart EC [91] | No basal PD-L1 expression | Enhanced after in vivo activation by CD8+ T cells | Mouse |
| HUVEC [92] | Low basal PD-L1 expression | Enhanced by IFN-y and -α, TNF-α, CD4+ T cells | Human |
| Brain EC [93] | No basal expression | Enhanced by TNF-α and IFN-γ | Human |
| HUVEC [94] | No basal expression | Enhanced by IFN-γ but not by TNF-α | Human |
| HUVEC [95] | No basal expression | Enhanced by coculture with CD4+CD25+foxp3+ regulatory T cells | Human |
| Heart and Brain EC [96] | Basal PD-L1 expression in heart EC | Enhanced PD-L1 expression on brain ECs in vivo by autoimmune encephalomyelitis | Mouse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veluswamy, P.; Wacker, M.; Scherner, M.; Wippermann, J. Delicate Role of PD-L1/PD-1 Axis in Blood Vessel Inflammatory Diseases: Current Insight and Future Significance. Int. J. Mol. Sci. 2020, 21, 8159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218159
Veluswamy P, Wacker M, Scherner M, Wippermann J. Delicate Role of PD-L1/PD-1 Axis in Blood Vessel Inflammatory Diseases: Current Insight and Future Significance. International Journal of Molecular Sciences. 2020; 21(21):8159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218159
Chicago/Turabian StyleVeluswamy, Priya, Max Wacker, Maximilian Scherner, and Jens Wippermann. 2020. "Delicate Role of PD-L1/PD-1 Axis in Blood Vessel Inflammatory Diseases: Current Insight and Future Significance" International Journal of Molecular Sciences 21, no. 21: 8159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218159