Brain Distribution of Dual ABCB1/ABCG2 Substrates Is Unaltered in a Beta-Amyloidosis Mouse Model

 , , , and
, , , and
Abstract
:1. Introduction
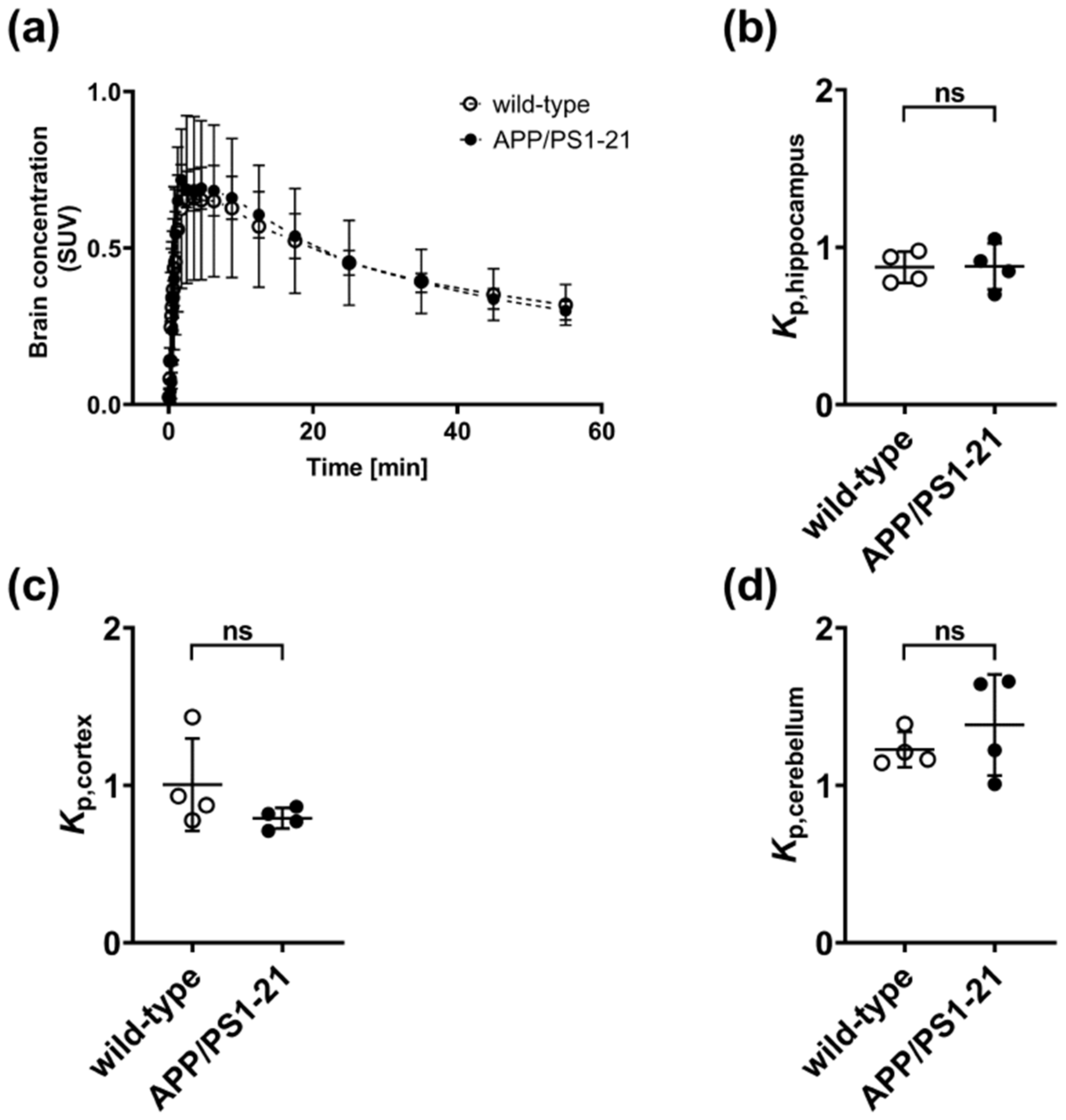
2. Results
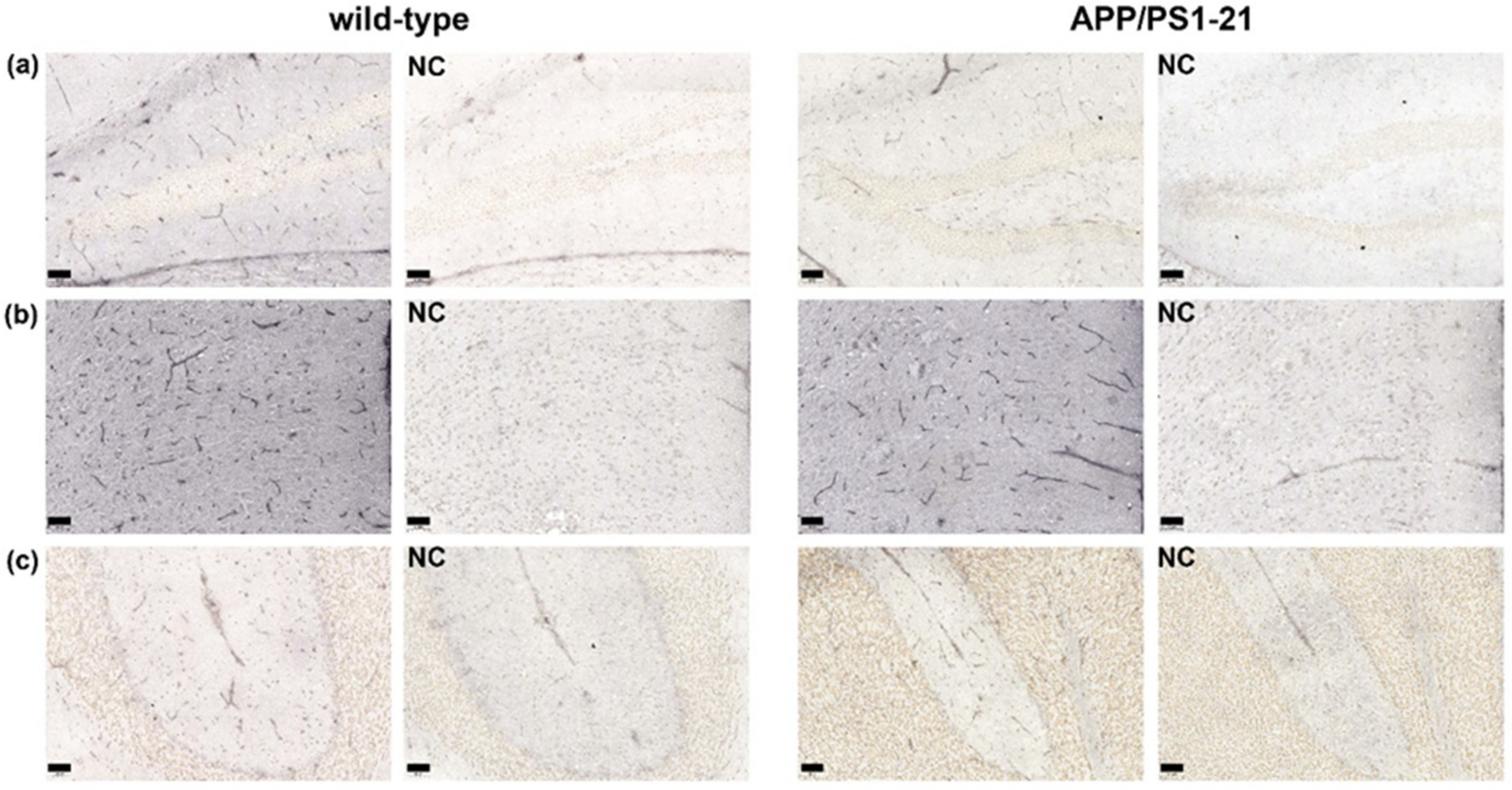
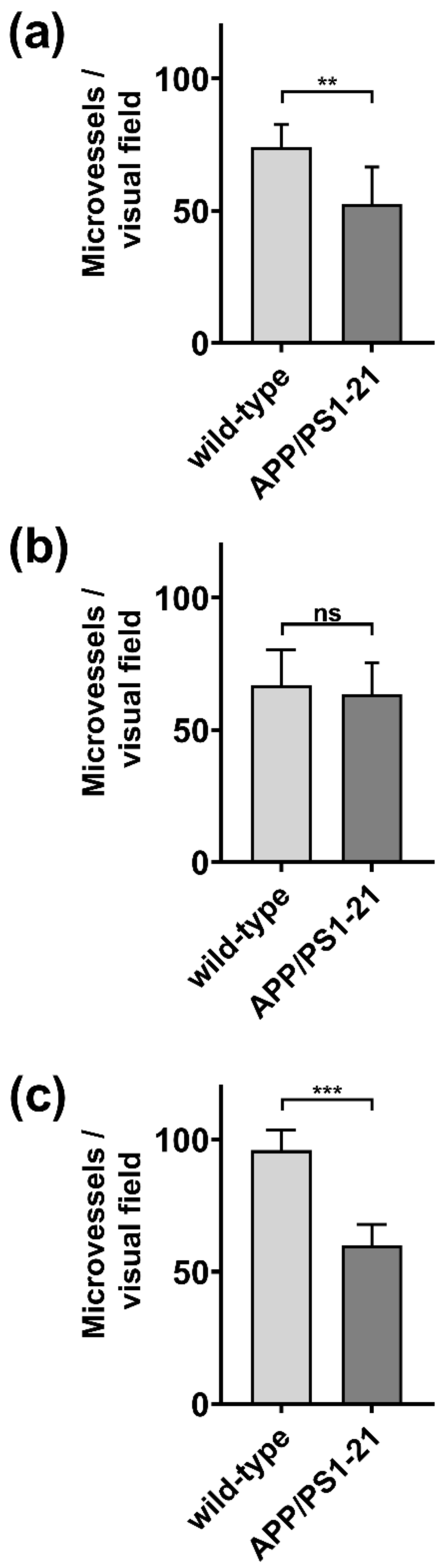
2.1. Immunohistochemistry
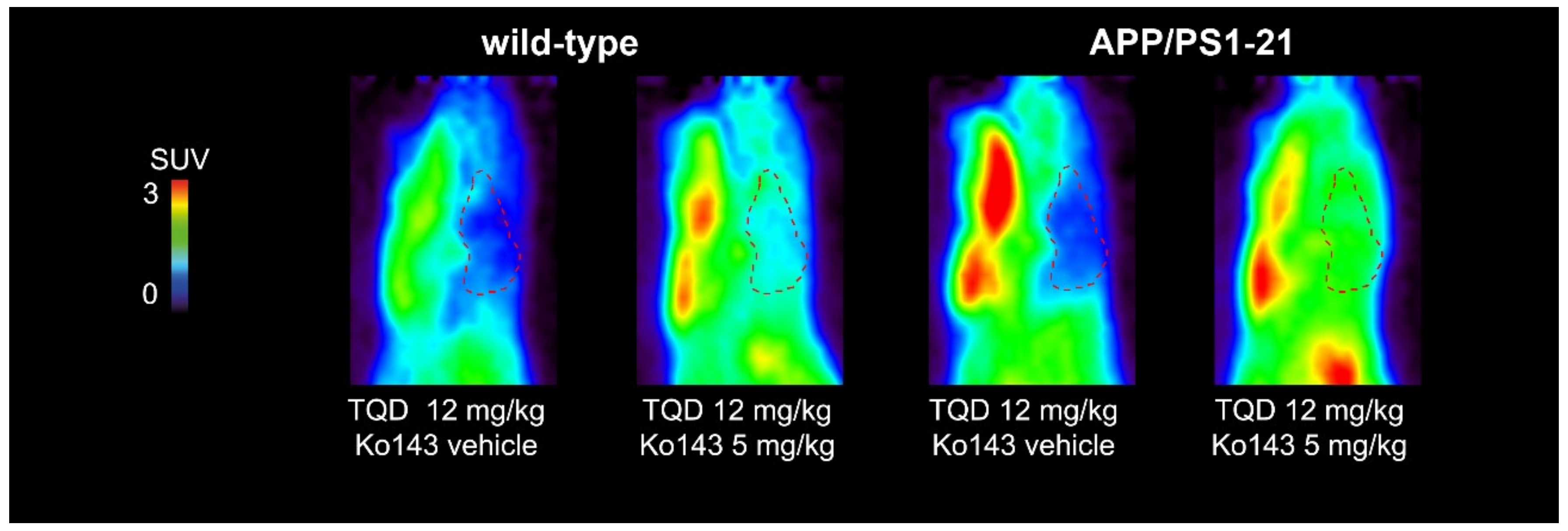
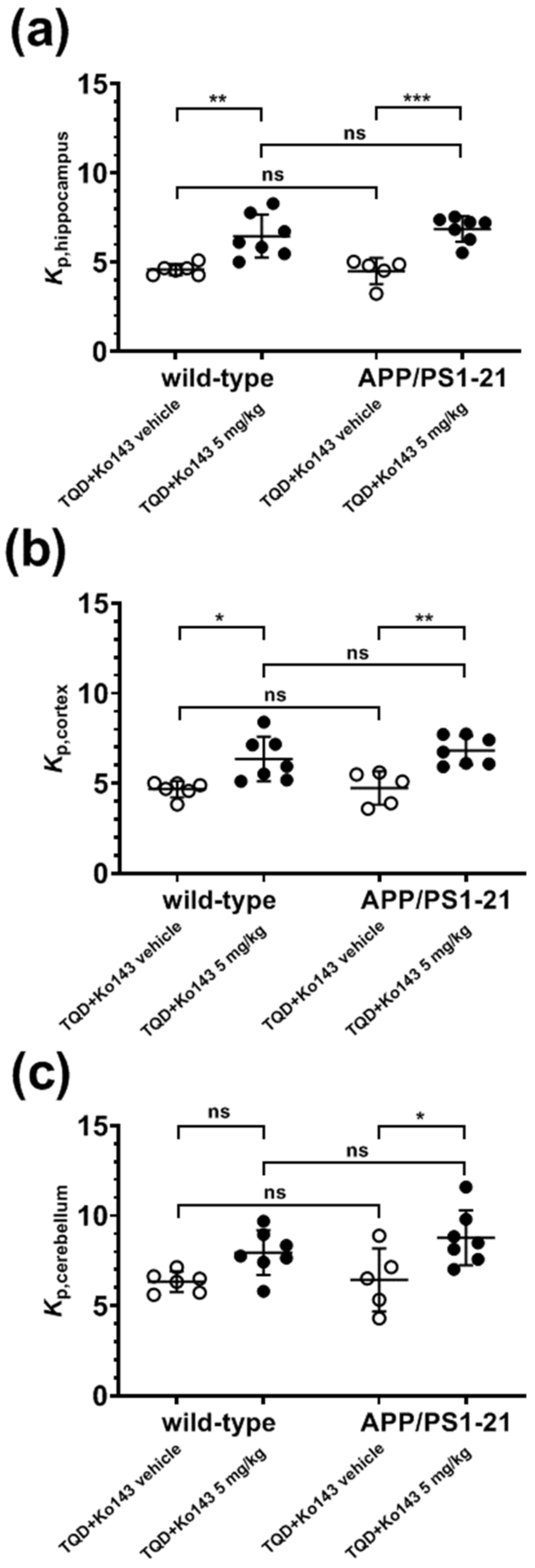
2.2. [11C]Tariquidar PET
2.3. [11C]Erlotinib PET
3. Discussion
4. Materials and Methods
4.1. General
4.2. Radiotracer Synthesis and Formulation
4.3. Animals
4.4. Experimental Design and Pretreatment
4.5. PET Imaging Procedure
4.6. PET Data Analysis
4.7. Immunohistochemistry
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | Adenosine triphopshate-binding cassette |
| ABCB1 | ABC subfamily B member 1, also known as P-glycoprotein (P-gp) |
| ABCG2 | ABC subfamily G member 2, also known as breast cancer resistance protein (BCRP) |
| AD | Alzheimer’s disease |
| Aβ | Beta-amyloid |
| BBB | Blood-brain barrier |
| Kp,brain | Brain-to-plasma radioactivity concentration ratio |
| PET | Positron Emission Tomography |
| SUV | Standardized uptake value |
| TAC | Time-activity curve |
References
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar]
- Pahnke, J.; Wolkenhauer, O.; Krohn, M.; Walker, L.C. Clinico-pathologic function of cerebral ABC transporters—implications for the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 396–405. [Google Scholar]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar]
- Storck, S.E.; Hartz, A.M.S.; Bernard, J.; Wolf, A.; Kachlmeier, A.; Mahringer, A.; Weggen, S.; Pahnke, J.; Pietrzik, C.U. The concerted amyloid-beta clearance of LRP1 and ABCB1/P-gp across the blood-brain barrier is linked by PICALM. Brain. Behav. Immun. 2018, 73, 21–33. [Google Scholar]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.M.; Sharom, F.J.; Reiner, P.B. beta-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar]
- Kuhnke, D.; Jedlitschky, G.; Grube, M.; Krohn, M.; Jucker, M.; Mosyagin, I.; Cascorbi, I.; Walker, L.C.; Kroemer, H.K.; Warzok, R.W.; et al. MDR1-P-glycoprotein (ABCB1) mediates transport of Alzheimer’s amyloid-beta peptides--implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 2007, 17, 347–353. [Google Scholar]
- Hartz, A.M.; Miller, D.S.; Bauer, B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol. Pharmacol. 2010, 77, 715–723. [Google Scholar]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar]
- Wijesuriya, H.C.; Bullock, J.Y.; Faull, R.L.; Hladky, S.B.; Barrand, M.A. ABC efflux transporters in brain vasculature of Alzheimer’s subjects. Brain Res. 2010, 1358, 228–238. [Google Scholar]
- Kannan, P.; Schain, M.; Kretzschmar, W.W.; Weidner, L.; Mitsios, N.; Gulyas, B.; Blom, H.; Gottesman, M.M.; Innis, R.B.; Hall, M.D.; et al. An automated method measures variability in P-glycoprotein and ABCG2 densities across brain regions and brain matter. J. Cereb. Blood Flow Metab. 2017, 37, 2062–2075. [Google Scholar]
- Carrano, A.; Snkhchyan, H.; Kooij, G.; van der Pol, S.; van Horssen, J.; Veerhuis, R.; Hoozemans, J.; Rozemuller, A.; de Vries, H.E. ATP-binding cassette transporters P-glycoprotein and breast cancer related protein are reduced in capillary cerebral amyloid angiopathy. Neurobiol. Aging 2014, 35, 565–575. [Google Scholar]
- van Assema, D.M.; Lubberink, M.; Bauer, M.; van der Flier, W.M.; Schuit, R.C.; Windhorst, A.D.; Comans, E.F.; Hoetjes, N.J.; Tolboom, N.; Langer, O.; et al. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain 2012, 135 (Pt 1), 181–189. [Google Scholar]
- Deo, A.K.; Borson, S.; Link, J.M.; Domino, K.; Eary, J.F.; Ke, B.; Richards, T.L.; Mankoff, D.A.; Minoshima, S.; O’Sullivan, F.; et al. Activity of P-glycoprotein, a beta-amyloid transporter at the blood-brain barrier, is compromised in patients with mild Alzheimer disease. J. Nucl. Med. 2014, 55, 1106–1111. [Google Scholar]
- Zoufal, V.; Mairinger, S.; Brackhan, M.; Krohn, M.; Filip, T.; Sauberer, M.; Stanek, J.; Wanek, T.; Tournier, N.; Bauer, M.; et al. Imaging P-glycoprotein induction at the blood-brain barrier of a beta-amyloidosis mouse model with 11C-metoclopramide PET. J. Nucl. Med. 2020, 61, 1050–1057. [Google Scholar]
- Qosa, H.; Abuznait, A.H.; Hill, R.A.; Kaddoumi, A. Enhanced brain amyloid-beta clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer’s disease. J. Alzheimers Dis. 2012, 31, 151–165. [Google Scholar]
- Durk, M.R.; Han, K.; Chow, E.C.; Ahrens, R.; Henderson, J.T.; Fraser, P.E.; Pang, K.S. 1alpha,25-Dihydroxyvitamin D3 reduces cerebral amyloid-beta accumulation and improves cognition in mouse models of Alzheimer’s disease. J. Neurosci. 2014, 34, 7091–7101. [Google Scholar]
- Brenn, A.; Grube, M.; Jedlitschky, G.; Fischer, A.; Strohmeier, B.; Eiden, M.; Keller, M.; Groschup, M.H.; Vogelgesang, S. St. John’s Wort reduces beta-amyloid accumulation in a double transgenic Alzheimer’s disease mouse model-role of P-glycoprotein. Brain Pathol. 2014, 24, 18–24. [Google Scholar]
- Agarwal, S.; Hartz, A.M.; Elmquist, W.F.; Bauer, B. Breast cancer resistance protein and P-glycoprotein in brain cancer: Two gatekeepers team up. Curr. Pharm. Des. 2011, 17, 2793–2802. [Google Scholar]
- Durmus, S.; Hendrikx, J.J.; Schinkel, A.H. Apical ABC transporters and cancer chemotherapeutic drug disposition. Adv. Cancer Res. 2015, 125, 1–41. [Google Scholar]
- Vogelgesang, S.; Warzok, R.W.; Cascorbi, I.; Kunert-Keil, C.; Schroeder, E.; Kroemer, H.K.; Siegmund, W.; Walker, L.C.; Pahnke, J. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2004, 1, 121–125. [Google Scholar]
- Xiong, H.; Callaghan, D.; Jones, A.; Bai, J.; Rasquinha, I.; Smith, C.; Pei, K.; Walker, D.; Lue, L.F.; Stanimirovic, D.; et al. ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Abeta(1-40) peptides. J. Neurosci. 2009, 29, 5463–5475. [Google Scholar]
- Tai, L.M.; Loughlin, A.J.; Male, D.K.; Romero, I.A. P-glycoprotein and breast cancer resistance protein restrict apical-to-basolateral permeability of human brain endothelium to amyloid-beta. J. Cereb. Blood Flow Metab. 2009, 29, 1079–1083. [Google Scholar]
- Do, T.M.; Noel-Hudson, M.S.; Ribes, S.; Besengez, C.; Smirnova, M.; Cisternino, S.; Buyse, M.; Calon, F.; Chimini, G.; Chacun, H.; et al. ABCG2- and ABCG4-mediated efflux of amyloid-beta peptide 1-40 at the mouse blood-brain barrier. J. Alzheimers Dis. 2012, 30, 155–166. [Google Scholar]
- Storelli, F.; Billington, S.; Kumar, A.R.; Unadkat, J.D. Abundance of P-glycoprotein and other drug transporters at the human blood-brain barrier in Alzheimer’s Disease: A quantitative targeted proteomic study. Clin. Pharmacol. Ther. 2020. [Google Scholar] [CrossRef]
- Al-Majdoub, Z.M.; Al Feteisi, H.; Achour, B.; Warwood, S.; Neuhoff, S.; Rostami-Hodjegan, A.; Barber, J. Proteomic quantification of human blood-brain barrier SLC and ABC transporters in healthy individuals and dementia patients. Mol. Pharm. 2019, 16, 1220–1233. [Google Scholar]
- Wanek, T.; Mairinger, S.; Langer, O. Radioligands targeting P-glycoprotein and other drug efflux proteins at the blood–brain barrier. J. Labelled Comp. Radiopharm. 2013, 56, 68–77. [Google Scholar]
- Wanek, T.; Kuntner, C.; Bankstahl, J.P.; Mairinger, S.; Bankstahl, M.; Stanek, J.; Sauberer, M.; Filip, T.; Erker, T.; Müller, M.; et al. A novel PET protocol for visualization of breast cancer resistance protein function at the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 2002–2011. [Google Scholar]
- Dallas, S.; Salphati, L.; Gomez-Zepeda, D.; Wanek, T.; Chen, L.; Chu, X.; Kunta, J.; Mezler, M.; Menet, M.C.; Chasseigneaux, S.; et al. Generation and characterization of a breast cancer resistance protein humanized mouse model. Mol. Pharmacol. 2016, 89, 492–504. [Google Scholar]
- Bauer, M.; Römermann, K.; Karch, R.; Wulkersdorfer, B.; Stanek, J.; Philippe, C.; Maier-Salamon, A.; Haslacher, H.; Jungbauer, C.; Wadsak, W.; et al. Pilot PET study to assess the functional interplay between ABCB1 and ABCG2 at the human blood-brain barrier. Clin. Pharmacol. Ther. 2016, 100, 131–141. [Google Scholar]
- Bankstahl, J.P.; Bankstahl, M.; Römermann, K.; Wanek, T.; Stanek, J.; Windhorst, A.D.; Fedrowitz, M.; Erker, T.; Müller, M.; Löscher, W.; et al. Tariquidar and elacridar are dose-dependently transported by p-glycoprotein and bcrp at the blood-brain barrier: A small-animal positron emission tomography and in vitro study . Drug Metab. Dispos. 2013, 41, 754–762. [Google Scholar]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jaggi, F.; Wolburg, H.; Gengler, S.; et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 2006, 7, 940–946. [Google Scholar]
- Zoufal, V.; Wanek, T.; Krohn, M.; Mairinger, S.; Filip, T.; Sauberer, M.; Stanek, J.; Pekar, T.; Bauer, M.; Pahnke, J.; et al. Age dependency of cerebral P-glycoprotein function in wild-type and APPPS1 mice measured with PET. J. Cereb. Blood Flow Metab. 2020, 40, 150–162. [Google Scholar]
- Allen, J.D.; van Loevezijn, A.; Lakhai, J.M.; van der Valk, M.; van Tellingen, O.; Reid, G.; Schellens, J.H.; Koomen, G.J.; Schinkel, A.H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 2002, 1, 417–425. [Google Scholar]
- Traxl, A.; Wanek, T.; Mairinger, S.; Stanek, J.; Filip, T.; Sauberer, M.; Müller, M.; Kuntner, C.; Langer, O. Breast cancer resistance protein and P-glycoprotein influence in vivo disposition of 11C-erlotinib. J. Nucl. Med. 2015, 56, 1930–1936. [Google Scholar]
- Linder, M.; Glitzner, E.; Srivatsa, S.; Bakiri, L.; Matsuoka, K.; Shahrouzi, P.; Dumanic, M.; Novoszel, P.; Mohr, T.; Langer, O.; et al. EGFR is required for FOS-dependent bone tumor development via RSK2/CREB signaling. EMBO Mol. Med. 2018, 10, e9408. [Google Scholar] [CrossRef]
- Kodaira, H.; Kusuhara, H.; Ushiki, J.; Fuse, E.; Sugiyama, Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J. Pharmacol. Exp. Ther. 2010, 333, 788–796. [Google Scholar]
- Hartz, A.M.; Zhong, Y.; Wolf, A.; LeVine, H., 3rd; Miller, D.S.; Bauer, B. Abeta40 reduces P-glycoprotein at the blood-brain barrier through the ubiquitin-proteasome pathway. J. Neurosci. 2016, 36, 1930–1941. [Google Scholar]
- Zoufal, V.; Mairinger, S.; Krohn, M.; Wanek, T.; Filip, T.; Sauberer, M.; Stanek, J.; Kuntner, C.; Pahnke, J.; Langer, O. Measurement of cerebral ABCC1 transport activity in wild-type and APP/PS1-21 mice with positron emission tomography. J. Cereb. Blood Flow Metab. 2019, 40, 954–965. [Google Scholar]
- Li, H.C.; Chen, P.Y.; Cheng, H.F.; Kuo, Y.M.; Huang, C.C. In vivo visualization of brain vasculature in Alzheimer’s Disease mice by high-frequency micro-Doppler imaging. IEEE Trans. Biomed. Eng. 2019, 66, 3393–3401. [Google Scholar]
- Bauer, M.; Wulkersdorfer, B.; Karch, R.; Philippe, C.; Jager, W.; Stanek, J.; Wadsak, W.; Hacker, M.; Zeitlinger, M.; Langer, O. Effect of P-glycoprotein inhibition at the blood-brain barrier on brain distribution of (R)-[11C]verapamil in elderly vs. young subjects. Br. J. Clin. Pharmacol. 2017, 83, 1991–1999. [Google Scholar]
- Bankstahl, J.P.; Bankstahl, M.; Kuntner, C.; Stanek, J.; Wanek, T.; Meier, M.; Ding, X.; Müller, M.; Langer, O.; Löscher, W. A novel PET imaging protocol identifies seizure-induced regional overactivity of P-glycoprotein at the blood-brain barrier. J. Neurosci. 2011, 31, 8803–8811. [Google Scholar]
- Feldmann, M.; Asselin, M.-C.; Liu, J.; Wang, S.; McMahon, A.; Anton-Rodriguez, J.; Walker, M.; Symms, M.; Brown, G.; Hinz, R.; et al. P-glycoprotein expression and function in patients with temporal lobe epilepsy: A case-control study. Lancet Neurol 2013, 12, 777–785. [Google Scholar]
- Shin, J.W.; Chu, K.; Shin, S.A.; Jung, K.H.; Lee, S.T.; Lee, Y.S.; Moon, J.; Lee, D.Y.; Lee, J.S.; Lee, D.S.; et al. Clinical applications of simultaneous PET/MR imaging using (R)-[11C]-verapamil with cyclosporin A: Preliminary results on a surrogate marker of drug-resistant epilepsy. Am. J. Neuroradiol. 2016, 37, 600–606. [Google Scholar]
- Mehta, D.C.; Short, J.L.; Nicolazzo, J.A. Altered brain uptake of therapeutics in a triple transgenic mouse model of Alzheimer’s disease. Pharm. Res. 2013, 30, 2868–2879. [Google Scholar]
- Gustafsson, S.; Lindström, V.; Ingelsson, M.; Hammarlund-Udenaes, M.; Syvänen, S. Intact blood-brain barrier transport of small molecular drugs in animal models of amyloid beta and alpha-synuclein pathology. Neuropharmacology 2018, 128, 482–491. [Google Scholar]
- Cheng, Z.; Zhang, J.; Liu, H.; Li, Y.; Zhao, Y.; Yang, E. Central nervous system penetration for small molecule therapeutic agents does not increase in multiple sclerosis- and Alzheimer’s disease-related animal models despite reported blood-brain barrier disruption. Drug Metab. Dispos. 2010, 38, 1355–1361. [Google Scholar]
- Kalvass, J.C.; Polli, J.W.; Bourdet, D.L.; Feng, B.; Huang, S.M.; Liu, X.; Smith, Q.R.; Zhang, L.K.; Zamek-Gliszczynski, M.J. Why clinical modulation of efflux transport at the human blood-brain barrier is unlikely: The ITC evidence-based position. Clin. Pharmacol. Ther. 2013, 94, 80–94. [Google Scholar]
- Wanek, T.; Römermann, K.; Mairinger, S.; Stanek, J.; Sauberer, M.; Filip, T.; Traxl, A.; Kuntner, C.; Pahnke, J.; Bauer, F.; et al. Factors governing p-glycoprotein-mediated drug-drug interactions at the blood-brain barrier measured with positron emission tomography. Mol. Pharm. 2015, 12, 3214–3225. [Google Scholar]
- Fox, E.; Bates, S.E. Tariquidar (XR9576): A P-glycoprotein drug efflux pump inhibitor. Expert Rev. Anticancer Ther. 2007, 7, 447–459. [Google Scholar]
- Bauer, F.; Kuntner, C.; Bankstahl, J.P.; Wanek, T.; Bankstahl, M.; Stanek, J.; Mairinger, S.; Dörner, B.; Löscher, W.; Müller, M.; et al. Synthesis and in vivo evaluation of [11C]tariquidar, a positron emission tomography radiotracer based on a third-generation P-glycoprotein inhibitor. Bioorg. Med. Chem. 2010, 18, 5489–5497. [Google Scholar]
- Philippe, C.; Mairinger, S.; Pichler, V.; Stanek, J.; Nics, L.; Mitterhauser, M.; Hacker, M.; Wanek, T.; Langer, O.; Wadsak, W. Comparison of fully-automated radiosyntheses of [11C]erlotinib for preclinical and clinical use starting from in target produced [11C]CO2 or [11C]CH4. EJNMMI Radiopharm. Chem. 2018, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Loening, A.M.; Gambhir, S.S. AMIDE: A free software tool for multimodality medical image analysis. Mol. Imaging 2003, 2, 131–137. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [11C]Tariquidar Study | Animal Group | ||
| Pretreatment | Wild-Type | APP/PS1-21 | |
| Tariquidar (12 mg/kg) and Ko143 vehicle | n | 10 (4) 1 | 7 (2) |
| Age [days] 2 | 189 ± 4 | 188 ± 1 | |
| Body weight [g] 2 | 26.7 ± 2.1 | 27.9 ± 3.2 | |
| Injected activity [MBq] 3 | 28 ± 6 | 25 ± 3 | |
| Tariquidar (12 mg/kg) and Ko143 (5 mg/kg) | n | 11 (4) | 12 (5) |
| Age [days] | 181 ± 5 | 183 ± 6 | |
| Body weight [g] | 28.9 ± 3.3 | 25.3 ± 2.4 | |
| Injected activity [MBq] | 24 ± 7 | 23 ± 10 | |
| [11C]Erlotinib Study | Animal Group | ||
| Wild-Type | APP/PS1-21 | ||
| n | 5 (1) | 5 (1) | |
| Age [days] | 176 ± 1 | 179 ± 2 | |
| Body weight [g] | 25.1 ± 1.2 | 25.2 ± 4.3 | |
| Injected activity [MBq] | 19 ± 3 | 26 ± 4 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wanek, T.; Zoufal, V.; Brackhan, M.; Krohn, M.; Mairinger, S.; Filip, T.; Sauberer, M.; Stanek, J.; Pekar, T.; Pahnke, J.; et al. Brain Distribution of Dual ABCB1/ABCG2 Substrates Is Unaltered in a Beta-Amyloidosis Mouse Model. Int. J. Mol. Sci. 2020, 21, 8245. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218245
Wanek T, Zoufal V, Brackhan M, Krohn M, Mairinger S, Filip T, Sauberer M, Stanek J, Pekar T, Pahnke J, et al. Brain Distribution of Dual ABCB1/ABCG2 Substrates Is Unaltered in a Beta-Amyloidosis Mouse Model. International Journal of Molecular Sciences. 2020; 21(21):8245. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218245
Chicago/Turabian StyleWanek, Thomas, Viktoria Zoufal, Mirjam Brackhan, Markus Krohn, Severin Mairinger, Thomas Filip, Michael Sauberer, Johann Stanek, Thomas Pekar, Jens Pahnke, and et al. 2020. "Brain Distribution of Dual ABCB1/ABCG2 Substrates Is Unaltered in a Beta-Amyloidosis Mouse Model" International Journal of Molecular Sciences 21, no. 21: 8245. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218245