Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer

 , , and
, , and
Abstract
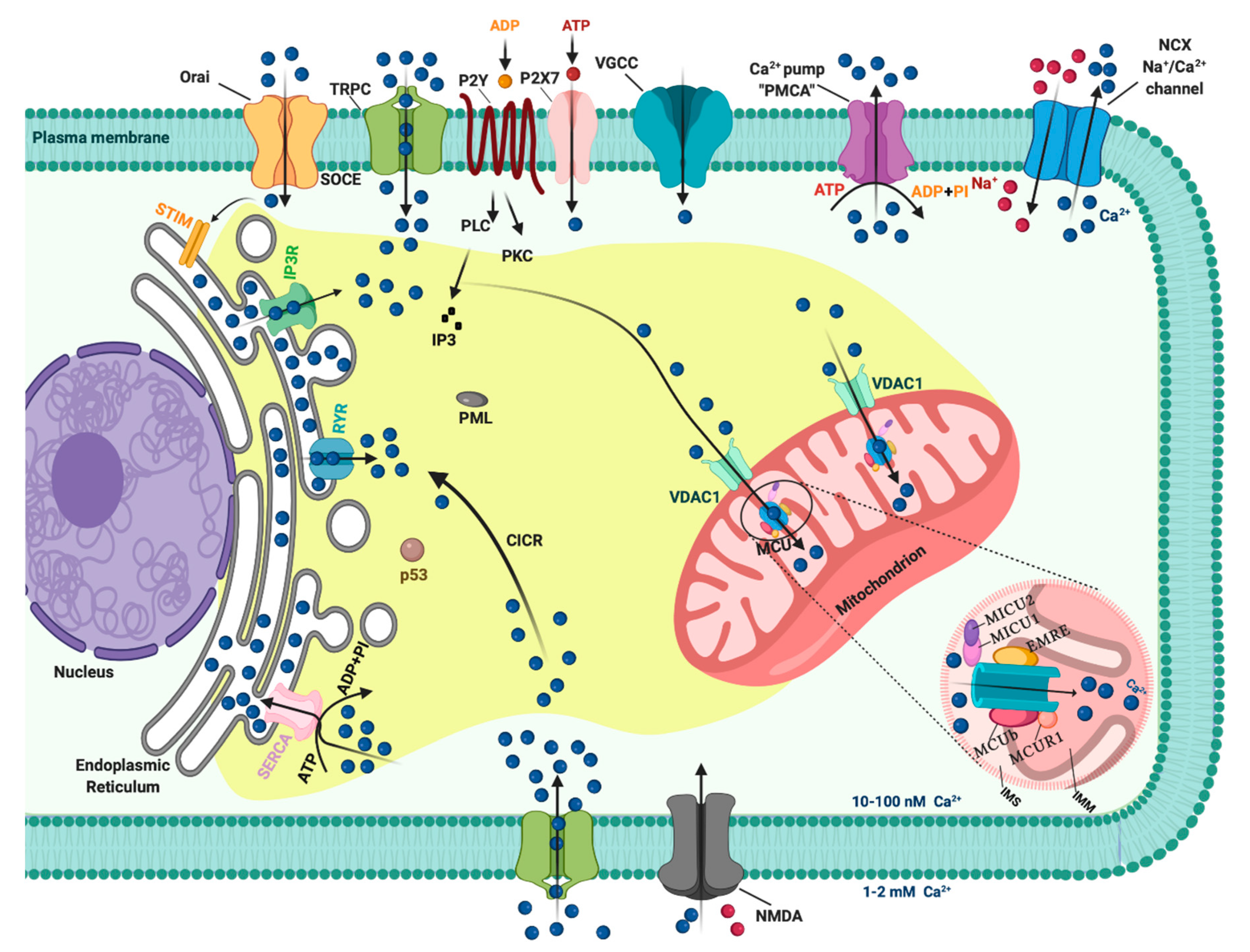
:1. Introduction: A General Overview of Ca2+ Signaling
2. Ca2+ Homeostasis during Cell Cycle and Tumor Growth
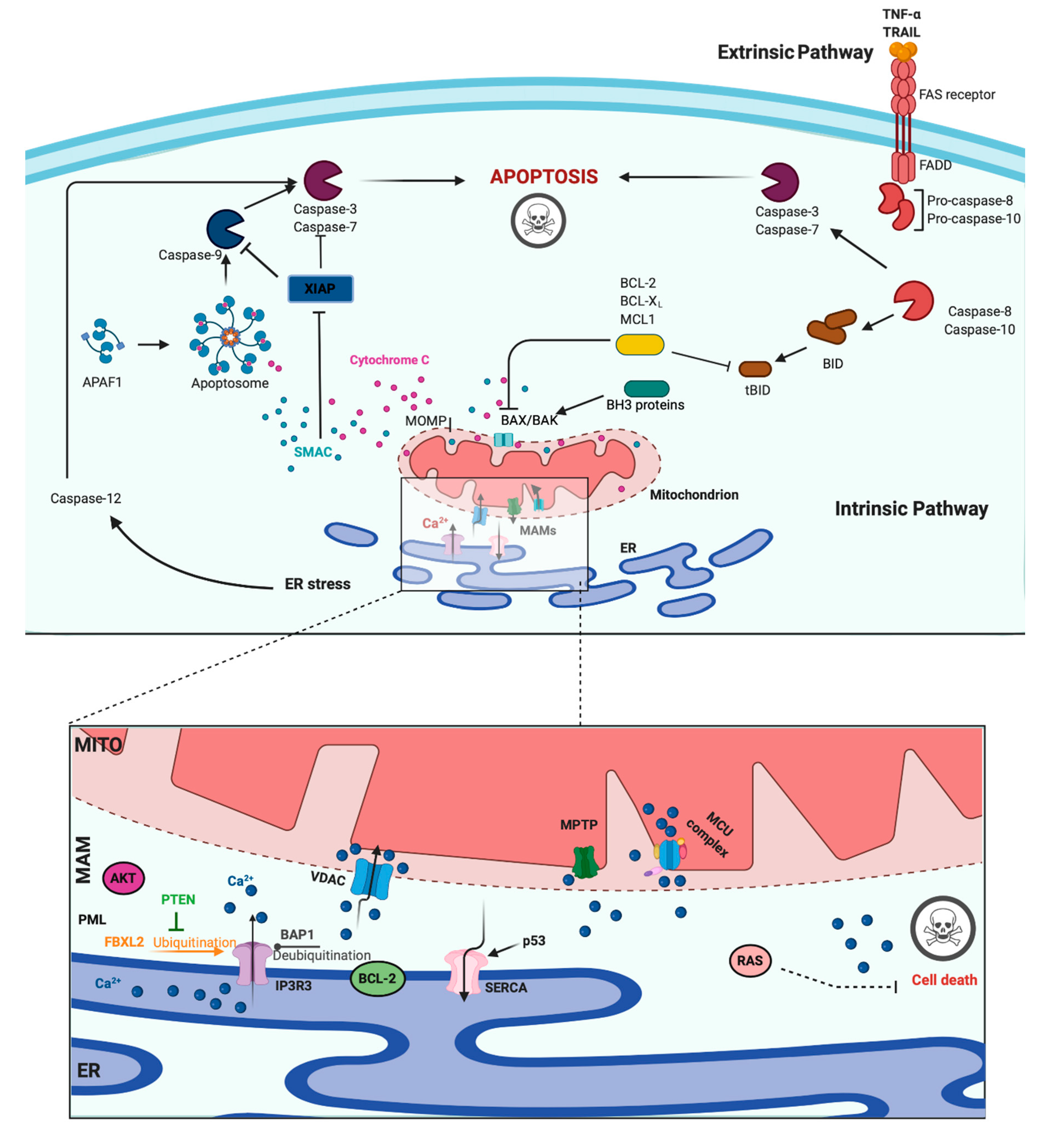
3. Role of Ca2+ in Apoptosis and Cancer
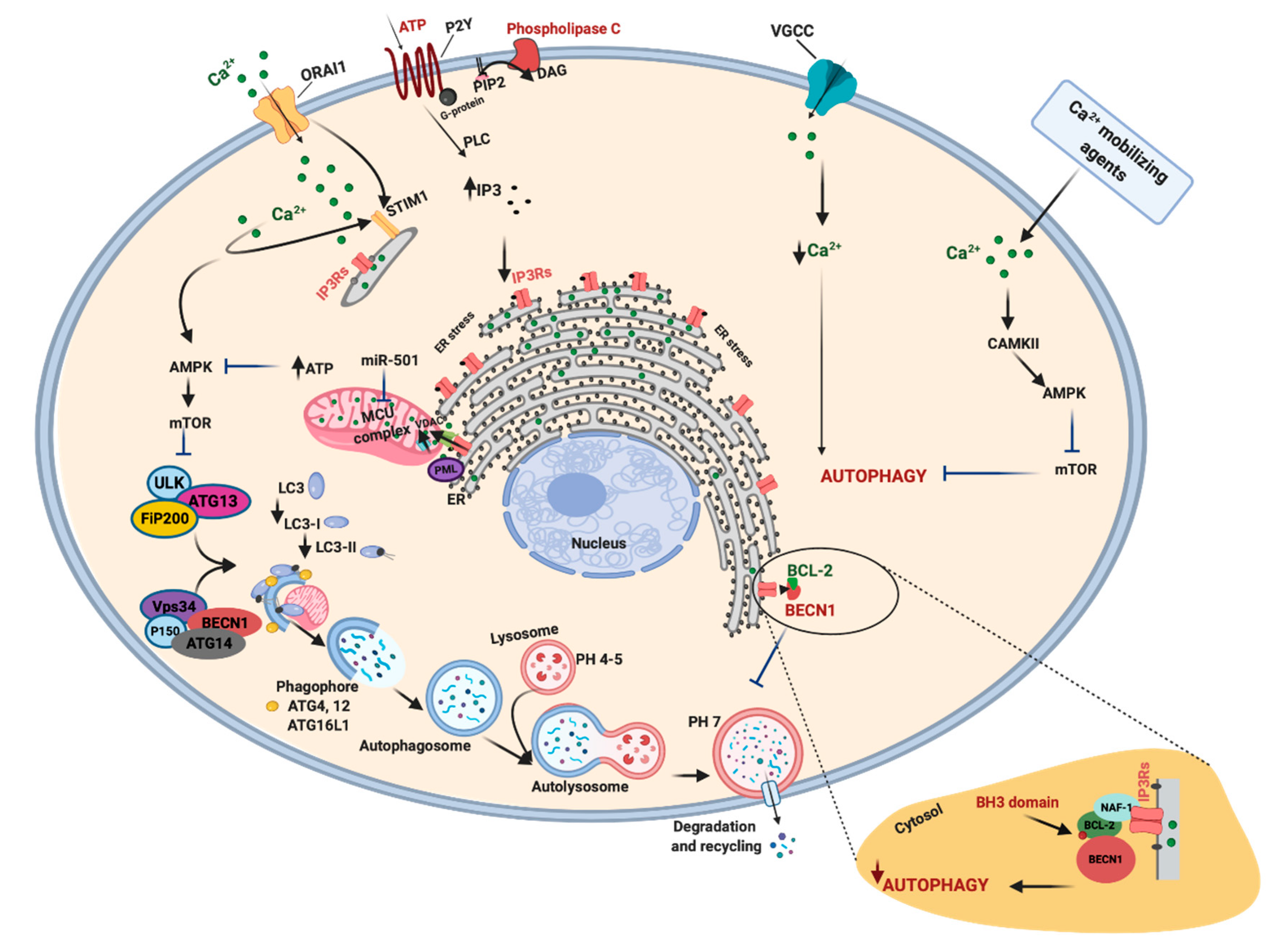
4. The Regulation of Autophagy by Calcium Signals and Its Involvement in Cancer
5. New Strategies for Ca2+ Signaling in Cancer Therapy: Ca2+ Channels and Pumps as Targets
5.1. TRP Channels
5.2. VGCCs and Purinergic P2 Receptors
5.3. SOCE Machinery Proteins (ORAI and STIM)
5.4. IP3Rs and Ca2+-ATPases
5.5. MCU and VDAC
6. Plan of Action in Cancer Therapy: Intersection between Cancer Therapies with Ca2+ Signaling
7. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| APs | autophagosomes |
| ATG | autophagy-related |
| BAP1 | BRCA1-associated protein 1 |
| BCL-2 | B-cell lymphoma 2 |
| BECN1 | coiled-coil, moesin-like BCL2 interacting protein |
| Ca2+ | calcium |
| Cyt-C | cytochrome C |
| CAM | calmodulin |
| CAMK | Ca2+/Calmodulin Dependent Protein Kinases |
| CaN | calcineurin |
| CDK | cyclin-dependent protein kinases |
| DISC | death-inducing signaling complex |
| ER | endoplasmic reticulum |
| HVA | high voltage activated |
| IP3Rs | inositol 1,4,5-triphosphate receptors |
| LC3 | microtubule-associated proteins 1A/1B light chain 3 |
| LVA | low voltage activated |
| MAMs | mitochondria associated membranes |
| MAPK | mitogen-activated protein kinase |
| MCU | mitochondrial calcium uniporter |
| MPT | mitochondrial permeability transition |
| mTOR | mammalian target of rapamycin |
| NCX | Na+/Ca2+ exchanger |
| NFAT | nuclear factor of activated T-cell |
| NMDA | N-methyl-d-aspartate receptor |
| PCD | programmed cell death |
| PD-1 | programmed death-1 |
| PI3K | phosphatidylinositol 3-kinase |
| PI3P | phosphatidylinositol-3-phosphate |
| PM | plasma membrane |
| PMCA | plasma membrane Ca2+ transport ATPase |
| PML | promyelocytic leukemia protein |
| PTEN | protein phosphatase and tensin homolog |
| RHEB | RAS homolog enriched in brain |
| RIPK | receptor-interacting serine/threonine-protein kinase |
| RyRs | ryanodine receptors |
| SERCA | sarcoendoplasmic reticulum Ca2+-ATPase |
| SOCE | store-operated Ca2+ entry |
| SPCA | secretory protein calcium ATPase |
| TFEB | transcription factor EB |
| TIL | tumor-infiltrating T lymphocytes |
| TRPML1 | transient receptor potential mucolipin 1 |
| TRPC | transient receptor channels |
| ULK | Unc-51 like autophagy activating kinase |
| V-ATPase | vacuolar-type ATPase |
| VDAC | Voltage-dependent anion channels |
| VGCC | voltage-gated Ca2+ channels |
References
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607. [Google Scholar] [PubMed]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca2+ Fluxes and Cancer. Mol. Cell 2020, 78, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Bultynck, G. Fundamentals of Cellular Calcium Signaling: A Primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, P.G.; Rao, A. Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun. 2015, 460, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Kavalali, E.T. Neuronal Ca2+ signalling at rest and during spontaneous neurotransmission. J. Physiol. 2020, 598, 1649–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [Green Version]
- Vallese, F.; Barazzuol, L.; Maso, L.; Brini, M.; Cali, T. ER-Mitochondria Calcium Transfer, Organelle Contacts and Neurodegenerative Diseases. Adv. Exp. Med. Biol. 2020, 1131, 719–746. [Google Scholar] [CrossRef] [PubMed]
- Gaspers, L.D.; Bartlett, P.J.; Politi, A.; Burnett, P.; Metzger, W.; Johnston, J.; Joseph, S.K.; Hofer, T.; Thomas, A.P. Hormone-induced calcium oscillations depend on cross-coupling with inositol 1,4,5-trisphosphate oscillations. Cell Rep. 2014, 9, 1209–1218. [Google Scholar] [CrossRef] [Green Version]
- Zalk, R.; Clarke, O.B.; des Georges, A.; Grassucci, R.A.; Reiken, S.; Mancia, F.; Hendrickson, W.A.; Frank, J.; Marks, A.R. Structure of a mammalian ryanodine receptor. Nature 2015, 517, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandecaetsbeek, I.; Vangheluwe, P.; Raeymaekers, L.; Wuytack, F.; Vanoevelen, J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb. Perspect. Biol. 2011, 3, a004184. [Google Scholar] [CrossRef] [Green Version]
- Bruce, J.I.E. Metabolic regulation of the PMCA: Role in cell death and survival. Cell Calcium 2018, 69, 28–36. [Google Scholar] [CrossRef]
- Cali, T.; Brini, M.; Carafoli, E. The PMCA pumps in genetically determined neuronal pathologies. Neurosci. Lett. 2018, 663, 2–11. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Trebak, M.; Perocchi, F.; Khananshvili, D.; Sekler, I. Crosslink between calcium and sodium signalling. Exp. Physiol. 2018, 103, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Pizzo, P. Highlighting the endoplasmic reticulum-mitochondria connection: Focus on Mitofusin 2. Pharmacol. Res. 2018, 128, 42–51. [Google Scholar] [CrossRef]
- Garrity, A.G.; Wang, W.; Collier, C.M.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 2016, 5, e15887. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Waller-Evans, H. Lysosomal Ca2+ Homeostasis and Signaling in Health and Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035311. [Google Scholar] [CrossRef]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Russa, A.D.; Maesawa, C.; Satoh, Y. Spontaneous [Ca2+]i oscillations in G1/S phase-synchronized cells. J. Electron Microsc. 2009, 58, 321–329. [Google Scholar] [CrossRef]
- Patel, R.; Holt, M.; Philipova, R.; Moss, S.; Schulman, H.; Hidaka, H.; Whitaker, M. Calcium/calmodulin-dependent phosphorylation and activation of human Cdc25-C at the G2/M phase transition in HeLa cells. J. Biol. Chem. 1999, 274, 7958–7968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heim, A.; Tischer, T.; Mayer, T.U. Calcineurin promotes APC/C activation at meiotic exit by acting on both XErp1 and Cdc20. EMBO Rep. 2018, 19, e46433. [Google Scholar] [CrossRef]
- Rao, A. Signaling to gene expression: Calcium, calcineurin and NFAT. Nat. Immunol. 2009, 10, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Kahl, C.R.; Means, A.R. Calcineurin regulates cyclin D1 accumulation in growth-stimulated fibroblasts. Mol. Biol. Cell 2004, 15, 1833–1842. [Google Scholar] [CrossRef] [Green Version]
- Keith, C.; DiPaola, M.; Maxfield, F.R.; Shelanski, M.L. Microinjection of Ca++-calmodulin causes a localized depolymerization of microtubules. J. Cell Biol. 1983, 97, 1918–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinsmore, J.H.; Sloboda, R.D. Calcium and calmodulin-dependent phosphorylation of a 62 kd protein induces microtubule depolymerization in sea urchin mitotic apparatuses. Cell 1988, 53, 769–780. [Google Scholar] [CrossRef]
- Chircop, M.; Malladi, C.S.; Lian, A.T.; Page, S.L.; Zavortink, M.; Gordon, C.P.; McCluskey, A.; Robinson, P.J. Calcineurin activity is required for the completion of cytokinesis. Cell. Mol. Life Sci. 2010, 67, 3725–3737. [Google Scholar] [CrossRef]
- Yokokura, S.; Yurimoto, S.; Matsuoka, A.; Imataki, O.; Dobashi, H.; Bandoh, S.; Matsunaga, T. Calmodulin antagonists induce cell cycle arrest and apoptosis in vitro and inhibit tumor growth in vivo in human multiple myeloma. BMC Cancer 2014, 14, 882. [Google Scholar] [CrossRef] [Green Version]
- Machaca, K. Ca2+ signaling, genes and the cell cycle. Cell Calcium 2011, 49, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Seas, A.; Kiyani, M.; Ji, K.S.Y.; Bell, H.N. A temporal examination of calcium signaling in cancer- from tumorigenesis, to immune evasion, and metastasis. Cell Biosci. 2018, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, M.; Kischel, P.; Hague, F.; Ahidouch, A.; Benzerdjeb, N.; Sevestre, H.; Penner, R.; Ouadid-Ahidouch, H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochim. Biophys. Acta 2013, 1833, 752–760. [Google Scholar] [CrossRef] [Green Version]
- Ahlin, C.; Lundgren, C.; Embretsen-Varro, E.; Jirstrom, K.; Blomqvist, C.; Fjallskog, M. High expression of cyclin D1 is associated to high proliferation rate and increased risk of mortality in women with ER-positive but not in ER-negative breast cancers. Breast Cancer Res. Treat. 2017, 164, 667–678. [Google Scholar] [CrossRef]
- Arnold, A.; Papanikolaou, A. Cyclin D1 in breast cancer pathogenesis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 4215–4224. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Savage, K.; Lambros, M.B.; James, M.; Steele, D.; Jones, R.L.; Dowsett, M. Cyclin D1 protein overexpression and CCND1 amplification in breast carcinomas: An immunohistochemical and chromogenic in situ hybridisation analysis. Mod. Pathol. 2006, 19, 999–1009. [Google Scholar] [CrossRef] [Green Version]
- Phan, N.N.; Wang, C.Y.; Chen, C.F.; Sun, Z.; Lai, M.D.; Lin, Y.C. Voltage-gated calcium channels: Novel targets for cancer therapy. Oncol. Lett. 2017, 14, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Pushparaj, C.; Bahi, N.; Sorolla, A.; Herreros, J.; Pamplona, R.; Vilella, R.; Matias-Guiu, X.; Marti, R.M.; Canti, C. Functional expression of voltage-gated calcium channels in human melanoma. Pigment Cell Melanoma Res. 2012, 25, 200–212. [Google Scholar] [CrossRef]
- Lehen’kyi, V.; Flourakis, M.; Skryma, R.; Prevarskaya, N. TRPV6 channel controls prostate cancer cell proliferation via Ca2+/NFAT-dependent pathways. Oncogene 2007, 26, 7380–7385. [Google Scholar] [CrossRef] [Green Version]
- Parmer, T.G.; Ward, M.D.; Hait, W.N. Effects of rottlerin, an inhibitor of calmodulin-dependent protein kinase III, on cellular proliferation, viability, and cell cycle distribution in malignant glioma cells. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1997, 8, 327–334. [Google Scholar]
- Parmer, T.G.; Ward, M.D.; Yurkow, E.J.; Vyas, V.H.; Kearney, T.J.; Hait, W.N. Activity and regulation by growth factors of calmodulin-dependent protein kinase III (elongation factor 2-kinase) in human breast cancer. Br. J. Cancer 1999, 79, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Zhang, J.; Ma, X.; Kim, B.W.; Wang, H.; Li, J.; Pan, Y.; Xu, Y.; Ding, L.; Yang, L.; et al. Stabilization of the c-Myc Protein by CAMKIIgamma Promotes T Cell Lymphoma. Cancer Cell 2017, 32, 115–128.e117. [Google Scholar] [CrossRef] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Malik, A.; Kanneganti, T.D. Inflammasome activation and assembly at a glance. J. Cell Sci. 2017, 130, 3955–3963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef]
- Danese, A.; Marchi, S.; Vitto, V.A.M.; Modesti, L.; Leo, S.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Cancer-Related Increases and Decreases in Calcium Signaling at the Endoplasmic Reticulum-Mitochondria Interface (MAMs). Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Patergnani, S.; Baldassari, F.; De Marchi, E.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; Pinton, P. Methods to monitor and compare mitochondrial and glycolytic ATP production. Methods Enzymol. 2014, 542, 313–332. [Google Scholar] [CrossRef]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Kerkhofs, M.; Bittremieux, M.; Morciano, G.; Giorgi, C.; Pinton, P.; Parys, J.B.; Bultynck, G. Emerging molecular mechanisms in chemotherapy: Ca2+ signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 2018, 9, 334. [Google Scholar] [CrossRef]
- Vervliet, T.; Parys, J.B.; Bultynck, G. Bcl-2 proteins and calcium signaling: Complexity beneath the surface. Oncogene 2016, 35, 5079–5092. [Google Scholar] [CrossRef] [Green Version]
- Pinton, P.; Ferrari, D.; Magalhaes, P.; Schulze-Osthoff, K.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J. Cell Biol. 2000, 148, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.P.; Aromolaran, A.S.; Bultynck, G.; Zhong, F.; Li, X.; McColl, K.; Matsuyama, S.; Herlitze, S.; Roderick, H.L.; Bootman, M.D.; et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2′s inhibition of apoptotic calcium signals. Mol. Cell 2008, 31, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Pihan, P.; Carreras-Sureda, A.; Hetz, C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017, 24, 1478–1487. [Google Scholar] [CrossRef] [Green Version]
- Um, H.D. Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: A review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget 2016, 7, 5193–5203. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Rimessi, A.; Marchi, S.; Patergnani, S.; Pinton, P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene 2014, 33, 2329–2340. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Marinello, M.; Bononi, A.; Bonora, M.; Giorgi, C.; Rimessi, A.; Pinton, P. Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis. 2012, 3, e304. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Poletti, F.; Giorgi, C.; Pandolfi, P.P.; Pinton, P. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013, 20, 1631–1643. [Google Scholar] [CrossRef] [Green Version]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Pandolfi, P.P. The role of PML in the control of apoptotic cell fate: A new key player at ER-mitochondria sites. Cell Death Differ. 2011, 18, 1450–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Giorgi, C.; Maniero, S.; Missiroli, S.; Maniscalco, P.; Bononi, I.; Martini, F.; Cavallesco, G.; Tognon, M.; Pinton, P. The endoplasmic reticulum mitochondrial calcium cross talk is downregulated in malignant pleural mesothelioma cells and plays a critical role in apoptosis inhibition. Oncotarget 2015, 6, 23427–23444. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Bonora, M.; Pinton, P. Inside the tumor: p53 modulates calcium homeostasis. Cell Cycle 2015, 14, 933–934. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Cho, K.J.; Naji, A.K.; Clemons, K.N.; Wong, C.O.; Villanueva, M.; Gregory, S.; Karagas, N.E.; Tan, L.; Liang, H.; et al. HRAS-driven cancer cells are vulnerable to TRPML1 inhibition. EMBO Rep. 2019, 20, e46685. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Tomassoni, D.; Rossi, F.; Cardinali, C.; Santoni, M.; Arcella, A.; Oliva, M.A.; Santoni, A.; et al. Overexpression of transient receptor potential mucolipin-2 ion channels in gliomas: Role in tumor growth and progression. Oncotarget 2016, 7, 43654–43668. [Google Scholar] [CrossRef] [Green Version]
- Giatromanolaki, A.; Kalamida, D.; Sivridis, E.; Karagounis, I.V.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer 2015, 90, 98–105. [Google Scholar] [CrossRef]
- Kauffman, E.C.; Ricketts, C.J.; Rais-Bahrami, S.; Yang, Y.; Merino, M.J.; Bottaro, D.P.; Srinivasan, R.; Linehan, W.M. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat. Rev. Urol. 2014, 11, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Marchand, B.; Arsenault, D.; Raymond-Fleury, A.; Boisvert, F.M.; Boucher, M.J. Glycogen synthase kinase-3 (GSK3) inhibition induces prosurvival autophagic signals in human pancreatic cancer cells. J. Biol. Chem. 2015, 290, 5592–5605. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Jia, X.; Wang, K.; Zhao, N. High expression of TFEB is associated with aggressive clinical features in colorectal cancer. Oncotargets Ther. 2018, 11, 8089–8098. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Sbano, L.; Bonora, M.; Marchi, S.; Baldassari, F.; Medina, D.L.; Ballabio, A.; Giorgi, C.; Pinton, P. TFEB-mediated increase in peripheral lysosomes regulates store-operated calcium entry. Sci. Rep. 2017, 7, 40797. [Google Scholar] [CrossRef]
- Marchi, S.; Lupini, L.; Patergnani, S.; Rimessi, A.; Missiroli, S.; Bonora, M.; Bononi, A.; Corra, F.; Giorgi, C.; De Marchi, E.; et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. 2013, 23, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Z.; Chen, K.H.; DasGupta, A.; Potus, F.; Dunham-Snary, K.; Bonnet, S.; Tian, L.; Fu, J.; Breuils-Bonnet, S.; Provencher, S.; et al. MicroRNA-138 and MicroRNA-25 Down-regulate Mitochondrial Calcium Uniporter, Causing the Pulmonary Arterial Hypertension Cancer Phenotype. Am. J. Respir. Crit. Care Med. 2017, 195, 515–529. [Google Scholar] [CrossRef]
- Leidinger, P.; Backes, C.; Dahmke, I.N.; Galata, V.; Huwer, H.; Stehle, I.; Bals, R.; Keller, A.; Meese, E. What makes a blood cell based miRNA expression pattern disease specific?—A miRNome analysis of blood cell subsets in lung cancer patients and healthy controls. Oncotarget 2014, 5, 9484–9497. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Nakaoka, T.; Saito, H. microRNA-34a as a Therapeutic Agent against Human Cancer. J. Clin. Med. 2015, 4, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Lee, J.; Seo, H.H.; Lee, C.Y.; Yoo, K.J.; Kim, S.M.; Lee, S.; Hwang, K.C.; Choi, E. Na+-Ca2+ exchanger targeting miR-132 prevents apoptosis of cardiomyocytes under hypoxic condition by suppressing Ca2+ overload. Biochem. Biophys. Res. Commun. 2015, 460, 931–937. [Google Scholar] [CrossRef]
- Chaudhuri, A.D.; Choi, D.C.; Kabaria, S.; Tran, A.; Junn, E. MicroRNA-7 Regulates the Function of Mitochondrial Permeability Transition Pore by Targeting VDAC1 Expression. J. Biol. Chem. 2016, 291, 6483–6493. [Google Scholar] [CrossRef] [Green Version]
- Zaglia, T.; Ceriotti, P.; Campo, A.; Borile, G.; Armani, A.; Carullo, P.; Prando, V.; Coppini, R.; Vida, V.; Stolen, T.O.; et al. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. USA 2017, 114, E9006–E9015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Pinton, P. Mitophagy and mitochondrial balance. Methods Mol. Biol. 2015, 1241, 181–194. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef]
- Xu, H.D.; Qin, Z.H. Beclin 1, Bcl-2 and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.L.; Wu, Y.Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [Green Version]
- Chapel, A.; Kieffer-Jaquinod, S.; Sagne, C.; Verdon, Q.; Ivaldi, C.; Mellal, M.; Thirion, J.; Jadot, M.; Bruley, C.; Garin, J.; et al. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol. Cell. Proteom. 2013, 12, 1572–1588. [Google Scholar] [CrossRef] [Green Version]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Fainardi, E.; Casetta, I.; Granieri, E.; Pugliatti, M.; Pinton, P. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J. Neuroinflamm. 2019, 16, 131. [Google Scholar] [CrossRef]
- Patergnani, S.; Castellazzi, M.; Bonora, M.; Marchi, S.; Casetta, I.; Pugliatti, M.; Giorgi, C.; Granieri, E.; Pinton, P. Autophagy and mitophagy elements are increased in body fluids of multiple sclerosis-affected individuals. J. Neurol. Neurosurg. Psychiatry 2018, 89, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Xue, J.; Patergnani, S.; Giorgi, C.; Suarez, J.; Goto, K.; Bononi, A.; Tanji, M.; Novelli, F.; Pastorino, S.; Xu, R.; et al. Asbestos induces mesothelial cell transformation via HMGB1-driven autophagy. Proc. Natl. Acad. Sci. USA 2020, 117, 25543–25552. [Google Scholar] [CrossRef]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafa, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427. [Google Scholar] [CrossRef] [Green Version]
- Vega-Rubin-de-Celis, S. The Role of Beclin 1-Dependent Autophagy in Cancer. Biology 2019, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, M.; Li, H.; Frank, J.A.; Dai, L.; Liu, H.; Zhang, Z.; Wang, C.; Chen, G. Autophagy inhibition by sustained overproduction of IL6 contributes to arsenic carcinogenesis. Cancer Res. 2014, 74, 3740–3752. [Google Scholar] [CrossRef] [Green Version]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Altman, B.J.; Jacobs, S.R.; Mason, E.F.; Michalek, R.D.; MacIntyre, A.N.; Coloff, J.L.; Ilkayeva, O.; Jia, W.; He, Y.W.; Rathmell, J.C. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011, 30, 1855–1867. [Google Scholar] [CrossRef] [Green Version]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef] [Green Version]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Ma, X.H.; Piao, S.; Wang, D.; McAfee, Q.W.; Nathanson, K.L.; Lum, J.J.; Li, L.Z.; Amaravadi, R.K. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 3478–3489. [Google Scholar] [CrossRef] [Green Version]
- Lazova, R.; Camp, R.L.; Klump, V.; Siddiqui, S.F.; Amaravadi, R.K.; Pawelek, J.M. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santanam, U.; Banach-Petrosky, W.; Abate-Shen, C.; Shen, M.M.; White, E.; DiPaola, R.S. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016, 30, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016, 12, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.; Thorburn, A. Modulation of pediatric brain tumor autophagy and chemosensitivity. J. Neuro Oncol. 2012, 106, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, P.J.; McCloskey, K.D. CaV channels and cancer: Canonical functions indicate benefits of repurposed drugs as cancer therapeutics. Eur. Biophys. J. 2016, 45, 621–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmohsen, K.; Srikantan, S.; Tominaga, K.; Kang, M.J.; Yaniv, Y.; Martindale, J.L.; Yang, X.; Park, S.S.; Becker, K.G.; Subramanian, M.; et al. Growth inhibition by miR-519 via multiple p21-inducing pathways. Mol. Cell. Biol. 2012, 32, 2530–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Mathiasen, I.S.; Sergeev, I.N.; Bastholm, L.; Elling, F.; Norman, A.W.; Jaattela, M. Calcium and calpain as key mediators of apoptosis-like death induced by vitamin D compounds in breast cancer cells. J. Biol. Chem. 2002, 277, 30738–30745. [Google Scholar] [CrossRef] [Green Version]
- Decuypere, J.P.; Welkenhuyzen, K.; Luyten, T.; Ponsaerts, R.; Dewaele, M.; Molgo, J.; Agostinis, P.; Missiaen, L.; De Smedt, H.; Parys, J.B.; et al. Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy 2011, 7, 1472–1489. [Google Scholar] [CrossRef] [Green Version]
- Sakaki, K.; Wu, J.; Kaufman, R.J. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J. Biol. Chem. 2008, 283, 15370–15380. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.H.; Shih, Y.L.; Ko, W.C.; Wei, Y.H.; Shih, C.M. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell. Mol. Life Sci. 2008, 65, 3640–3652. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Guzzo, S.; Mangolini, A.; dell’Atti, L.; Pinton, P.; Aguiari, G. The induction of AMPK-dependent autophagy leads to P53 degradation and affects cell growth and migration in kidney cancer cells. Exp. Cell Res. 2020, 395, 112190. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Novel frontiers in calcium signaling: A possible target for chemotherapy. Pharmacol. Res. 2015, 99, 82–85. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Elzamzamy, O.M.; Penner, R.; Hazlehurst, L.A. The Role of TRPC1 in Modulating Cancer Progression. Cells 2020, 9, 388. [Google Scholar] [CrossRef] [Green Version]
- Hantute-Ghesquier, A.; Haustrate, A.; Prevarskaya, N.; Lehen’kyi, V. TRPM Family Channels in Cancer. Pharmaceuticals 2018, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.M. TRPV6 as a Target for Cancer Therapy. J. Cancer 2020, 11, 374–387. [Google Scholar] [CrossRef] [Green Version]
- Santoni, G.; Maggi, F.; Morelli, M.B.; Santoni, M.; Marinelli, O. Transient Receptor Potential Cation Channels in Cancer Therapy. Med. Sci. 2019, 7, 108. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.L.; Barszczyk, A.; Turlova, E.; Deurloo, M.; Liu, B.; Yang, B.B.; Rutka, J.T.; Feng, Z.P.; Sun, H.S. Inhibition of TRPM7 by carvacrol suppresses glioblastoma cell proliferation, migration and invasion. Oncotarget 2015, 6, 16321–16340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolcher, A.; Patnaik, A.; Papadopoulos, K.; Mays, T.; Stephan, T.; Humble, D.; Frohlich, M.; Sims, R. 376 Preliminary results from a Phase 1 study of D-3263 HCl, a TRPM8 calcium channel agonist, in patients with advanced cancer. EJC Suppl. 2010, 8, 119. [Google Scholar] [CrossRef]
- Yamada, T.; Ueda, T.; Shibata, Y.; Ikegami, Y.; Saito, M.; Ishida, Y.; Ugawa, S.; Kohri, K.; Shimada, S. TRPV2 activation induces apoptotic cell death in human T24 bladder cancer cells: A potential therapeutic target for bladder cancer. Urology 2010, 76, 509.e1–509.e7. [Google Scholar] [CrossRef]
- Fu, S.; Hirte, H.; Welch, S.; Ilenchuk, T.T.; Lutes, T.; Rice, C.; Fields, N.; Nemet, A.; Dugourd, D.; Piha-Paul, S.; et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Antal, L.; Martin-Caraballo, M. T-type Calcium Channels in Cancer. Cancers 2019, 11, 134. [Google Scholar] [CrossRef] [Green Version]
- Lara, R.; Adinolfi, E.; Harwood, C.A.; Philpott, M.; Barden, J.A.; Di Virgilio, F.; McNulty, S. P2X7 in Cancer: From Molecular Mechanisms to Therapeutics. Front. Pharmacol. 2020, 11, 793. [Google Scholar] [CrossRef]
- Gilbert, S.M.; Gidley Baird, A.; Glazer, S.; Barden, J.A.; Glazer, A.; Teh, L.C.; King, J. A phase I clinical trial demonstrates that nfP2X7 -targeted antibodies provide a novel, safe and tolerable topical therapy for basal cell carcinoma. Br. J. Dermatol. 2017, 177, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.P.; Zhang, X.X.; Jiang, S.H.; Tao, L.Y.; Li, Q.; Zhu, L.L.; Yang, M.W.; Huo, Y.M.; Jiang, Y.S.; Tian, G.A.; et al. Targeting Purinergic Receptor P2Y2 Prevents the Growth of Pancreatic Ductal Adenocarcinoma by Inhibiting Cancer Cell Glycolysis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1318–1330. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, S.B.; Monteith, G.R. ORAI channels and cancer. Cell Calcium 2018, 74, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanwar, J.; Arora, S.; Motiani, R.K. Orai3: Oncochannel with therapeutic potential. Cell Calcium 2020, 90, 102247. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting Stim and Orai Proteins as an Alternative Approach in Anticancer Therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef]
- Latour, S.; Zanese, M.; Le Morvan, V.; Vacher, A.M.; Menard, N.; Bijou, F.; Durrieu, F.; Soubeyran, P.; Savina, A.; Vacher, P.; et al. Role of Calcium Signaling in GA101-Induced Cell Death in Malignant Human B Cells. Cancers 2019, 11, 291. [Google Scholar] [CrossRef] [Green Version]
- Rezuchova, I.; Hudecova, S.; Soltysova, A.; Matuskova, M.; Durinikova, E.; Chovancova, B.; Zuzcak, M.; Cihova, M.; Burikova, M.; Penesova, A.; et al. Type 3 inositol 1,4,5-trisphosphate receptor has antiapoptotic and proliferative role in cancer cells. Cell Death Dis. 2019, 10, 186. [Google Scholar] [CrossRef] [Green Version]
- Akl, H.; Monaco, G.; La Rovere, R.; Welkenhuyzen, K.; Kiviluoto, S.; Vervliet, T.; Molgo, J.; Distelhorst, C.W.; Missiaen, L.; Mikoshiba, K.; et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013, 4, e632. [Google Scholar] [CrossRef]
- Kang, S.S.; Han, K.S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Gelebart, P.; Kovacs, T.; Brouland, J.P.; van Gorp, R.; Grossmann, J.; Rivard, N.; Panis, Y.; Martin, V.; Bredoux, R.; Enouf, J.; et al. Expression of endomembrane calcium pumps in colon and gastric cancer cells. Induction of SERCA3 expression during differentiation. J. Biol. Chem. 2002, 277, 26310–26320. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Li, A.; Li, W.; Cai, P.; Yang, B.; Zhang, M.; Gu, Y.; Shu, Y.; Sun, Y.; Shen, Y.; et al. Novel role of Sarco/endoplasmic reticulum calcium ATPase 2 in development of colorectal cancer and its regulation by F36, a curcumin analog. Biomed. Pharmacother. 2014, 68, 1141–1148. [Google Scholar] [CrossRef]
- Bruce, J.I.E.; James, A.D. Targeting the Calcium Signalling Machinery in Cancer. Cancers 2020, 12, 2351. [Google Scholar] [CrossRef]
- Varga, K.; Hollosi, A.; Paszty, K.; Hegedus, L.; Szakacs, G.; Timar, J.; Papp, B.; Enyedi, A.; Padanyi, R. Expression of calcium pumps is differentially regulated by histone deacetylase inhibitors and estrogen receptor alpha in breast cancer cells. BMC Cancer 2018, 18, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruschoff, J.H.; Brandenburger, T.; Strehler, E.E.; Filoteo, A.G.; Heinmoller, E.; Aumuller, G.; Wilhelm, B. Plasma membrane calcium ATPase expression in human colon multistep carcinogenesis. Cancer Investig. 2012, 30, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Curry, M.C.; Roberts-Thomson, S.J.; Monteith, G.R. Plasma membrane calcium ATPases and cancer. BioFactors 2011, 37, 132–138. [Google Scholar] [CrossRef]
- Sritangos, P.; Pena Alarcon, E.; James, A.D.; Sultan, A.; Richardson, D.A.; Bruce, J.I.E. Plasma Membrane Ca2+ ATPase Isoform 4 (PMCA4) Has an Important Role in Numerous Hallmarks of Pancreatic Cancer. Cancers 2020, 12, 218. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef]
- Mahalingam, D.; Peguero, J.; Cen, P.; Arora, S.P.; Sarantopoulos, J.; Rowe, J.; Allgood, V.; Tubb, B.; Campos, L. A Phase II, Multicenter, Single-Arm Study of Mipsagargin (G-202) as a Second-Line Therapy Following Sorafenib for Adult Patients with Progressive Advanced Hepatocellular Carcinoma. Cancers 2019, 11, 833. [Google Scholar] [CrossRef] [Green Version]
- Peterkova, L.; Kmonickova, E.; Ruml, T.; Rimpelova, S. Sarco/Endoplasmic Reticulum Calcium ATPase Inhibitors: Beyond Anticancer Perspective. J. Med. Chem. 2020, 63, 1937–1963. [Google Scholar] [CrossRef]
- Muscella, A.; Calabriso, N.; Vetrugno, C.; Fanizzi, F.P.; De Pascali, S.A.; Storelli, C.; Marsigliante, S. The platinum (II) complex [Pt(O,O’-acac)(gamma-acac)(DMS)] alters the intracellular calcium homeostasis in MCF-7 breast cancer cells. Biochem. Pharmacol. 2011, 81, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.A.; Crowther, C.M.; Andrus, M.B.; Kenealey, J.D. Resveratrol derivatives increase cytosolic calcium by inhibiting plasma membrane ATPase and inducing calcium release from the endoplasmic reticulum in prostate cancer cells. Biochem. Biophys. Rep. 2019, 19, 100667. [Google Scholar] [CrossRef]
- Vultur, A.; Gibhardt, C.S.; Stanisz, H.; Bogeski, I. The role of the mitochondrial calcium uniporter (MCU) complex in cancer. Pflug. Arch. Eur. J. Physiol. 2018, 470, 1149–1163. [Google Scholar] [CrossRef]
- Mazure, N.M. VDAC in cancer. Biochim. Biophys. Acta. Bioenerg. 2017, 1858, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Shteinfer-Kuzmine, A.; Amsalem, Z.; Arif, T.; Zooravlov, A.; Shoshan-Barmatz, V. Selective induction of cancer cell death by VDAC1-based peptides and their potential use in cancer therapy. Mol. Oncol. 2018, 12, 1077–1103. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.D.; Xia, X.; Lv, X.F.; Yu, B.X.; Yuan, J.N.; Mai, X.Y.; Shang, J.Y.; Zhou, J.G.; Liang, S.J.; Pang, R.P. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell. Mol. Med. 2017, 21, 904–915. [Google Scholar] [CrossRef]
- Can, G.; Akpinar, B.; Baran, Y.; Zhivotovsky, B.; Olsson, M. 5-Fluorouracil signaling through a calcium-calmodulin-dependent pathway is required for p53 activation and apoptosis in colon carcinoma cells. Oncogene 2013, 32, 4529–4538. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Wen, N.; Xia, M.; Zhang, Y.U.; Liu, W.; Xu, Y.E.; Sun, L. Calcium efflux from the endoplasmic reticulum regulates cisplatin-induced apoptosis in human cervical cancer HeLa cells. Oncol. Lett. 2016, 11, 2411–2419. [Google Scholar] [CrossRef] [PubMed]
- Abdoul-Azize, S.; Dubus, I.; Vannier, J.P. Improvement of dexamethasone sensitivity by chelation of intracellular Ca2+ in pediatric acute lymphoblastic leukemia cells through the prosurvival kinase ERK1/2 deactivation. Oncotarget 2017, 8, 27339–27352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdoul-Azize, S.; Buquet, C.; Vannier, J.P.; Dubus, I. Pyr3, a TRPC3 channel blocker, potentiates dexamethasone sensitivity and apoptosis in acute lymphoblastic leukemia cells by disturbing Ca2+ signaling, mitochondrial membrane potential changes and reactive oxygen species production. Eur. J. Pharmacol. 2016, 784, 90–98. [Google Scholar] [CrossRef]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Boehmerle, W.; Splittgerber, U.; Lazarus, M.B.; McKenzie, K.M.; Johnston, D.G.; Austin, D.J.; Ehrlich, B.E. Paclitaxel induces calcium oscillations via an inositol 1,4,5-trisphosphate receptor and neuronal calcium sensor 1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 18356–18361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Bonora, M.; Missiroli, S.; Poletti, F.; Ramirez, F.G.; Morciano, G.; Morganti, C.; Pandolfi, P.P.; Mammano, F.; Pinton, P. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget 2015, 6, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.F.; Zhao, P.; Luo, X.; Xu, J.C.; Xue, L.; Zhou, Q.; Xiong, M.; Shen, J.; Peng, Y.B.; Yu, M.F.; et al. Chloroquine inhibits Ca2+ permeable ion channels-mediated Ca2+ signaling in primary B lymphocytes. Cell Biosci. 2017, 7, 28. [Google Scholar] [CrossRef]
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T.; et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018, 9, 873. [Google Scholar] [CrossRef] [Green Version]
- Jang, E.; Kim, I.Y.; Kim, H.; Lee, D.M.; Seo, D.Y.; Lee, J.A.; Choi, K.S.; Kim, E. Quercetin and chloroquine synergistically kill glioma cells by inducing organelle stress and disrupting Ca2+ homeostasis. Biochem. Pharmacol. 2020, 178, 114098. [Google Scholar] [CrossRef]
- Vu, H.T.; Kobayashi, M.; Hegazy, A.M.; Tadokoro, Y.; Ueno, M.; Kasahara, A.; Takase, Y.; Nomura, N.; Peng, H.; Ito, C.; et al. Autophagy inhibition synergizes with calcium mobilization to achieve efficient therapy of malignant gliomas. Cancer Sci. 2018, 109, 2497–2508. [Google Scholar] [CrossRef]
- Chen, X.; Pan, X.; Zhang, W.; Guo, H.; Cheng, S.; He, Q.; Yang, B.; Ding, L. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm. Sin. B 2020, 10, 723–733. [Google Scholar] [CrossRef]
- Newton, H.S.; Gawali, V.S.; Chimote, A.A.; Lehn, M.A.; Palackdharry, S.M.; Hinrichs, B.H.; Jandarov, R.; Hildeman, D.; Janssen, E.M.; Wise-Draper, T.M.; et al. PD1 blockade enhances K+ channel activity, Ca2+ signaling, and migratory ability in cytotoxic T lymphocytes of patients with head and neck cancer. J. Immunother. Cancer 2020, 8, e000844. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Xu, S.; Li, Y.F.; Lam, K.P. Adaptor protein DOK3 promotes plasma cell differentiation by regulating the expression of programmed cell death 1 ligands. Proc. Natl. Acad. Sci. USA 2014, 111, 11431–11436. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Zhong, S.; Ma, Z.; Kong, H.; Medvec, A.; Ahmed, R.; Freeman, G.J.; Krogsgaard, M.; Riley, J.L. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc. Natl. Acad. Sci. USA 2013, 110, E2480–E2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grolez, G.P.; Hammadi, M.; Barras, A.; Gordienko, D.; Slomianny, C.; Volkel, P.; Angrand, P.O.; Pinault, M.; Guimaraes, C.; Potier-Cartereau, M.; et al. Encapsulation of a TRPM8 Agonist, WS12, in Lipid Nanocapsules Potentiates PC3 Prostate Cancer Cell Migration Inhibition through Channel Activation. Sci. Rep. 2019, 9, 7926. [Google Scholar] [CrossRef]
- Dubois, C.; Vanden Abeele, F.; Sehgal, P.; Olesen, C.; Junker, S.; Christensen, S.B.; Prevarskaya, N.; Moller, J.V. Differential effects of thapsigargin analogues on apoptosis of prostate cancer cells: Complex regulation by intracellular calcium. FEBS J. 2013, 280, 5430–5440. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. The SERCA pump as a therapeutic target: Making a “smart bomb” for prostate cancer. Cancer Biol. Ther. 2005, 4, 14–22. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Channel/Transporter/Pump | Compound | Cancer |
|---|---|---|
| TRPCs | 20-GPPD | Colorectal |
| SKF96365 | Glioblastoma | |
| Carvacrol | Glioma | |
| D-3263 | Prostate, colon, breast, lung, pancreas, leiomyosarcoma, and Kaposi’s sarcoma | |
| Capsaicin | Prostate | |
| Cannabidiol | Bladder | |
| SOR-C13 | Ovarian and prostate | |
| Dexamethasone | Leukemia | |
| VGGCs | Mibefradil | Esophageal, colon, glioblastoma, and ovarian |
| NNC-55-096 | Glioblastoma and ovarian | |
| Purinergic P2 receptors | BIL010t | Basal cell carcinoma |
| BIL06v | Advanced or metastatic solid tumors | |
| AR-C118925XX | Pancreatic ductal adenocarcinoma | |
| ORAI and STIM | SKF96365 | Breast |
| DPB-162AE/-163AE | Colon and glioma | |
| ML-9 | Prostate | |
| GA101/obinutuzumab | Non-Hodgkin lymphoma and leukemia | |
| 5-Fluorouracil | Hepatocarcinoma | |
| SERCA | Mipsargargin | Prostate cancers, glioblastoma, kidney, and hepatocellular carcinoma |
| Curcumin | Breast, lung, ovarian, and colon | |
| PMCA | Pt(O,O0-acac)(γ-acac)(DMS) | Breast |
| Resveratrol | Prostate | |
| IP3R3 | Paclitaxel | Ovarian, breast, neck and head |
| VDAC | R-Tf-D-LP4 | Glioblastoma, lung, and breast |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218323
Patergnani S, Danese A, Bouhamida E, Aguiari G, Previati M, Pinton P, Giorgi C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. International Journal of Molecular Sciences. 2020; 21(21):8323. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218323
Chicago/Turabian StylePatergnani, Simone, Alberto Danese, Esmaa Bouhamida, Gianluca Aguiari, Maurizio Previati, Paolo Pinton, and Carlotta Giorgi. 2020. "Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer" International Journal of Molecular Sciences 21, no. 21: 8323. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218323